

Abstract
Complement activation contributes to the pathogenesis of numerous inflammatory and autoimmune diseases. Therapeutic complement inhibitors have proven effective in several of these diseases and have now entered clinical use. Complement activation has multiple different biologic effects, however, and the currently available drugs can have undesirable side-effects, such as an increased risk of infection. Several different complement inhibitors have been developed that bind to target molecules, thereby concentrating the drug at a specific anatomic site. This approach appears to be both more effective than untargeted drugs and to have fewer side effects. In this article we review different targeting strategies that have been developed and the evidence supporting the use and benefits of targeted drugs.
Keywords: Complement, therapeutics, targeted, inflammation
1. Introduction
The complement cascade has long been recognized as a key part of the innate immune system. Complement activation provides an early line of defense against invasive pathogens, triggering a rapid inflammatory response as well as modulating a subsequent adaptive immune response. Recent work has also revealed an important role for the complement system in other physiologic and pathophysiologic processes [1]. For example, complement-mediated injury to host cells is a critical (and “druggable”) component of injury in many inflammatory diseases. In contrast to these inflammatory effects, complement activation also helps to clear damaged cells and debris without inflammation, and it plays an important role in the development and regeneration of some tissues. Furthermore, there is cross-talk between the complement system and other physiologic systems, such as the coagulation cascade and the kallikrein-kinin system. Thus, an important challenge for complement inhibitory therapeutics is to block its deleterious effects without impairing its beneficial functions. One approach to achieve this is with site-targeted complement inhibitors. These drugs block complement activation at specific anatomic sites without blocking systemic activity or activation elsewhere in the body.
2. The local character of complement activation
The various functions of the complement cascade depend on specific protein-protein interactions that trigger its activation on pathogens, damaged cells and tissues, and cancerous cells. The ability to distinguish foreign or altered surfaces from normal host cells allow the system to target specific surfaces with minimal damage to healthy host tissue. To accomplish this, the three activation pathways are initiated by specific molecular interactions involving the binding of C1q, MBL or C3b to target ligands/surfaces [2]. Healthy host cells also express regulatory proteins that limit activation on the cell surface, and the degree of complement activation on a particular surface is determined by the combination of these “positive” and “negative” factors.
Even though complement proteins circulate throughout the body in plasma (and to a lesser degree in lymphatic fluid [3]), activation of the system is a local phenomenon that is determined by the particular microenvironment. One example of this occurs at the interface between T cells and antigen presenting cells (APCs). When ACPs interact with cognate T cells, co-stimulatory signals induce the cells to produce C3, factor B, and factor D [4]. At the same time, expression of the regulatory protein decay accelerating factor (DAF) is reduced. These responses promote alternative pathway activation at the APC-T cell interface, and increase T cell proliferation [5]. Thus, T cells and APCs can modulate activation by local molecular changes, even though the cells are constantly exposed to complement proteins in plasma.
In addition to complement activation on cell surfaces, activation can also occur in solution (fluid phase) and intracellularly (Figure 1). The alternative pathway is continually activated in the fluid phase by the hydrolysis of the C3 protein, a process called tickover. This generates an active form of C3 called C3(H2O) that can bind to factor B and generate a soluble C3 convertase [C3(H2O)Bb]. This convertase generates C3b which remains in solution or binds to hydroxyl or amide groups on nearby surfaces. Some conditions increase fluid phase activation of complement. C3 nephritic factor, for example, protects soluble C3bBb from inactivation and can cause consumption of C3 [6]. Although C3 nephritic factor and fluid phase complement activation are associated with some diseases, the degree to which fluid phase activation contributes to tissue injury is not clear [7].
Figure 1. Sites of complement activation and inhibition.
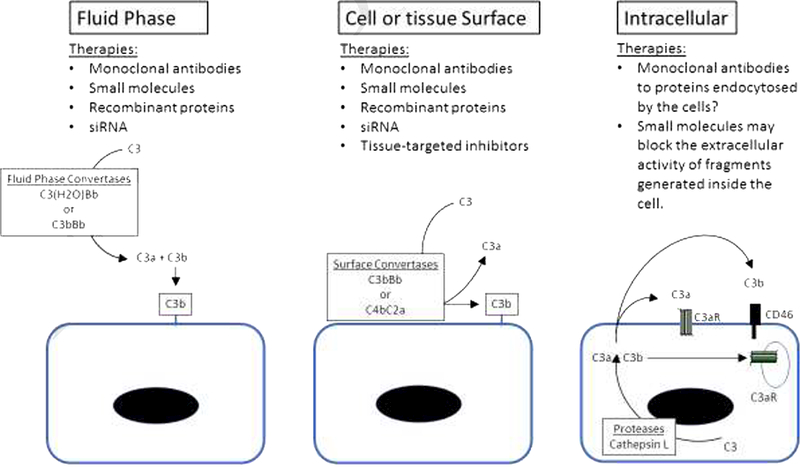
Complement activation can occur in the fluid phase or on the surface of cells and tissues. Monoclonal antibodies, small molecules, and recombinant proteins have access to complement proteins in plasma and can block fluid phase activation. siRNA can reduce levels of complement proteins in plasma and decrease fluid phase activation. These same strategies also block complement activation on cell and tissue surfaces. Targeted inhibitors block activation in the fluid phase while they are still in the plasma, and effectively block the activity of convertases on cell and tissue surfaces. Monoclonal antibodies may prevent cells from endocytosing complement proteins. Small molecules may block the extracellular activity of complement fragments that are generated inside cells.
More recently, activation of complement proteins within cells has been found to be important for a number of cellular processes. In resting T cells, cathepsin L cleaves C3 and generates C3a and C3b [8]. C3a can activate the C3a receptor (C3aR) within the cell. T cell activation also causes transport of C3a and C3b outside of the cell where they serve as ligands for extracellular C3aR and CD46, respectively. Furthermore, cleavage of C5 in T cells generates C5a that signals through intracellular C5aR1 [9], a process critical to the Th1 response. An intracellular complement system recycling pathway for fluid phase C3(H2O) that represents a source of C3a has also been described [10]. Some complement therapeutics, such as monoclonal antibodies or recombinant proteins, probably do not block these intracellular processes. As new complement inhibitory molecules are developed, it will be important to determine the degree to which they block intracellular, cell surface, and fluid phase complement. However, the pathophysiologic role of intracellular complement proteins still needs to be resolved, and the importance of blocking the intracellular fragments is incompletely understood.
3. Complement inhibitors currently in clinical use
Two therapeutic complement inhibitors are currently in clinical use. C1 esterase inhibitors are approved for treatment of patients with deficiency of this endogenous serine protease inhibitor, and have been tested in additional complement-mediated diseases [11]. A monoclonal antibody to C5 (eculizumab) was approved by the FDA in 2007 for treatment of paroxysmal nocturnal hemoglobinuria. Subsequently it was approved for atypical hemolytic uremic syndrome and for adult patients with generalized myasthenia gravis (gMG) who are anti-acetylcholine receptor antibody-positive. Ongoing clinical trials are testing the efficacy of eculizumab in additional diseases [12].
The established role of complement in a large number of diseases and an increasing understanding of complement-mediated pathogenic mechanisms has generated interest in the development and commercialization of new drugs to modulate the complement system. Several recent papers have reviewed clinical trials that have been completed or that are underway for these drugs [12–14]. Many different types of molecule are being tested, including monoclonal antibodies to complement proteins, small molecule inhibitors of complement proteases or fragments, purified and recombinant complement inhibitory proteins, and siRNA to knock down production of complement proteins. Among the different drugs in development, some target specific complement activation pathways. For example, there are monoclonal antibodies that specifically block components of the classical pathway (C1s), the lectin pathway (MASP-2), and the alternative pathway (factor B or factor D) [12, 15]. Based on the efficacy of eculizumab, new drugs that block C5 cleavage are also in development [12].
4. Advantages of site-targeted complement inhibition
The currently approved complement therapeutics, as well as the vast majority of complement inhibitory drugs in development cause, and indeed rely on, systemic inhibition of the complement system. There remain, however, concerns and obstacles with regard to the therapeutic application of systemic complement inhibition. One issue, which depends on the complement target, is that many complement proteins circulate at high levels (e.g. > 1 mg/ml for C3) and have rapid turnover, thus necessitating the use of high doses of inhibitor.
Expression of complement proteins can increase as a stress response or in response to inhibition, and complement proteins can also be expressed locally within tissues. Thus, the target abundance can change over time and can vary between locations. In addition to outnumbering the inhibitor, target abundance can also affect the efficacy of a drug. For example, it was recently discovered that C5 inhibition with monoclonal antibodies can be incomplete at sites of high C3b density [16]. A systemically applied complement inhibitor may therefore have limited bioavailability and insufficient efficacy at sites of disease or injury. In this context, we have shown that targeting a complement inhibitor specifically to sites of complement activation (C3d deposition) can increase its efficacy between 10–20 fold as compared to a systemic or untargeted counterpart [17].
A long-recognized risk of complement inhibition, and in particular systemic complement inhibition, is an increased risk of infectious complications. The increased risk of infection can be inferred from patients with congenital deficiencies of complement proteins [18]. Patients with genetic deficiencies of the terminal complement proteins (C5-C9) are at increased risk of Neisserial infections. This is also the primary infectious complication of therapeutic C5 blockade, and meningococcal vaccination is required for all patients receiving eculizumab [19]. Patients treated with eculizumab are at increased risk of infection even if they are immunized, however, and some providers continue antibiotic prophylaxis as long as patients are receiving active treatment [20]. Consideration of infectious complications is most relevant for chronic complement inhibitor treatment, but even short-term interference of complement-dependent host defense poses potential risks. Short-term risks will be magnified if the patient is immunocompromised, either as a result of co-administration of an immunosuppressive drug, or as a result of disease or injury (e.g. acute infection is a significant risk factor for patients with ischemic or traumatic injury to the central nervous system). Of note, in contrast to an untargeted C3 inhibitor or C3 deficiency, C3 inhibition targeted to a site of complement activation does not increase susceptibility to polymicrobial sepsis or to Streptococcus pneumonia infection (see below). Complement also plays important roles in shaping an adaptive immune response [1], and systemic complement inhibition also has the potential to modulate autoimmune, alloimmune and anti-tumor responses.
Complement has many other protective and homeostatic functions, including a role in tissue repair and regeneration [21–23]. By tagging dead and dying cells for clearance, complement promotes the resolution of inflammation and provides a permissive environment for tissue repair/remodeling. Complement also promotes tissue repair and regeneration through direct signaling mechanisms mediated by complement activation products. For example, C3a and/or C5a play important roles in repair and regenerative mechanisms, such as promoting neurogenesis and neuroblast migration after brain injury [23], and in the priming of hepatocyte regeneration after liver injury or resection [24, 25]. Targeted or pathway specific drugs may be able to parse the various physiologic effects of complement in damaged organs [22]. Another important homeostatic function of complement is the opsonization and catabolism of circulating immune complexes. Interference with this process may exacerbate pathology in immune complex-associated diseases such as systemic lupus erythematosus (SLE), even though complement is also implicated in promoting tissue injury in SLE.
In general, a site-targeted complement inhibitor will optimally have a short circulatory half-life with an extended half-life at the target tissue. Supported by experimental data, such a design feature will increase inhibitor bioavailability and durability at a localized site of disease or injury, while minimizing disruption of normal and protective physiological functions of complement. The short circulatory half-life is likely a key reason why targeted complement inhibition does not appear to interfere with complement-dependent protective mechanisms, such as host defense [26] or immune complex clearance [27], in which bacterial replication and immune complex formation is a dynamic ongoing process. In the same way, acute administration of targeted complement inhibition did not interfere with post-injury repair and regeneration in the brain, a process that engages sub-acutely after brain injury [23]. Thus, complement activation at later time points after injury that occurs as part of a homeostatic or reparatory process, will be less impeded by a targeted inhibitor.
Lastly, one other potential advantage of a targeted complement inhibitor is that the targeting moiety itself may also contribute to protective activity (i.e. a dual functioning drug). We describe below different targeting moieties that in addition to delivering a complement inhibitor, can by themselves modulate either autoantibody production or the binding of pathogenic IgM that can trigger complement activation.
These advantages of targeted complement inhibition will not apply equally to all diseases. For example, complement inhibitors are currently being tested for opthalmologic diseases using intra-vitreal injection [28]. This approach limits the drug to the site of injury and should have fewer systemic effects than intravenous administration. The risks of systemic complement inhibition are also probably less important when used for acute indications. For example, complement inhibition during hemodialysis treatments, which are typically four hour treatments, should not impair most physiologic functions of the complement system [29]. Finally, some drugs block specific activation fragments (e.g. C5a receptor antagonists). Drugs with a narrow mechanism of action, such as this, might not benefit from a targeted approach.
5. Pharmacodynamics of targeted complement inhibitors
Eculizumab is dosed to achieve complete inhibition of the complement cascade throughout the plasma, and adequate dosing can be monitored by measurement of the CH50. Full complement inhibition is reflected by nearly complete suppression of the CH50 (<5%) [30], although activation may still proceed at the tissue level. Drug levels can be monitored, and some studies have aimed at levels of 50–100 μg/mL [31]. The pharmacokinetics and pharmacodynamics of other monoclonal antibodies can similarly be monitored by testing patient serum in complement activation assays [32]. Unfortunately, the CH50 or other activation assays are unlikely to accurately reflect how well a targeted drug blocks complement activation within tissues. At a high enough serum level these drugs could fully block all complement activity, similar to untargeted drugs, but this would undermine some of the advantages of a targeted drug (see below). It is possible, though, that targeted drugs will be more effective than untargeted drugs within tissues, irrespective of serum inhibition. The pharmacodynamics of a targeted drug will depend on tissue penetration and retention at the target site. Monoclonal antibodies to complement proteins, in contrast, may not need good tissue penetrance since they bind their targets in the plasma, potentially blocking the supply of proteins necessary for activation within tissues.
The level of C3 in plasma is commonly measured as a biomarker of complement consumption, and decreased C3 levels sometimes reflect disease activity [33]. Pre-clinical data suggests that complement inhibition within tissues by targeted drugs increases plasma C3 levels, although this may also have been due to complement inhibition in the fluid phase by the unbound drug [34]. Alternative methods of monitoring a targeted complement inhibition include measurement of complement activation fragments in plasma or staining of biopsy tissue for complement deposits. Complement activation in diseased organs often generates elevated levels of complement activation fragments in plasma, such as increased sC5b-9. The levels of these fragments could be used to monitor the PK/PD of a targeted drug, although they are not always reliable indicators of tissue complement activation [35]. Tissue biopsy is probably the most accurate way to confirm complement inhibition in a specific tissue. Biopsies are invasive procedures, though, limiting the number of biopsies that can be performed. Furthermore, organs such as the brain or heart are not easily accessible for biopsy. Non-invasive methods of measuring tissue complement deposits are in development [36]. These imaging methods could be very useful for monitoring the response to targeted complement inhibitors
6. Antibody-targeted complement inhibitors.
Engineered proteins have been developed in which the binding regions of various antibodies have been used deliver a complement regulator to a specific tissue or to a specific marker of inflammation. The first examples of this approach were chimeric proteins that contained the antigen binding region of an IgG specific for 5-dimethylamino-naphthalene-1-sulfonyl (Dansyl) linked to a soluble form of CD59 [37] or DAF [38]. CD59 is an endogenous complement regulator protein that prevents membrane attack complex (MAC) formation on cell membranes. The proteins blocked the lysis of Chinese hamster ovary cells expressing the target antigen but did not block lysis of cells that did not express the antigen. Dansyl did not play a physiologic role in this experiment, but rather was used to provide proof-of-concept for a targeted inhibitor.
6.1. Complement inhibitors targeted to specific tissue epitopes.
Using an antibody that binds to epitopes in the rat glomeruli and kidney tubules, chimeric proteins were generated linking a single chain variable fragment (scFv) form of the antibody to complement regulatory fragments of either Crry or CD59 [39]. Crry is a rodent protein that has both C3 convertase decay accelerating activity and cofactor activity for Factor I cleavage of C3b. As a result, it inhibits C3 convertase activity induced by activation through all three pathways.
decay accelerating activity and cofactor activity. It inhibits C3 activation through all three pathways. Biodistribution studies showed that the drugs accumulated in rat kidneys following intravenous injection. Using a model of chemically induced kidney injury, the authors demonstrated that the drugs prevented complement activation in the kidneys and reduced injury, whereas untargeted Crry did not. Furthermore, the scFv-targeted inhibitors had short circulatory half-lives (~30 minutes) and did not cause systemic complement inhibition. In another study, investigators linked the complement regulatory region of Crry to a scFv specific for glycophorin A, a glycoprotein expressed on erythrocytes [40]. This agent reduced hemolysis of Crry-deficient erythrocytes transferred into wild-type animals. These two studies illustrate that complement activation can be inhibited on specific cell types using inhibitory drugs targeted to epitopes expressed on the cell surface.
6.2. Complement inhibitors targeted to markers of injury or inflammation.
The body recognizes damaged tissue through several mechanisms of tissue surveillance. Tissue injury causes release of damage-associated molecular patterns (DAMPs), generation or exposure of neo-epitopes, and induces expression of adhesion molecules to facilitate trafficking of immune cells to the affected tissue. These molecular signatures of injury have been utilized to deliver complement inhibitors to damaged tissue.
Localized delivery of complement inhibition to sites of adhesion molecule expression was one of the first site-targeting strategies investigated. Adhesion molecules are upregulated on activated endothelium, and the sLex carbohydrate moiety binds to P and E selectins. The decoration of soluble CR1 (sCR1) with sialyl Lewisx (sLex) moieties served to deliver sCR1 to inflamed endothelium in a rat model of lung injury [41] and a mouse model of ischemic stroke [42]. In the respective models there was localization of sCR1 sLex to the vasculature of either the lung or brain, and sCR1 sLex provided an enhanced protective effect relative to sCR1. Importantly, the efficacy of sCR1 sLex was still dependent on systemic complement inhibition. sCR1sLex, however, was not protective in a nonhuman primate model of reperfused stroke [43].
Investigators have identified natural antibody IgM molecules that bind to neoepitopes expressed in damaged tissues [44]. These natural antibodies are specific for areas of cell injury, although they do not seem to be restricted to injury of particular organs. Targeted complement inhibitors that incorporate scFvs derived from several of these natural antibodies have been developed and tested. B4 and C2 are natural antibody clones that have been found to bind injury epitopes in several different tissues [45, 46]. A targeted inhibitor consisting of B4 scFv linked to Crry (B4-Crry) specifically bound to endothelial cells in ischemic heart allografts and protected the grafts from post-transplant IRI [47]. This agent had two protective effects; it competitively inhibited endogenous pathogenic natural antibody from binding the tissue (as demonstrated by use of the B4scFv targeting moiety alone), and it reduced complement activation in the tissue. B4-Crry also reduced tissue damage in a model of spinal cord injury [48] and ischemic stroke (Science Transl. Med,. In press), and B4scFv reduced injury following hepatic ischemia and reperfusion [49]. These latter studies support the use of this targeting strategy across different organ systems.
7. Complement receptor (CR)-2-targeted complement inhibitors
Although complement activation occurs downstream of other molecular events, such as binding of natural antibody, it covalently marks the affected tissues with C3 molecules. Tissue-bound C3 fragments can, therefore, be utilized as markers of tissue injury. By far the most extensively studied strategy of targeted complement inhibition is CR2-mediated delivery of a complement inhibitor to sites of complement activation. Following C3 cleavage into C3b and C3a, C3b covalently attaches to cell surface molecules through amide or ester bonds. It is then cleaved into iC3b and subsequently C3dg and C3d (73). Whereas the half-life of C3b is short, C3d remains membrane-bound for an extended period. iC3b, C3dg and C3d are ligands for CR2, a receptor expressed predominantly on B cells and dendritic cells. CR2 is an ~145 Kd type I transmembrane protein consisting of 15 extracellular SCRs, a transmembrane domain and a short intracellular tail [50, 51]. A high affinity interaction with C3dg and C3d occurs in the first 2 N-terminal SCR domains of CR2, although there is data that suggest SCR domains 3 and 4 help to promote the interaction between CR2 and its C3 ligands [52].
7.1. Design of CR2-targeted drugs.
The work described below utilized C3d-targeted complement inhibitors prepared by linking the 4 N-terminal SCR domains of CR2 with complement inhibitory domains of different complement inhibitors; namely the extracellular domains of DAF or Crry, the extracellular domain of CD59 (inhibits membrane attack complex formation), or the 5 N-terminal SCR domains of factor H (inhibitor of the alternative pathway). A comprehensive list of studies that have been undertaken using CR2 targeted complement inhibitors is presented in Table 1. Below we discuss selected studies that illustrate therapeutic benefits of this targeting approach, as well as its use to investigate complement-dependent mechanisms in clinically relevant settings of complement inhibition.
Table 1. Disease models in which CR2-targeted drugs have been tested.
| Model | Test agent | Reference |
|---|---|---|
| Stroke | CR2-Crry and CR2-fH | [45, 84, 85] |
| Traumatic brain injury | CR2-Crry, CR2-fH and CR2-CD59 | [57, 59] |
| Spinal cord injury | CR2-Crry | [86] |
| Experimental autoimmune encephalomyelitis (MS) | CR2-Crry and CR2-fH | [87] |
| Post-transplant cardiac IRI | CR2-Crry and CR2-fH | [63] |
| Vascularized composite allograft transplantation with and without immunosuppression | CR2-Crry | [64] |
| Donor brain death and cardiac IRI | CR2-Crry | [66] |
| Liver transplantation (lean and steatotic grafts), hepatic IRI and liver regeneration | CR2-Crry and CR2-CD59 | [22, 25, 88] |
| LPS/D-GalN-induced fulminant hepatic failure | CR2-fH | [89] |
| Graft ischemia in rejection of orthotopic tracheal transplants | CR2-Crry | [65] |
| Kidney transplantation and delayed graft function | TT30 (humanized CR2-fH) | [90] |
| Intestine IRI and secondary lung injury | CR2-Crry and CR2-fH | [26, 53] |
| C3 glomerulopathy | CR2-fH | [34] |
| Lupus | CR2-Crry, CR2-fH and sCR2 | [27, 60, 61] |
| Arthritis (CIA and CAIA) | CR2-Crry and CR2-fH | [91, 92] |
| Age related macular degeneration (including gene therapy approach) | CR2-Crry, CR2-fH, CR2-CD59 and TT30 (humanized CR2-fH) | [58, 83, 93, 94] |
| Smoke induced ocular injury | CR2-fH | [95] |
| Hemorrhage | CR2-fH | [96] |
| PNH erythrocytes | CR2-fH | [55, 69] |
| Preeclampsia | CR2-Crry | [97] |
| Atherosclerosis | CR2-Crry | [98] |
| DSS colitis (including oral delivery of CR2-Crry) | CR2-Crry and CR2-fH | [99–101] |
| Paraquat-induced lung injury | CR2-Crry and CR2-fH | [102] |
| PMN recruitment in response to adenovirus and effect on eliminating virus-containing cells | CR2-Crry and CR2-fH | [103] |
| Polymicrobial sepsis | CR2-Crry | [26] |
| In vitro cell targeting and activity, in vivo targeting to kidney in lupus model | Human CR2-DAF and CR2-CD59 | [17] |
7.2. In vitro efficacy studies.
CR2 linked complement inhibitors were originally validated using human protein constructs and in vitro assay systems [17]. These studies demonstrated that CR2-DAF and CR2-CD59 bound to CHO cells and erythrocytes opsonized with C3, and that both fusion proteins were significantly more effective than soluble DAF or soluble CD59 at protecting cells from complement attack. Also noteworthy, and something that has seldom been considered in subsequent studies, was that the CR2 fusion proteins also interfered with the interaction between the immune cell receptor CR3 (CD11b/18) and its complement C3 ligands, representing an additional potential anti-inflammatory mechanism of CR2 fusion proteins.
7.2. Efficacy of CR2-targeted drugs using in vivo models.
Initial investigations of the CR2 targeting strategy in experimental paradigms utilized the murine construct, CR2-Crry. In a mouse model of intestinal ischemia reperfusion injury, it was shown that CR2-Crry was at least 10-fold more effective than its untargeted counterpart (Crry-Ig) at providing protection from both intestinal and remote complement-dependent inflammation and injury [26]. CR2-Crry specifically targeted to sites of complement deposition, had a much shorter circulatory half-life than Crry-Ig, and had minimum impact on serum complement activity at a therapeutic dose. Furthermore, unlike Crry-Ig, a therapeutic dose of CR2-Crry had no effect on susceptibility to infection in a model of polymicrobial sepsis.
In subsequent studies, the targeted murine complement inhibitors CR2-fH and CR2-CD59 were prepared and characterized in several experimental models. The initial characterization of CR2-fH was also performed in a murine model of intestine ischemia reperfusion injury [53]. Similar to CR2-Crry, CR2-fH targeted to sites of C3 deposition and was able to provide complete protection from both local and remote inflammation and injury. An important role for the alternative pathway in intestine ischemia-reperfusion injury (IRI) had been established using complement deficient mice [54], and the studies with CR2-fH demonstrated alternative pathway dependence in a clinically relevant model. The binding and complement inhibitory activity of CR2-fH was shown to be dependent on the CR2-C3d interaction. In this context, it is curious that serum factor H fails to provide protection against intestine IRI, even though it contains endogenous C3b/C3d binding sites and is present in serum at much higher concentrations than administered CR2-fH. The reason for this is unknown, but there is a complex and incompletely understood relationship between the functional activity of factor H and its interaction with polyanions and C3 at cell surfaces. The discrepancy in activities may also be due to differences in ligand binding affinity and/or the flexibility of the two molecules with regard to binding and the orientation of the inhibitory domain relative to cell surface convertases. It is interesting that a recombinant truncated form of factor H (“miniFH”) that incorporated the N-terminal inhibitory domain and C-terminal C3 binding domains of native factor H was more effective than CR2-fH at controlling complement on PNH erythrocytes [55]. In the same study, it was also reported that CR2-fH and miniFH had minimal effect on serum killing of bacteria, in contrast to eculizumab.
The murine CR2-CD59 (CD59a) construct was first tested in a study investigating the role of complement in liver injury and regeneration [22]. In this study, CR2-CD59 and CR2-Crry effectively and similarly protected against hepatic IRI, indicating that the MAC is the primary mediator of injury, further supported by the fact that CR2-CD59 had minimal effect on C3d deposition and C5a generation. On the other hand, after partial hepatectomy (with or without accompanying ischemia, the former being the more common occurrence clinically) CR2-Crry increased injury and impaired regeneration, whereas CR2-CD59 protected the remnant liver from injury and promoted hepatocyte proliferation and regeneration. C3a and C5a are known to be essential for liver regeneration [25, 56], and unlike CR2-Crry, CR2-CD59 acts downstream of the generation of these anaphylatoxins and did not interfere with hepatocyte proliferation. Thus, CR2-CD59 dissected the complement pathway to optimize outcome for treating a clinical situation in which complement has dueling roles.
CR2-CD59 has also been characterized in murine models of traumatic brain injury (TBI) [57] and laser-induced choroidal neovascularization (a model of the exudative form of age-related macular degeneration) [58]. In the latter study, CR2-CD59 was shown to specifically target the site of laser-induced injury following intraperitoneal (systemic) injection, and to significantly reduce lesion size and fluid accumulation, thus implicating a role for the MAC in this model. The MAC had also been thought to be the primary contributor to complement-mediated post-TBI pathology and was thought to represent a therapeutic target for reducing secondary injury after TBI. However, a recent study using a controlled cortical impact model of TBI found that whereas CR2-CD59 provided protection from injury and post-TBI deficits acutely, inhibition of C3 activation with either CR2-Crry or CR2-fH was required to protect against chronic inflammation, ongoing neuronal loss, and long-term functional and cognitive decline [57]. Furthermore, CR2-Crry and CR2-fH provided similar levels of protection, indicating a key role for the alternative pathway in propagating chronic post-TBI pathology. In the above studies, a single injection of inhibitor administered either 1 or 12 hours after TBI was protective. CR2-fH has also been shown to improve acute post-TBI outcomes in a closed head injury model [59]. Traumatic brain injury and stroke share similar features of secondary inflammation and injury, and it is interesting that unlike the findings with TBI, whereas a single CR2-fH treatment provided sustained protection into the subacute phase after stroke (middle cerebral artery occlusion and reperfusion), CR2-Crry treatment was only protective acutely. Mice treated with CR2-Crry showed an evolution of brain injury and declining behavioral scores in the subacute phase relative to mice treated with CR2-fH [23]. One possible explanation for this finding is that CR2-Crry, unlike CR2-fH, completely blocked C5a generation in the brain, and the anaphylatoxins have been reported to have neuroprotective roles and to promote neurogenesis, both basal and following injury [23].
Spontaneous models of lupus (both MRL/lpr and NZB/W F1) are other models in which alternative pathway inhibition with CR2-fH provided a better outcome than inhibition of all pathways with CR2-Crry [60, 61]. These studies also addressed long-term complement inhibitor treatment and the durability of tissue targeted complement inhibition. In an initial study, MRL/lpr mice were treated with CR2-Crry once a week for 8 weeks, with treatment beginning after the development of proteinuria [27]. CR2-Crry provided significant benefits in terms of clinical and pathological readouts of renal disease, as well as survival. Furthermore, a previous study demonstrated that treatment of MRL/lpr mice with Crry-Ig, an untargeted counterpart of CR2-Crry, reduced proteinuria and maintained renal function, but did not improve survival or glomerular pathologic disease [62]. Of additional importance, Crry-Ig was administered at a dose of 3 mg every other day to maintain systemic complement inhibition and therapeutic effect, whereas CR2-Crry provided superior protection at a dose of 0.25 mg administered once a week. A biodistribution study using radiolabeled CR2-Crry administered to MRL/lpr mice with ongoing disease determined a tissue (kidney) half-life of approximately 24 hours, and CR2-Crry was still detectable in kidneys 7 days after injection, co-localized with C3d [27]. CR2-Crry and CR2-fH have circulatory half-lives in C57BL/6 mice of 8.8 hours [26] and 8.7 hours [53], respectively. Subsequent studies compared the effects of CR2-Crry and CR2-fH in the MRL/lpr [60] and NZB/W F1 [61] models of lupus. While both inhibitors were protective and there were some minor variations in outcome measures between the two models, overall CR2-fH provided significantly better protection from disease activity (glomerulonephritis and proteinuria) and renal deposition of IgG than CR2-Crry. Also, whereas both targeted inhibitors reduced serum levels of circulating immune complexes in the MRL/lpr model (and unlike Crry-Ig that resulted in increased levels), only CR2-fH reduced autoantibody levels [60]. It is also noteworthy that in both models the soluble CR2-targeting vehicle alone significantly reduced autoantibody levels and reduced renal deposition of IgG and complement. Taken together, the above data provides further support for the dual role of complement in lupus and indicate distinct roles for alternative and classical/lectin pathways of complement in disease expression. In this regard, the classical pathway has a distinct role in the catabolism of immune complexes and in resolving inflammation via the clearance of apoptotic cells, which represent protective functions in lupus. In addition, the CR2 targeting moiety appears to contribute to therapeutic activity by modulating autoimmunity, likely through interruption of endogenous CR2 function on B cells and/or follicular dendritic cells.
One model in which CR2-Crry proved more efficacious than CR2-fH was a model of acute vascular (antibody mediated) rejection in which hearts from C3H mice were transplanted into Balb/C mice (unpublished data). A single injection of either inhibitor immediately post-transplant significantly increased graft survival, but multiple injections every 3 days after transplantation further increased survival of grafts only in CR2-Crry treated mice. Grafts in mice treated with either a single or multiple dose of CR2-fH were rejected with an acute vascular rejection phenotype. However, for mice treated with multiple doses of CR2-Crry in which rejection was significantly delayed, the rejection phenotype had changed to acute cellular rejection. Thus, CR2-Crry, but not the alternative pathway inhibitor, continued to provide protection against antibody (classical pathway)-mediated rejection, but only until the induction of a cell mediated response.
CR2-Crry and CR2-fH have also been compared in a model of post-transplant cardiac IRI [63]. The two inhibitors were similarly efficacious at reducing myocardial damage, although some parameters of inflammation were significantly lower in grafts from CR2-Crry mice compared to CR2-fH treated mice. In another transplant model, acute CR2-Crry treatment protected vascularized composite (limb) allografts from IRI in both muscle and skin compartments, and prolonged graft survival [64]. Furthermore, acute CR2-Crry treatment significantly prolonged allograft survival when combined with an otherwise subtherapeutic dose of cyclosporine A, as compared to either treatment alone. In this regard, strategies to minimize immunosuppression are particularly relevant for vascularized composite allograft transplantation, since the surgery is usually performed in nonlife-threatening situations and where recipients require aggressive and lifelong immunosuppression that can lead to serious side effects. Using a mouse model of orthotopic tracheal transplantation, CR2-Crry has also been shown to attenuate microvascular injury that is associated with transplant ischemia and chronic rejection [65]. Donor brain death can immunologically prime donor organs and exacerbate ischemia reperfusion injury in the recipient, and CR2-Crry treatment of the recipient has been shown to significantly reduce brain death-exacerbated cardiac IRI [66].
7.4. CR2-targeted inhibitors to increase inflammation.
In contrast to the above studies, CR2-Crry has also been used with the goal of enhancing inflammation rather than dampening an inflammatory response. Compared to localized fractionated radiation therapy alone, combined CR2-Crry treatment and radiation therapy of subcutaneous murine lymphoma significantly and markedly reduced tumor growth and improved survival [67]. Complement plays a role in the clearance of apoptotic cells, and the therapeutic effect of CR2-Crry was associated with impaired clearance of radiation-induced apoptotic cancer cells and increased inflammation within the tumor environment. Failure to clear apoptotic cells is associated with inflammation and formation of an immunostimulatory environment, and the anti-tumor effect of CR2-Crry when combined with radiation therapy was shown to be dependent on the modulation of a CD8+ T cell immune response. It has also been reported that the local production of C3a and C5a is essential for a tumor response to radiotherapy and the stimulation of T cell immunity [68].
7.5. Human studies.
A humanized version of CR2-fH, termed TT30, was shown to bind PNH erythrocytes and completely inhibit hemolysis in a dose dependent manner [69]. TT30 was shown to be safe and non-immunogenic in a phase 1 trial for PNH. Although not yet published, the results of this study have been presented (http://www.bloodjournal.org/content/126/23/2137?sso-checked=true) This study showed that TT30 effectively inhibited alternative pathway activity, and decreased lactate dehydrogenase levels (LDH, a biomarker of hemolysis) in PNH patients. In spite of these interesting results, further clinical trials of this drug have not been publicly announced.
8. Other Targeting Strategies.
A derivative of sCR1 consisting of the 3 N-terminal SCRs was shown to target to cell membranes in a non-specific manner by the attachment of a membrane inserting myristoylated peptide [70]. Although soluble CR1(SCR1–3) was itself significantly less active than full length sCR1, the membrane inserting derivative (APT070) was 100-fold more active than its unmodified counterpart in complement inhibition assays. Intra-articular injection APT070 in a rat model of antigen-induced arthritis resulted in its retention on cell membranes within the joint, and it provided significantly more protection than its untargeted counterpart [71]. APT070 has also been shown to be protective in models of intestinal ischemia-reperfusion injury [72] and Miller Fisher syndrome [73]. An interesting application of this targeting approach is graft protection during cold storage, and it has been shown that perfusion of donor rat kidneys with APT070 reduced tubular injury and increased graft survival after cold ischemia and transplantation [74]. A clinical trial is currently underway to see if this agent protects kidney allografts from ischemic injury. CD59 has been similarly tagged and shown insert into erythrocyte membranes and protect them from complement-mediated lysis in vitro and in vivo [75].
As discussed above, TT30 utilizes the C3d binding region of CR2 to target the complement inhibitory region of factor H to sites of C3d deposition. The native Factor H protein binds to C3b at its amino terminus and to C3d at its carboxy terminus. The truncated form of factor H (miniFH, discussed above) incorporates both of these C3 fragment binding sites. It is believed that the compact structure of this protein may make the carboxy region more accessible for iC3b/C3d binding, enhancing its ability to target to sites of iC3b/C3d deposition [76].
In addition to the TBI studies with CR2-targeted inhibitors described above, CD59 has also been targeted to the post-TBI injury site by linking it to complement receptor of the Ig superfamily (CRIg) [77]. Ligands for CRIg are C3b and iC3b, and a CD59-CRIg fusion protein was shown to bind C3b coated surfaces in vitro, and to significantly reduce MAC formation and improve acute pathological and functional outcome measures in a closed head injury model of TBI. In another approach, a neutralizing anti-C5 human antibody was coupled with a cyclic RGD peptide (termed Ergidina) and shown to preferentially localize to ischemic endothelial cells in a rat model of renal IRI [78]. Ergidina ameliorated renal injury and inhibited complement at the tissue level without affecting circulating levels of C5. The inhibitor also bound ex vivo to surgically removed kidneys during cold ischemic storage, suggesting it can be applied in an organ perfusion approach to prevent post-transplant IRI. Organ perfusion with inhibitors during cold storage can result in localized delivery but does not necessarily rely on a specific targeting approach. Similarly, localized inhibition of complement has been achieved by delivery of untargeted complement inhibitors directly to anatomical sites, such as the eye [28], joint [79, 80] and lung [81].
A less conventional approach to provide localized complement inhibition involved the construction of a pro-drug, in which DAF and CD59 were attached to an antibody Fc region by means of a linker incorporating a cleavage site for matrix metalloproteases and/or aggrecanases [82]. Cleavage of the linker by certain enzymes that are present in high concentrations at sites of inflammation was shown to release an active form of the inhibitor. Importantly, prior to cleavage the fusion protein had only limited complement inhibitory activity, thus minimally impacting systemic complement activity. Although the proteins were not characterized in an in vivo model, enzymes present in supernatants of cytokine stimulated chondrocytes, or synovial fluid itself, were both shown to effectively release active DAF from Fc fusion proteins.
Finally, adeno-associated virus (AAV) mediated delivery of CR2-fH has been investigated in a mouse model of choroidal neovascularization [83]. Subretinal gene delivery one month before laser-induced injury significantly reduced choroidal neovascularization and reduced injury-associated C3a production. Gene therapy also delivered similar levels of CR2-fH to the retinal pigmented epithelium/choroid as treatment by intravenous injection in the same model.
9. Conclusions and future directions.
Numerous pre-clinical studies have demonstrated that targeted complement inhibitors are more effective than their untargeted homologues, and that the targeted drugs may have a lower risk of infection. Some of the targeted drugs have been developed to bind markers expressed on specific tissues, and others employ targeting moieties that bind to more general markers of inflammation or injury. The localized character of complement activation lends itself to a targeted approach. Furthermore, the complement cascade has multiple different biologic functions, and sometimes complement fragments can elicit opposing effects. For example, complement activation can promote both tissue injury and tissue repair, and it can both enhance and suppress adaptive immune responses. Drugs that block specific complement effects or whose function is restricted in time and space may enable clinicians to block the deleterious effects of complement while leaving beneficial effects intact. The targeting strategies developed for complement inhibitory drugs can also be employed for delivery of other drugs.
Highlights.
Therapeutic complement inhibitors are approved for treatment of several diseases, and new drugs are currently under development
Targeted complement inhibitors can reduce tissue inflammation without blocking the complement cascade systemically
Targeted complement inhibitors may be more effective than untargeted drugs, and may have fewer side effects
Acknowledgements
This work was supported by National Institutes of Health Grants DK113586 and DK076690 (JMT), and R21NS097653, RO1 DK102912, UO1AI132894 (ST), and the Department of Veterans Affairs RX001141 (ST).
Abbreviations:
- MBL
mannose binding lectin
- APC
antigen presenting cell
- DAF
decay accelerating factor
- C3aR
C3a receptor
- SLE
systemic lupus erythematosus
- MAC
membrane attack complex
- IRI
ischemia-reperfusion injury
- scFv
single chain variable fragment
- sCR1
soluble CR1
- DAMP
damage-associated molecular pattern
- CRIg
complement receptor of the Ig superfamily
Footnotes
Conflicts of Interest
Both authors receive royalties from Alexion Pharmaceuticals, Inc. Both authors are also consultants for AdMIRx, Inc., a company developing complement inhibitors. They also hold stock and will receive royalty income from AdMIRx.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References.
- [1].Hajishengallis G, Reis ES, Mastellos DC, Ricklin D, Lambris JD, Novel mechanisms and functions of complement, Nat Immunol, 18 (2017) 1288–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT, Complement System Part I -Molecular Mechanisms of Activation and Regulation, Frontiers in immunology, 6 (2015) 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Olszewski WL, Engeset A, Haemolytic complement in peripheral lymph of normal men, Clin Exp Immunol, 32 (1978) 392–398. [PMC free article] [PubMed] [Google Scholar]
- [4].Heeger PS, Lalli PN, Lin F, Valujskikh A, Liu J, Muqim N, Xu Y, Medof ME, Decay-accelerating factor modulates induction of T cell immunity, J Exp Med, 201 (2005) 1523–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS, Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis, Blood, 112 (2008) 1759–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Daha MR, Fearon DT, Austen KF, C3 nephritic factor (C3NeF): stabilization of fluid phase and cell-bound alternative pathway convertase, J Immunol, 116 (1976) 1–7. [PubMed] [Google Scholar]
- [7].Zipfel PF, Skerka C, Chen Q, Wiech T, Goodship T, Johnson S, Fremeaux-Bacchi V, Nester C, Cordoba SR, Noris M, Pickering M, Smith R, The role of complement in C3 glomerulopathy, Mol Immunol, DOI 10.1016/j.molimm.2015.03.012(2015). [DOI] [PubMed] [Google Scholar]
- [8].Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, Subias M, Pickering MC, Drouet C, Meri S, Arstila TP, Pekkarinen PT, Ma M, Cope A, Reinheckel T, Rodriguez de Cordoba S, Afzali B, Atkinson JP, Kemper C, Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation, Immunity, 39 (2013) 1143–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Arbore G, West EE, Spolski R, Robertson AA, Klos A, Rheinheimer C, Dutow P, Woodruff TM, Yu ZX, O’Neill LA, Coll RC, Sher A, Leonard WJ, Kohl J, Monk P, Cooper MA, Arno M, Afzali B, Lachmann HJ, Cope AP, Mayer-Barber KD, Kemper C, T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4(+) T cells, Science, 352 (2016) aad1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Elvington M, Liszewski MK, Bertram P, Kulkarni HS, Atkinson JP, A C3(H20) recycling pathway is a component of the intracellular complement system, J Clin Invest, 127 (2017) 970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Viglietti D, Gosset C, Loupy A, Deville L, Verine J, Zeevi A, Glotz D, Lefaucheur C, C1-Inhibitor in Acute Antibody-Mediated Rejection Non-Responsive to Conventional Therapy in Kidney Transplant Recipients: A Pilot Study, Am J Transplant, 16 (2015) 1596–1603. [DOI] [PubMed] [Google Scholar]
- [12].Ricklin D, Barratt-Due A, Mollnes TE, Complement in clinical medicine: Clinical trials, case reports and therapy monitoring, Mol Immunol, 89 (2017) 10–21. [DOI] [PubMed] [Google Scholar]
- [13].Holers VM, Tomlinson S, Kulik L, Atkinson C, Rohrer B, Banda N, Thurman JM, New therapeutic and diagnostic opportunities for injured tissue-specific targeting of complement inhibitors and imaging modalities, Seminars in immunology, 28 (2016) 260–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hawksworth OA, Li XX, Coulthard LG, Wolvetang EJ, Woodruff TM, New concepts on the therapeutic control of complement anaphylatoxin receptors, Mol Immunol, DOI 10.1016/j.molimm.2017.05.015(2017). [DOI] [PubMed] [Google Scholar]
- [15].Thurman JM, Many drugs for many targets: novel treatments for complement-mediated glomerular disease, Nephrol Dial Transplant, 32 (2017) Î57–Î64. [DOI] [PubMed] [Google Scholar]
- [16].Harder MJ, Kuhn N, Schrezenmeier H, Hochsmann B, von Zabern I, Weinstock C, Simmet T, Ricklin D, Lambris JD, Skerra A, Anliker M, Schmidt CQ, Incomplete inhibition by eculizumab: mechanistic evidence for residual C5 activity during strong complement activation, Blood, 129 (2017) 970–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Song H, He C, Knaak C, Guthridge JM, Holers VM, Tomlinson S, Complement receptor 2-mediated targeting of complement inhibitors to sites of complement activation, J Clin Invest, 111 (2003)1875–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Skattum L, van Deuren M, van der Poll T, Truedsson L, Complement deficiency states and associated infections, Mol Immunol, 48 (2011) 1643–1655. [DOI] [PubMed] [Google Scholar]
- [19].Benamu E, Montoya JG, Infections associated with the use of eculizumab: recommendations for prevention and prophylaxis, Curr Opin Infect Dis, 29 (2016) 319–329. [DOI] [PubMed] [Google Scholar]
- [20].McNamara LA, Topaz N, Wang X, Hariri S, Fox L, MacNeil JR, High Risk for Invasive Meningococcal Disease Among Patients Receiving Eculizumab (Soliris) Despite Receipt of Meningococcal Vaccine, MMWR Morb Mortal Wkly Rep, 66 (2017) 734–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rutkowski MJ, Sughrue ME, Kane AJ, Ahn BJ, Fang S, Parsa AT, The complement cascade as a mediator of tissue growth and regeneration, Inflamm Res, 59 (2010) 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Marshall KM, He S, Zhong Z, Atkinson C, Tomlinson S, Dissecting the complement pathway in hepatic injury and regeneration with a novel protective strategy, J Exp Med, 211 (2014) 1793–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Alawieh A, Elvington A, Zhu H, Yu J, Kindy MS, Atkinson C, Tomlinson S, Modulation of post-stroke degenerative and regenerative processes and subacute protection by site-targeted inhibition of the alternative pathway of complement, J Neuroinflammation, 12 (2015) 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Markiewski MM, Mastellos D, Tudoran R, DeAngelis RA, Strey CW, Franchini S, Wetsel RA, Erdei A, Lambris JD, C3a and C3b activation products of the third component of complement (C3) are critical for normal liver recovery after toxic injury, J Immunol, 173 (2004) 747–754. [DOI] [PubMed] [Google Scholar]
- [25].He S, Atkinson C, Qiao F, Cianflone K, Chen X, Tomlinson S, A complement-dependent balance between hepatic ischemia/reperfusion injury and liver regeneration in mice, J Clin Invest, 119 (2009) 2304–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Atkinson C, Song H, Lu B, Qiao F, Burns TA, Holers VM, Tsokos GC, Tomlinson S, Targeted complement inhibition by C3d recognition ameliorates tissue injury without apparent increase in susceptibility to infection, J Clin Invest, 115 (2005) 2444–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Atkinson C, Qiao F, Song H, Gilkeson GS, Tomlinson S, Low-dose targeted complement inhibition protects against renal disease and other manifestations of autoimmune disease in MRL/lpr mice, J Immunol, 180 (2008) 1231–1238. [DOI] [PubMed] [Google Scholar]
- [28].Yaspan BL, Williams DF, Holz FG, Regillo CD, Li Z, Dressen A, van Lookeren Campagne M, Le KN, Graham RR, Beres T, Bhangale TR, Honigberg LA, Smith A, Henry EC, Ho C, Strauss EC, Investigators MS, Targeting factor D of the alternative complement pathway reduces geographic atrophy progression secondary to age-related macular degeneration, Science translational medicine, 9 (2017). [DOI] [PubMed] [Google Scholar]
- [29].Reis ES, DeAngelis RA, Chen H, Resuello RR, Ricklin D, Lambris JD, Therapeutic C3 inhibitor Cp40 abrogates complement activation induced by modern hemodialysis filters, Immunobiology, 220 (2015) 476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jodele S, Fukuda T, Vinks A, Mizuno K, Laskin BL, Goebel J, Dixon BP, Teusink A, Pluthero FG, Lu L, Licht C, Davies SM, Eculizumab therapy in children with severe hematopoietic stem cell transplantation-associated thrombotic microangiopathy, Biol Blood Marrow Transplant, 20 (2014) 518–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fakhouri F, Hourmant M, Campistol JM, Cataland SR, Espinosa M, Gaber AO, Menne J, Minetti EE, Provot F, Rondeau E, Ruggenenti P, Weekers LE, Ogawa M, Bedrosian CL, Legendre CM, Terminal Complement Inhibitor Eculizumab in Adult Patients With Atypical Hemolytic Uremic Syndrome: A Single-Arm, Open-Label Trial, Am J Kidney Dis, 68 (2016) 84–93. [DOI] [PubMed] [Google Scholar]
- [32].Frazer-Abel A, Sepiashvili L, Mbughuni MM, Willrich MA, Overview of Laboratory Testing and Clinical Presentations of Complement Deficiencies and Dysregulation, Adv Clin Chem, 77 (2016) 1–75. [DOI] [PubMed] [Google Scholar]
- [33].Birmingham DJ, Irshaid F, Nagaraja HN, Zou X, Tsao BP, Wu H, Yu CY, Hebert LA, Rovin BH, The complex nature of serum C3 and C4 as biomarkers of lupus renal flare, Lupus, 19 (2010) 1272–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ruseva MM, Peng T, Lasaro MA, Bouchard K, Liu-Chen S, Sun F, Yu ZX, Marozsan A, Wang Y, Pickering MC, Efficacy of Targeted Complement Inhibition in Experimental C3 Glomerulopathy, J Am Soc Nephrol, 27 (2016) 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bu F, Meyer NC, Zhang Y, Borsa NG, Thomas C, Nester C, Smith RJ, Soluble c5b-9 as a biomarker for complement activation in atypical hemolytic uremic syndrome, Am J Kidney Dis, 65 (2015) 968–969. [DOI] [PubMed] [Google Scholar]
- [36].Sargsyan SA, Serkova NJ, Renner B, Hasebroock KM, Larsen B, Stoldt C, McFann K, Pickering MC, Thurman JM, Detection of glomerular complement C3 fragments by magnetic resonance imaging in murine lupus nephritis, Kidney Int, 81 (2012) 152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhang HF, Yu J, Bajwa E, Morrison SL, Tomlinson S, Targeting of functional antibody-CD59 fusion proteins to a cell surface, J Clin Invest, 103 (1999) 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhang H, Lu S, Morrison SL, Tomlinson S, Targeting of functional antibody-decay-accelerating factor fusion proteins to a cell surface, J Biol Chem, 276 (2001) 27290–27295. [DOI] [PubMed] [Google Scholar]
- [39].He C, Imai M, Song H, Quigg RJ, Tomlinson S, Complement inhibitors targeted to the proximal tubule prevent injury in experimental nephrotic syndrome and demonstrate a key role for c5b-9, J Immunol, 174 (2005) 5750–5757. [DOI] [PubMed] [Google Scholar]
- [40].Spitzer D, Unsinger J, Mao D, Wu X, Molina H, Atkinson JP, In vivo correction of complement regulatory protein deficiency with an inhibitor targeting the red blood cell membrane, J Immunol, 175 (2005) 7763–7770. [DOI] [PubMed] [Google Scholar]
- [41].Mulligan MS, Warner RL, Rittershaus CW, Thomas LJ, Ryan US, Foreman KE, Crouch LD, Till GO, Ward PA, Endothelial targeting and enhanced antiinflammatory effects of complement inhibitors possessing sialyl Lewisx moieties, J Immunol, 162 (1999) 4952–4959. [PubMed] [Google Scholar]
- [42].Huang J, Kim LJ, Mealey R, Marsh HC Jr., Zhang Y, Tenner AJ, Connolly ES Jr., Pinsky DJ, Neuronal protection in stroke by an sLex-glycosylated complement inhibitory protein, Science, 285 (1999) 595–599. [DOI] [PubMed] [Google Scholar]
- [43].Mocco J, Mack WJ, Ducruet AF, King RG, Sughrue ME, Coon AL, Sosunov SA, Sciacca RR, Zhang Y, Marsh HC Jr., Pinsky DJ, Connolly ES Jr., Preclinical evaluation of the neuroprotective effect of soluble complement receptor type 1 in a nonhuman primate model of reperfused stroke, J Neurosurg, 105 (2006) 595–601. [DOI] [PubMed] [Google Scholar]
- [44].Fleming SD, Shea-Donohue T, Guthridge JM, Kulik L, Waldschmidt TJ, Gipson MG, Tsokos GC, Holers VM, Mice deficient in complement receptors 1 and 2 lack a tissue injury-inducing subset of the natural antibody repertoire, J Immunol, 169 (2002) 2126–2133. [DOI] [PubMed] [Google Scholar]
- [45].Elvington A, Atkinson C, Kulik L, Zhu H, Yu J, Kindy MS, Holers VM, Tomlinson S, Pathogenic natural antibodies propagate cerebral injury following ischemic stroke in mice, J Immunol, 188 (2012) 1460–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kulik L, Fleming SD, Moratz C, Reuter JW, Novikov A, Chen K, Andrews KA, Markaryan A, Quigg RJ, Silverman GJ, Tsokos GC, Holers VM, Pathogenic natural antibodies recognizing annexin IV are required to develop intestinal ischemia-reperfusion injury, J Immunol, 182 (2009) 5363–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Atkinson C, Qiao F, Yang X, Zhu P, Reaves N, Kulik L, Goddard M, Holers VM, Tomlinson S, Targeting pathogenic postischemic self-recognition by natural IgM to protect against posttransplantation cardiac reperfusion injury, Circulation, 131 (2015) 1171–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Narang A, Qiao F, Atkinson C, Zhu H, Yang X, Kulik L, Holers VM, Tomlinson S, Natural IgM antibodies that bind neoepitopes exposed as a result of spinal cord injury , drive secondary injury by activating complement, J Neuroinflammation, 14 (2017) 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Marshall K, Jin J, Atkinson C, Alawieh A, Qiao F, Lei B, Chavin KD, He S, Tomlinson S, Natural immunoglobulin M initiates an inflammatory response important for both hepatic ischemia reperfusion injury and regeneration in mice, Hepatology, DOI 10.1002/hep.29512(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Weis JJ, Toothaker LE, Smith JA, Weis JH, Fearon DT, Structure of the human B lymphocyte receptor for C3d and the Epstein-Barr virus and relatedness to other members of the family of C3/C4 binding proteins, J Exp Med, 167 (1988) 1047–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Carel JC, Frazier B, Ley TJ, Holers VM, Analysis of epitope expression and the functional repertoire of recombinant complement receptor 2 (CR2/CD21) in mouse and human cells, J Immunol, 143 (1989) 923–930. [PubMed] [Google Scholar]
- [52].Carel JC, Myones BL, Frazier B, Holers VM, Structural requirements for C3d,g/Epstein-Barr virus receptor (CR2/CD21) ligand binding, internalization, and viral infection, J Biol Chem, 265 (1990) 12293–12299. [PubMed] [Google Scholar]
- [53].Huang Y, Qiao F, Atkinson C, Holers VM, Tomlinson S, A novel targeted inhibitor of the alternative pathway of complement and its therapeutic application in ischemia/reperfusion injury, J Immunol, 181 (2008) 8068–8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Stahl GL, Xu Y, Hao L, Miller M, Buras JA, Fung M, Zhao H, Role for the alternative complement pathway in ischemia/reperfusion injury, Am J Pathol, 162 (2003) 449–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Schmidt CQ, Harder MJ, Nichols EM, Hebecker M, Anliker M, Hochsmann B, Simmet T, Csincsi AI, Uzonyi B, Pappworth IY, Ricklin D, Lambris JD, Schrezenmeier H, Jozsi M, Marchbank KJ, Selectivity of C3-opsonin targeted complement inhibitors: A distinct advantage in the protection of erythrocytes from paroxysmal nocturnal hemoglobinuria patients, Immunobiology, 221 (2016) 503–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Strey CW, Markiewski M, Mastellos D, Tudoran R, Spruce LA, Greenbaum LE, Lambris JD, The proinflammatory mediators C3a and C5a are essential for liver regeneration, J Exp Med, 198 (2003) 913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Alawieh A, Langley EF, Weber S, Adkins D, Tomlinson S, Identifying the role of complement in triggering neuroinflammation after traumatic brain injury, J Neurosci, DOI 10.1523/JNEUROSCI.2197-17.2018(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Schnabolk G, Beon MK, Tomlinson S, Rohrer B, New Insights on Complement Inhibitor CD59 in Mouse Laser-Induced Choroidal Neovascularization: Mislocalization After Injury and Targeted Delivery for Protein Replacement, J Ocul Pharmacol Ther, 33 (2017) 400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Rich MC, Keene CN, Neher MD, Johnson K, Yu ZX, Ganivet A, Holers VM, Stahel PF, Site-targeted complement inhibition by a complement receptor 2-conjugated inhibitor (mTT30) ameliorates post-injury neuropathology in mouse brains, Neurosci Lett, 617 (2016) 188–194. [DOI] [PubMed] [Google Scholar]
- [60].Sekine H, Kinser TT, Qiao F, Martinez E, Paulling E, Ruiz P, Gilkeson GS, Tomlinson S, The benefit of targeted and selective inhibition of the alternative complement pathway for modulating autoimmunity and renal disease in MRL/lpr mice, Arthritis Rheum, 63 (2011) 1076–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sekine H, Ruiz P, Gilkeson GS, Tomlinson S, The dual role of complement in the progression of renal disease in NZB/W F(1) mice and alternative pathway inhibition, Mol Immunol, 49 (2011) 317–323. [DOI] [PubMed] [Google Scholar]
- [62].Bao L, Haas M, Kraus DM, Hack BK, Rakstang JK, Holers VM, Quigg RJ, Administration of a soluble recombinant complement C3 inhibitor protects against renal disease in MRL/lpr mice, J Am Soc Nephrol, 14 (2003) 670–679. [DOI] [PubMed] [Google Scholar]
- [63].Atkinson C, He S, Morris K, Qiao F, Casey S, Goddard M, Tomlinson S, Targeted complement inhibitors protect against posttransplant cardiac ischemia and reperfusion injury and reveal an important role for the alternative pathway of complement activation, J Immunol, 185 (2010) 7007–7013. [DOI] [PubMed] [Google Scholar]
- [64].Zhu P, Bailey SR, Lei B, Paulos CM, Atkinson C, Tomlinson S, Targeted Complement Inhibition Protects Vascularized Composite Allografts From Acute Graft Injury and Prolongs Graft Survival When Combined With Subtherapeutic Cyclosporine A Therapy, Transplantation, 101 (2017) e75–e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Khan MA, Jiang X, Dhillon G, Beilke J, Holers VM, Atkinson C, Tomlinson S, Nicolls MR, CD4+ T cells and complement independently mediate graft ischemia in the rejection of mouse orthotopic tracheal transplants, Circ Res, 109 (2011) 1290–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Atkinson C, Floerchinger B, Qiao F, Casey S, Williamson T, Moseley E, Stoica S, Goddard M, Ge X, Tullius SG, Tomlinson S, Donor brain death exacerbates complement-dependent ischemia/reperfusion injury in transplanted hearts, Circulation, 127 (2013) 1290–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Elvington M, Scheiber M, Yang X, Lyons K, Jacqmin D, Wadsworth C, Marshall D, Vanek K, Tomlinson S, Complement-dependent modulation of antitumor immunity following radiation therapy, Cell reports, 8 (2014) 818–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Surace L, Lysenko V, Fontana AO, Cecconi V, Janssen H, Bicvic A, Okoniewski M, Pruschy M, Dummer R, Neefjes J, Knuth A, Gupta A, van den Broek M, Complement is a central mediator of radiotherapy-induced tumor-specific immunity and clinical response, Immunity, 42 (2015) 767–777. [DOI] [PubMed] [Google Scholar]
- [69].Risitano AM, Notaro R, Pascariello C, Sica M, del Vecchio L, Horvath CJ, Fridkis-Hareli M, Selleri C, Lindorfer MA, Taylor RP, Luzzatto L, Holers VM, The complement receptor 2/factor H fusion protein TT30 protects paroxysmal nocturnal hemoglobinuria erythrocytes from complement-mediated hemolysis and C3 fragment, Blood, 119 (2012) 6307–6316. [DOI] [PubMed] [Google Scholar]
- [70].Smith RA, Targeting anticomplement agents, Biochemical Society transactions, 30 (2002) 1037–1041. [DOI] [PubMed] [Google Scholar]
- [71].Linton SM, Williams AS, Dodd I, Smith R, Williams BD, Morgan BP, Therapeutic efficacy of a novel membrane-targeted complement regulator in antigen-induced arthritis in the rat, Arthritis Rheum, 43 (2000) 2590–2597. [DOI] [PubMed] [Google Scholar]
- [72].Souza DG, Esser D, Bradford R, Vieira AT, Teixeira MM, APT070 (Mirococept), a membrane-localised complement inhibitor, inhibits inflammatory responses that follow intestinal ischaemia and reperfusion injury, Br J Pharmacol, 145 (2005) 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Halstead SK, Humphreys PD, Goodfellow JA, Wagner ER, Smith RA, Willison HJ, Complement inhibition abrogates nerve terminal injury in Miller Fisher syndrome, Ann Neurol, 58 (2005) 203–210. [DOI] [PubMed] [Google Scholar]
- [74].Patel H, Smith RA, Sacks SH, Zhou W, Therapeutic strategy with a membrane-localizing complement regulator to increase the number of usable donor organs after prolonged cold storage, J Am Soc Nephrol, 17 (2006) 1102–1111. [DOI] [PubMed] [Google Scholar]
- [75].Fraser DA, Harris CL, Williams AS, Mizuno M, Gallagher S, Smith RA, Morgan BP, Generation of a recombinant, membrane-targeted form of the complement regulator CD59: activity in vitro and in vivo, J Biol Chem, 278 (2003) 48921–48927. [DOI] [PubMed] [Google Scholar]
- [76].Schmidt CQ, Lambris JD, Ricklin D, Protection of host cells by complement regulators, Immunol Rev, 274 (2016) 152–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ruseva MM, Ramaglia V, Morgan BP, Harris CL, An anticomplement agent that homes to the damaged brain and promotes recovery after traumatic brain injury in mice, Proc Natl Acad Sci U S A, 112 (2015) 14319–14324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Durigutto P, Sblattero D, Biffi S, De Maso L, Garrovo C, Baj G, Colombo F, Fischetti F, Di Naro AF, Tedesco F, Macor P, Targeted Delivery of Neutralizing Anti-C5 Antibody to Renal Endothelium Prevents Complement-Dependent Tissue Damage, Frontiers in immunology, 8 (2017) 1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Mizuno M, Nishikawa K, Morgan BP, Matsuo S, Comparison of the suppressive effects of soluble CR1 and C5a receptor antagonist in acute arthritis induced in rats by blocking of CD59, Clin Exp Immunol, 119 (2000) 368–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Banda NK, Acharya S, Scheinman RI, Mehta G, Coulombe M, Takahashi M, Sekine H, Thiel S, Fujita T, Holers VM, Mannan-Binding Lectin-Associated Serine Protease 1/3 Cleavage of Pro-Factor D into Factor D In Vivo and Attenuation of Collagen Antibody-Induced Arthritis through Their Targeted Inhibition by RNA Interference-Mediated Gene Silencing, J Immunol, 197 (2016) 3680–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Cheng Q, Patel K, Lei B, Rucker L, Allen DP, Zhu P, Vasu C, Martins PN, Goddard M, Nadig SN, Atkinson C, Donor Pre-treatment with nebulized complement C3a Receptor antagonist mitigates brain-death induced immunological injury post-lung transplant, Am J Transplant, DOI 10.1111/ajt.14717(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Harris CL, Fraser DA, Morgan BP, Tailoring anti-complement therapeutics, Biochemical Society transactions, 30 (2002) 1019–1026. [DOI] [PubMed] [Google Scholar]
- [83].Schnabolk G, Parsons N, Obert E, Annamalai B, Nasarre C, Tomlinson S, Lewin AS, Rohrer B, Delivery of CR2-fH Using AAV Vector Therapy as Treatment Strategy in the Mouse Model of Choroidal Neovascularization, Mol Ther Methods Clin Dev, 9 (2018) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Atkinson C, Zhu H, Qiao F, Varela JC, Yu J, Song H, Kindy MS, Tomlinson S, Complement-dependent P-selectin expression and injury following ischemic stroke, J Immunol, 177 (2006) 7266–7274. [DOI] [PubMed] [Google Scholar]
- [85].Alawieh A, Tomlinson S, Injury site-specific targeting of complement inhibitors for treating stroke, Immunol Rev, 274 (2016) 270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Qiao F, Atkinson C, Song H, Pannu R, Singh I, Tomlinson S, Complement plays an important role in spinal cord injury and represents a therapeutic target for improving recovery following trauma, Am J Pathol, 169 (2006) 1039–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Hu X, Tomlinson S, Barnum SR, Targeted inhibition of complement using complement receptor 2-conjugated inhibitors attenuates EAE, Neurosci Lett, 531 (2012) 35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].He S, Atkinson C, Evans Z, Ellett JD, Southwood M, Elvington A, Chavin KD, Tomlinson S, A role for complement in the enhanced susceptibility of steatotic livers to ischemia and reperfusion injury, J Immunol, 183 (2009) 4764–4772. [DOI] [PubMed] [Google Scholar]
- [89].Sun S, Guo Y, Zhao G, Zhou X, Li J, Hu J, Yu H, Chen Y, Song H, Qiao F, Xu G, Yang F, Wu Y, Tomlinson S, Duan Z, Zhou Y, Complement and the alternative pathway play an important role in LPS/D-GalN-induced fulminant hepatic failure, PloS one, 6 (2011) e26838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Yu ZX, Qi S, Lasaro MA, Bouchard K, Dow C, Moore K, Wu Z, Barama A, Xu J, Johnson K, Marozsan AJ, Wang Y, Targeting Complement Pathways During Cold Ischemia and Reperfusion Prevents Delayed Graft Function, Am J Transplant, 16 (2016) 2589–2597. [DOI] [PubMed] [Google Scholar]
- [91].Banda NK, Levitt B, Glogowska MJ, Thurman JM, Takahashi K, Stahl GL, Tomlinson S, Arend WP, Holers VM, Targeted inhibition of the complement alternative pathway with complement receptor 2 and factor H attenuates collagen antibody-induced arthritis in mice, J Immunol, 183 (2009) 5928–5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Song H, Qiao F, Atkinson C, Holers VM, Tomlinson S, A complement C3 inhibitor specifically targeted to sites of complement activation effectively ameliorates collagen-induced arthritis in DBA/1J mice, J Immunol, 179 (2007) 7860–7867. [DOI] [PubMed] [Google Scholar]
- [93].Rohrer B, Long Q, Coughlin B, Wilson RB, Huang Y, Qiao F, Tang PH, Kunchithapautham K, Gilkeson GS, Tomlinson S, A targeted inhibitor of the alternative complement pathway reduces angiogenesis in a mouse model of age-related macular degeneration, Invest Ophthalmol Vis Sci, 50 (2009) 3056–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Rohrer B, Long Q, Coughlin B, Renner B, Huang Y, Kunchithapautham K, Ferreira VP, Pangburn MK, Gilkeson GS, Thurman JM, Tomlinson S, Holers VM, A targeted inhibitor of the complement alternative pathway reduces RPE injury and angiogenesis in models of age-related macular degeneration, Adv Exp Med Biol, 703 (2010) 137–149. [DOI] [PubMed] [Google Scholar]
- [95].Woodell A, Jones BW, Williamson T, Schnabolk G, Tomlinson S, Atkinson C, Rohrer B, A Targeted Inhibitor of the Alternative Complement Pathway Accelerates Recovery From Smoke-Induced Ocular Injury, Invest Ophthalmol Vis Sci, 57 (2016) 1728–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Hylton DJ, Hoffman SM, Van Rooijen N, Tomlinson S, Fleming SD, Macrophage-produced IL-12p70 mediates hemorrhage-induced damage in a complement-dependent manner, Shock, 35 (2011) 134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Qing X, Redecha PB, Burmeister MA, Tomlinson S, D’Agati VD, Davisson RL, Salmon JE, Targeted inhibition of complement activation prevents features of preeclampsia in mice, Kidney Int, 79 (2011) 331–339. [DOI] [PubMed] [Google Scholar]
- [98].Liu F, Wu L, Wu G, Wang C, Zhang L, Tomlinson S, Qin X, Targeted mouse complement inhibitor CR2-Crry protects against the development of atherosclerosis in mice, Atherosclerosis, 234 (2014) 237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Schepp-Berglind J, Atkinson C, Elvington M, Qiao F, Mannon P, Tomlinson S, Complement-dependent injury and protection in a murine model of acute dextran sulfate sodium-induced colitis, J Immunol, 188 (2012) 6309–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Elvington M, Schepp-Berglind J, Tomlinson S, Regulation of the alternative pathway of complement modulates injury and immunity in a chronic model of dextran sulphate sodium-induced colitis, Clin Exp Immunol, 179 (2015) 500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Elvington M, Blichmann P, Qiao F, Scheiber M, Wadsworth C, Luzinov I, Lucero J, Vertegel A, Tomlinson S, A novel protocol allowing oral delivery of a protein complement inhibitor that subsequently targets to inflamed colon mucosa and ameliorates murine colitis, Clin Exp Immunol, 177 (2014) 500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Sun S, Wang H, Zhao G, An Y, Guo Y, Du L, Song H, Qiao F, Yu H, Wu X, Atkinson C, Jiang S, Tomlinson S, Zhou Y, Complement inhibition alleviates paraquat-induced acute lung injury, American journal of respiratory cell and molecular biology, 45 (2011) 834–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Di Paolo NC, Baldwin LK, Irons EE, Papayannopoulou T, Tomlinson S, Shayakhmetov DM, IL-1alpha and complement cooperate in triggering local neutrophilic inflammation in response to adenovirus and eliminating virus-containing cells, PLoS Pathog, 10 (2014) e1004035. [DOI] [PMC free article] [PubMed] [Google Scholar]