

Abstract
The Nrf2 antioxidant transcription factor promotes redox homeostasis in part through reciprocal signaling between neurons and neighboring cells, but the signals involved in intertissue signaling in response to Nrf2 activation are not well defined. In Caenorhabditis elegans, activation of SKN-1/Nrf2 in the intestine negatively regulates neuropeptide secretion from motor neurons. Here, we show that sphingosine kinase (SPHK-1) functions downstream of SKN-1/Nrf2 in the intestine to regulate neuropeptide secretion from motor neurons during the oxidative stress response in C. elegans hermaphrodites. SPHK-1 localizes to mitochondria in the intestine and SPHK-1 mitochondrial localization and kinase activity are essential for its function in regulating motor neuron function. SPHK-1 is recruited to mitochondria from cytosolic pools and its mitochondrial abundance is negatively regulated by acute or chronic SKN-1 activation. Finally, the regulation of motor function by SKN-1 requires the activation of the p38 MAPK cascade in the intestine and occurs through controlling the biogenesis or maturation of dense core vesicles in motor neurons. These findings show that the inhibition of SPHK-1 in the intestine by SKN-1 negatively regulates neuropeptide secretion from motor neurons, revealing a new mechanism by which SPHK-1 signaling mediates its effects on neuronal function in response to oxidative stress.
SIGNIFICANCE STATEMENT Neurons are highly susceptible to damage by oxidative stress, yet have limited capacity to activate the SKN-1/Nrf2 oxidative stress response, relying instead on astrocytes to provide redox homeostasis. In Caenorhabditis elegans, intertissue signaling from the intestine plays a key role in regulating neuronal function during the oxidative stress response. Here, through a combination of genetic, behavioral, and fluorescent imaging approaches, we found that sphingosine kinase functions in the SKN-1/Nrf2 pathway in the intestine to regulate neuropeptide biogenesis and secretion in motor neurons. These results implicate sphingolipid signaling as a new component of the oxidative stress response and suggest that C. elegans may be a genetically tractable model to study non-cell-autonomous oxidative stress signaling to neurons.
Keywords: mitochondria, motor neuron, neuropeptide, Nrf2/SKN-1, oxidative stress, sphingosine kinase
Introduction
Multicellular organisms encounter environmental and endogenous stress imposed by their ever-changing surroundings and have evolved complex stress response strategies to repair cellular damage and restore homeostasis. The nervous system plays a central role in adaptive responses to stress and bidirectional communication between neurons and target cells plays an important role in regulating proteostasis, immunity, development, lifespan, and redox homeostasis (Prahlad and Morimoto, 2011; Taylor et al., 2014). The signaling mechanisms underlying stress responses and how these signals are regulated by stress are not well defined.
The antioxidant response plays a crucial role in the maintenance of cellular redox homeostasis in the face of reactive oxygen species and is controlled by the transcription factor Nrf2 in mammals and SKN-1 in Caenorhabditis elegans. SKN-1/Nrf2 activation promotes organismal survival, longevity, xenobiotic detoxification, and pathogen resistance by directing the expression of a cascade of antioxidant, anti-inflammatory, and detoxification enzymes in response to oxidative stress (Ma, 2013). Nrf2 is normally targeted for ubiquitin-mediated degradation by its endogenous inhibitor Keap1. Under conditions of oxidative stress, SKN-1/Nrf2 is stabilized and translocates into the nucleus, where it binds antioxidant response elements in enhancer regions of hundreds of target genes, including glutathione-synthesizing enzymes, thioredoxins, and proteosomal subunits (Park et al., 2009).
Neurons are particularity susceptible to damage by oxidative stress due to the high metabolic demands required for neuronal function. However, Nrf2 activation is notably weak or absent in neurons (Baxter and Hardingham, 2016). Despite this, mature neurons maintain redox homeostasis over an organism's lifetime, largely by receiving antioxidant support from surrounding glial cells through intercellular signaling. In neuronal cocultures, activation of Nrf2 in astrocytes confers non-cell-autonomous neuroprotection from oxidative stress, in part through the release of glutathione from astrocytes (Liddell, 2017). In vivo, selective overexpression of Nrf2 in astrocytes in animal models of neurodegenerative diseases is sufficient to confer neuroprotection (Vargas et al., 2008; Chen et al., 2009; Gan et al., 2012), though the intercellular signals involved are not known. In C. elegans, SKN-1 can regulate neuronal function, survival, and longevity by non-cell-autonomous mechanisms. For example, selective activation of SKN-1 in the intestine negatively regulates neurotransmitter release from motor neurons (Staab et al., 2013). In addition, SKN-1 may function non-cell-autonomously to regulate synapse structure (Staab et al., 2014) and to inhibit dopamine neuron degeneration in a model for Parkinson's disease (Vanduyn et al., 2010).
Sphingosine kinases (SphKs) are conserved lipid kinases that catalyze the conversion of sphingosine (SPH) to sphingosine-1-phosphate (S1P), which are potent bioactive lipids with important functions in survival, differentiation, migration, and trafficking in diverse cell types (Hannun and Obeid, 2008). SphK also has roles in the nervous system in regulating synaptic plasticity and neurotransmitter release (Okada et al., 2009). SphK is highly enriched on a variety of cellular membranes, including sites of endocytosis, synapses, and mitochondria (Strub et al., 2011; Shen et al., 2014; Lima et al., 2017). SphK activity is largely controlled by the regulation of its recruitment to cellular membranes from cytosolic pools by a variety of extracellular and intracellular factors. S1P can function either as a lipid second messenger or as a secreted signal, where it activates cell surface S1P receptors. In C. elegans, the sole SphK ortholog, SPHK-1, regulates locomotion, axon repair, and acetylcholine release (Chan et al., 2012; Chisholm et al., 2016; McCulloch et al., 2017).
In this study, we report a function for SPHK-1 in the intestine in regulating the release of neuropeptides from motor neurons during the oxidative stress response. We show that activation of SKN-1 in the intestine negatively regulates neuropeptide release via the inhibition of S1P production by mitochondrially associated SPHK-1. Our results reveal a new role for sphingolipid signaling in the SKN-1 pathway and suggest that regulating neuronal function through intercellular signaling may be a critical component of the oxidative stress response.
Materials and Methods
C. elegans strains.
All strains used in this study were maintained at 20°C on nematode growing medium (NGM) seeded with OP50 bacteria following standard methods. Young adult hermaphrodites were used for all experiments unless otherwise noted. The following strains were provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health–National Center for Research Resources: skn-1(zu67) (WB catalog #EU1, RRID:WB-STRAIN:EU1), pmk-1(km25) (WB catalog #KU25, RRID:WB-STRAIN:KU25), nsy-1(ag3) (WB catalog #AU3, RRID:WB-STRAIN:AU3), sek-1(km4) (WB catalog #KU4, RRID:WB-STRAIN:KU4), egl-30(ad806) (WB catalog #DA1084, RRID:WB-STRAIN:DA1084), skn-1(lax188) (WormBase ID: WBVar01474253), egl-3(nr2090) (WormBase ID: WBVar00091400), tph-1(mg280) (WB catalog #MT15434, RRID:WB-STRAIN:MT15434), asna-1(ok938) (WormBase ID: WBVar00092209), aex-5(sa23) (WB catalog #JT23, RRID:WB-STRAIN:JT23), unc-25(e156) (WB catalog #CB156, RRID:WB-STRAIN:CB156), eat-4(ky5) (WB catalog #MT6308, RRID:WB-STRAIN:MT6308), aex-4(sa22) (WB catalog #JT5244, RRID:WB-STRAIN:JT5244), pho-4(ok583) (WormBase ID: WBVar00091869), tdc-1(n3419) (WB catalog #MT13113, RRID:WB-STRAIN:MT13113), cat-2(e1112) (WB catalog #CB1112, RRID:WB-STRAIN:CB1112), unc-17(e113) (WB catalog #CB113, RRID:WB-STRAIN:CB113), and pkc-1(nu448) (WB catalog #KP2342, RRID:WB-STRAIN:KP2342), unc-73(e936) (WB catalog #CB936, RRID:WB-STRAIN:CB936). The wdr-23(tm1817) (WormBase ID: WBVar00250781) and snt-2(tm1711) (WormBase ID: WBVar00250682) strains were provided by National BioResource Project (Japan). The wild-type reference strain was N2 Bristol (WB catalog #N2_(ancestral), RRID:WB-STRAIN:N2_(ancestral)). All strains used were outcrossed at least four times, except sphk-1(ok1097) (WB catalog #VC916, RRID:WB-STRAIN:VC916), which was outcrossed 12 times. The strain information is available at http://www.wormbase.org.
Molecular biology.
Genes were cloned from C. elegans cDNA from wild-type worms and then inserted into pPD49.26 using standard molecular biology techniques. Promoter DNA fragments were amplified from mixed-stage genomic DNA. The following plasmids were generated and used: pSK3[Prab-3::pmk-1], pSK4[Pges-1::pmk-1], pSK9[Pges-1::sphk-1::gfp], pSK26[Pges-1::tomm-20::mCherry], pSK28[Pges-1::sphk-1(ΔCaM)::gfp], pSK29[Pges-1::sphk-1(KD)::gfp].
Transgenic lines.
Transgenic strains were generated by injecting expression constructs (10–25 ng/μl) and the coinjection marker KP#708 (Pttx-3::rfp, 40 ng/μl) or KP#1106 (Pmyo-2::gfp, 10 ng/μl) into N2 or mutants. Microinjection was performed following standard techniques as described previously (Mello et al., 1991). At least three lines for each transgene were tested and a representative transgene was used for the further experiments. The following transgenic arrays were made and used: vjEx903[Pges-1::pmk-1], vjEx900[Prab-3::pmk-1], vjEx920[Pges-1::sphk-1::gfp], vjEx1058[Pges-1::sphk-1(KD)::gfp], vjEx1025[Pges-1::sphk-1(ΔCaM)::gfp], vjEx661[Pges-1::invom::rfp]. The following integrated transgenes were used in this study: vjIs28 (Punc-17::ins-22::yfp), vjIs30 (Punc-17::nlp-21::yfp), vjIs61 (Punc-17::gfp::snb-1), vjIs138(Pges-1::sphk-1::gfp), vjIs148(Pges-1::tomm-20::mCherry).
Microscopy and analysis.
Fluorescence microscopy experiments were performed following previous methods (Chan et al., 2012). Briefly, L4 stage or young adult worms were immobilized by using 2,3-butanedione monoxime (BDM, 30 mg/ml; Sigma-Aldrich) in M9 buffer and then mounted on 2% agarose pads for imaging. For quantification of synaptic vesicle (SV) and dense core vesicle (DCV) fluorescence, the dorsal nerve cord near the posterior gonadal bend of young adult worms was imaged. To measure the fluorescence intensity of DCV marker in coelomocytes, L4 worms were used. To image and quantify the fluorescence intensity of SPHK-1::GFP and mito-markers, posterior intestinal cells were selected as a representative region because of the mosaic expression of transgenes throughout the intestinal cells. For fluorescence microscopy experiments, images were captured with a Nikon eclipse 90i microscope equipped with a Nikon PlanApo 60× or 100× objective (numerical aperture = 1.4) and a Photometrics Coolsnap ES2 camera. MetaMorph 7.0 software (Universal Imaging/Molecular Devices) was used to capture serial image stacks and the maximum intensity projection was used for analysis of the dorsal cords. Line scans of the maximum intensity projection image were also recorded using MetaMorph. The fluorescence intensity values were then quantified using Puncta 6.0 software written with Igor Pro (Wavemetrics) as described previously (Staab et al., 2013). To equalize the absolute fluorescence levels between samples within the same experimental set, intensity quantification analysis was performed on the same day. Otherwise, fluorescence values were normalized to the values of 0.5 mm FluoSphere beads (Invitrogen) captured during each imaging session. Based on the expression pattern of mitochondrial marker TOMM-20::mCherry, the “net”-like pattern of fluorescence was considered to be mitochondria. For the mitochondrial occupancy analysis, animals showing a net-like pattern of fluorescence in >66%, 66–10%, or <10% of intestinal cells were sorted into the “high (H),” “medium (M),” or “low (L)” categories, respectively.
RNA interference (RNAi).
Feeding RNAi knock-down assays were performed following established protocols (Kamath and Ahringer, 2003). Briefly, gravid adult animals were placed on RNAi plates (1 mm IPTG, 25 mg/ml carbenicillin) seeded with HT115(DE3) bacteria transformed with L4440 vector containing fragments of knock-down genes or empty L4440 vector as a control for 4 h to obtain synchronized worm populations. Young adult animals were examined 3–4 d later. For intestinal-specific RNAi, eggs from gravid MG168 (intestine-specific RNAi strain, sid-1(qt9); vha-6::sid-1) (Khanna et al., 2014) adults were collected for 4 h on RNAi plates seeded with HT115(DE3) bacteria harboring the respective RNAi clones to obtain age-matched worm populations. After 3 d, aldicarb assays were performed by transferring adults to aldicarb plates. Each candidate gene was tested once in experimental duplicate and genes that showed aldicarb phenotypes were subjected to further screening up to five additional times.
Aldicarb and survival assays.
For paralysis assay using aldicarb or levamisole, paralyzed young adult animals were counted every 10–15 min starting from 40–60 min after placing worms on aldicarb plate. Then, 1 mm aldicarb or 200 μM levamisole (Sigma-Aldrich) was used for all paralysis experiments. At least two to three replicates of at least 20 worms per strain were placed on NGM plates supplemented with aldicarb or levamisole and paralyzed worms on each plate were counted to obtain percentage of paralyzed worms at each time point. The strains were blinded to scorer and performed in duplicate or triplicate at least two times of repetition for each analysis. For oxidative stressor treatment, young adult animals were placed on NGM plates supplemented with 5 mm sodium arsenite (RICCA Chemical), 200 μm juglone (Millipore), or 10 mm paraquat (ACROS organic) for 4 h before aldicarb assay. For H2O2 (Sigma-Aldrich) treatment, strains were incubated in a drop of 5 or 7 mm H2O2 in M9 buffer for 2 h; the supernatant was then removed and worms were placed on NGM plates for 2 h before aldicarb assay. NGM Plates containing drugs were freshly made 1 d before assays. For arsenite survival assays, young adult animals were placed onto NGM plates containing 4 mm arsenite for 48 h. The percentage of surviving animals was counted after indicated hours over the course of 48 h.
Statistical analysis.
For all assays, the Student's t test (two-tailed) was used to determine the statistical significance. Exact p-values for each result are noted in the Results section. p-values <0.0001 are represented as p < 0.0001. Error bars in the figures indicate ± SEM. The numbers of animals tested are indicated in each figure. All experiments were conducted at least twice with experimental duplicates or triplicates.
Results
Acute activation of intestinal SKN-1 by oxidative stress regulates motor neuron function
To investigate the mechanism by which intestinal SKN-1 regulates motor function, we first determined whether acute changes in SKN-1 activity can affect aldicarb responsiveness. Aldicarb is an acetylcholine esterase inhibitor and aldicarb treatment causes acetylcholine accumulation in synaptic clefts at neuromuscular junctions (NMJs), which leads to tonic muscle contraction and eventual paralysis. Animals with reduced acetylcholine release from motor neurons take longer to paralyze due to a delay in aldicarb-induced acetylcholine accumulation in synaptic clefts (Mahoney et al., 2006). To increase SKN-1 activity, we exposed animals to arsenite for various lengths of time. Arsenite is a mitochondrial toxin that leads to ROS production and arsenite treatment leads to robust SKN-1 activation and SKN-1 target gene expression in intestinal cells (Wu et al., 2016). We found that arsenite treatment for as little as 4 h caused a dramatic delay in aldicarb-induced paralysis compared with nontreated controls, referred to hereafter as aldicarb resistance (t(2) = −903, p < 0.0001 at 120 min, two-sample t test; Fig. 1A). Increasing arsenite treatment times to 6 or 14 h did not increase aldicarb resistance further (t(2) = 31, p = 0.001 at 120 min, two-sample t test and Staab et al., 2013; Fig. 1A). In C. elegans, which lacks a clear Keap1 ortholog, SKN-1 activity is repressed by the CUL-4 E3 ubiquitin ligase complex subunit WDR-23, which functions to keep SKN-1 activity low during nonstressed conditions by negatively regulating SKN-1 activation in intestinal cells (Choe et al., 2009). The magnitude of aldicarb resistance following arsenite treatment was similar to that seen when SKN-1 was chronically activated in wdr-23 mutants (Staab et al., 2013). However, decreasing treatment time to 2.5 h diminished aldicarb resistance and treatment for 1 h had no effect on aldicarb responsiveness (2.5 h: t(2) = −16.97, p = 0.003 1 h: t(2) = −0.45, p = 0.700 at 120 min, two-sample t test; Fig. 1A). Four-hour treatment with H2O2, juglone, or paraquat, which increase ROS signaling and activate SKN-1 (Hoeven et al., 2011; Staab et al., 2014; Raynes et al., 2017), also induced aldicarb resistance to similar extents as arsenite treatment (H2O2: t(4) = 6.38, p = 0.003 paraquat: t(2) = 7.27, p = 0.018 juglone: t(2) = 10.13, p = 0.009 at 90 min, two-sample t test; Fig. 1B). The aldicarb resistance caused by 4 h arsenite, paraquat, juglone, or H2O2 treatment was completely dependent upon SKN-1 H2O2: t(4) = 0.03, p = 0.975 paraquat: t(2) = 1.27, p = 0.33 juglone: t(2) = 5.98, p = 0.02 at 90 min (Figure 1B); t(4) = −3.90, p = 0.02 at 120 (Figure 1C), two-sample t test; Fig. 1B). Aldicarb resistance can arise from defects in motor neuron function or muscle excitability (Sieburth et al., 2005). Arsenite or H2O2 treatment did not alter rates of paralysis to the muscle agonist levamisole t(4) = −0.02, p = 0.981 at 120 min (Figure 1D); 5 μM H2O2: t(4) = −0.27, p = 0.796 at 120 min, 7 μM H2O2: t(4) = −1.54, p = 0.198 at 120 min (Figure 1E), two-sample t test; Fig. 1D). These results indicate that acute increases in ROS levels may not impact muscle excitability, but instead lead to defects in motor neuron function via a mechanism that is likely to depend upon the transcription of SKN-1 targets.
Figure 1.

Acute SKN-1 activation negatively regulates motor neuron function. A, Time course of aldicarb-induced paralysis of wild-type (wt) animals following treatment with arsenite (As) for the indicated number of hours. After 90 min in the presence of aldicarb, ∼70% of untreated animals were paralyzed. Arsenite treatment for at least 2.5 h before the assay elicited significant reduction in the percentage of animals paralyzed by aldicarb. B, Percentage of wild-type or skn-1 mutant animals paralyzed by aldicarb after 90 min following 4 h treatment with the ROS generators juglone, paraquat, or H2O2. ROS generator-induced aldicarb resistance was blocked in skn-1 mutants. C, Time course of aldicarb-induced paralysis of wild-type (wt) or skn-1 mutants treated with arsenite for 4 h. D, E, Time course of levamisole-induced paralysis of wild-type (wt) animals treated with arsenite or the indicated concentrations of H2O2 for 4 h. Numbers of animals tested is indicated. Error bars indicate ± SEM. Student's t test, **p < 0.01, ***p < 0.001.
p38 MAPK pathway functions in the intestine to regulate oxidative stress-induced aldicarb resistance
The activation of SKN-1 and Nrf2 by ROS is regulated by multiple intracellular signal transduction pathways (Inoue et al., 2005; Tullet et al., 2008; Kawli et al., 2010; Leung et al., 2014). A p38 MAPK signaling cascade composed of NSY-1/MAPKKK, SEK-1/MAPKK, and PMK-1/p38 MAPK pathway functions in the intestine to phosphorylate SKN-1 and promote SKN-1 nuclear translocation in response to oxidative stress (Inoue et al., 2005). SKN-1 is also activated by XREP-4, an F-box protein that activates SKN-1 through its interaction with the SKR-1 E3 ubiquitin ligase (Wu et al., 2017). We found that pmk-1-null mutants displayed similar rates of aldicarb-induced paralysis as wild-type controls in the absence of stress (t(2) = −0.71, p = 0.550 at 105 min, two-sample t test; Fig. 2A). However, pmk-1 mutants treated with arsenite failed to become more aldicarb resistant than untreated controls (t(3) = 0.56, p = 0.611 at 105 min, two-sample t test; Fig. 2A). sek-1 and nsy-1 mutants also failed to become aldicarb resistant following arsenite treatment (sek-1: t(4) = 1.04, p = 0.353 nsy-1: t(4) = −0.07, p = 0.944, two-sample t test; Fig. 2B). In contrast, xrep-4 mutants displayed robust responsiveness to arsenite treatment, becoming as resistant to aldicarb-induced paralysis as wild-type controls (t(2) = 22.88, p = 0.002, two-sample t test; Fig. 2B). Therefore, the PMK-1/p38 MAPK signaling pathway, but not XREP-4, is required for aldicarb resistance caused by arsenite.
Figure 2.

The p38 MAPK pathway functions in the intestine to promote oxidative stress-induced aldicarb resistance. Rates of paralysis of the indicated strains when exposed to aldicarb are shown. A, Time course of aldicarb-induced paralysis of wild-type (wt) animals or pmk-1 mutants following treatment with arsenite. B, Percentage of animals of the indicated genotypes paralyzed by aldicarb after 90 min following 4 h treatment with arsenite. sek-1 and nsy-1 mutants failed to display arsenite-induced aldicarb resistance, whereas xrep-4 mutants showed arsenite-induced aldicarb resistance. C, Time course of aldicarb-induced paralysis of wild-type (wt) animals or transgenic pmk-1 mutants following treatment with arsenite. Intestine rescue denotes pmk-1 mutants expressing transgenes containing pmk-1 cDNA under control of the intestine-specific ges-1 promoter. Neuron rescue denotes pmk-1 mutants expressing transgenes containing pmk-1 cDNA under control of the pan-neuronal promoter, rab-3. Arsenite treated pmk-1 mutants overexpressing intestinal pmk-1 cDNA are resistant to aldicarb. Numbers of animals tested are indicated. Error bars indicate ± SEM. Student's t test, **p < 0.01, ***p < 0.001.
PMK-1 functions in both the intestine to activate SKN-1 and in the nervous system to regulate sensory neuron development (Sagasti et al., 2001; Tanaka-Hino et al., 2002). Expression of wild-type pmk-1 cDNA in the intestine (under the ges-1 promoter) fully restored arsenite-induced aldicarb resistance to pmk-1 mutants (t(2) = 4.89, p = 0.039 at 100 min, two-sample t test; Fig. 2C). In contrast, pmk-1 cDNA expression in the nervous system (under the rab-3 promoter) failed to restore arsenite-induced aldicarb resistance to pmk-1 mutants (t(2) = 0.45, p = 0.697 at 100 min, two-sample t test; Fig. 2C). PMK-1 overexpression in the intestine in the absence of PMK-1 in other tissues was sufficient to cause aldicarb resistance (t(2) = 6.13, p = 0.026 at 80 min, two-sample t test; Fig. 2C). Therefore, loss of PMK-1 signaling in the intestine inhibits stress-induced aldicarb resistance, whereas restricted overexpression of PMK-1 in the intestine can induce aldicarb resistance. These results suggest that arsenite activates SKN-1 by a mechanism that requires PMK-1/p38 MAPK signaling and that SKN-1 activation negatively regulates motor neuron function.
SKN-1 activation in the intestine inhibits DCV biogenesis
Aldicarb resistance can arise from defects in either acetylcholine release or neuropeptide release from motor neurons (Richmond and Jorgensen, 1999; Sieburth et al., 2007). We determined the effect of arsenite treatment on the distribution of fluorescently labeled SVs and DCVs in motor neurons. SNB-1/synaptobrevin is a transmembrane SV-associated v-SNARE (Nonet et al., 1998) and GFP::SNB-1 fusion proteins expressed in cholinergic motor neurons adopt a punctate pattern of fluorescence along the lengths of motor axons (Fig. 3A). Defects in SV cycling at synapses lead to changes in the average fluorescence intensities of axonal GFP::SNB-1 puncta (Sieburth et al., 2005). We found that the GFP::SNB-1 punctal fluorescence intensity and number of GFP::SNB-1 puncta in motor axons were not significantly different in animals treated with arsenite for 4 h compared with untreated controls [puncta fluorescence: p = 0.157, degree of freedom (df) = 54 puncta interval: p = 0.069, df = 54, t test; Fig. 3A], suggesting that acute oxidative stress does not impact SV release.
Figure 3.
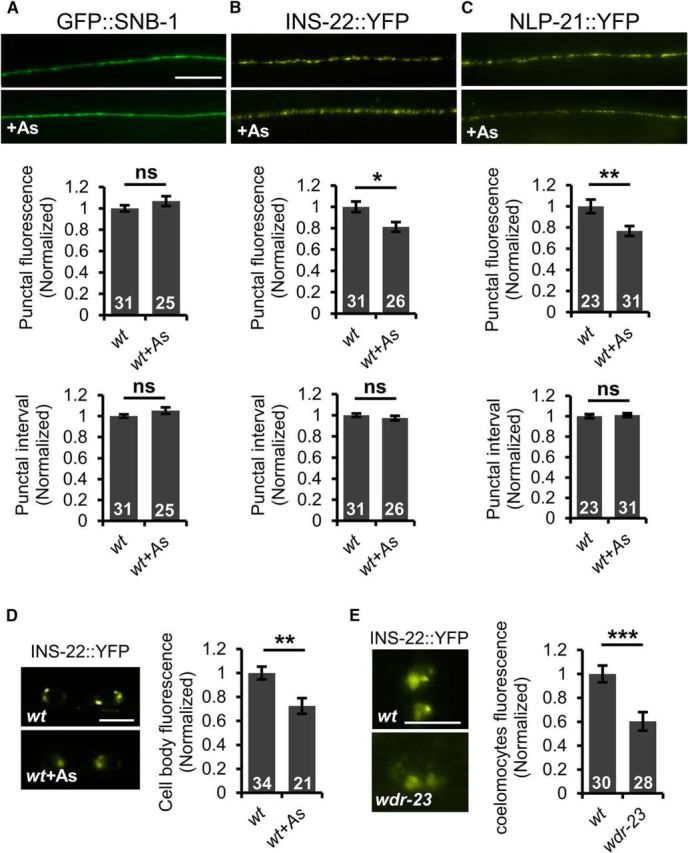
SKN-1 activation inhibits DCV biogenesis and secretion. A, Top, Representative images of the distribution of the synaptic vesicle protein GFP::SNB-1/synaptobrevin driven by the unc-17 promoter in cholinergic motor neurons of the dorsal cord in young adult wild-type animals in the absence or presence of arsenite (As). Middle, Quantification of the punctal fluorescence of GFP::SNB-1. Bottom, Quantification of punctal interval of GFP::SNB-1. B, C, Top, Representative images of the distribution of INS-22::YFP or NLP-21::YFP driven by the unc-17 promoter in the dorsal nerve cord in young adults in the absence or presence of arsenite. Middle, Quantification of the punctal fluorescence of INS-22::YFP and NLP-21::YFP. Bottom, Punctal interval of INS-22::YFP and NLP-21::YFP in the absence or presence of arsenite. D, Left, Representative images of motor neuron cell body fluorescence in wild-type adults expressing INS-22::YFP in the absence or presence of arsenite. Right, Quantification of INS-22::YFP pixel intensity in the cell body of motor neurons in the absence or presence of arsenite. E, Left, Representative images of coelomocyte fluorescence in L4 stage wild-type or wdr-23 mutants expressing INS-22::YFP respectively. Right, Quantification of INS-22::YFP pixel intensity of wild-type or wdr-23 mutants, respectively. Numbers of animals tested are indicated in white. Scale bar, 10 μm. Error bars indicate ± SEM. Student's t test, *p < 0.05, **p < 0.01, ***p < 0.001.
C. elegans cholinergic neurons also secrete the neuropeptides NLP-21 and INS-22, which are FMRF amide-related peptide (FaRP) and an insulin-like growth factor, respectively. Neuropeptides are packaged into immature DCVs at the Golgi complex, which undergo maturation and trafficking to release sites in the soma and axons, where they await release by calcium-dependent exocytosis (Gondré-Lewis et al., 2012). NLP-21::YFP and INS-22::YFP fusion proteins adopt a punctate pattern of localization in somas and axons (Sieburth et al., 2007; Fig. 3B–D). Defects in DCV biogenesis or secretion result in alterations in axonal punctal fluorescence intensity in motor neurons (Sieburth et al., 2007; Speese et al., 2007; Hao et al., 2012; Hoover et al., 2014). We found that INS-22::YFP and NLP-21::YFP punctal fluorescence intensity in axons of the dorsal nerve cord was reduced by 19% and 24%, respectively, in animals treated with arsenite compared with untreated controls (p = 0.014, df = 55 t test; Fig. 3B; p = 0.007, df = 52, t test; Fig. 3C). However, the number of DCV puncta in axons remained unchanged (p = 0.420, df = 55, t test; Fig. 3B; p = 0.904, df = 52, t test; Fig. 3C). INS-22::YFP fluorescence was also detected in the somas of motor neurons. INS-22::YFP fluorescence in motor neuron somas was significantly reduced following arsenite treatment (t(108) = 3.25, p = 0.002, two-sample t test; Fig. 3D). The reduction in neuropeptide fluorescence cannot be accounted for by reduced transgene expression because arsenite treatment did not reduce fluorescence of GFP::SNB-1, which was expressed under the same (unc-17) promoter (Fig. 3A).
The reduction in INS-22::YFP fluorescence intensity in both the axons and somas of motor neurons indicates that arsenite treatment may inhibit the biogenesis or maturation of DCVs. If this is the case, then we predict that SKN-1 activation in the intestine should impair neuropeptide secretion. To measure neuropeptide secretion, we examined the fluorescence intensity of scavenger cells (coelomocytes) in animals expressing INS-22::YFP in motor neurons. Once secreted, neuropeptides are internalized into endocytic compartments of coelomocytes and the fluorescence intensity of coelomocytes in animals expressing fluorescently tagged neuropeptides in neurons provides a measure of the rate of neuropeptide secretion (Sieburth et al., 2007; Hao et al., 2012). Because arsenite treatment inhibits coelomocyte uptake (data not shown), we instead examined neuropeptide secretion in wdr-23 mutants, in which SKN-1 is chronically activated, and in pmk-1 mutants, in which skn-1 activation is compromised. We observed a significant reduction in INS-22::YFP fluorescence in coelomocytes in wdr-23 mutants, consistent with our previous findings (t(56) = 3.81, p = 0.0003, two-sample t test; Fig. 3E) (Staab et al., 2013). Examination of INS-22::YFP axonal distribution in pmk-1 mutants revealed that INS-22::YFP punctal fluorescence in axons was significantly decreased in pmk-1 mutants compared with wild-type controls (p < 0.0001, df = 51, t test; Fig. 4A), suggesting that PMK-1 has a previously undescribed function in regulating DCV accumulation in axons in the absence of stress. Following arsenite treatment, we observed no further reduction in peak INS-22::YFP fluorescence in axons of pmk-1 mutants (p = 0.255, df = 53, t test; Fig. 4A). Expression of pmk-1 cDNA in the intestines of pmk-1 mutants (using the ges-1 promoter) fully restored arsenite responsiveness to pmk-1 mutants to wild-type levels (wild-type: p < 0.0001, df = 46 pmk-1; intestine rescue: p = 0.0004, df = 49, t test; Fig. 4A). The punctal fluorescence intensity was reduced by 30% following arsenite treatment, which is similar to the reduction seen in arsenite-treated wild-type controls (Fig. 4B). Intestinal pmk-1 expression had no effect on INS-22::YFP punctal intensity in pmk-1 mutants in the absence of stress compared with wild-type controls (p < 0.0001, df = 52, t test; Fig. 4A). Together, these results indicate that SKN-1 activation in the intestine results in the inhibition of neuropeptide secretion from motor neurons via a mechanism involving the inhibition of DCV biogenesis or maturation.
Figure 4.

Intestinal PMK-1/p38 MAPK mediates the oxidative stress-induced decrease of DCVs in motor axons. A, Left, Representative images of the distribution of the proneuropeptide INS-22::YFP driven by the unc-17 promoter in cholinergic motor neurons in young adult wild-type, pmk-1 mutants, or pmk-1 mutants expressing pmk-1 cDNA under the intestine-specific promoter ges-1 in the presence or absence of arsenite (As). Right, Quantification of the puncta fluorescence of INS-22::YFP of the indicated strains in the absence or presence of arsenite. B, Fold change of INS-22::YFP punctal fluorescence following arsenite treatment of the indicated strains. Numbers of animals tested are indicated in white. Scale bar, 10 μm. Error bars indicate ± SEM. Student's t test, **p < 0.01, ***p < 0.001.
Sphingosine kinase is required for oxidative stress-induced aldicarb resistance
Our findings suggest that intestinal SKN-1 controls an intertissue signaling pathway originating from the intestine that acts upon the nervous system to regulate neuropeptide secretion during the oxidative stress response. To identify the components of the signaling pathway, we first examined mutants defective in biosynthesis and/or secretion of signals known to be released from the intestine for their ability to block arsenite-induced aldicarb resistance. The regulated release of protons, neuropeptides, and insulins from the basolateral surface of the intestine into the body cavity (pseudocelom), control worm behavior and development (Kao et al., 2007; Beg et al., 2008; Mahoney et al., 2008; Wang et al., 2013). We found that mutants that impair the release of these signals (aex-4/SNAP-23, aex-5/CPE, snt-2/synaptotagmin, asna-1/ATPase, or pbo-4/sodium/proton exchanger) displayed normal responses to arsenite on our assays (wild-type: t(4) = 7.64, p = 0.001 at 105 min aex-4: t(2) = 27.16, p = 0.001 at 105 min aex-5: t(4) = 7.45, p = 0.001 at 225 min snt-2: t(4) = 4.99, p = 0.007 at 135 min asna-1: t(3) = 3.77, p = 0.033 at 105 min pbo-4: t(2) = 4.33, p = 0.049 at 105 min, two-sample t test; Table 1). We also found that mutants defective in the biosynthesis and/or secretion of known neurotransmitters and neuromodulators, including serotonin (tph-1/tryptophan hydroxylase) (Berendzen et al., 2016), dopamine (cat-2/tyrosine hydroxylase) (Lints and Emmons, 1999), tyramine/octopamine (tdc-1/tyrosine decarboxylase) (Alkema et al., 2005), acetylcholine (unc-17/VChAT) (Lickteig et al., 2001), glutamate (eat-4/VGAT) (Lee et al., 1999), GABA (unc-25/glutamic acid decarboxylase) (Jin et al., 1999), and neuropeptide (egl-3/prohormone convertase and pkc-1/protein kinase C) (Sieburth et al., 2007; Hung et al., 2014), displayed normal responses to arsenite in our aldicarb assays (tph-1: t(2) = 9.62, p = 0.011 at 135 min cat-2: t(4) = 8.60, p = 0.001 at 135 min tdc-1: t(4) = 32.02, p < 0.0001 at 120 min unc-17: t(4) = 9.63, p = 0.0007 at 240 min eat-4: t(2) = 4.89, p = 0.039 at 105 min unc-25: t(4) = 8.71, p = 0.001 at 60 min egl-3: t(4) = 4.05, p = 0.015 at 420 min pkc-1: t(2) = 6.41, p = 0.024 at 120 min, two-sample t test; Table 1). We were unable to examine the importance of glutathione signaling because mutants in glutathione biosynthetic genes (gcs-1/ gamma-glutamine cysteine synthetase or gss-1/ glutathione synthetase) die before reaching adulthood (www.wormbase.org). These results suggest that a previously uncharacterized, intestinally derived signal may mediate the effects of SKN-1 activation on motor neuron function.
Table 1.
Mutants defective in biosynthesis and/or secretion of secreted signals and SPHK-1 synapse targeting were tested for arsenite (As)-induced aldicarb resistance
| Strain | Gene name | Function | Paralyzed on aldicarb (%) |
|||||
|---|---|---|---|---|---|---|---|---|
| −As | ±SEM | +As | ±SEM | Time (min) | Reference | |||
| Wild-type | n/a | n/a | 90.6 | ±3.8 | 40.9 | ±4.5 | 105 | n/a |
| aex-4 | SNAP23/synaptosome associated protein 23 | In intestine to release neuropeptide | 70.7 | ±0.6 | 11.6 | ±1.7 | 105 | Mahoney et al., 2008 |
| aex-5 | CPE/calcium-dependent serine endoproteinases | In intestine to release neuropeptide | 91.7 | ±1.7 | 54.4 | ±4.7 | 225 | Mahoney et al., 2008 |
| snt-2 | Synaptotagmin | In intestine to release neuropeptide | 81.5 | ±2.8 | 50.6 | ±5.1 | 135 | Wang et al., 2013 |
| asna-1 | ASNA1/ATPase | In intestine to release insulin | 91.7 | ±6.0 | 60.7 | ±2.9 | 105 | Kao et al., 2007 |
| pbo-4 | NHE/Na+/H + ion exchanger | In intestine to release proton | 72.5 | ±6.1 | 40.0 | ±0.0 | 105 | Beg et al., 2008 |
| tph-1 | Tryptophan hydroxylase | Serotonin secretion | 66.0 | ±0.6 | 32.6 | ±3.4 | 135 | Berendzen et al., 2016 |
| cat-2 | Tyrosine hydroxylase | Dopamine secretion | 59.2 | ±4.7 | 18.0 | ±1.0 | 135 | Lints and Emmons, 1999 |
| tdc-1 | Tyrosine decarboxylase | Tyramine/octopamine secretion | 94.6 | ±1.5 | 11.6 | ±2.1 | 120 | Alkema et al., 2005 |
| unc-17 | VChAT/ synaptic vesicle acetylcholine transporter | Acetylcholine secretion | 72.3 | ±3.1 | 38.2 | ±1.8 | 240 | Lickteig et al., 2001 |
| eat-4 | VGAT/glutamate transporter | Glutamate secretion | 76.0 | ±3.3 | 29.9 | ±7.0 | 105 | Lee et al., 1999 |
| unc-25 | Glutamic acid decarboxylase | GABA secretion | 93.4 | ±1.5 | 70.0 | ±3.3 | 60 | Jin et al., 1999 |
| egl-3 | Prohormone convertase | Neuropeptide secretion | 87.1 | ±3.1 | 65.6 | ±4.3 | 420 | Hung et al., 2014 |
| pkc-1 | PKC/protein kinase C | Neuropeptide secretion | 67.5 | ±7.5 | 18.6 | ±1.3 | 120 | Sieburth et al., 2007 |
| egl-30 | Heterotrimeric G protein alpha subunit Gq | sphk-1 effector | 68.0 | ±0.0 | 5.7 | ±3.8 | 280 | Chan et al., 2012 |
| unc-73 | GNEF/guanine nucleotide exchange factor | sphk-1 effector | 62.5 | ±8.3 | 13.7 | ±1.7 | 60 | Chan et al., 2012 |
| sphk-1 | SphK/sphingosine kinase | Sphingosine-1-phosphate synthesis | 38.1 | ±2.7 | 65.9 | ±2.6 | 135 | Chan et al., 2012 |
Percentage of animals paralyzed at the indicated time point in the absence or presence of As is indicated. The results for RNAi screening of additional genes are given in Table 1-1
RNAi screening of genes that alter the aldicarb response using MGH168 strain (intestine specific RNAi strain). Download Table 1-1, XLSX file (25.5KB, xlsx)
We reasoned that components of the intestine-to-neuron signaling pathway could be identified by screening for genes that alter aldicarb sensitivity when selectively knocked down in the intestine. Accordingly, we conducted a pilot feeding RNAi screen of 153 genes previously shown to regulate aldicarb responsiveness, but the mechanism of action of which is not well understood (Sieburth et al., 2005). We used the strain MGH168, which allows efficient RNAi in the intestine, but not in other tissues, including the nervous system (Khanna et al., 2014). As negative controls, we randomly included in our screen seven genes known to function exclusively in the nervous system to promote acetylcholine release from motor neurons and the RNAi knock-down of which causes aldicarb resistance (unc-36, unc-2, unc-17, unc-18, unc-13, unc-10, and snb-1; Sieburth et al., 2005). None of these seven genes scored as positive in our screen (Table 1-1). Among the 153 genes screened, we identified one candidate, sphk-1, that conferred significant and reproducible aldicarb resistance when knocked down selectively in the intestine (Table 1-1).
SPHK-1 functions in motor neurons to promote acetylcholine release (Chan et al., 2012), but its role in the intestine or in the antioxidant pathway had not been investigated previously. sphk-1-null mutants displayed significant resistance to aldicarb-induced paralysis (t(4) = 13.58, p = 0.0002 at 120 min, two-sample t test; Fig. 5A) (Chan et al., 2012). If SPHK-1 functions in the antioxidant pathway, then we predict that pharmacological or genetic activation of SKN-1 should not enhance the aldicarb resistance of sphk-1 mutants. In agreement with this, we found that the aldicarb resistance of sphk-1 mutants was not enhanced by arsenite treatment, but instead was slightly suppressed (t(4) = −7.04, p = 0.002 at 120 min, two-sample t test; Fig. 5A). Similarly, the aldicarb resistance of sphk-1 mutants was not enhanced, but instead slightly suppressed, by RNAi knock-down of wdr-23 (t(4) = −3.08, p = 0.036, two-sample t test; Fig. 5B) or by a constitutively active skn-1 mutation, skn-1(lax188gf) (Paek et al., 2012) (t(4) = −2.22, p = 0.090, two-sample t test; Fig. 5C) (Staab et al., 2013). In addition, overexpression of PMK-1 in the intestine, which causes aldicarb resistance (Fig. 2C), was unable to enhance the aldicarb resistance of sphk-1 mutants (t(4) = −0.80, p = 0.469, two-sample t test; Fig. 5D). Finally, the paraquat- or juglone-induced aldicarb resistance, which was dependent upon SKN-1, was not enhanced by sphk-1 mutations (wild-type paraquat: t(2) = 4.90, p = 0.039 juglone: t(2) = 13.69, p = 0.005 sphk-1 mutant paraquat: t(2) = 0.56, p = 0.627 juglone: t(2) = −3.52, p = 0.071, two-sample t test; Fig. 5E). egl-30/Gαq and unc-73/Trio GEF function in motor neurons to activate SPHK-1 (Chan et al., 2012); however, egl-30 or unc-73 mutants became significantly more aldicarb resistant following arsenite treatment than untreated controls (egl-30: t(4) = 16.19, p < 0.0001 at 280 min unc-73: t(2) = 8.89, p = 0.012 at 60 min, two-sample t test; Table 1). Together, these observations reveal a function for SPHK-1 in SKN-1-mediated regulation of aldicarb responsiveness that may be distinct from its function in promoting acetylcholine release from motor neurons.
Figure 5.
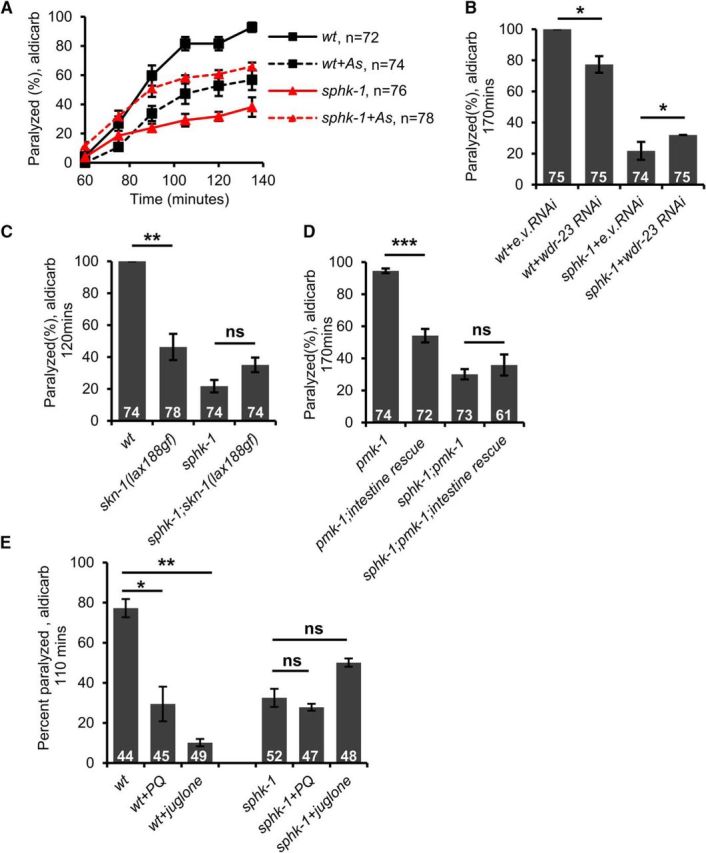
sphk-1 mutants block SKN-1-mediated aldicarb resistance. Rates of aldicarb-induced paralysis of indicated strains following exposure to arsenite (As). A, Wild-type (wt) animals treated with arsenite were resistant to the paralytic effects of aldicarb. sphk-1 mutants were resistant to aldicarb but exhibited sensitivity to aldicarb when exposed to arsenite. B, Knock-down of wdr-23 by RNAi increased the aldicarb resistance in wild-type controls but not in sphk-1 mutants. C, Ability of constitutively active skn-1(lax188gf) mutation to cause aldicarb resistance was blocked by sphk-1 mutants. D, Overexpression of pmk-1 cDNA in the intestine of pmk-1 mutants caused aldicarb resistance in sphk-1(+) but not sphk-1(null) background. E, Percentage of wild-type and sphk-1 mutants paralyzed by aldicarb after 110 min following 4 h treatment with juglone and paraquat. Numbers of animals tested are indicated. Error bars indicate ± SEM. Student's t test, *p < 0.05, **p < 0.01, ***p < 0.001.
We next examined neuropeptide secretion from motor neurons in sphk-1 mutants and found that sphk-1 mutants expressing INS-22::YFP displayed significantly reduced punctal fluorescence in the dorsal nerve cord compared with wild-type controls (p < 0.0001, df = 52, t test; Fig. 6A). In addition, the coelomocyte fluorescence of sphk-1 mutants expressing INS-22::YFP was reduced by nearly 40% (t(53) = 3.06, p = 0.004, two-sample t test; Fig. 6B). These results indicate that SPHK-1 promotes neuropeptide secretion from motor neurons under normal conditions. Following arsenite treatment, INS-22::YFP punctal fluorescence of sphk-1 mutants did not undergo any further reduction (p = 0.375, df = 42, t test; Fig. 6A), suggesting that SPHK-1 is required for the effects of oxidative stress on neuropeptide secretion. These results are consistent with a model whereby SPHK-1 functions to promote neuropeptide secretion from motor neurons under nonstressed conditions and that oxidative stress blocks neuropeptide secretion by inhibiting the function of SPHK-1.
Figure 6.
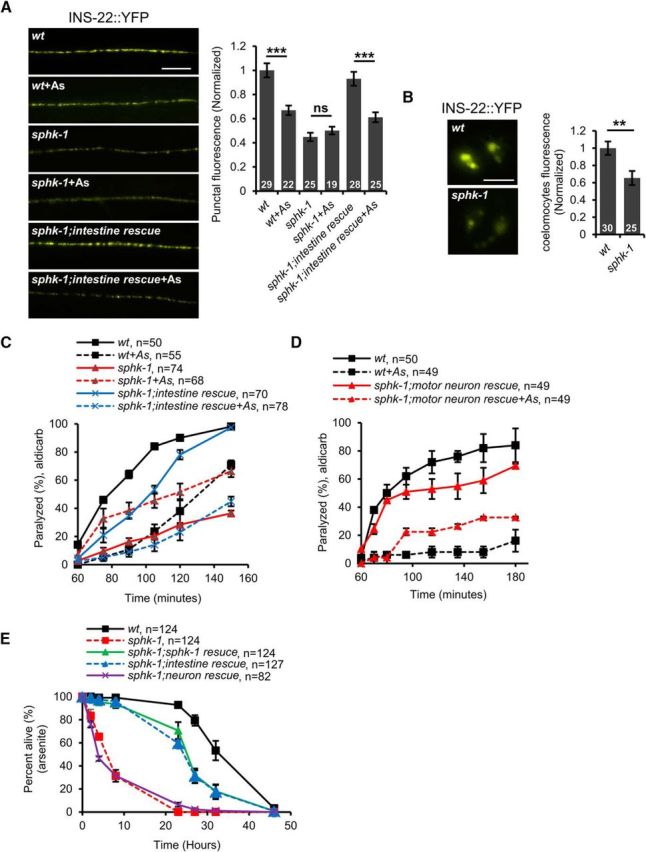
SPHK-1 functions in the intestine to regulate neuropeptide secretion. A, Left, Representative images of the distribution of INS-22::YFP fluorescence in cholinergic motor neurons of the dorsal nerve cord in the indicated strains. Intestine rescue denotes sphk-1 mutants expressing sphk-1 cDNA under the intestine-specific promoter ges-1. Right, Quantification of INS-22::YFP punctal fluorescence in the indicated strains in the absence or presence of arsenite (As). B, Left, Representative images of coelomocyte fluorescence in L4 stage wild-type or sphk-1 mutants expressing INS-22::YFP. Right, Quantification of fluorescence intensity in coelomocytes of L4 stage wild-type or sphk-1 mutants. Numbers of animals tested are indicated in white. C, Time course of aldicarb-induced paralysis of the indicated strains. Intestine rescue refers to sphk-1 mutants expressing sphk-1 cDNA transgenes driven by the intestine-specific promoter, ges-1. D, Time course of aldicarb-induced paralysis of the indicated strains. Motor neuron rescue refers to sphk-1 mutants expressing sphk-1 cDNA transgenes driven by the motor-neuron-specific promoter unc-129. E, Survival rate curves of indicated strains on plates containing 4 mM arsenite. Numbers of animals tested are indicated. Scale bar, 10 μm. Error bars indicate ± SEM. Student's t test, **p < 0.01, ***p < 0.001.
SPHK-1 functions in the intestine to regulate aldicarb responsiveness and neuropeptide secretion
To determine the site of action of SPHK-1 in the oxidative stress response, we expressed sphk-1 cDNA transgenes under tissue-specific promoters. As expected, expression of sphk-1 cDNA in the nervous system was able to partially rescue the aldicarb resistance of sphk-1, confirming the role of sphk-1 in neurons to promote acetylcholine release (Fig. 6D) (Chan et al., 2012). However, we found that neuronal sphk-1 cDNA expression was not able to fully restore arsenite-induced aldicarb resistance to sphk-1 mutants (Fig. 6D). Conversely, sphk-1 cDNA expression in the intestine only partially rescued aldicarb resistance of sphk-1 mutants (Fig. 6C), but fully restored arsenite-induced aldicarb resistance to sphk-1 mutants (Fig. 6C). sphk-1 cDNA expression in the intestines of sphk-1 mutants restored INS-22::YFP punctal fluorescence back to wild-type levels (p = 0.220, df = 55, t test; Fig. 6A). Intestinal sphk-1 cDNA also restored normal arsenite responsiveness to sphk-1 mutants: arsenite was able to reduce INS-22::YFP punctal fluorescence by 35% compared with nontreated controls (p = 0.0001, df = 51, t test; Fig. 6A).
Prolonged arsenite exposure causes toxicity and populations of sphk-1 mutants exhibited significantly reduced survival over 48 h compared with wild-type controls (Fig. 6E). The expression of sphk-1 cDNA in the intestine rescued the reduced survival of sphk-1 mutants in the presence of arsenite. In contrast, the expression of sphk-1 cDNA in the nervous system did not rescue the survival defects of sphk-1 mutants (Fig. 6E). These results indicate that SPHK-1 functions in the intestine to promote DCV secretion from motor neurons, aldicarb resistance, and survival in response to oxidative stress, revealing roles for SPHK-1 that are distinct from its role in neurons in promoting acetylcholine release (Chan et al., 2012). These results also suggest that the negative regulation of DCV secretion from motor neurons by stress occurs through the SKN-1-mediated inhibition of SPHK-1 function in the intestine.
Stress-regulated SPHK-1 association with intestinal mitochondria regulates aldicarb responsiveness
How might SKN-1 regulate SPHK-1 function in the intestine? One possibility is that the activation of SKN-1 leads to the downregulation of sphk-1 expression. SKN-1 activation is not likely to downregulate the expression of sphk-1 because transcriptome profiling reveals that sphk-1 levels are not reduced in wdr-23 mutants compared with wild-type controls (Staab et al., 2013). Another possibility is that SKN-1 may regulate SPHK-1 protein function. The activity of sphingosine kinase is primarily controlled by regulating its targeting to cellular membranes from cytosolic pools (Pitson et al., 2003; Stahelin et al., 2005; Alemany et al., 2007; Pitson et al., 2011). To determine whether SKN-1 regulates the targeting of SPHK-1 to membranes, we first examined the distribution of SPHK-1::GFP fusion proteins overexpressed from extrachromosomal arrays in the intestine. SPHK-1::GFP fusion proteins are functional because they fully rescue the aldicarb defects of sphk-1 mutants, suggesting that the localization pattern of SPHK-1::GFP should reflect that of endogenous SPHK-1 (Chan and Sieburth, 2012). We found that SPHK-1::GFP was not evenly distributed in intestinal cells, but instead adopted a “net”-like localization pattern throughout the cytoplasm. This pattern was similar to that observed for the outer membrane mitochondrial marker TOMM-20::mCherry (Fig. 7A) (Palikaras et al., 2015). Colocalization studies revealed strong colocalization of SPHK-1::GFP with TOMM-20::mCherry in the intestine (Fig. 7A). Colocalization was also seen between SPHK-1::GFP and an inverted outer membrane mitochondrial marker (INVOM::RFP; Staab et al., 2013) (Fig. 7A).
Figure 7.
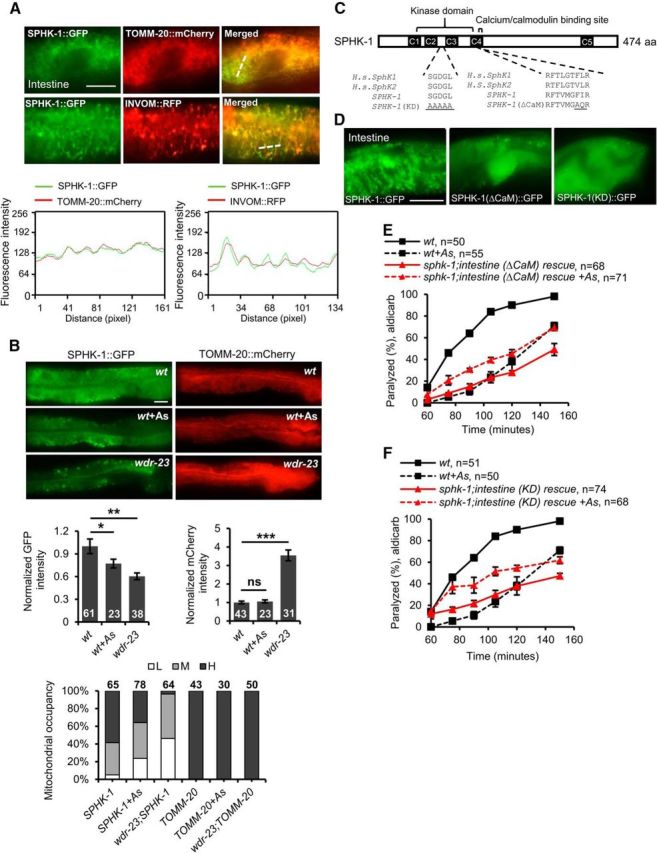
Mitochondrial accumulation of intestinal SPHK-1::GFP is negatively regulated by SKN-1 activation. A, Top, Representative images showing the colocalization of SPHK-1::GFP fusion proteins and mitochondria marker TOMM-20::mCherry and INVOM::RFP driven by the ges-1 promoter in the intestine of wild-type animals. Bottom, Alignment of cross-sectioned (dotted line) fluorescence intensity curve graph of GFP and mCherry/RFP in intestine. B, Top, Representative images of SPHK-1::GFP or TOMM-20::mCherry intestinal localization in wild-type controls following arsenite (As) treatment or in the wdr-23 mutants. Middle, Average GFP or mCherry intestinal fluorescence intensity in the indicated strains. Bottom, Mitochondria occupancy rates show the percentage of animals expressing H (high), M (medium), or L (low) levels of SPHK-1::GFP or TOMM-20::mCherry in the intestines of the indicated strains. H indicates a net-like fluorescence pattern is observed in >66% of intestinal cells, M indicates a net-like fluorescence pattern is observed in 10–66% of intestinal cells, and L indicates a net-like fluorescence pattern is observed in <10% of intestinal cells. C, Predicted domain structure of SPHK-1 showing conservation of the kinase domain and CaM-binding domain with human (H.s.) SphK proteins. The amino acids mutated to generate KD and (ΔCaM) SPHK-1 variants are underlined. D, Representative image of wild-type expressing SPHK-1::GFP or SPHK-1(ΔCaM)::GFP or SPHK-1(KD)::GFP in intestine. E, F, Time course of aldicarb-induced paralysis of the indicated strains. sphk-1 mutants expressing SPHK-1(KD) or SPHK-1(ΔCaM) in the intestine were not responsive to arsenite treatment. Numbers of animals tested are indicated. Scale bar, 10 μm. Error bars indicate ± SEM. Student's t test, *p < 0.05, **p < 0.01, ***p < 0.001.
SPHK-1::GFP mitochondrial fluorescence intensity was significantly reduced in wdr-23 mutants or following arsenite treatment (wdr-23: t(76) = −3.34, p = 0.001 arsenite: t(42) = 2.05, p = 0.047, two-sample t test; Fig. 7B). The reduction in fluorescence was not a consequence of reduced mitochondrial mass in the intestine or reduced expression of the sphk-1::gfp transgene because we saw no decrease in fluorescence of TOMM-20::mCherry (expressed under the same ges-1 promoter) in wdr-23 mutants or following arsenite treatment (wdr-23: t(52) = −7.17, p < 0.0001 arsenite: t(42) = −0.50, p = 0.62, two-sample t test; Fig. 7B). Together, these results suggest that SKN-1 activation negatively regulates SPHK-1 mitochondrial abundance in the intestine, possibly by regulating its translocation from cytosolic pools to mitochondrial membranes.
Sphingosine kinase contains a conserved calcium-/calmodulin (CaM)-binding motif that mediates the regulated targeting of SphK to cellular membranes (Fig. 7C) (Young et al., 2003; Sutherland et al., 2006; Jarman et al., 2010). To determine whether the association of SPHK-1 with mitochondria is important for its function in the stress response, we generated transgenic sphk-1 mutants expressing a version of SPHK-1 in the intestine which the calcium-/CaM-binding domain had been mutated [SPHK-1(ΔCaM)::GFP] (Fig. 7C). SPHK-1(ΔCaM)::GFP adopted a diffuse pattern of fluorescence in the intestine consistent with a cytosolic, but not mitochondrial association (Fig. 7D). sphk-1 mutants expressing SPHK-1(ΔCaM)::GFP in the intestine did not become aldicarb resistant in response to arsenite, but rather become slightly aldicarb sensitive, unlike sphk-1 mutants expressing wild-type SPHK-1::GFP (t(4) = −4.33, p = 0.012 at 105 min, two-sample t test; Fig. 7E). We found that kinase-dead SPHK-1 [SPHK-1(KD)::GFP] adopted a more diffuse pattern of fluorescence throughout the intestinal cells similar to SPHK-1(ΔCaM)::GFP transgenes (Fig. 7D). sphk-1 mutants expressing SPHK-1(KD)::GFP did not become aldicarb resistant, but rather become slightly aldicarb sensitive, after treatment with arsenite (t(4) = −3.82, p = 0.018 at 105 min, two-sample t test; Fig. 7F). Together, our observations suggest that the generation of S1P from SPH by SPHK-1 on mitochondrial membranes in the intestine is critical for stress-induced aldicarb resistance. We propose a model in which S1P production on mitochondrial membranes either directly or indirectly promotes neuropeptide secretion from motor neurons under nonstressed conditions. Following acute stress, intestinal SKN-1 activation negatively regulates SPHK-1 translocation to mitochondria and a reduction of S1P production, which subsequently blocks neuropeptide secretion from motor neurons (Fig. 8).
Figure 8.

Working model for how SKN-1-regulated SPHK-1 signaling regulates neuropeptide secretion. A, During normal conditions, SKN-1 levels are kept low by WDR-23, expression of SKN-1 targets is low, and SPHK-1 abundance on the outer mitochondrial membrane is high. S1P generated by SPHK-1 in the intestine directly or indirectly promotes neuropeptide secretion from motor neurons. B, During high oxidative stress conditions, PMK-1 promotes SKN-1 nuclear translocation, SKN-1 target genes are expressed, and SPHK-1 mitochondrial abundance is low. Decreased generation of S1P by SPHK-1 leads to a reduction in neuropeptide secretion from motor neurons.
Discussion
Our study reveals a central role for intestinal SKN-1 activation in reducing neuropeptide secretion from motor neurons by inhibiting the biogenesis or maturation of DCVs. We found that SPHK-1 on mitochondrial membranes in the intestine promotes neuropeptide release from motor neurons and that SKN-1 activation leads to a reduction in SPHK-1 mitochondrial abundance and the subsequent inhibition of neuropeptide release. We propose that S1P production by SPHK-1 may provide a link between the oxidative stress response in the intestine and motor function.
Effects of oxidative stress on neuropeptide release
DCVs are generated at the Golgi apparatus as immature DCVs, which subsequently undergo a maturation process through membrane remodeling events and cargo processing (Gondré-Lewis et al., 2012). The reduction in DCV abundance in both somas and axons that we observed following arsenite treatment is strikingly similar to the DCV defects reported in rab-2 and tbc-8 mutants (Edwards et al., 2009; Sumakovic et al., 2009; Hannemann et al., 2012). rab-2 encodes a small GTPase of the Ras superfamily and tbc-8 encodes a GTPase-activating protein that may inactivate RAB-2. RAB-2 and TCB-8 are localized to the Golgi–endosomal system, where they are required to retain neuropeptides in maturing DCVs in the somas. We speculate that SKN-1 signaling may inhibit DCV biogenesis or maturation by a mechanism that involves the regulation of RAB-2 activity in the soma.
We uncovered two functions for PMK-1 p38 MAPK signaling in regulating DCV abundance in motor neurons. First, we found that PMK-1 functions to increases DCV abundance in motor neurons in the absence of stress. MAPK signaling regulates DCV capture at release sites (Bharat et al., 2017) and is implicated in activity-dependent potentiation of DCV release (Park et al., 2006), raising the possibility that PMK-1 may regulate DCV abundance via a similar mechanism. We also found that intestinal PMK-1 activity was both necessary for SKN-1-mediated inhibition of neuropeptide secretion and sufficient to cause aldicarb resistance. Therefore, the strength of PMK-1 p38 MAPK signaling in the intestine may be an important determinant of the efficacy of the intestine-to-motor neuron signaling pathway.
Why would the downregulation of neuropeptide secretion from neurons by SKN-1 activation be beneficial to an organism? The nervous system is particularly susceptible to oxidative damage due in part to the high metabolic demands required for neuronal function, yet neurons have limited intrinsic capability to neutralize ROS (Halliwell, 2006). We propose that downregulation of DCV biogenesis by SKN-1 activation may serve two functions during the oxidative stress response. First, it may be an effective way to reduce metabolic demands of the nervous system during periods of stress. Once released from motor neurons, neuropeptides can function in an autocrine manner to increase acetylcholine release from NMJs during locomotion (Steuer Costa et al., 2017). Therefore, the inhibition of neuropeptide release by SKN-1 activation may indirectly reduce energy consumption (and ROS generation) associated with SV release and muscle contraction. Second, specific neuropeptides with release that is regulated by SKN-1 may themselves function as neuroendocrine signals on target tissues to relay the oxidative stress response throughout the organism. C. elegans encodes >100 neuropeptide-like and insulin-like genes that make hundreds of mature peptides. Neuropeptide release has been shown to activate the mitochondrial unfolded protein response (Shao et al., 2016), the heat-shock protein response (Prahlad et al., 2008), and the pathogen response (Zhang et al., 2005) in distal tissues. Identification of the specific neuropeptides that may function to relay the oxidative stress response in distal tissues will be of interest.
Mitochondrial function of sphingosine kinase
Our results examining the localization of SPHK-1::GFP indicate that SKN-1 regulation of SPHK-1 abundance on mitochondria occurs via a mechanism that is likely to involve controlling its translocation to mitochondria from cytosolic pools. First, SPHK-1::GFP fusion proteins occupy an area on the outer edge of mitochondria (Chan and Sieburth, 2012), consistent with outer mitochondrial membrane localization. Second, SPHK-1::GFP fusion proteins lacking key residues in the CaM site, which is critical for membrane targeting, are no longer enriched on mitochondrial membranes and fail to function in the stress response (Fig. 7D,E). Finally, either pharmacological or genetic activation of SKN-1 significantly reduces SPHK-1 abundance on mitochondrial membranes without detectably altering mitochondrial morphology (Fig. 7B). The 40% reduction in SPHK-1::GFP abundance in wdr-23 mutants is likely to be an underestimate because the mitochondrial mass in the intestine was increased in wdr-23 mutants (Fig. 7B). The increase in mitochondrial mass seen in wdr-23 mutants is consistent with reports that Nrf2 knock-out reduces mitochondrial content, whereas activation of Nrf2 promotes mitochondrial biogenesis (Shen et al., 2008; Zhang et al., 2013). SPHK-1::GFP also localizes to mitochondria in muscle cells, but in neurons, SPHK-1::GFP localizes to the plasma membrane at synapses (Chan and Sieburth, 2012), suggesting cell-specific and/or context-dependent regulation of SPHK-1 localization.
Mammalian SphK1 is translocated to specialized sites of the plasma membrane, ER, and Golgi from cytoplasmic pools via a variety of agonists and growth factors (Pitson, 2011). SphK1 recruitment to plasma membranes is positively regulated by MAPK, which phosphorylates SphK1 on Ser225, and by CIB1 (calcium and integrin-binding protein1), which promotes calcium-dependent SphK1 translocation through interactions with the CaM-binding domain of SphK1 (Pitson et al., 2003). Our data do not rule out the possibility that SPHK-1 abundance on mitochondria may (also) be regulated by ubiquitin-mediated degradation. Interestingly, WDR-23, which is a component of the CUL-4 E3 ubiquitin ligase complex, localizes to mitochondria (Staab et al., 2013), raising the possibility that WDR-23 CUL-4 complex participates in regulating mitochondrial SPHK-1. Nonetheless, our study provides the first example to our knowledge that sphingosine kinase abundance on mitochondrial membranes is subject to regulation.
Signaling mechanisms of S1P
Our results show that the generation of S1P from SPH by SPHK-1 on mitochondria is important for its function in regulating motor function. The generation of S1P by SphK is the last step in an enzymatic cascade that converts sphingomyelin to S1P. Ceramidase, which converts ceramide to SPH, associates with mitochondria in a dynamic fashion, accumulating on the outer membrane of mitochondria during apoptosis and mitophagy (Novgorodov et al., 2011; Hernández-Corbacho et al., 2017). SphK2 is enriched in mitochondrial fractions in HeLa cells, where it catalyzes S1P production from SPH to regulate respiration (Strub et al., 2011). Therefore, the spatial and temporal regulation of the enzymes in the sphingolipid cascade is likely to be critical in precisely regulating the production of S1P on mitochondria.
Our screen for diffusible factors that mediate intestine-to-neuron signaling ruled out the involvement of known classical neurotransmitters, neuromodulators, and neuropeptides in stress-regulated motor function. In principle, S1P itself could be the signal or S1P may function via a less direct mechanism involving regulation of an unidentified diffusible factor or factors. S1P is known to be secreted from a number of cell types and S1P concentrations are high in the circulation (Blaho and Hla, 2014; Nagahashi et al., 2014). Secreted S1P signals via a family of five S1PR/EDG G-protein-coupled receptors (Rosen et al., 2013), which have important roles in neuronal development, neuroprotection, and neuronal function (Chi and Nicol, 2010). The C. elegans genome does not encode obvious S1PR/EDG homologs (Hobert et al., 2013), so if the S1P generated by SPHK-1 in the intestine is secreted, then it would presumably activate a structurally distinct but functionally similar type of receptor. S1P also functions as an intracellular signaling molecule that can directly associate with and modulate the activities of cytoplasmic proteins, including the E3 ubiquitin ligase TRAF2, histone deacetylases (HDAC1 and HDAC2), and the mitochondrial mitophagy protein prohibitin (Hait et al., 2009; Alvarez et al., 2010; Wei et al., 2017). Identifying the intracellular or extracellular targets of S1P will help to elucidate the molecular mechanisms by which S1P regulates presynaptic function in response to stress.
Footnotes
This work was supported by the National Institutes of Health–National Institute of Neurological Disorders and Stroke (Grants NS071085 and NS099414). Some strains were provided by the Caenorhabditis Genetics Center, which is funded by NIH Office of Research Infrastructure Programs (Grant P40 OD010440). We thank members of the Sieburth laboratory for critical reading of the manuscript and P. Sternberg for the INVOM:RFP strain.
The authors declare no competing financial interests.
References
- Alemany R, van Koppen CJ, Danneberg K, Ter Braak M, Meyer Zu Heringdorf D (2007) Regulation and functional roles of sphingosine kinases. Naunyn Schmiedebergs Arch Pharmacol 374:413–428. 10.1007/s00210-007-0132-3 [DOI] [PubMed] [Google Scholar]
- Alkema MJ, Hunter-Ensor M, Ringstad N, Horvitz HR (2005) Tyramine functions independently of octopamine in the Caenorhabditis elegans nervous system. Neuron 46:247–260. 10.1016/j.neuron.2005.02.024 [DOI] [PubMed] [Google Scholar]
- Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, Maceyka M, Jiang H, Luo C, Kordula T, Milstien S, Spiegel S (2010) Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 465:1084–1088. 10.1038/nature09128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter PS, Hardingham GE (2016) Adaptive regulation of the brain's antioxidant defences by neurons and astrocytes. Free Radic Biol Med 100:147–152. 10.1016/j.freeradbiomed.2016.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg AA, Ernstrom GG, Nix P, Davis MW, Jorgensen EM (2008) Protons act as a transmitter for muscle contraction in C. elegans. Cell 132:149–160. 10.1016/j.cell.2007.10.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendzen KM, Durieux J, Shao LW, Tian Y, Kim HE, Wolff S, Liu Y, Dillin A (2016) Neuroendocrine coordination of mitochondrial stress signaling and proteostasis. Cell 166:1553–1563.e10. 10.1016/j.cell.2016.08.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharat V, Siebrecht M, Burk K, Ahmed S, Reissner C, Kohansal-Nodehi M, Steubler V, Zweckstetter M, Ting JT, Dean C (2017) Capture of dense core vesicles at synapses by JNK-dependent phosphorylation of synaptotagmin-4. Cell Rep 21:2118–2133. 10.1016/j.celrep.2017.10.084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaho VA, Hla T (2014) An update on the biology of sphingosine 1-phosphate receptors. J Lipid Res 55:1596–1608. 10.1194/jlr.R046300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JP, Sieburth D (2012) Localized sphingolipid signaling at presynaptic terminals is regulated by calcium influx and promotes recruitment of priming factors. J Neurosci 32:17909–17920. 10.1523/JNEUROSCI.2808-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JP, Hu Z, Sieburth D (2012) Recruitment of sphingosine kinase to presynaptic terminals by a conserved muscarinic signaling pathway promotes neurotransmitter release. Genes Dev 26:1070–1085. 10.1101/gad.188003.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PC, Vargas MR, Pani AK, Smeyne RJ, Johnson DA, Kan YW, Johnson JA (2009) Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson's disease: critical role for the astrocyte. Proc Natl Acad Sci U S A 106:2933–2938. 10.1073/pnas.0813361106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi XX, Nicol GD (2010) The sphingosine 1-phosphate receptor, S1PR(1), plays a prominent but not exclusive role in enhancing the excitability of sensory neurons. J Neurophysiol 104:2741–2748. 10.1152/jn.00709.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisholm AD, Hutter H, Jin Y, Wadsworth WG (2016) The genetics of axon guidance and axon regeneration in Caenorhabditis elegans. Genetics 204:849–882. 10.1534/genetics.115.186262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe KP, Przybysz AJ, Strange K (2009) The WD40 repeat protein WDR-23 functions with the CUL4/DDB1 ubiquitin ligase to regulate nuclear abundance and activity of SKN-1 in Caenorhabditis elegans. Mol Cell Biol 29:2704–2715. 10.1128/MCB.01811-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SL, Charlie NK, Richmond JE, Hegermann J, Eimer S, Miller KG (2009) Impaired dense core vesicle maturation in Caenorhabditis elegans mutants lacking Rab2. J Cell Biol 186:881–895. 10.1083/jcb.200902095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan L, Vargas MR, Johnson DA, Johnson JA (2012) Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J Neurosci 32:17775–17787. 10.1523/JNEUROSCI.3049-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondré-Lewis MC, Park JJ, Loh YP (2012) Cellular mechanisms for the biogenesis and transport of synaptic and dense-core vesicles. Int Rev Cell Mol Biol 299:27–115. 10.1016/B978-0-12-394310-1.00002-3 [DOI] [PubMed] [Google Scholar]
- Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S, Spiegel S (2009) Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325:1254–1257. 10.1126/science.1176709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B. (2006) Oxidative stress and neurodegeneration: where are we now? J Neurochem 97:1634–1658. 10.1111/j.1471-4159.2006.03907.x [DOI] [PubMed] [Google Scholar]
- Hannemann M, Sasidharan N, Hegermann J, Kutscher LM, Koenig S, Eimer S (2012) TBC-8, a putative RAB-2 GAP, regulates dense core vesicle maturation in Caenorhabditis elegans. PLoS Genet 8:e1002722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9:139–150. 10.1038/nrm2329 [DOI] [PubMed] [Google Scholar]
- Hao Y, Hu Z, Sieburth D, Kaplan JM (2012) RIC-7 promotes neuropeptide secretion. PLoS Genet 8:e1002464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Corbacho MJ, Salama MF, Canals D, Senkal CE, Obeid LM (2017) Sphingolipids in mitochondria. Biochim Biophys Acta 1862:56–68. 10.1016/j.bbalip.2016.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O. (2013) The neuronal genome of Caenorhabditis elegans. WormBook 13:1–106. 10.1895/wormbook.1.161.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeven Rv, McCallum KC, Cruz MR, Garsin DA (2011) Ce-Duox1/BLI-3 generated reactive oxygen species trigger protective SKN-1 activity via p38 MAPK signaling during infection in C. elegans. PLoS Pathog 7:e1002453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover CM, Edwards SL, Yu SC, Kittelmann M, Richmond JE, Eimer S, Yorks RM, Miller KG (2014) A novel CaM kinase II pathway controls the location of neuropeptide release from Caenorhabditis elegans motor neurons. Genetics 196:745–765. 10.1534/genetics.113.158568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung WL, Wang Y, Chitturi J, Zhen M (2014) A Caenorhabditis elegans developmental decision requires insulin signaling-mediated neuron-intestine communication. Development 141:1767–1779. 10.1242/dev.103846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Hisamoto N, An JH, Oliveira RP, Nishida E, Blackwell TK, Matsumoto K (2005) The C. elegans p38 MAPK pathway regulates nuclear localization of the transcription factor SKN-1 in oxidative stress response. Genes Dev 19:2278–2283. 10.1101/gad.1324805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarman KE, Moretti PA, Zebol JR, Pitson SM (2010) Translocation of sphingosine kinase 1 to the plasma membrane is mediated by calcium- and integrin-binding protein 1. J Biol Chem 285:483–492. 10.1074/jbc.M109.068395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Jorgensen E, Hartwieg E, Horvitz HR (1999) The Caenorhabditis elegans gene unc-25 encodes glutamic acid decarboxylase and is required for synaptic transmission but not synaptic development. J Neurosci 19:539–548. 10.1523/JNEUROSCI.19-02-00539.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Ahringer J (2003) Genome-wide RNAi screening in Caenorhabditis elegans. Methods 30:313–321. 10.1016/S1046-2023(03)00050-1 [DOI] [PubMed] [Google Scholar]
- Kao G, Nordenson C, Still M, Rönnlund A, Tuck S, Naredi P (2007) ASNA-1 positively regulates insulin secretion in C. elegans and mammalian cells. Cell 128:577–587. 10.1016/j.cell.2006.12.031 [DOI] [PubMed] [Google Scholar]
- Kawli T, Wu C, Tan MW (2010) Systemic and cell intrinsic roles of gqalpha signaling in the regulation of innate immunity, oxidative stress, and longevity in Caenorhabditis elegans. Proc Natl Acad Sci U S A 107:13788–13793. 10.1073/pnas.0914715107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna A, Johnson DL, Curran SP (2014) Physiological roles for mafr-1 in reproduction and lipid homeostasis. Cell Rep 9:2180–2191. 10.1016/j.celrep.2014.11.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RY, Sawin ER, Chalfie M, Horvitz HR, Avery L (1999) EAT-4, a homolog of a mammalian sodium-dependent inorganic phosphate cotransporter, is necessary for glutamatergic neurotransmission in Caenorhabditis elegans. J Neurosci 19:159–167. 10.1523/JNEUROSCI.19-01-00159.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung CK, Hasegawa K, Wang Y, Deonarine A, Tang L, Miwa J, Choe KP (2014) Direct interaction between the WD40 repeat protein WDR-23 and SKN-1/Nrf inhibits binding to target DNA. Mol Cell Biol 34:3156–3167. 10.1128/MCB.00114-14 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lickteig KM, Duerr JS, Frisby DL, Hall DH, Rand JB, Miller DM 3rd (2001) Regulation of neurotransmitter vesicles by the homeodomain protein UNC-4 and its transcriptional corepressor UNC-37/groucho in Caenorhabditis elegans cholinergic motor neurons. J Neurosci 21:2001–2014. 10.1523/JNEUROSCI.21-06-02001.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddell JR. (2017) Are astrocytes the predominant cell type for activation of Nrf2 in aging and neurodegeneration? Antioxidants (Basel) 6: E65. 10.3390/antiox6030065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima S, Milstien S, Spiegel S (2017) Sphingosine and sphingosine kinase 1 involvement in endocytic membrane trafficking. J Biol Chem 292:3074–3088. 10.1074/jbc.M116.762377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lints R, Emmons SW (1999) Patterning of dopaminergic neurotransmitter identity among Caenorhabditis elegans ray sensory neurons by a TGFbeta family signaling pathway and a hox gene. Development 126:5819–5831. [DOI] [PubMed] [Google Scholar]
- Ma Q. (2013) Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53:401–426. 10.1146/annurev-pharmtox-011112-140320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney TR, Luo S, Nonet ML (2006) Analysis of synaptic transmission in Caenorhabditis elegans using an aldicarb-sensitivity assay. Nat Protoc 1:1772–1777. 10.1038/nprot.2006.281 [DOI] [PubMed] [Google Scholar]
- Mahoney TR, Luo S, Round EK, Brauner M, Gottschalk A, Thomas JH, Nonet ML (2008) Intestinal signaling to GABAergic neurons regulates a rhythmic behavior in Caenorhabditis elegans. Proc Natl Acad Sci U S A 105:16350–16355. 10.1073/pnas.0803617105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch KA, Qi YB, Takayanagi-Kiya S, Jin Y, Cherra SJ 3rd (2017) Novel mutations in synaptic transmission genes suppress neuronal hyperexcitation in Caenorhabditis elegans. G3 (Bethesda) 7:2055–2063. 10.1534/g3.117.042598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V (1991) Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J 10:3959–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahashi M, Takabe K, Terracina KP, Soma D, Hirose Y, Kobayashi T, Matsuda Y, Wakai T (2014) Sphingosine-1-phosphate transporters as targets for cancer therapy. BioMed Res Int 2014:651727. 10.1155/2014/651727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet ML, Saifee O, Zhao H, Rand JB, Wei L (1998) Synaptic transmission deficits in Caenorhabditis elegans synaptobrevin mutants. J Neurosci 18:70–80. 10.1523/JNEUROSCI.18-01-00070.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novgorodov SA, Wu BX, Gudz TI, Bielawski J, Ovchinnikova TV, Hannun YA, Obeid LM (2011) Novel pathway of ceramide production in mitochondria: thioesterase and neutral ceramidase produce ceramide from sphingosine and acyl-CoA. J Biol Chem 286:25352–25362. 10.1074/jbc.M110.214866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T, Kajimoto T, Jahangeer S, Nakamura S (2009) Sphingosine kinase/sphingosine 1-phosphate signalling in central nervous system. Cell Signal 21:7–13. 10.1016/j.cellsig.2008.07.011 [DOI] [PubMed] [Google Scholar]
- Paek J, Lo JY, Narasimhan SD, Nguyen TN, Glover-Cutter K, Robida-Stubbs S, Suzuki T, Yamamoto M, Blackwell TK, Curran SP (2012) Mitochondrial SKN-1/Nrf mediates a conserved starvation response. Cell Metab 16:526–537. 10.1016/j.cmet.2012.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palikaras K, Lionaki E, Tavernarakis N (2015) Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521:525–528. 10.1038/nature14300 [DOI] [PubMed] [Google Scholar]
- Park SK, Tedesco PM, Johnson TE (2009) Oxidative stress and longevity in Caenorhabditis elegans as mediated by SKN-1. Aging Cell 8:258–269. 10.1111/j.1474-9726.2009.00473.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YS, Jun DJ, Hur EM, Lee SK, Suh BS, Kim KT (2006) Activity-dependent potentiation of large dense-core vesicle release modulated by mitogen-activated protein kinase/extracellularly regulated kinase signaling. Endocrinology 147:1349–1356. 10.1210/en.2005-0959 [DOI] [PubMed] [Google Scholar]
- Pitson SM. (2011) Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem Sci 36:97–107. 10.1016/j.tibs.2010.08.001 [DOI] [PubMed] [Google Scholar]
- Pitson SM, Moretti PA, Zebol JR, Lynn HE, Xia P, Vadas MA, Wattenberg BW (2003) Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J 22:5491–5500. 10.1093/emboj/cdg540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitson SM, Powell JA, Bonder CS (2011) Regulation of sphingosine kinase in hematological malignancies and other cancers. Anticancer Agents Med Chem 11:799–809. 10.2174/187152011797655078 [DOI] [PubMed] [Google Scholar]
- Prahlad V, Morimoto RI (2011) Neuronal circuitry regulates the response of Caenorhabditis elegans to misfolded proteins. Proc Natl Acad Sci U S A 108:14204–14209. 10.1073/pnas.1106557108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahlad V, Cornelius T, Morimoto RI (2008) Regulation of the cellular heat shock response in Caenorhabditis elegans by thermosensory neurons. Science 320:811–814. 10.1126/science.1156093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynes R, Juarez C, Pomatto LC, Sieburth D, Davies KJ (2017) Aging and SKN-1-dependent loss of 20S proteasome adaptation to oxidative stress in C. elegans. J Gerontol A Biol Sci Med Sci 72:143–151. 10.1093/gerona/glw093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Jorgensen EM (1999) One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat Neurosci 2:791–797. 10.1038/12160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen H, Stevens RC, Hanson M, Roberts E, Oldstone MB (2013) Sphingosine-1-phosphate and its receptors: structure, signaling, and influence. Ann Rev Biochem 82:637–662. 10.1146/annurev-biochem-062411-130916 [DOI] [PubMed] [Google Scholar]
- Sagasti A, Hisamoto N, Hyodo J, Tanaka-Hino M, Matsumoto K, Bargmann CI (2001) The CaMKII UNC-43 activates the MAPKKK NSY-1 to execute a lateral signaling decision required for asymmetric olfactory neuron fates. Cell 105:221–232. 10.1016/S0092-8674(01)00313-0 [DOI] [PubMed] [Google Scholar]
- Shao LW, Niu R, Liu Y (2016) Neuropeptide signals cell non-autonomous mitochondrial unfolded protein response. Cell Res 26:1182–1196. 10.1038/cr.2016.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Giordano F, Wu Y, Chan J, Zhu C, Milosevic I, Wu X, Yao K, Chen B, Baumgart T, Sieburth D, De Camilli P (2014) Coupling between endocytosis and sphingosine kinase 1 recruitment. Nat Cell Biol 16:652–662. 10.1038/ncb2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Liu K, Tian C, Yang L, Li X, Ren J, Packer L, Cotman CW, Liu J (2008) R-alpha-lipoic acid and acetyl-L-carnitine complementarily promote mitochondrial biogensis in murine 3T3–L1 adipocytes. Diabetologia 51:165–174. 10.1007/s00125-007-0852-4 [DOI] [PubMed] [Google Scholar]
- Sieburth D, Ch'ng Q, Dybbs M, Tavazoie M, Kennedy S, Wang D, Dupuy D, Rual JF, Hill DE, Vidal M, Ruvkun G, Kaplan JM (2005) Systematic analysis of genes required for synapse structure and function. Nature 436:510–517. 10.1038/nature03809 [DOI] [PubMed] [Google Scholar]
- Sieburth D, Madison JM, Kaplan JM (2007) PKC-1 regulates secretion of neuropeptides. Nat Neurosci 10:49–57. 10.1038/nn1810 [DOI] [PubMed] [Google Scholar]
- Speese S, Petrie M, Schuske K, Ailion M, Ann K, Iwasaki K, Jorgensen EM, Martin TF (2007) UNC-31 (CAPS) is required for dense-core vesicle but not synaptic vesicle exocytosis in Caenorhabditis elegans. J Neurosci 27:6150–6162. 10.1523/JNEUROSCI.1466-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staab TA, Evgrafov O, Egrafov O, Knowles JA, Sieburth D (2014) Regulation of synaptic nlg-1/neuroligin abundance by the skn-1/Nrf stress response pathway protects against oxidative stress. PLoS Genet 10:e1004100. 10.1371/journal.pgen.1004100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staab TA, Griffen TC, Corcoran C, Evgrafov O, Knowles JA, Sieburth D (2013) The conserved SKN-1/Nrf2 stress response pathway regulates synaptic function in Caenorhabditis elegans. PLoS Genet 9:e1003354. 10.1371/journal.pgen.1003354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahelin RV, Hwang JH, Kim JH, Park ZY, Johnson KR, Obeid LM, Cho W (2005) The mechanism of membrane targeting of human sphingosine kinase 1. J Biol Chem 280:43030–43038. 10.1074/jbc.M507574200 [DOI] [PubMed] [Google Scholar]
- Steuer Costa W, Yu SC, Liewald JF, Gottschalk A (2017) Fast cAMP modulation of neurotransmission via neuropeptide signals and vesicle loading. Curr Biol 27:495–507. 10.1016/j.cub.2016.12.055 [DOI] [PubMed] [Google Scholar]
- Strub GM, Paillard M, Liang J, Gomez L, Allegood JC, Hait NC, Maceyka M, Price MM, Chen Q, Simpson DC, Kordula T, Milstien S, Lesnefsky EJ, Spiegel S (2011) Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J 25:600–612. 10.1096/fj.10-167502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumakovic M, Hegermann J, Luo L, Husson SJ, Schwarze K, Olendrowitz C, Schoofs L, Richmond J, Eimer S (2009) UNC-108/RAB-2 and its effector RIC-19 are involved in dense core vesicle maturation in Caenorhabditis elegans. J Cell Biol 186:897–914. 10.1083/jcb.200902096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland CM, Moretti PA, Hewitt NM, Bagley CJ, Vadas MA, Pitson SM (2006) The calmodulin-binding site of sphingosine kinase and its role in agonist-dependent translocation of sphingosine kinase 1 to the plasma membrane. J Biol Chem 281:11693–11701. 10.1074/jbc.M601042200 [DOI] [PubMed] [Google Scholar]
- Tanaka-Hino M, Sagasti A, Hisamoto N, Kawasaki M, Nakano S, Ninomiya-Tsuji J, Bargmann CI, Matsumoto K (2002) SEK-1 MAPKK mediates Ca2+ signaling to determine neuronal asymmetric development in Caenorhabditis elegans. EMBO Rep 3:56–62. 10.1093/embo-reports/kvf001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC, Berendzen KM, Dillin A (2014) Systemic stress signalling: understanding the cell non-autonomous control of proteostasis. Nat Rev Mol Cell Biol 15:211–217. 10.1038/nrm3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tullet JM, Hertweck M, An JH, Baker J, Hwang JY, Liu S, Oliveira RP, Baumeister R, Blackwell TK (2008) Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 132:1025–1038. 10.1016/j.cell.2008.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanduyn N, Settivari R, Wong G, Nass R (2010) SKN-1/Nrf2 inhibits dopamine neuron degeneration in a Caenorhabditis elegans model of methylmercury toxicity. Toxicol Sci 118:613–624. 10.1093/toxsci/kfq285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA (2008) Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci 28:13574–13581. 10.1523/JNEUROSCI.4099-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Girskis K, Janssen T, Chan JP, Dasgupta K, Knowles JA, Schoofs L, Sieburth D (2013) Neuropeptide secreted from a pacemaker activates neurons to control a rhythmic behavior. Curr Biol 23:746–754. 10.1016/j.cub.2013.03.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Chiang WC, Sumpter R Jr, Mishra P, Levine B (2017) Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 168:224–238.e10. 10.1016/j.cell.2016.11.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CW, Deonarine A, Przybysz A, Strange K, Choe KP (2016) The Skp1 homologs SKR-1/2 are required for the Caenorhabditis elegans SKN-1 antioxidant/detoxification response independently of p38 MAPK. PLoS Genet 12:e1006361. 10.1371/journal.pgen.1006361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CW, Wang Y, Choe KP (2017) F-box protein XREP-4 is a new regulator of the oxidative stress response in Caenorhabditis elegans. Genetics 206:859–871. 10.1534/genetics.117.200592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KW, Willets JM, Parkinson MJ, Bartlett P, Spiegel S, Nahorski SR, Challiss RA (2003) Ca2+/calmodulin-dependent translocation of sphingosine kinase: role in plasma membrane relocation but not activation. Cell Calcium 33:119–128. 10.1016/S0143-4160(02)00205-1 [DOI] [PubMed] [Google Scholar]
- Zhang YK, Wu KC, Klaassen CD (2013) Genetic activation of Nrf2 protects against fasting-induced oxidative stress in livers of mice. PLoS One 8:e59122. 10.1371/journal.pone.0059122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lu H, Bargmann CI (2005) Pathogenic bacteria induce aversive olfactory learning in Caenorhabditis elegans. Nature 438:179–184. 10.1038/nature04216 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
RNAi screening of genes that alter the aldicarb response using MGH168 strain (intestine specific RNAi strain). Download Table 1-1, XLSX file (25.5KB, xlsx)