

Abstract
Neuromodulation mediated by metabotropic glutamate receptors (mGluRs) regulates many brain functions. However, the functions of mGluRs in the auditory system under normal and diseased states are not well understood. The medial nucleus of the trapezoid body (MNTB) is a critical nucleus in the auditory brainstem nuclei involved in sound localization. In addition to the classical calyx excitatory inputs, MNTB neurons also receive synaptic inhibition and it remains entirely unknown how this inhibition is regulated. Here, using whole-cell voltage clamp in brain slices, we investigated group I mGluR (mGluR I)-mediated modulation of the glycinergic and GABAergic inputs to MNTB neurons in both WT mice and a fragile X syndrome (FXS) mouse model (both sexes) in which the fragile X mental retardation gene 1 is knocked out (Fmr1 KO), causing exaggerated activity of mGluR I and behavioral phenotypes. Activation of mGluR I by (RS)-3,5-dihydroxyphenylglycine (3,5-DHPG) increased the frequency and amplitude of glycinergic spontaneous IPSCs (sIPSCs) in both WT and Fmr1 KO neurons in a voltage-gated sodium channel-dependent fashion, but did not modulate glycinergic evoked IPSCs (eIPSCs). In contrast, 3,5-DHPG did not affect GABAergic sIPSCs, but did suppress eIPSCs in WT neurons via endocannabinoid signaling. In the KO, the effect of 3,5-DHPG on GABAergic eIPSCs was highly variable, which supports the notion of impaired GABAergic signaling in the FXS model. The differential modulation of sIPSC and eIPSC and differential modulation of glycinergic and GABAergic transmission suggest distinct mechanisms responsible for spontaneous and evoked release of inhibitory transmitters and their modulation through the mGluR I signaling pathway.
SIGNIFICANCE STATEMENT Neurons communicate with each other through the release of neurotransmitters, which assumes two basic modes, spontaneous and evoked release. These two release modes are believed to function using the same vesicle pool and machinery. Recent works have challenged this dogma, pointing to distinct vesicle release mechanisms underlying the two release modes. Here, we provide the first evidence in the central auditory system supporting this novel concept. We discovered neural-transmitter- and release-mode-specific neuromodulation of inhibitory transmission by metabotropic glutamate receptors and revealed part of the signaling pathways underlying this differential modulation. The results establish the foundation for a multitude of directions to study physiological significance of different release modes in auditory processing.
Keywords: GABA, glycine, IPSC, mGluR, MNTB, neuromodulation
Introduction
Since the discovery of spontaneous release of neurotransmitters (Fatt and Katz, 1950, 1952), it has been believed that these synaptic responses are simply induced stochastically and independently of presynaptic action potentials (APs) and that the spontaneous release serves no physiologically relevant function. Additionally, it was believed that the same pool of synaptic vesicles was used in both spontaneous and evoked neurotransmitter release. Recent works challenge these views (Kavalali, 2015) and support the alternative notions that spontaneous synaptic events serve physiological roles in maintaining protein synthesis in dendritic spines (McKinney et al., 1999; Sutton et al., 2004) and that the vesicle pool responsible for spontaneous release may be different from the pool for evoked release (Sara et al., 2005; Glitsch, 2006; Atasoy et al., 2008; Chung et al., 2010; Deng et al., 2010). These different modes of transmitter release may act as separate signaling pathways via activation of nonoverlapping postsynaptic receptors (Atasoy et al., 2008; Sara et al., 2011). In the central auditory system, such a distinction of spontaneous and evoked transmission at the level of synaptic vesicle pools or release machinery has not been reported.
We address this issue by investigating metabotropic glutamate receptor (mGluR) neuromodulation of synaptic inhibition in the medial nucleus of the trapezoid body (MNTB). Several distinct features of MNTB establish the nucleus as an ideal model for this study. First, the MNTB plays an important integral role in many auditory circuits (Thompson and Schofield, 2000), especially for sound localization (Grothe et al., 2010). Understanding neuromodulation in MNTB therefore has critical implications for multiple aspects of auditory processing. Second, MNTB neurons receive both synaptic excitation and inhibition (Awatramani et al., 2004, 2005; Mayer et al., 2014), with the inhibitory inputs arising from both extrinsic and intrinsic sources (Guinan and Li, 1990; Kuwabara et al., 1991; Smith et al., 1998; Albrecht et al., 2014; Dondzillo et al., 2016). There is a known role for group I mGluR (mGluR I) modulation of the excitatory input (Kushmerick et al., 2004). We thus hypothesized that inhibition to the MNTB is also subject to mGluR I modulation because most neurons receive equalizing excitatory and inhibit inputs (Wehr and Zador, 2003; Xue et al., 2014; Froemke, 2015) and the balance of excitation and inhibition is a critical feature of sound localizing circuits (Magnusson et al., 2008). Third, the synaptic inhibition MNTB receives is both glycinergic and GABAergic (Awatramani et al., 2004, 2005; Albrecht et al., 2014; Mayer et al., 2014), rendering study of transmitter-specific modulation in the same neuron possible. Finally, we investigated this modulation in a fragile X Syndrome (FXS) mouse model in which fragile X mental retardation protein (FMRP) is knocked out. Due to the exaggerated mGluR I activity that underlies this disease (Bear et al., 2004; D'Antoni et al., 2014) and the associated auditory deficits such as hyperexcitability, altered frequency tuning, and impaired neural plasticity in auditory neurons (Kim et al., 2013; Rotschafer and Razak, 2013, 2014; Rotschafer et al., 2015; Garcia-Pino et al., 2017), we propose that dysfunctional mGluR modulation may partially account for the hearing abnormalities in FXS. Additionally, recent studies have shown that there is a strong expression of FMRP in MNTB neurons (Zorio et al., 2017) and there is altered topographic distribution of inhibitory inputs to MNTB in FXS mice (McCullagh et al., 2017), reflecting the critical interaction between FMRP and mGluRs in MNTB and the necessity for studying fine-tuned mGluR modulation of the inhibitory input in FXS.
Materials and Methods
Animals.
All animal procedures were approved by the Institutional Animal Care and Use Committee at the Northeast Ohio Medical University (NEOMED) and were performed in accordance with the National Institutes of Health's policies on animal use. WT and Fmr1 KO mice (with a background of C57BL/6J; RRID:IMSR_JAX:000664) were purchased from The Jackson Laboratory and bred at NEOMED. Genotype was confirmed with standard PCR protocol provided by The Jackson Laboratory. All mice were housed in a vivarium with a normal light/dark cycle (12 h light/12 h dark).
Slice preparation and in vitro whole-cell recordings.
Coronal brainstem slices (250 μm in thickness) were prepared from postnatal day 12 (P12)–P25 and P125 mice of both sexes, as described previously with minor modifications (Curry and Lu, 2016). Mice were deeply anesthetized with isoflurane and rapidly decapitated. The brainstem was removed and sliced under warm (35°C) artificial CSF (ASCF) containing the following (in mm): 250 glycerol, 3 KCl, 1.2 KH2PO4, 20 NaHCO3, 3 HEPES, 1.2 CaCl2, 5 MgCl2, and 10 glucose, pH 7.4 (when gassed with 95% O2 and 5% CO2). Slices were incubated in an interface chamber at 34–36°C for >1 h in normal ACSF containing the following (in mm): 130 NaCl, 20 NaHCO3, 3 KCl, 2.4 CaCl2, 1.3 MgSO4, 1.2 KH2PO4, and 10 glucose, pH 7.4. For recording, slices were transferred to a 0.5 ml chamber mounted on a Zeiss Axioskop 2 FS Plus microscope with a 40× water-immersion objective and infrared differential interference contrast optics. The chamber was continuously superfused with ACSF (2–5 ml/min) by gravity.
Patch pipettes were drawn on a PP-830 microelectrode puller (Narishige) to a 1–2 μm tip diameter using borosilicate glass micropipettes (inner diameter, 0.84 mm; outer diameter, 1.5 mm; World Precision Instruments). The electrodes had resistances between 3 and 6 MΩ when filled with a solution containing the following (in mm): 105 Cs-methanesulfonate, 35 CsCl, 5 EGTA, 10 HEPES, 1 MgCl2, 4 ATP-Mg, 0.46 GTP-Na, 5 QX-314, with pH 7.2, adjusted with CsOH and osmolarity ∼290 mOsm/L. The liquid junction potential was 10 mV and data were corrected accordingly. Voltage-clamp experiments were performed with an AxoPatch 200B amplifier (Molecular Devices). Recordings were performed under near physiological temperatures (34–36°C) and were obtained at a holding potential of −70 mV. Only cells with series resistances <20 MΩ were used and the series resistance was compensated by 70–80%. Data were low-pass filtered at 5 kHz and digitized with a Data Acquisition Interface ITC-18 (InstruTech) at 50 kHz. Recording protocols were written and run using the acquisition and analysis software AxoGraph X (AxoGraph Scientific).
In all recordings, IPSCs were isolated pharmacologically with an antagonist for AMPARs (50 μm DNQX) and NMDARs (100 μm APV). Glycinergic and GABAergic currents were pharmacologically separated by bath application of the GABAA receptor (GABAAR) antagonist gabazine (10 μm) and glycine receptor antagonist strychnine (1 μm), respectively. All chemicals were from Sigma-Aldrich except for gabazine (SR95531) and (RS)-3,5-dihydroxyphenylglycine [(RS)-3,5-DHPG], which were from Tocris Bioscience, and DNQX, which was from Abcam. 3,5-DHPG was prepared in ACSF at a working concentration of 200 μm, which is at least 3-fold higher than its EC50 (0.7–60 μm depending on animal tissues) (Cartmell and Schoepp, 2000). This was expected to achieve a saturating concentration and thus full activation of group I mGluRs in our experiments.
Synaptic stimulation experiments [evoked IPSC (eIPSC) recording].
Extracellular stimulation was performed using concentric bipolar electrodes with a tip core diameter of 127 μm (World Precision Instruments). The stimulating electrode was placed using a NMN-25 Micromanipulator (Narishige) and was positioned lateral and ventral to the MNTB to activate the inhibitory afferent fibers presumably originating from the ventral NTB (VNTB). The stimulus intensity at which maximal response was elicited was chosen to perform experiments with a paired-pulse paradigm in which a pair of identical stimuli were given at a time interval of 10–20 ms at a frequency of 0.1 Hz. The mGluR I agonist 3,5-DHPG (200 μm) was bath applied to only one recorded cell per brain slice to avoid contamination. Other pharmacological agents were also bath applied. Typically, responses were averaged from a minimum of six eIPSC traces per condition for further data analyses.
Spontaneous IPSC (sIPSC) recording.
sIPSCs are defined as recorded events that occur in the absence of external (electrical) stimulation and therefore may consist of both mIPSCs, which are AP-independent events, and additional events, which may be AP dependent, resulting from intrinsic circuit activity. During sIPSC recording, mGluR I agonist 3,5-DHPG (200 μm) was bath applied for 2–5 min to one recorded cell per slice. mIPSCs were recorded in the presence of tetrodotoxin (TTX, 1 μm), a blocker for voltage-gated Na+ channels. In a subset of experiments, additional antagonists were bath applied in the presence of 3,5-DHPG: mGluR1a antagonist (LY36785, 200 μm); mGluR5 antagonist [2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP), 10 μm]. sIPSCs were detected by a template function using a function for product of exponentials, f(t) = [1 − exp(−t/rise time)] × exp(−t/decay tau), where t is time and tau is the time constant. The values of the parameters for the glycinergic template are as follows: amplitude of −50 pA, rise time of 0.5 ms, decay tau of 1 ms, with a template baseline of 2 ms and a template length of 5 ms. For the GABAergic template, values are as follows: amplitude of −50 pA, rise time of 0.5 ms, decay tau of 4 ms, with a template baseline of 2 ms and a template length of 15 ms. These parameters were determined based on an average of visually detected synaptic events. The detection threshold is threefold the noise SD, which detects most of the events with the least number of false positives. The average of detected events for each cell was obtained using AxoGraph to measure amplitude and decay tau.
Experimental design and statistical analysis.
For statistical analysis of sIPSCs, only cells with differences of at least ± 10% of control (baseline) values are included and are referred to as responding cells. sIPSC frequency, amplitude, and decay tau were typically averaged from 60 s periods from each condition: control, drug, and wash. GABAergic sIPSCs were sampled from a larger time window to compensate for low sIPSC frequency. For eIPSC experiments, the peak amplitude of eIPSCs was measured after each stimulus. Averages were obtained from the first 2 min of the control period, the last minute of the mGluR I agonist application (3,5-DHPG) and the last minute of the wash period, which was a minimum of 5 min after agonist application. eIPSC amplitude was normalized for individual experiments by dividing the peak amplitude of individual eIPSCs by the average control eIPSC amplitude. IPSC data were submitted to a two-way mixed-model ANOVA, with genotype as the between-subjects factor and drug condition (control, 3,5-DHPG, wash) as the within-subjects factor. If sphericity was violated, the Greenhouse–Geisser correction was applied when ε was <0.75 and the Huynh–Feldt correction was applied when ε was >0.75. For analysis of data from WT only, a repeated-measures ANOVA (RM-ANOVA) was used to compare IPSC properties across drug conditions. For significant differences observed in a two-way or RM-ANOVA, a Bonferroni-corrected paired comparison was conducted for individual sample comparisons. Paired t test was also used for one experiment as indicated in the results. Mean ± SEM values are reported, with p < 0.05 being considered statistically significant. Statistical analyses were performed using SPSS (RRID:SCR_002865) and GraphPad Prism (RRID:SCR_002798).
Results
Activation of mGluR I increases frequency and amplitude of glycinergic sIPSCs
We first examined the effects of mGluR I activation on the sIPSCs from principal neurons in the MNTB in the presence of antagonists for ionotropic glutamate receptors (100 μm APV, 50 μm DNQX). Because MNTB neurons receive both GABAergic and glycinergic inputs, glycinergic sIPSCs were pharmacologically isolated with a GABAAR antagonist (gabazine, 10 μm). sIPSCs were recorded under three conditions: control, mGluR I agonist 3,5-DHPG (200 μm), and wash (Fig. 1). For the majority of cells tested, activation of mGluR I with bath application of 3,5-DHPG (200 μm) increased the frequency of glycinergic sIPSCs (WT: 26 of 31, KO: 19 of 22 cells), resulting in the maximal frequencies as high as 14.3 Hz in WT and 28.6 Hz in Fmr1 KO neurons during 3,5-DHPG application. The increased sIPSC frequency was not always reversible and could persist >15 min after 3,5-DHPG application (Fig. 1A) and the modulatory effects seemed stronger in Fmr1 KO neurons (Fig. 1B–E).
Figure 1.

Activation of mGluR I increases frequency and amplitude of glycinergic sIPSCs. A, 3,5-DHPG produced a burst of large glycinergic sIPSCs in a WT MNTB neuron. The instantaneous frequency of sIPSCs (indicated by the colored bar below the original recording) reaches the highest during 3,5-DHPG application and lasts for several minutes after washout of the drug. B, C, Sample sIPSC traces from a WT neuron and Fmr1 KO neuron showing a tendency of more dramatic effects of 3,5-DHPG on the amplitude of sIPSCs in the KO neuron. D, E, 3,5-DHPG application increased the frequency and amplitude of glycinergic sIPSCs in both WT and Fmr1 KO. Averaged sIPSCs are shown in red and the population of sIPSCs in black. Under 3,5-DHPG, the distribution of sIPSC amplitude is distinct between the WT and Fmr1 KO sample cells, with the Fmr1 KO distribution displaying greater bimodality, suggesting multiple vesicular release in the KO neuron. Dashed vertical line indicates maximum sIPSC amplitude under control condition. F, Frequency of glycinergic sIPSCs was significantly increased during 3,5-DHPG compared with control (ctrl) in both WT and KO neurons. However, no significant difference in the mean frequency of sIPSCs between WT (n = 26) and Fmr1 KO (n = 19) was observed. G, Mean glycinergic sIPSC amplitude also significantly increased during 3,5-DHPG compared with control, but did not significantly differ between WT (n = 26) and Fmr1 KO (n = 19). Data were analyzed by 2-way mixed-design ANOVA. For this and subsequent figures, means ± SEM are shown; *p < 0.05, **p < 0.01, and ***p < 0.001.
There was a significant effect of the drug condition on glycinergic sIPSC frequency such that sIPSC frequency was significantly greater under 3,5-DHPG than control (F(1,42) = 39.246, p < 0.001, partial η2 = 0.483; Fig. 1F). Although sIPSC mean frequency appeared to be elevated under 3,5-DHPG in Fmr1 KO neurons (DHPG: 7.12 ± 1.27 Hz, n = 19) compared with WT neurons (DHPG: 4.52 ± 1.10 Hz, n = 26), there was no significant effect of genotype, indicating that sIPSC frequency was similar between WT and Fmr1 KO (F(1,42) = 2.167, p = 0.148, partial η2 = 0.049). No statistically significant interaction was found between the drug condition and mouse genotype on glycinergic sIPSC frequency (F(1,42) = 2.584, p = 0.115, partial η2 = 0.058).
For the majority of cells tested, 3,5-DHPG (200 μm) also increased the amplitude of glycinergic sIPSCs (WT: 22 of 30, KO: 13 of 22 cells). sIPSC amplitude was significantly greater under 3,5-DHPG than control (F(1,33) = 14.030, p < 0.001, partial η2 = 0.298; Fig. 1G). Mean sIPSC amplitude increased from 68 ± 8 pA (ctrl) to 110 ± 25 pA (DHPG) in WT, and from 72 ± 10 pA (control) to 151 ± 32 pA (DHPG) in Fmr1 KO neurons. However, there was no significant effect of genotype, indicating that mean sIPSC amplitude was similar between WT and Fmr1 KO (F(1,33) = 0.783, p = 0.383, partial η2 = 0.023) and no statistically significant interaction was found between the drug condition and mouse genotype on glycinergic sIPSC amplitude (F(1,33) = 1.289, p = 0.264, partial η2 = 0.038). Not only did the mean sIPSC amplitude increase during 3,5-DHPG application, but large glycinergic sIPSCs (>1 nA) were observed in some neurons, strongly indicating multivesicular release (Fig. 1E) and suggesting greater excitability of the presynaptic inhibitory terminals in the Fmr1 KO. Overall, the increase of glycinergic sIPSC frequency and amplitude under mGluR I activation suggests that the spontaneous glycinergic transmission in the MNTB is facilitated by mGluR I.
mGluR I modulation of glycinergic sIPSCs persists throughout development
Modulation of synaptic excitation by group II and III mGluRs in the MNTB has been shown to be prominent during development around hearing onset followed by a functional downregulation in mature animals (Takahashi et al., 1996; Leão and Von Gersdorff, 2002; Renden et al., 2005). However, there is evidence that mGluR I may not be as developmentally restricted (Kushmerick et al., 2004). Therefore, to determine whether the modulatory effect of mGluR I is developmentally dependent, the change in frequency and amplitude of glycinergic sIPSCs during 3,5-DHPG application was compared between three age groups: P14–P17 (WT: n = 8, KO: n = 9), P18–P21 (WT: n = 12, KO: n = 9), and P22+ (WT: n = 10, KO: n = 4). These age groups reflect general periods of developmental change after hearing onset. In rat, MNTB neurons receive GABAergic input by P5–P7 and the glycinergic component emerges by P8–P12 (Awatramani et al., 2005). Expression of the mature form of the glycine receptor containing the α1 subunit peaks around P21 in the MNTB (Friauf et al., 1997; Piechotta et al., 2001), which coincides with the development of large-magnitude glycinergic inputs capable of suppressing excitation (Awatramani et al., 2004). Therefore, sIPSCs were compared from mice in the first half (P14–P17) and second half (P18–P21) of the third postnatal week, during which time the glycinergic component undergoes rapid development, as well as from mice in the fourth postnatal week and beyond (P22+), which represents the period in which the glycinergic component stabilizes and can functionally counteract excitatory inputs.
During 3,5-DHPG application, frequency of glycinergic sIPSCs significantly increased in all three age groups in both WT and Fmr1 KO compared with control (F(1,40) = 32.118, p < 0.001, partial η2 = 0.445) without significant differences between age groups (P14–P17, P18–P21, and P22+) (F(2,40) = 0.571, p = 0.570, partial η2 = 0.028) and no interaction between age group and genotype was detected, indicating that sIPSC frequency was similar across age groups in both WT and Fmr1 KO neurons (WT: P14–P17: 6.19 ± 1.40 Hz, n = 9; P18–P21: 3.05 ± 1.04 Hz, n = 9; P22+: 3.87 ± 1.05 Hz, n = 8; KO: P14–P17: 7.46 ± 3.30 Hz, n = 8; P18–P21: 6.45 ± 2.14 Hz, n = 9; P22+: 6.85 ± 2.31 Hz, n = 3; Fig. 2A). This suggests that mGluR I modulation of glycinergic sIPSC frequency persists throughout development and may persist in the mature auditory system. Indeed, increased glycinergic frequency during mGluR I activation was observed in WT mice as old as P125 (data not shown). Elevation of the mGluR I modulation on glycinergic sIPSC frequency in the Fmr1 KO is consistent with the “mGluR theory” of FXS, in which loss of FMRP results in exaggerated functions of mGluRs (Bear et al., 2004).
Figure 2.
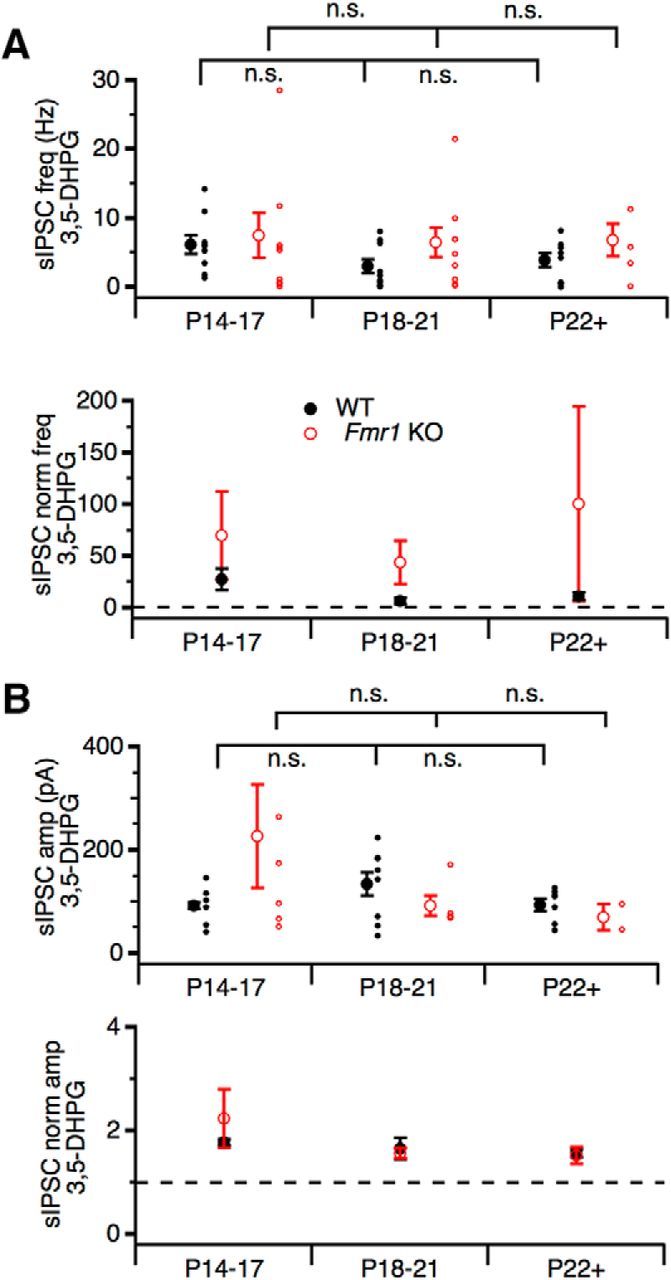
mGluR I modulation of glycinergic sIPSCs persists throughout development after hearing onset. A, Population averages of mean glycinergic sIPSC frequency (top) and normalized sIPSC frequency (bottom) during 3,5-DHPG application do not significantly differ between genotypes or age groups, but tend to be elevated in Fmr1 KO compared with WT in all age groups (P14–17, WT: n = 8, KO: n = 9; P18–21, WT: n = 12, KO: n = 9; and P22+, WT: n = 10, KO: n = 4). B, Population averages of mean glycinergic sIPSC amplitude (top) and normalized sIPSC amplitude (bottom) during 3,5-DHPG application do not significantly differ between genotypes or age groups. n.s., Not significant.
Mean sIPSC amplitude during 3,5-DHPG application also did not significantly differ between genotypes (F(1,28) = 0.225, p = 0.639, partial η2 = 0.008) or age groups (F(2,28) = 0.955, p = 0.397, partial η2 = 0.064) (Fig. 2B). This suggests that mean glycinergic sIPSC amplitudes were similar between genotypes and age groups during mGluR I activation and no strong developmental effect was observed. In Fmr1 KO, mean glycinergic sIPSC amplitude during 3,5-DHPG was greater than WT at P14–P17 with large variations (Fmr1 KO: 227 ± 101 pA, n = 6; WT: 92 ± 6 pA, n = 6; Fig. 2B), but, in general, mean sIPSC amplitude remained stable throughout the third postnatal week and beyond. This may suggest different mechanisms underlying the modulation of glycinergic sIPSC frequency and amplitude by mGluR I.
3,5-DHPG modulation of glycinergic sIPSCs is dependent on voltage-gated sodium channels and mGluR1 and mGluR5
To ensure that the sIPSC events observed under 3,5-DHPG application were glycinergic, strychnine (1 μm), a glycine receptor antagonist, was bath applied after verification of responding cells in WT. Strychnine abolished all events during 3,5-DHPG application, confirming that the synaptic events that 3,5-DHPG increased are glycinergic sIPSCs (control: 1.09 ± 0.89 Hz; DHPG: 8.05 ± 2.51 Hz; DHPG + strychnine: 0 ± 0 Hz; for normalized frequency, DHPG: 39.60 ± 11.80, F(2,10) = 10.99, p = 0.003, n = 6; Fig. 3A–C). The sIPSC amplitude exhibited consistent alterations (control: 34 ± 6 pA; DHPG: 67 ± 13 pA; DHPG + strychnine: 0 ± 0 pA; for normalized amplitude, DHPG: 2.56 ± 1.98, F(2,10) = 7.608, p = 0.0098, n = 6). To determine whether 3,5-DHPG affects glycinergic miniature release, a NaV channel antagonist (1 μm), was bath applied for 5 min after the increased frequency was established. In the presence of TTX, the frequency and amplitude of glycinergic sIPSCs returned to baseline levels (control: 0.72 ± 0.30 Hz; DHPG: 4.15 ± 1.27 Hz; DHPG + TTX: 0.59 ± 0.26 Hz; for normalized frequency, DHPG: 12.14 ± 5.69; DHPG + TTX: 1.01 ± 0.15, F(2,14) = 4.176, p = 0.0378, n = 8; Fig. 3D–G). The amplitude showed similar alterations (control: 73 ± 10 pA; DHPG: 123 ± 45 pA; DHPG + TTX: 50 ± 5 pA; for normalized amplitude, DHPG: 1.53 ± 0.31, DHPG + TTX: 0.71 ± 0.06, F(2,14) = 4.94, p = 0.0238, n = 8). These results support that the 3,5-DHPG modulation is NaV channel dependent and that glycinergic miniature release is not significantly affected by 3,5-DHPG.
Figure 3.
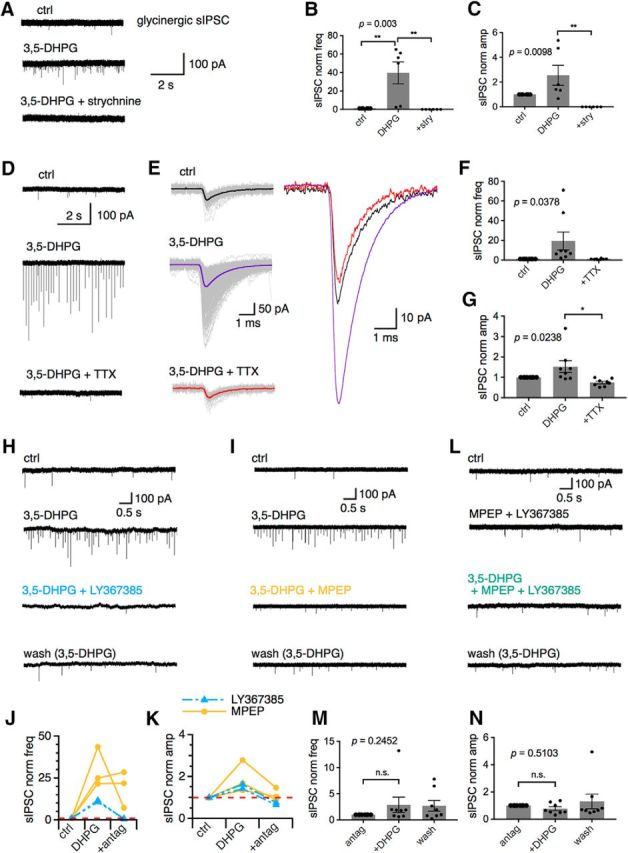
MGluR I modulation of glycinergic sIPSCs is NaV channel dependent and both mGluR1 and mGluR5 are involved in modulation of glycinergic sIPSCs, with large variations in individual cells. A–C, Bath application of a glycine receptor agonist (strychnine, 1 μm) abolished all the spontaneous events observed during 3,5-DHPG (n = 6). D, Application of TTX (1 μm) abolished the large sIPSCs observed under 3,5-DHPG. The remaining inward currents are mIPSCs. E, Individual (gray) and averaged (colored; superimposed at a larger scale on the right) sIPSC traces from three conditions in D. sIPSCs under control (ctrl) and TTX (3,5-DHPG + TTX) are similar in amplitude and frequency. F, G, TTX significantly reduced the effects of 3,5-DHPG on sIPSCs (n = 8). H–K, Respective antagonist for mGluR1a (LY367385, 200 μm) and mGluR5 (MPEP, 10 μm) alone largely but not completely reversed the increased frequency and amplitude induced by 3,5-DHPG. L–N, Preincubation of brain slices with both the mGluR1a and mGluR5 antagonists prevented 3,5-DHPG modulation (n = 8). stry, Strychnine; antag, antagonist.
Group I mGluRs include two members, mGluR1 and mGluR5, and 3,5-DHPG activates both. To determine whether mGluR1 and mGluR5 have distinct modulatory roles for the increased glycinergic sIPSCs, an mGluR1a antagonist LY367385 (200 μm) and mGluR5 antagonist MPEP (10 μm) were separately applied during the application of 3,5-DHPG. Antagonists, but not agonists, for specific mGluRs were used here because an mGluR1a agonist is not commercially available and the mGluR5 agonist 2-chloro-5-hydroxyphenylglycine (CHPG) has relatively weak potency and rapid desensitization (Homayoun and Moghaddam, 2010). Both LY367385 (Fig. 3H) and MPEP (Fig. 3I) reduced the frequency of glycinergic sIPSCs during 3,5-DHPG application, with the mGluR1a antagonist being more effective compared with the mGluR5 antagonist (Fig. 3J). The increased amplitude of glycinergic sIPSCs during 3,5-DHPG was also reduced back to control levels by the mGluR1a antagonist and the mGluR5 antagonist (Fig. 3K). Given that mGluRs can activate a variety of downstream signaling pathways, it is possible that antagonism of the mGluR I after receptor activation may not effectively abolish the observed modulation. Therefore, slices were preincubated with a combination of LY367385 (200 μm) and MPEP (10 μm) for at least 15 min before 3,5-DHPG application (Fig. 3L). Preincubation with both antagonists did not seem to produce an additive effect. It prevented the increased frequency of glycinergic sIPSCs during 3,5-DHPG application (mGluR I antagonists: 0.51 ± 0.26 Hz; DHPG + mGluR I antagonists: 2.19 ± 1.81 Hz; wash with mGluR I antagonists: 1.37 ± 1.05 Hz; for normalized frequency, DHPG + mGluR I antagonists: 2.87 ± 1.49; wash with mGluR I antagonists: 2.72 ± 1.01, F(2,14) = 1.557, p = 0.2452, n = 8; Fig. 3M) and prevented the increase in glycinergic sIPSC amplitude (mGluR I antagonists: 63 ± 15 pA; DHPG + mGluR I antagonists: 50 ± 18 pA; wash with mGluR I antagonists: 63 ± 21 pA; for normalized amplitude, DHPG + mGluR I antagonists: 0.76 ± 0.14, wash with mGluR I antagonists: 1.30 ± 0.54, F(2,14) = 0.7062, p = 0.5103, n = 8; Fig. 3N). These results suggest that the modulation by 3,5-DHPG may occur through activation of both the mGluR1 and mGluR5 pathways, although each group I member may have distinct roles. Due to the high spontaneous spiking activity in MNTB neurons (Blosa et al., 2015), it is conceivable that mGluRs are activated by ambient glutamate and exert a tonic modulation of neurotransmission. Our preliminary in vitro results did not readily detect such endogenous activity of mGluR I on inhibitory responses (data not shown). Even after blocking glutamate reuptake to enhance glutamate accumulation and activation of mGluRs, Renden et al. (2005) observed no evidence of mGluR I activation at the calyx. This is possibly because mGluR I have a lower affinity for glutamate (compared with the other mGluRs) and may be activated only under heightened input conditions (Cartmell and Schoepp, 2000).
Lack of 3,5-DHPG effect on glycinergic eIPSCs
Given the robust modulation of glycinergic sIPSCs by 3,5-DHPG that was NaV channel dependent, we hypothesized that glycinergic eIPSCs would also be subject to mGluR I modulation. To evoke eIPSCs, a concentric bipolar electrode was positioned lateral and ventral to the MNTB to activate the inhibitory afferent fibers originating from the VNTB. The glycinergic component was pharmacologically isolated by bath application of the GABAAR antagonist gabazine (10 μm). Although application of 3,5-DHPG resulted in modulation of glycinergic sIPSCs, surprisingly, there was no significant effect of drug condition on glycinergic eIPSC amplitude in the sample WT and KO neurons (Fig. 4A,B). For WT neurons, mean glycinergic eIPSC amplitude ranged from 72 to 3703 pA (n = 10) and bifurcated into responses >2 nA (3.11 ± 0.44 nA, n = 3) and smaller responses <1 nA (262 ± 42 pA, n = 7). Fmr1 KO eIPSC amplitudes did not show this bifurcation and averaged 817 ± 142 pA (n = 5), suggesting differences in the level of recruitment of glycinergic afferent fibers between cells. To control for these differences in baseline eIPSC amplitude, eIPSC amplitude was normalized to the control condition. In WT, no modulation of eIPSC normalized amplitude between control and 3,5-DHPG was observed (n = 10) (Fig. 4C). In Fmr1 KO neurons, normalizing the amplitude of glycinergic eIPSCs to the control condition shows a different pattern of responses from WT, with a small transient initial depression of eIPSC amplitude and an enhancement of eIPSC amplitude during the wash period (Fig. 4D). However, there was no significant effect of drug condition (F(2,26) = 1.335, p = 0.281, partial η2 = 0.093) or genotype (F(1,13) = 0.116, p = 0.739, partial η2 = 0.009) on normalized glycinergic eIPSC amplitude (Fig. 4F). Consistently, 3,5-DHPG did not change the paired pulse ratio (PPR) (F(2,26) = 0.954, p = 0.374, partial η2 = 0.068; Fig. 4G) or decay tau (F(1,13) = 0.49, p = 0.348, partial η2 = 0.068; Fig. 4H) in WT or Fmr1 KO neurons. The fact that the effect of 3,5-DHPG on glycinergic sIPSCs was blocked by TTX but no effect of 3,5-DHPG on eIPSCs was observed suggests that 3,5-DHPG depolarized the glycinergic terminals, activating TTX-sensitive Nav currents associated with the spontaneous but not the evoked glycine neurotransmission. In summary, activation of mGluR I differentially modulate spontaneous versus evoked glycinergic transmission in MNTB neurons.
Figure 4.

Lack of 3,5-DHPG modulation of glycinergic eIPSCs. A, B, Amplitude of individual (top) and averaged glycinergic eIPSCs (bottom) remained relatively constant during 3,5-DHPG application compared with control in both WT and Fmr1 KO neurons. C, Normalized glycinergic eIPSC amplitude is not affected by 3,5-DHPG in WT neurons (n = 10). D, In Fmr1 KO neurons, normalized glycinergic eIPSC amplitude is not affected by 3,5-DHPG, although eIPSC amplitude was briefly suppressed within the first minute of 3,5-DHPG application and showed some facilitation during wash (n = 5). No significant differences between drug conditions or genotypes were observed for mean glycinergic eIPSC amplitude (E), normalized amplitude (F), PPR (G), or decay time constant (tau) (H) (WT: n = 10; Fmr1 KO: n = 5).
Different effects of 3,5-DHPG on GABAergic sIPSCs and eIPSCs
Because MNTB also receives GABAergic inhibition, we further examined the effects of 3,5-DHPG on spontaneous and evoked GABAergic transmission. GABAergic IPSCs were pharmacologically isolated (100 μm APV, 50 μm DNQX, 1 μm strychnine). Interestingly, GABAergic sIPSCs were not affected by 3,5-DHPG in their frequency in either WT neurons (control: 1.19 ± 0.61 Hz; DHPG: 0.39 ± 0.21 Hz; wash: 0.14 ± 0.05 Hz, F(2,22) = 1.82, p = 0.1856, n = 9; Fig. 5) or KO neurons (control: 1.23 ± 0.94 Hz; DHPG: 1.47 ± 1.28 Hz; wash: 1.26 ± 0.97 Hz, F(2,14) = 0.0146, p = 0.9856, n = 6). For unknown reasons, a rundown of the frequency of the GABAergic sIPSCs was observed for WT neurons. Mean GABAergic sIPSC amplitude was not changed by 3,5-DHPG in either WT neurons (control: 40 ± 9 pA; DHPG: 42 ± 11 pA; wash: 35 ± 6 pA, F(2,22) = 0.1197, p = 0.8878, n = 9) or KO neurons (control: 49 ± 6 pA; DHPG: 48 ± 5 pA; wash: 44 ± 5 pA, F(2,14) = 0.3028, p = 0.7435, n = 6). We did not examine developmental changes in the modulation of GABAergic sIPSCs because they are much less frequent and weaker than the glycinergic sIPSCs, especially in older animals, rendering analyses of modulatory effects unreliable.
Figure 5.

3,5-DHPG does not change GABAergic sIPSCs. A, B, Sample traces of GABAergic sIPSCs from a WT (A) and Fmr1 KO (B) neuron. The detected events (gray) and the averaged sIPSCs (thicker traces) from each drug condition are shown on the right. C, Frequency of GABAergic sIPSCs was not changed during 3,5-DHPG compared with control (ctrl) in both WT (n = 9) and KO (n = 6) neurons. A rundown of sIPSC frequency in WT was observed. D, Mean GABAergic sIPSC amplitude was also not changed by 3,5-DHPG.
We next investigated the effect of mGluR I activation on GABAergic eIPSCs. The stimulation paradigm was the same as in Figure 4 except that the GABAergic component was pharmacologically isolated by bath application of strychnine (1 μm). Application of 3,5-DHPG (200 μm) resulted in a significant interaction between drug condition (control, 3,5-DHPG, wash) and mouse genotype (F(2,22) = 3.825, p = 0.038, partial η2 = 0.258; Fig. 6A–C). This indicates that the effect of mGluR I activation differed in WT and Fmr1 KO neurons. The mean amplitude of GABAergic eIPSCs in WT was significantly different between drug conditions (control: 152 ± 38 pA; DHPG: 88 ± 35 pA; wash: 98 ± 20 pA; p = 0.006, n = 5), but mean amplitude did not significantly differ in Fmr1 KO neurons (control: 148 ± 22 pA; DHPG: 149 ± 30 pA; wash: 143 ± 28 pA; p = 0.923 n = 8), meaning that, on average 3,5-DHPG depresses GABAergic eIPSC amplitude in WT, but not Fmr1 MNTB neurons.
Figure 6.

Activation of mGluR I suppressed GABAergic eIPSCs in WT and produced variable effects in Fmr1 KO neurons. A, Application of 3,5-DHPG (200 μm) suppressed the GABAergic eIPSC in a WT neuron. B, In a Fmr1 KO neuron, the GABAergic eIPSC amplitude was not affected by 3,5-DHPG. Averaged eIPSCs from the paired pulse stimulus (bottom) show eIPSCs under control (black) and 3,5-DHPG (purple) conditions. C, Mean GABAergic eIPSC amplitude was significantly reduced by 3,5-DHPG in WT (n = 6) but not Fmr1 KO neurons (n = 8). D, Normalized GABAergic eIPSC amplitude for individual neurons show a consistent suppression of eIPSC amplitude in WT (n = 6), but in Fmr1 KO, the effect of 3,5-DHPG is highly variable (n = 8). Decay tau (E) and PPR (F) did not significantly differ between drug conditions or genotypes. *p < 0.05.
Analysis of the normalized GABAergic eIPSC amplitude shows a consistent depression of eIPSC amplitude during 3,5-DHPG application in all WT neurons (Fig. 6D). In Fmr1 KO neurons, there was a heterogeneous effect of mGluR I activation, with 50% (4 of 8) of neurons showing depression and 50% (4/8) showing enhancement of normalized GABAergic eIPSC amplitude. The normalized data support that mGluR I modulate GABAergic eIPSCs in both WT and Fmr1 KO, but that the modulatory effect is altered in Fmr1 KO, which could possibly result in dysfunctional GABAergic transmission. Furthermore, no significant difference in decay tau was observed between drug conditions (control, 3,5-DHPG) (F(1,7) = 0.116, p = 0.744, partial η2 = 0.016) or genotype (F(1,7) = 0.599, p = 0.464, partial η2 = 0.079) (Fig. 6E). Additionally, no significant difference in PPR was observed between drug conditions (control, 3,5-DHPG, wash) (F(1,11) = 0.029, p = 0.927, partial η2 = 0.003) or genotype (F(1,11) = 1.367, p = 0.261, partial η2 = 0.111) (Fig. 6F). However, PPR was slightly elevated under 3,5-DHPG in WT compared with control PPR (control: 1.34 ± 0.17; DHPG: 1.52 ± 0.20, n = 6).
Given that 3,5-DHPG is known to result in the release of endocannabinoids and inhibit the release of glutamate from the calyx of Held (Kushmerick et al., 2004), we hypothesized that our observed suppression of GABAergic eIPSCs by 3,5-DHPG could also be a result of activation of endocannabinoid receptor type 1 (CB1R) on the inhibitory terminals. Therefore, we used bath application of the CB1R antagonist AM251 (5 μm) to determine whether this pathway is involved in the mGluR I suppression of GABAergic eIPSCs in WT neurons. Consistent with the data in Figure 6, 3–5-DHPG significantly suppressed the GABAergic eIPSCs. Application of AM251 at least partially blocked the depression induced by 3,5-DHPG, resulting in nonsignificant changes in eIPSC amplitude during and 5 min after wash of 3,5-DHPG (control: 312 ± 78 pA; DHPG: 220 ± 74 pA; AM251+DHPG: 250 ± 79 pA; DHPG: 243 ± 78 pA; F(3,15) = 3.761, p = 0.034, n = 6; Fig. 7A–C). Additionally, preincubation with AM251 (5 μm) for 5 min was sufficient to prevent 3,5-DHPG depression of the eIPSC amplitude (AM251: 204 ± 54 pA; AM251+DHPG: 200 ± 56 pA; DHPG: 195 ± 81 pA; F(2,29) = 0.0046, p = 9954, n = 12; Fig. 7D,E). These results suggest that the modulation of evoked GABAergic transmission by mGluR I is mediated via the endocannabinoid pathway. To test whether there is a tonic CB1R activity on GABAergic transmission, we used paired t test to compare the eIPSC amplitude between control and AM251 application, and found no significant changes (control: 226 ± 53 pA, AM251: 239 ± 60 pA, t(9) = 0.5788, p = 0.5769, n = 10; Fig. 7F), suggesting an absence of tonic endocannabinoid signaling at the MNTB that suppresses GABAergic transmission.
Figure 7.
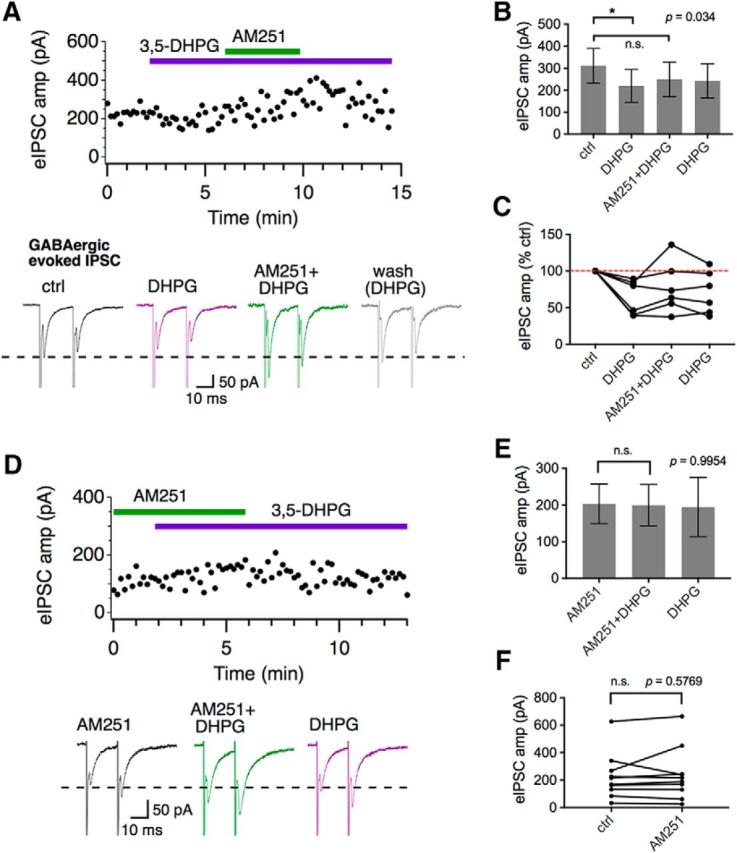
Modulation of GABAergic eIPSCs in WT by 3,5-DHPG is CB1R dependent. A, Application of a CB1R antagonist AM251 (5 μm) blocked the 3,5-DHPG-induced suppression of GABAergic eIPSCs. Averaged eIPSC traces (bottom) show that AM251 recovered eIPSC amplitude above control levels in this neuron. B, C, 3,5-DHPG significantly suppressed eIPSCs (n = 6). AM251 partially blocked this effect, resulting in slightly increased eIPSCs that lasted for the time period of the experiment (5 min wash with 3,5-DHPG). D, E, Preincubation with AM251 prevented 3,5-DHPG-induced suppression of the GABAergic eIPSCs. Application of 3,5-DHPG after AM251 had no effects on GABAergic eIPSCs (n = 12). F, AM251 alone did not significantly change the amplitude of GABAergic eIPSCs (paired t test, n = 10).
Discussion
Our results revealed a novel phenomenon that mGluR I exerted differential modulation of spontaneous versus evoked inhibitory transmission at MNTB. Further, mGluR modulation of the glycinergic inhibition is complementary in release modes to that of the GABAergic inhibition. Therefore, the modulation of synaptic inhibition by mGluR I in MNTB is both transmitter and release mode dependent (Fig. 8A). Finally, the modulation exhibited subtle differences between WT and FXS animals.
Figure 8.
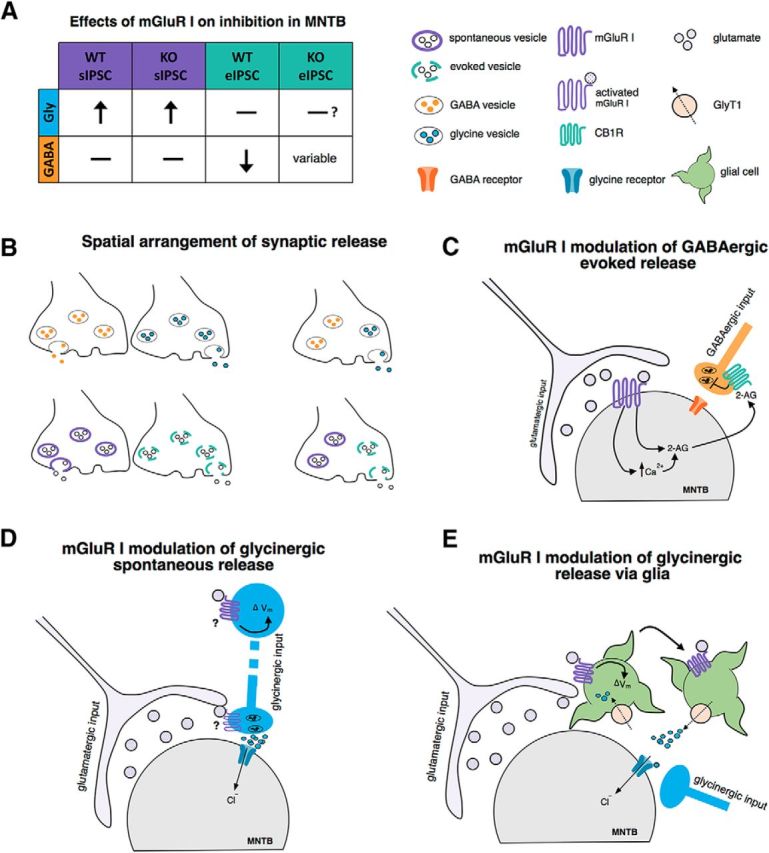
Hypothetical mechanisms underlying differential mGluR I modulation of synaptic inhibition in MNTB neurons. A, Summary of the effects of mGluR I activation on synaptic inhibition in the MNTB. Activation of mGluR I significantly increased the frequency and amplitude of glycinergic sIPSCs in both WT and Fmr1 KO neurons without affecting GABAergic sIPSCs in either genotype. For evoked inhibition, mGluR I did not modulate glycinergic transmission. Evoked GABAergic transmission was significantly suppressed in WT neurons by mGluR I, but the effect in Fmr1 KO neurons was highly variable, suggesting dysfunction of the mGluR and GABA signaling pathways in KO neurons. B, Spatial arrangement of inhibitory terminals for different neurotransmitters and release modes. The different synaptic inputs could be spatially segregated (top left) or assume cotransmission within a single synapse (top right). Similarly, for the same neurotransmitter, the vesicle pools for spontaneous versus evoked release could be segregated (left bottom) or exist in the same terminal (right bottom). The differential modulation of glycine and GABA release could arise from differential mGluR expression and differential targeting of specific release pools by mGluRs. C, Proposed mechanism of mGluR I-induced suppression of evoked GABAergic transmission. Our results show that the suppression of GABAergic eIPSCs was reversed by blocking CB1R of the endocannabinoid signaling pathway. Given that activation of postsynaptic mGluR I is known to suppress the glutamatergic transmission at MNTB through retrograde endocannabinoid signaling, it is possible that the regulation of glutamate and GABA release by mGluR I shares a common pathway through the production and release of an endocannabinoid (2-AG, 2-arachidonoyglycerol). D, mGluR I activation may drive the spontaneous release of glycine by depolarizing the membrane potential (Vm) at the soma or synaptic terminal of the presynaptic inhibitory neuron, resulting in increased neurotransmitter release. Based on our results, this release is NaV channel dependent, but not necessarily AP dependent. E, Alternatively, the modulation of spontaneous glycinergic activity may be a result of segregation of cell type and occur at the level of glia. Many glial cells express the Na+- and Cl−-dependent GlyT1, which are often positioned near glycinergic terminals and function to clear glycine via reuptake of the transmitter. Because the reversal potential (Erev) of GlyT1 is near the resting membrane potential, depolarization of the glial cell can result in reversed function of GlyT1, which allows for glycine to be released back into the extrasynaptic space, producing glycinergic sIPSCs.
Differential modulation of spontaneous and evoked neurotransmission
Multiple mechanisms could underlie the separation and differential modulation of spontaneous and evoked release. Different synapses of the same neurotransmitter are biased toward different release modes and can therefore be physically segregated onto the same postsynaptic cell (Sara et al., 2005; Atasoy et al., 2008). MNTB receives inhibition from multiple sources, rendering physical segregation of synapses with different release modes and diverse vesicle pools possible. Differences in calcium dependence (Xu et al., 2009; Tsintsadze et al., 2017), SNARE protein components (Raingo et al., 2012; Ramirez et al., 2012), SNARE machinery (Schoch et al., 2001; Bronk et al., 2007), and biochemical properties of synaptic vesicles (Chung et al., 2010; Li et al., 2017) can all explain the separation of spontaneous and evoked transmitter release without mutual exclusion.
The enhancement of spontaneous glycine release by mGluR I is likely caused by presynaptic membrane depolarization. Regulation of Nav conductance is a potential mechanism because the effect was blocked by TTX. A TTX-sensitive persistent Nav current (Leão et al., 2005), if present on the inhibitory terminals innervating MNTB, could be responsible for the membrane depolarization leading to enhanced spontaneous glycine release. Indeed, activation of mGluR I has been linked to the enhancement of such a Nav current by shifting the activation voltage to more negative levels (Dong and Ennis, 2014). Further, activation of Nav channels can cause increased transmitter release even without spike generation (Ermolyuk et al., 2013). Additionally, the increased spontaneous release may also occur through mGluR I-induced increases in intracellular Ca2+ concentrations (Mannaioni et al., 2001). The protein kinase C (PKC) pathway may also be involved because PKC activators enhance glutamate release (Hori et al., 1999) and mGluR I activates PKC (Niswender and Conn, 2010). These processes may intimately interact, with the activation of Nav channels being a necessary step leading to enhanced glycine release. The observation that the glycinergic eIPSCs were not affected suggests that mGluR I activate the Nav current associated with the spontaneous, but not the evoked glycine neurotransmission.
The differential effects of mGluR I on glycinergic sIPSCs and eIPSCs suggest that mGluR I can selectively modulate the spontaneous but not the evoked vesicle pools of glycine. The Nav conductance increased by mGluR I may be selectively linked to the release pool and machinery for sIPSCs, similar to the action of adrenoreceptors on synaptic inhibition in sensorimotor cortex (Bennett et al., 1998). Alternatively, the evoked response may capture a different population of glycinergic inputs than the inputs for the spontaneous release. VNTB neurons are heterogeneous and it is unknown whether mGluR I expression varies between different cell types. Differential expression of mGluR I on VNTB neurons could give rise to the distinct effect on glycinergic sIPSCs versus eIPSCs if different VNTB cell types are recruited for the evoked response. Additionally, because sIPSCs arise from presumably all the functional inhibitory synapses impinging on MNTB neurons whereas eIPSCs are evoked from only one of these sources in our experiments, there exists the possibility that the lack of mGluR I modulation of eIPSCs could be because the modulation is input specific.
Differential modulation of glycinergic and GABAergic inputs
The cellular events required for mGluR modulation of inhibitory transmission to occur include spillover of glutamate and activation of mGluRs and signaling pathways leading to modulation of the release machinery (Schoepp, 2001). In MNTB, the presence and strength of mGluR modulation on each inhibitory neurotransmitter system are expected to be dependent on the spatial arrangement of these synapses and whether cotransmission or co-release occurs. The differential modulation suggests distinct mechanisms that allow for the targeted modulation either at the level of a single synapse (Melom et al., 2013; Peled et al., 2014) or between different synaptic terminals (Fig. 8B). In WT neurons, activation of mGluR I resulted in a consistent depression of GABAergic eIPSCs without affecting glycinergic eIPSCs via the endocannabinoid signaling pathway (Fig. 8C). The release of endocannobinoids and activation of CB1Rs presynaptically is a common mechanism regulating synaptic transmission in the brain. In the auditory brainstem, the synaptic inputs subject to such regulation vary depending on the neural circuits. Activation of CB1Rs suppresses glutamatergic transmission in MNTB (Kushmerick et al., 2004) and chicken cochlear nucleus (Stincic and Hyson, 2011). In the mammalian cochlear nucleus, CB1R activation suppresses glutamatergic transmission without affecting inhibitory transmission mediated by glycine and GABA (Sedlacek et al., 2011), whereas in the superior olivary complex, both glutamatergic and glycinergic inputs are suppressed (Trattner et al., 2013). Our results showing suppression of evoked GABA but not glycine release add another layer of complexity for CB1R modulation, pointing to the necessity for in-depth investigation of the anatomy and function of these receptors in various neural circuits. Via a different Nav-dependent mechanism, mGluR I modulates glycinergic but not GABAergic sIPSCs (Fig. 8D). This suggests that the synapses that spontaneously release GABA or co-release glycine and GABA are not affected. Alternatively, vesicles containing glycine may be distinctly affected by mGluR I, bypassing the GABA release pathway even at the same inhibitory input.
The mixed sIPSCs in our recordings did not show strong evidence of co-release because the decay of most mixed sIPSCs could be well fit with a single exponential function (n = 11, data not shown). However, even if these two inhibitory neurotransmitters are co-released, differential reuptake of glycine and GABA via NaV-dependent mechanisms could still underlie differential modulation of sIPSCs mediated by glycine versus GABA. This could occur if the glycine reuptake transporter 2 (GlyT2), but not the GABA transporter (GAT), is affected by mGluR I. GlyT2 is a high-affinity, Na+-coupled glycine uptake transporter expressed in glycinergic neuronal terminals (Aubrey et al., 2007) that can regulate GABA and glycine vesicular content (Rousseau et al., 2008). The mGluR I-mediated increase in Na+ influx may drive a large reuptake of glycine, but not GABA. If more glycine is available in the terminal than GABA, then the vesicles will be predominantly filled with glycine. It is then feasible that mGluR I could increase presynaptic release in a way that is biased toward glycine in a terminal where co-release exists. Finally, glial cells may be involved (Fig. 8E) because mGluR I interacts with glycine transporter 1 (GlyT1) in glia (Jursky and Nelson, 1996). Depolarization of astrocytes by mGluR I can reverse GlyT1 operation, promoting the efflux of glycine into the synaptic cleft, where it can activate glycine receptors on neuronal membranes (Roux et al., 2001; Aubrey et al., 2005).
Functional implications
Synaptic excitation has been shown to be modulated in MNTB by mGluR I, with suppression of synaptic evoked excitation (Kushmerick et al., 2004) and enhancement of spontaneous excitation (our observation). A matched mGluR modulation of synaptic inhibition in modulatory strength and direction would be critical for maintaining excitation/inhibition (E/I) balance. Here, we show that glycinergic sIPSCs are enhanced by mGluR I activation, but that glycinergic eIPSCs are not suppressed. However, the smaller GABAergic evoked component is suppressed by mGluR I activation. This suggests a complex interaction between excitation and inhibition in MNTB. The net effect of mGluRs on synaptic output of MNTB is ultimately dependent on the combined neuromodulatory actions by synaptically released glutamate on glutamatergic, glycinergic, and GABAergic inputs to MNTB. Therefore, consistent with the modern view that MNTB does not serve just as a fast “relay” station in the ascending auditory pathway (Kopp-Scheinpflug et al., 2003), synaptic integration in MNTB might be substantially dynamic because of the modulatory actions by mGluRs.
In the FXS mouse model, hyperexcitability of neurons is a general observation resulting from loss of E/I balance. A coherent modulation of synaptic excitation and inhibition is proposed to be one of the compensatory mechanisms initiated in response to the loss of FMRP regulation of synaptic proteins in an attempt to return the system back to normal levels (Contractor et al., 2015). Compensation at the circuit level by mGluR modulation of neurotransmission may reflect this general principle of maintenance of E/I balance. Therefore, the enhanced glycinergic sIPSCs (Fig. 1) and the heterogeneous modulation of GABAergic eIPSCs in the Fmr1 KO neurons by mGluRs (Fig. 6) suggest disruption of the function of the inhibitory systems and an attempt of the system to counteract the altered synaptic excitation. This raised the possibility that, to alleviate the deficits in FXS, the direction of manipulation of mGluR activity may need to vary depending on the brain areas. At the same synaptic input, persistent differential modulation of the evoked versus spontaneous release of inhibitory transmitters would keep synaptic strength in check and maintain a homeostatic condition for synaptic integration under the disordered state.
Footnotes
This work was supported by the National Institute on Deafness and other Communication Disorders–National Institutes of Health (Grant R01DC016054 to Y.L.) and by Institutional Bridge Funding (Y.L.). We thank Dr. Yang Xu and Dr. Yanqiao Zhang for assistance in genotyping animals and Dr. Chun-Jen Hsiao for comments.
The authors declare no competing financial interests.
References
- Albrecht O, Dondzillo A, Mayer F, Thompson JA, Klug A (2014) A inhibitory projections from the ventral nucleus of the trapezoid body to the medial nucleus of the trapezoid body in the mouse. Front Neural Circuits 8:83. 10.3389/fncir.2014.00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, Ertunc M, Moulder KL, Blackwell J, Chung C, Su J, Kavalali ET (2008) Spontaneous and evoked glutamate release activates two populations of NMDA receptors with limited overlap. J Neurosci 28:10151–10166. 10.1523/JNEUROSCI.2432-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubrey KR, Vandenberg RJ, Clements JD (2005) Dynamics of forward and reverse transport by the glial glycine transporter, GlyT1b. Biophys J 89:1657–1668. 10.1529/biophysj.105.061572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubrey KR, Rossi FM, Ruivo R, Alboni S, Bellenchi GC, Le Goff A, Gasnier B, Supplisson S (2007) The transporters GlyT2 and VIAAT cooperate to determine the vesicular glycinergic phenotype. J Neurosci 27:6273–6281. 10.1523/JNEUROSCI.1024-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awatramani GB, Turecek R, Trussell LO (2004) Inhibitory control at a synaptic relay. J Neurosci 24:2643–2647. 10.1523/JNEUROSCI.5144-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awatramani GB, Turecek R, Trussell LO (2005) Staggered development of GABAergic and glycinergic transmission in the MNTB. J Neurophysiol 93:819–828. 10.1152/jn.00798.2004 [DOI] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST (2004) The mGluR theory of fragile X mental retardation. Trends Neurosci 27:370–377. 10.1016/j.tins.2004.04.009 [DOI] [PubMed] [Google Scholar]
- Bennett BD, Huguenard JR, Prince DA (1998) Adrenergic modulation of GABAA receptor-mediated inhibition in rat sensorimotor cortex. J Neurophysiol 79:937–946. 10.1152/jn.1998.79.2.937 [DOI] [PubMed] [Google Scholar]
- Blosa M, Sonntag M, Jäger C, Weigel S, Seeger J, Frischknecht R, Seidenbecher CI, Matthews RT, Arendt T, Rübsamen R, Morawski M (2015) The extracellular matrix molecule brevican is an integral component of the machinery mediating fast synaptic transmission at the calyx of held. J Physiol 593:4341–4360. 10.1113/JP270849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronk P, Deák F, Wilson MC, Liu X, Südhof TC, Kavalali ET (2007) Differential effects of SNAP-25 deletion on Ca2+ -dependent and Ca2+ -independent neurotransmission. J Neurophysiol 98:794–806. 10.1152/jn.00226.2007 [DOI] [PubMed] [Google Scholar]
- Cartmell J, Schoepp DD (2000) Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem 75:889–907. [DOI] [PubMed] [Google Scholar]
- Chung C, Barylko B, Leitz J, Liu X, Kavalali ET (2010) Acute dynamin inhibition dissects synaptic vesicle recycling pathways that drive spontaneous and evoked neurotransmission. J Neurosci 30:1363–1376. 10.1523/JNEUROSCI.3427-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor A, Klyachko VA, Portera-Cailliau C (2015) Altered neuronal and circuit excitability in fragile X syndrome. Neuron 87:699–715. 10.1016/j.neuron.2015.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curry RJ, Lu Y (2016) Synaptic inhibition in avian interaural level difference sound localizing neurons. eNeuro 10.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Antoni S, Spatuzza M, Bonaccorso CM, Musumeci SA, Ciranna L, Nicoletti F, Huber KM, Catania MV (2014) Dysregulation of group-I metabotropic glutamate (mGlu) receptor mediated signaling in disorders associated with intellectual disability and autism. Neurosci Biobehav Rev 46:228–241. 10.1016/j.neubiorev.2014.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Xiao Z, Lei S (2010) Distinct modes of modulation of GABAergic transmission by group I metabotropic glutamate receptors in rat entorhinal cortex. Hippocampus 20:980–993. 10.1002/hipo.20697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondzillo A, Thompson JA, Klug A (2016) Recurrent inhibition to the medial nucleus of the trapezoid body in the Mongolian gerbil (Meriones unguiculatus). PLoS One 11:e0160241. 10.1371/journal.pone.0160241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong HW, Ennis M (2014) Activation of group I metabotropic glutamate receptors enhances persistent sodium current and rhythmic bursting in main olfactory bulb external tufted cells. J Neurophysiol 111:641–647. 10.1152/jn.00696.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermolyuk YS, Alder FG, Surges R, Pavlov IY, Timofeeva Y, Kullmann DM, Volynski KE (2013) Differential triggering of spontaneous glutamate release by P/Q-, N- and R-type Ca2+ channels. Nat Neurosci 16:1754–1763. 10.1038/nn.3563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatt P, Katz B (1950) Some observations on biological noise. Nature 166:597–598. 10.1038/166597a0 [DOI] [PubMed] [Google Scholar]
- Fatt P, Katz B (1952) Spontaneous subthreshold activity at motor nerve endings. J Physiol 117:109–128. [PMC free article] [PubMed] [Google Scholar]
- Friauf E, Hammerschmidt B, Kirsch J (1997) Development of adult-type inhibitory glycine receptors in the central auditory system of rats. J Comp Neurol 385:117–134. 10.1002/(SICI)1096-9861(19970818)385:1%3C117::AID-CNE7%3E3.0.CO%3B2-5 [DOI] [PubMed] [Google Scholar]
- Froemke RC. (2015) Plasticity of cortical excitatory-inhibitory balance. Annu Rev Neurosci 38:195–219. 10.1146/annurev-neuro-071714-034002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Pino E, Gessele N, Koch U (2017) Enhanced excitatory connectivity and disturbed sound processing in the auditory brainstem of fragile X mice. J Neurosci 37:7403–7419. 10.1523/JNEUROSCI.2310-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glitsch M. (2006) Selective inhibition of spontaneous but not Ca2+ -dependent release machinery by presynaptic group II mGluRs in rat cerebellar slices. J Neurophysiol 96:86–96. 10.1152/jn.01282.2005 [DOI] [PubMed] [Google Scholar]
- Grothe B, Pecka M, McAlpine D (2010) Mechanisms of sound localization in mammals. Physiol Rev 90:983–1012. 10.1152/physrev.00026.2009 [DOI] [PubMed] [Google Scholar]
- Guinan JJ Jr, Li RY (1990) Signal processing in brainstem auditory neurons which receive giant endings (calyces of Held) in the medial nucleus of the trapezoid body of the cat. Hear Res 49:321–334. 10.1016/0378-5955(90)90111-2 [DOI] [PubMed] [Google Scholar]
- Homayoun H, Moghaddam B (2010) Group 5 metabotropic glutamate receptors: role in modulating cortical activity and relevance to cognition. Eur J Pharmacol 639:33–39. 10.1016/j.ejphar.2009.12.042 [DOI] [PubMed] [Google Scholar]
- Hori T, Takai Y, Takahashi T (1999) Presynaptic mechanism for phorbol ester-induced synaptic potentiation. J Neurosci 19:7262–7267. 10.1523/JNEUROSCI.19-17-07262.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jursky F, Nelson N (1996) Developmental expression of the glycine transporters GlyT1 and GlyT2 in mouse brain. J Neurochem 67:336–344. [DOI] [PubMed] [Google Scholar]
- Kavalali ET. (2015) The mechanisms and functions of spontaneous neurotransmitter release. Nat Rev Neurosci 16:5–16. 10.1038/nrn3875 [DOI] [PubMed] [Google Scholar]
- Kim H, Gibboni R, Kirkhart C, Bao S (2013) Impaired critical period plasticity in primary auditory cortex of fragile X model mice. J Neurosci 33:15686–15692. 10.1523/JNEUROSCI.3246-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp-Scheinpflug C, Lippe WR, Dörrscheidt GJ, Rübsamen R (2003) The medial nucleus of the trapezoid body in the gerbil is more than a relay: comparison of pre- and postsynaptic activity. J Assoc Res Otolaryngol 4:1–23. 10.1007/s10162-002-2010-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushmerick C, Price GD, Taschenberger H, Puente N, Renden R, Wadiche JI, Duvoisin RM, Grandes P, Von Gersdorff H (2004) Retroinhibition of presynaptic Ca2+ currents by endocannabinoids released via postsynaptic mGluR activation at a calyx synapse. J Neurosci 24:5955–5965. 10.1523/JNEUROSCI.0768-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara N, DiCaprio RA, Zook JM (1991) Afferents to the medial nucleus of the trapezoid body and their collateral projections. J Comp Neurol 314:684–706. 10.1002/cne.903140405 [DOI] [PubMed] [Google Scholar]
- Leão RM, Von Gersdorff H (2002) Noradrenaline increases high-frequency firing at the calyx of held synapse during development by inhibiting glutamate release. J Neurophysiol 87:2297–2306. 10.1152/jn.2002.87.5.2297 [DOI] [PubMed] [Google Scholar]
- Leão RM, Kushmerick C, Pinaud R, Renden R, Li GL, Taschenberger H, Spirou G, Levinson SR, Von Gersdorff H (2005) Presynaptic Na+ channels: locus, development, and recovery from inactivation at a high-fidelity synapse. J Neurosci 25:3724–3738. 10.1523/JNEUROSCI.3983-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YC, Chanaday NL, Xu W, Kavalali ET (2017) Synaptotagmin-1- and synaptotagmin-7-dependent fusion mechanisms target synaptic vesicles to kinetically distinct endocytic pathways. Neuron 93:616–631.e3. 10.1016/j.neuron.2016.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson AK, Park TJ, Pecka M, Grothe B, Koch U (2008) Retrograde GABA signaling adjusts sound localization by balancing excitation and inhibition in the brainstem. Neuron 59:125–137. 10.1016/j.neuron.2008.05.011 [DOI] [PubMed] [Google Scholar]
- Mannaioni G, Marino MJ, Valenti O, Traynelis SF, Conn PJ (2001) Metabotropic glutamate receptors 1 and 5 differentially regulate CA1 pyramidal cell function. J Neurosci 21:5925–5934. 10.1523/JNEUROSCI.21-16-05925.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer F, Albrecht O, Dondzillo A, Klug A (2014) Glycinergic inhibition to the medial nucleus of the trapezoid body shows prominent facilitation and can sustain high levels of ongoing activity. J Neurophysiol 112:2901–2915. 10.1152/jn.00864.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullagh EA, Salcedo E, Huntsman MM, Klug A (2017) Tonotopic alterations in inhibitory input to the medial nucleus of the trapezoid body in a mouse model of fragile X syndrome. J Comp Neurol 525:3543–3562. 10.1002/cne.24290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney RA, Capogna M, Dürr R, Gähwiler BH, Thompson SM (1999) Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat Neurosci 2:44–49. 10.1038/4548 [DOI] [PubMed] [Google Scholar]
- Melom JE, Akbergenova Y, Gavornik JP, Littleton JT (2013) Spontaneous and evoked release are independently regulated at individual active zones. J Neurosci 33:17253–17263. 10.1523/JNEUROSCI.3334-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50:295–322. 10.1146/annurev.pharmtox.011008.145533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peled ES, Newman ZL, Isacoff EY (2014) Evoked and spontaneous transmission favored by distinct sets of synapses. Curr Biol 24:484–493. 10.1016/j.cub.2014.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piechotta K, Weth F, Harvey RJ, Friauf E (2001) Localization of rat glycine receptor α1 and α2 subunit transcripts in the developing auditory brainstem. J Comp Neurol 438:336–352. 10.1002/cne.1319 [DOI] [PubMed] [Google Scholar]
- Raingo J, Khvotchev M, Liu P, Darios F, Li YC, Ramirez DM, Adachi M, Lemieux P, Toth K, Davletov B, Kavalali ET (2012) VAMP4 directs synaptic vesicles to a pool that selectively maintains asynchronous neurotransmission. Nat Neurosci 15:738–745. 10.1038/nn.3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DM, Khvotchev M, Trauterman B, Kavalali ET (2012) Vti1a identifies a vesicle pool that preferentially recycles at rest and maintains spontaneous neurotransmission. Neuron 73:121–134. 10.1016/j.neuron.2011.10.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renden R, Taschenberger H, Puente N, Rusakov DA, Duvoisin R, Wang LY, Lehre KP, Von Gersdorff H (2005) Glutamate transporter studies reveal the pruning of metabotropic glutamate receptors and absence of AMPA receptor desensitization at mature calyx of held synapses. J Neurosci 25:8482–8497. 10.1523/JNEUROSCI.1848-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotschafer S, Razak K (2013) Altered auditory processing in a mouse model of fragile X syndrome. Brain Res 1506:12–24. 10.1016/j.brainres.2013.02.038 [DOI] [PubMed] [Google Scholar]
- Rotschafer SE, Razak KA (2014) Auditory processing in fragile X syndrome. Front Cell Neurosci 8:19. 10.3389/fncel.2014.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotschafer SE, Marshak S, Cramer KS (2015) Deletion of Fmr1 alters function and synaptic inputs in the auditory brainstem. PLoS One 10:e0117266. 10.1371/journal.pone.0117266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau F, Aubrey KR, Supplisson S (2008) The glycine transporter GlyT2 controls the dynamics of synaptic vesicle refilling in inhibitory spinal cord neurons. J Neurosci 28:9755–9768. 10.1523/JNEUROSCI.0509-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux MJ, Martínez-Maza R, Le Goff A, López-Corcuera B, Aragón C, Supplisson S (2001) The glial and the neuronal glycine transporters differ in their reactivity to sulfhydryl reagents. J Biol Chem 276:17699–17705. 10.1074/jbc.M009196200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sara Y, Virmani T, Deák F, Liu X, Kavalali ET (2005) An isolated pool of vesicles recycles at rest and drives spontaneous neurotransmission. Neuron 45:563–573. 10.1016/j.neuron.2004.12.056 [DOI] [PubMed] [Google Scholar]
- Sara Y, Bal M, Adachi M, Monteggia LM, Kavalali ET (2011) Use-dependent AMPA receptor block reveals segregation of spontaneous and evoked glutamatergic neurotransmission. J Neurosci 31:5378–5382. 10.1523/JNEUROSCI.5234-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch S, Deák F, Königstorfer A, Mozhayeva M, Sara Y, Südhof TC, Kavalali ET (2001) SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 294:1117–1122. 10.1126/science.1064335 [DOI] [PubMed] [Google Scholar]
- Schoepp DD. (2001) Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J Pharmacol Exp Ther 299:12–20. [PubMed] [Google Scholar]
- Sedlacek M, Tipton PW, Brenowitz SD (2011) Sustained firing of cartwheel cells in the dorsal cochlear nucleus evokes endocannabinoid release and retrograde suppression of parallel fiber synapses. J Neurosci 31:15807–15817. 10.1523/JNEUROSCI.4088-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PH, Joris PX, Yin TC (1998) Anatomy and physiology of principal cells of the medial nucleus of the trapezoid body (MNTB) of the cat. J Neurophysiol 79:3127–3142. 10.1152/jn.1998.79.6.3127 [DOI] [PubMed] [Google Scholar]
- Stincic TL, Hyson RL (2011) The localization and physiological effects of cannabinoid receptor 1 in the brain stem auditory system of the chick. Neuroscience 194:150–159. 10.1016/j.neuroscience.2011.05.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Wall NR, Aakalu GN, Schuman EM (2004) Regulation of dendritic protein synthesis by miniature synaptic events. Science 304:1979–1983. 10.1126/science.1096202 [DOI] [PubMed] [Google Scholar]
- Takahashi T, Forsythe ID, Tsujimoto T, Barnes-Davies M, Onodera K (1996) Presynaptic calcium current modulation by a metabotropic glutamate receptor. Science 274:594–597. 10.1126/science.274.5287.594 [DOI] [PubMed] [Google Scholar]
- Thompson AM, Schofield BR (2000) Afferent projections of the superior olivary complex. Microsc Res Tech 51:330–354. 10.1002/1097-0029(20001115)51:4%3C330::AID-JEMT4%3E3.0.CO%3B2-X [DOI] [PubMed] [Google Scholar]
- Trattner B, Berner S, Grothe B, Kunz L (2013) Depolarization-induced suppression of a glycinergic synapse in the superior olivary complex by endocannabinoids. J Neurochem 127:78–90. 10.1111/jnc.12369 [DOI] [PubMed] [Google Scholar]
- Tsintsadze T, Williams CL, Weingarten DJ, Von Gersdorff H, Smith SM (2017) Distinct actions of voltage-activated Ca2+ channel block on spontaneous release at excitatory and inhibitory central synapses. J Neurosci 37:4301–4310. 10.1523/JNEUROSCI.3488-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehr M, Zador AM (2003) Balanced inhibition underlies tuning and sharpens spike timing in auditory cortex. Nature 426:442–446. 10.1038/nature02116 [DOI] [PubMed] [Google Scholar]
- Xu J, Pang ZP, Shin OH, Südhof TC (2009) Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nat Neurosci 12:759–766. 10.1038/nn.2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Atallah BV, Scanziani M (2014) Equalizing excitation–inhibition ratios across visual cortical neurons. Nature 511:596–600. 10.1038/nature13321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorio DA, Jackson CM, Liu Y, Rubel EW, Wang Y (2017) Cellular distribution of the fragile X mental retardation protein in the mouse brain. J Comp Neurol 525:818–849. 10.1002/cne.24100 [DOI] [PMC free article] [PubMed] [Google Scholar]