

Abstract
Background
Classical Galactosaemia (CG) (OMIM #230400) is a rare inborn error of galactose metabolism caused by deficiency of the enzyme galactose-1-phosphate uridylyltransferase (GALT). Long-term complications persist in treated patients despite dietary galactose restriction with significant variations in outcomes suggesting epigenetic glycosylation influences. Primary Ovarian Insufficiency (POI) is a very significant complication affecting females with follicular depletion noted in early life. We studied specific glycan synthesis, leptin system and inflammatory gene expression in white blood cells as potential biomarkers of infertility in 54 adults with CG adults (27 females and 27 males) (age range 17–51 yr) on a galactose-restricted diet in a multi-site Irish and Dutch study. Gene expression profiles were tested for correlation with a serum Ultra-high Performance Liquid Chromatography (UPLC)-Immunoglobulin (IgG)-N-glycan galactose incorporation assay and endocrine measurements.
Results
Twenty five CG females (93%) had clinical and biochemical evidence of POI. As expected, the CG female patients, influenced by hormone replacement therapy, and the healthy controls of both genders showed a positive correlation between log leptin and BMI but this correlation was not apparent in CG males. The strongest correlations between serum leptin levels, hormones, G-ratio (galactose incorporation assay) and gene expression data were observed between leptin, its gene and G-Ratios data (rs = − 0.68) and (rs = − 0.94) respectively with lower circulating leptin in CG patients with reduced IgG galactosylation. In CG patients (males and females analysed as one group), the key glycan synthesis modifier genes MGAT3 and FUT8, which influence glycan chain bisecting and fucosylation and subsequent cell signalling and adhesion, were found to be significantly upregulated (p < 0.01 and p < 0.05) and also the glycan synthesis gene ALG9 (p < 0.01). Both leptin signalling genes LEP and LEPR were found to be upregulated (p < 0.01) as was the inflammatory genes ANXA1 and ICAM1 and the apoptosis gene SEPT4 (p < 0.01).
Conclusions
These results validate our previous findings and provide novel experimental evidence for dysregulation of genes LEP, LEPR, ANXA1, ICAM1 and SEPT4 for CG patients and combined with our findings of abnormalities of IgG glycosylation, hormonal and leptin analyses elaborate on the systemic glycosylation and cell signalling abnormalities evident in CG which likely influence the pathophysiology of POI.
Keywords: Classical galactosaemia, Infertility, Glycan modifier genes
Background
Classical galactosaemia (CG) (OMIM #230400) is a rare autosomal recessive inborn error of galactose metabolism caused by deficiency of the enzyme galactose-1-phosphate uridylyltransferase (GALT; EC 2.7.7.12). This condition occurs with a prevalence ranging from 1:16,000 to 1:60,000 in Europe and USA. Life-long galactose restricted diet is the only treatment currently available for this condition. Although this treatment is life-saving in the neonate, long-term complications including cognitive impairment, neurological and speech abnormalities, and fertility problems in female patients, persist in treated adult patients despite early diagnosis and initiation of treatment [1–8]. Ovarian damage and subfertility with primary ovarian insufficiency (POI) is a major complication for females causing a very significant disease burden. The exact timing of the ovarian insult and its pathophysiology remains poorly understood [3, 6].
A key limitation to improving care and personalised therapy for affected individuals for this rare disease is the lack of insight into the pathophysiology and the lack of reliable and accurate biomarkers that can predict the risk of developing disease complications and can monitor the outcome of therapeutic interventions.
Moreover, outcomes can differ even in siblings with the same GALT genotype, illustrating the complex nature of this condition with the presence of recognised significant epigenetic effects on the fundamental glycosylation pathways involved in galactosaemia [9–11].
The toxic build-up of the GALT substrate galactose-1-phosphate (Gal-1-P) and its metabolites are proposed to be central to the pathophysiology of the ongoing complications [6]. In the neonatal acute intoxicated phase, very high ambient levels of Gal-1-P can inhibit a number of metabolic processes. Elevated Gal-1-P is a known competitive substrate for inositol monophosphatase 1 (IMPase1) and various glycosyltransferases and also inhibits UDP-hexose pyrophosphorylases [6, 12]. In addition, over-restriction of galactose in the long-term may contribute to the disease phenotype by further depleting UDP-galactose in susceptible individuals, potentially disrupting glycosylation dependent pathways [13, 14]. Down-regulation of the key P13K/Akt signalling pathway has also been recently reported in the GALT deficient mouse model [15].
It has long been recognised that the measurement of red blood cell (RBC) Gal-1-P in neonatal blood samples from untreated patients prompts the initial treatment of the Gal-1-P intoxicated neonate and is an important diagnostic marker. However, monitoring RBC Gal-1-P and urinary galactitol concentrations have not generally been considered to be reliable prognostic indices of long-term outcomes [4, 13, 16, 17].
Previous investigations by our group have explored the mechanisms by which abnormal galactosylation of glycoproteins may contribute to the ongoing pathophysiology in galactosaemia and its complications. Of relevance to fertility, recent studies have not demonstrated any differences in Follicle stimulating hormone (FSH) glycosylation pattern or bioactivity in females with CG [18, 19]. A high percentage of females with CG have Anti-mullerian hormone (AMH) levels below the detection limit refecting a low ovarian reserve, however the glycosylation status of AMH has not been studied to date in CG [20]. IgG is the predominant circulating glycoprotein in serum and is very well studied [21]. We developed a glyco-analysis of Immunoglobulin G (IgG) and studied the incorporation of galactose into IgG in galactosaemia patients in comparison to healthy controls using an automated hydrophilic interaction ultra-high performance liquid chromatography (HILIC-UPLC) N-glycan analytical method for serum IgG, to monitor N-glycan processing defects in galactosaemia [22, 23].
We previously evaluated the impact of several genes on abnormal glycosylation in galactosaemia patients and noted significant altered expression of a number of relevant N-glycan biosynthesis genes in peripheral blood mononuclear cells (PBMCs) from adult galactosaemia patients involving four key N-glycan biosynthesis genes: ALG9, MGAT3, FUT8 and B4GALT1 which correlated with the IgG variant profiles also identified in the study [9]. Lending further weight to this finding, a large European population genome wide association study using liquid chromatography mass spectrometry (LC-MS) to measure IgG glycoprotein variant characteristics has recently demonstrated that polymorphisms of the glycan genes encoding the glycosyltransferases (ST6GAL1, B4GALT1, FUT8 and MGAT3) represent the most important loci associated with variation in IgG traits [24].
In addition to altered glycosylation, we previously identified a number of key central signalling pathways affected in T lymphocyte cell studies to include the unfolded protein response (UPR) pathway, the inositol signalling pathway, oxidative phosphorylation, and inflammatory pathways [25, 26]. We also observed dysregulation of pathways in leptin metabolism, a key hormone in the Hypothalamic-Pituitary-Gonadal (HPG) axis which stimulates release of luteinising hormone (LH), follicle stimulating hormone (FSH) and oestrogen [27, 28].
The majority of adult females with CG have POI which is a spectrum varying from absent or delayed pubertal development, primary amenorrhoea in adolescents, secondary amenorrhoea to irregular or premature menopause [29]. POI in females with CG is identifiable by elevated follicle stimulating hormone (FSH) and LH levels and decreased oestradiol levels [29]. FSH levels have been found to be significantly elevated from four months of life [6, 29, 30]. The precise timing of the severe decrease in primordial ovarian follicles and the absence of intermediate and Graafian follicles, which suggest a maturation arrest, is not clear [6, 29, 30]. A recent paper has suggested that follicles are maintained in early childhood, but commence depletion as early as the 1st year of life [31]. Different mechanisms have been proposed to explain these changes to include prenatal toxicity of galactose and its metabolites, including apoptosis, abnormal signalling pathways and abnormal gonadotropin function [29, 30, 32].
There is limited data available for assessing fertility in CG adult males, though studies have reported delayed onset of puberty [6]. In a study of 26 CG males, the prevalence of cryptorchidism was noted to be higher in CG males than in the general population and the CG patients had subtle decreases in testosterone, inhibin B and sperm concentration [33].
As it is not practical to study temporal ovarian or testicular gene expression in patients, in this study we have aimed to investigate the expression of key glycan and other relevant genes in GC patients in a cohort of patients from a three-site study, (Irish and two Dutch national galactosaemia cohorts) using accessible PBMCs (peripheral blood mononucleocytes) and correlated these findings with the patient endocrine markers and N-glycome glycosylation status as measured by the IgG N-glycome.
Results
Clinical Characterisation: Table 1 illustrates the study subject characteristics: age, gender and genotypes.
Table 1.
Patient Characteristics
| Group | Controls | Galactosaemia | |
|---|---|---|---|
| Ethnicity | Irish | Irish | Dutch |
| Patients | n = 16 | n = 36 | n = 18 |
| Age (years) | 20–40 | 17–51 | 18–47 |
| Gender | 9F, 7 M | 15F, 21 M | 12F, 6 M |
| Genotype (nucleotide annotation) | NA | 31: (c.563A > G/c.563A > G) 2: c.568A > G/c.580 T > C 1: c.563A > G/c.997C > T 1: c.563A > G/c.379.A > G) 1: c.563A > G/unknown |
10: (c.563A > G/c.563A > G) 3: (c.584 T > C/c.687G > T) 1: (c.563A > G/c.584 T > C) 1: (c.563A > G/c.400delT) 1: (c.563A > G/del exon 1_10) 1: (c.956A > C/c.956A > C) 1: Unknown |
| Genotype (protein variant) | NA | 31: p.Q188R/p.Q188R 2: Q188R/p.F194 L 1: p.Q188R/p.R333W 1: p.Q188R/p.K127E, 1: p.Q188R/unknown |
10: (p.Q188R/p.Q188R) 3: (p.L195P/p.K229 N) 1: (p.Q188R/p.L195P) 1: (p.Q188R/p.T134 fs) 1: (p.Q188R/del exon 1_10) 1: (c.956A > C/c.956A > C) 1: Unknown |
Endocrine studies
Patient FSH, LH, oestradiol, testosterone serum concentrations and reference intervals are illustrated in Table 2. Of the 27 CG females, 25 had clinical and biochemically documented POI (92.59%, age range 18–42 yr). 22 of these patients were on hormone replacement therapy (HRT).
Table 2.
Patient Hormone Results
| LH (IU/L) | FSH (IU/L) | Oestradiol (pmol/L) | LH (IU/L) | FSH (IU/L) | Testosterone (nmol/L)(Males only) | |
|---|---|---|---|---|---|---|
| Normal reference rangesa | F: 1.8–11.8 | F: 3.0–8.1 | F: 92–921 | 1.4–6.5 | 0–10.9 | 7.1–31.1 |
| M: 7.6–89.1 | M: 2.6–16.7 | M: 139–2382 | ||||
| L: 0.6–14.0 | L: 1.4–5.5 | L: 92–1145 | ||||
| CG Females (n = 24) | CG Males (n = 18) | |||||
| Median | 11.57 | 32.85 | 460 (n = 9) < 92 (n = 14) No data (n = 1) |
4.00 | 2.00 | 18.80 |
| Mean | 14.94 | 40.89 | 500.67 (n = 9) < 92 (n = 14) No data (n = 1) |
5.51 | 8.40 | 19.98 |
| Range | 0.5–40.50 | 0.5–120 | < 92–967 | 0.5–9.4 | 0.5–9.3 | 10–31.6 |
| SD | 38.43 | 12.52 | N/A | 6.10 | 2.521.47 | 5.49 |
aReference ranges are provided for healthy females and males for LH, FSH and oestradiol for comparison to CG patients, with the females divided into three menstrual stages: Follicular Phase (F), Mid Cycle Phase (M) and Luteal Phase (L). The normal reference ranges were provided from the Investigator site accredited diagnostic laboratory. For CG patients, median, mean and range data are provided with standard deviation (SD)-no menstrual stage data was available. Oestradiol levels less than 92 pmol/L were noted as undetectable according to the limits of the assay. Hormone data was available for 38 of the 54 patients
It was not possible to determine which phase of the menstrual cycle patients were in when the hormone samples were taken. Inappropriately raised FSH (> 20 IU) in the context of suppressed oestradiol in females below the age of 40 is an indicator of POI. Oestradiol results were available for 23 CG females, 14 of whom had low or undetectable levels (< 92 pmol/mL). All but one of these 14 females were on HRT. However synthetic oestradiol levels are not detected by the assay used. There were no significant hormone abnormalities noted in male CG patients (Table 2).
Table 3 illustrates the leptin data for available CG subjects (n = 37) and healthy controls (n = 20). Circulating serum leptin data from both male and female galactosaemia cohorts were determined. The mean serum leptin level was lower in both CG males and females in comparison to healthy controls although this only reached statistical significance in males (p < 0.03). There was no significant difference noted in the soluble leptin receptor (sObR) levels between CG patients and healthy controls in either gender group. Leptin levels corrected for BMI were correlated using linear regression analysis with the IgG galactose incorporation (G0/G1 and G0/G2) ratios and with LH, FSH levels for both genders, and with testosterone for males.
Table 3.
Serum Leptin levels in CG vs healthy controls and correlations with hormone, G-ratios and ANXAI gene expression
| Serum Leptina | |||||
|---|---|---|---|---|---|
| Gender | Group | n= | Mean ± SD | Range | p-value |
| Male | CG | 18 | 2.6 ± 1.86 | 0.10–6.60 | 0.031 |
| Healthy Control | 10 | 7.40 ± 5.60 | 0.70–18.80 | ||
| Female | CG | 19 | 13.22 ± 9.47 | 1.18–33.12 | 0.099 |
| Healthy Control | 10 | 20.10 ± 12.53 | 3.60–45.44 | ||
| Spearman Correlations - Serum Leptin Corrected for BMIb | |||||
| Variable 1 | Variable 2 | Gender | n= | p-value | Correlation Coefficient |
| Leptin, BMI corrected | G0/G1 | Male and Female | 26 | 0.0001 | − 0.681 |
| Leptin, BMI corrected | G0/G2 | Male and Female | 26 | 0.002 | − 0.588 |
| Leptin, BMI corrected | Testosterone | Male | 11 | 0.026 | 0.664 |
| Leptin, BMI corrected | FSH | Male and Female | 25 | 0.027 | 0.441 |
| Leptin, BMI corrected | ANXA1 | Female | 17 | 0.012 | 0.593 |
aLeptin data was available for 37 of 54 patients. The p-values of this section show results of the difference in means of serum leptin levels in CG vs healthy control (Mann Whitney U test)
bCorrelations between serum leptin corrected for BMI vs other variables in combined and separate gender cohorts
The Spearman correlations between abnormal N-glycosylation as measured by G ratios and leptin are shown in Table 3. For those CG subjects that had RBC Gal-1-P levels performed (n = 22), we identified a direct positive correlation between Gal-1-P and G-ratios in the combined group (rs = 0.699, p < 0.0005 [G0/G1], rs = 0.666, p < 0.005 [G0/G2] (data not shown).
We observed a strong negative correlation between leptin and G-ratios in the combined CG group, indicating lower circulating leptin in patients with reduced IgG galactosylation (Table 3). We also observed a significant correlation between BMI corrected leptin levels and testosterone levels in CG males with a less significant correlation evident with FSH levels in the CG combined group (males and females analysed together).
As shown in Fig. 1, LH levels correlated with G0/G1 and G0/G2 ratios (rs = 0.529, p < 0.05 and rs = 0.608, p < 0.01) respectively in CG females (Fig. 1). There was no significant correlation evident between FSH levels and G-ratios in either genders (data not shown).
Fig. 1.

Hormones, G Ratio and gene correlations in galactosaemia. The strength of the association is denoted by rs (Spearman’s ρ), 1 being a perfect positive correlation, − 1 being a perfect negative correlation. The p-value (2-tailed) for the association is indicated. The sloping line is the best fit line and indicates the direction of the correlation. a FSH levels correlate positively with BMI corrected leptin in the CG combined group (n = 28: rs = 0.445, p < 0.05). b ALG9, LEP and MGAT3 expression correlate negatively with G-ratios (G0/G1) in CG females (ALG9: n = 18, rs = − 0.600, p < 0.01. MGAT3: n = 13, rs = − 0.687, p < 0.05. LEP: n = 6, rs = − 0.943, p < 0.01). c LH levels correlate positively with G-ratios (G0/G1) in CG females (n = 18, rs = 0.529, p < 0.05). d LH levels correlate positively with G-ratios (G0/G2) in CG females (n = 18, rs = 0.608, p < 0.01)
Figure 1 illustrates a positive correlation between FSH and leptin in the combined group (leptin corrected for BMI: rs = 0.441, p < 0.05).
Leptin is known to correlate positively with BMI in both healthy males and females [32].
As shown in Fig. 2, the association between log leptin and BMI evident in CG females was absent in CG males (Fig. 2).
Fig. 2.
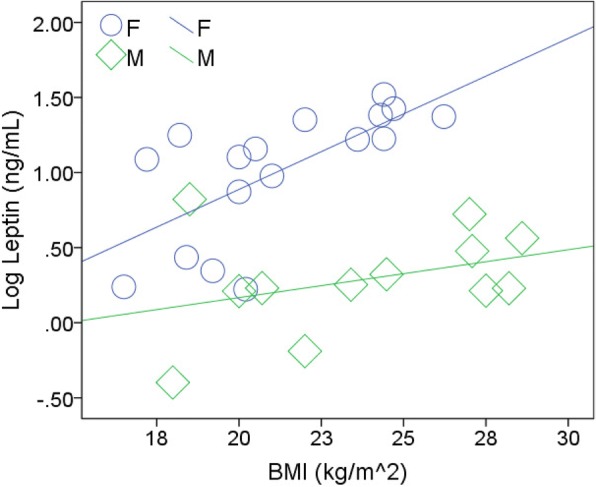
Correlation of log leptin and BMI in both CG males and females. Associations are measured with rs and p values as described previously. Serum leptin correlates positively with BMI in healthy males and females [32]. This correlation was apparent in the CG female cohort (n = 17, rs = 0.730, p < 0.005) but not in CG males (n = 12, rs = 0.301, NS)
Gene expression studies
In this current study we observed significant upregulation of the glycan synthesis genes, and inflammatory and leptin signaling genes; ALG9, ANXA1, FUT8, ICAM1, LEP, LEPR, MGAT3, SEPT4 and UGDH (Fig. 3) in PBMC cells of the CG combined group (both genders). The expression of the genes B4GALT1, MGAT1, UGP2 was not significantly altered in this group.
Fig. 3.

Boxplots of PBMC gene expression in CG combined group vs healthy controls (Ctrl). Each boxplot is titled with the relevant gene. The y-axis represents the 2-ΔCT value of gene expression (Applied Biosystems). The y-axis scale has been log transformed to the base 10 for clarity. Fold change (RQ) and 2ΔΔCT calculated with DataAssist (Applied Biosystems). Differences in expression between CG and Ctrl groups as calculated by The Mann Whitney U test, giving a p value which has been Benjamini-Hochberg False Discovery Rate (FDR) adjusted. Boxes indicate median (middle), 25th (bottom) and 75th (top) percentiles. Error bars indicate 1.5 times the interquartile range. Outliers are indicated with small circles or asterisks (extreme outliers). a ALG9 upregulated in CG vs Ctrl (n = 52 vs 16, RQ = 5.97, p < 0.005). b ANXA1 upregulated in CG vs Ctrl (n = 54 vs 16, RQ = 6.21, p < 0.01). (c) FUT8 upregulated in CG vs Ctrl (n = 50 vs 16, RQ = 4.13, p < 0.05). d ICAM1 upregulated in CG vs Ctrl (n = 53 vs 16, RQ = 4.79, p < 0.005). e LEP upregulated in CG vs Ctrl (n = 22 vs 10, RQ = 8.88, p < 0.005). f LEPR upregulated in CG vs Ctrl (n = 47 vs 16, RQ = 6.38, p < 0.0005). g MGAT3 upregulated in CG vs Ctrl (n = 47 vs 16, RQ = 6.10, p < 0.01). h SEPT4 upregulated in CG vs Ctrl (n = 52 vs 16, RQ = 3.62, p < 0.01). i UGDH upregulated in CG vs Ctrl (n = 46 vs 16, RQ = 2.66, p < 0.05). j UGP2 in CG vs Ctrl (n = 53 vs 16, not significant [NS]). k MGAT1 (n = 53 vs 16, NS). l B4GALT (n = 54 vs 16, NS)
The key dysregulated glycan synthesis genes (ALG9, MGAT3 and FUT8) correlate positively with each other (p < 0.05− 4) in CG which could be expected as they share the same glycan synthesis pathway.
MGAT3 was noted to correlate directly with the expression of the inflammatory/apoptosis genes ANXA1 and SEPT4 (p < 0.05− 4) in CG, (combined group). The two inflammatory markers (ANXA1 and ICAM1) are positively correlated (p < 0.05− 4) in CG. Of note, increased expression of FUT8 (indicating dysregulation of the core fucosylation pathway) was significantly correlated with LEPR expression (p < 0.0005) in CG.
There was no gender dependent difference in expression of any of the genes studied as determined by multivariate analysis of 2-ΔCT values. The same finding was apparent between males and females of the healthy control group.
Discussion
In this study, we sought firstly to validate identified glycan gene expression markers identified from our earlier studies in this larger multi-site study. We then aimed to introduce further novel inflammatory and putative fertility-linked expression markers and to correlate these expression profiles with the concurrent hormonal profiles of the study subjects with a view to further study the pathophysiology and fertility issues observed in CG.
ALG9 (the gene that encodes α-1,2-mannosyltransferase) was shown to be overexpressed in this larger male and female CG cohort (n = 54 Irish and Dutch subjects: 5.97 fold, p < 0.01) in agreement with our earlier study [9]. α-1,2-mannosyltransferase catalyses the transfer of mannose from Dol-P-Man to lipid-linked oligosaccharides in N-Glycan assembly. This enzyme is involved in the addition of the seventh and ninth mannose to the growing N-glycan chain. This enzyme may be overexpressed in galactosaemia as cellular stresses increase the rate of glycan assembly in the endoplasmic reticulum (ER) which leads to improved levels of mono- and di-galactose glycan species (G1 and G2) downstream of the processing chain in the Golgi. In our previous cell studies, we observed that the activity of this enzyme is very responsive to galactose intoxication [26].
MGAT3 (the gene that encodes β-1,4-mannosyl-glycoprotein 4-β-N-acetylglucosaminyltransferase) was the most significantly dysregulated glycan synthesis gene in this study, upregulated 6.1 fold in CG males & females vs controls (p < 0.01). The importance of this gene in N-glycosylation is demonstrated in the recent Genome-Wide Association Study (GWAS) [24] whereby variation in MGAT3 was observed to be significantly associated with IgG glycan variant phenotypes. MGAT3 is responsible for synthesis of complex hybrid type glycans in the endoplasmic reticulum. In the GWAS study, ratios of structures with bisecting GlcNAcs to structures without bisecting GlcNAcs were associated with SNPs at the MGAT3 locus. Abnormal expression of this gene in our earlier study was associated with decreased bisecting GlcNAcs (decreased core fucosylated, non-fucosylated and monogalactosylated glycans) in CG [9]. It is considered that bisecting GlcNAcs influence glycan processing and glycan adhesion [34].
We observed that FUT8 (that encodes alpha-1,6–fucosyltransferase) was upregulated in the CG combined group vs healthy controls (4.12 fold, p < 0.05)) in this study. This was more significant than in our previous smaller study and correlates with the findings of increased glycan core fucosylation, which we have observed previously [9]. The findings also indicate this gene as a significant modifier of glycan phenotype variation (ratios of fucosylated over non-fucosylated glycan structures) as noted in the GWAS study [24].
The group of Jumbo-Lucioni et al. have demonstrated in a Drosophila galactosaemia model that the UGDH (UDP-Glucose 6-dehydrogenase) gene is upregulated and considered to be a modifier/rescue glycosylation gene in drosophila [35]. We included this gene in our analysis and we also observed upregulation 2.6 fold in the CG combined group (p < 0.05). This change may be a response to reduced substrate from disruption of UDP-Glc turnover via the GALT enzyme in the Leloir pathway (Fig. 4). Another potential salvage gene that we included is UGP2 (UDP-glucose pyrophosphorylase 2). We did not note any significant difference in expression of this gene between CG patients and healthy controls.
Fig. 4.

Galactose metabolism, glycosylation and inflammatory cascades in galactosaemia. a In galactosaemia, the Leloir pathway is disrupted with the absence of GALT, leading to an increase of Gal-1-P and disruption of UDP-hexose turnover. b Increased Gal-1-P levels may lead to cellular toxicity and ER stress, with competitive inhibition of glycosyltransferases and inhibition of UDP-hexose pyrophosphorylases. T [6, 11–13]. Disrupted glycosylation may lead to an upregulation of glycan synthesis genes. Abnormal glycosylation of IgG may lead to an activated immune conformation [36] and increased expression of anti-inflammatory genes. Leptin expression, influenced by abnormal glycosylation, cross signals with the HPG axis and gonadal function [52, 53]
Of the glycan synthesis genes, ALG9 overexpression correlated with the overexpression also of the genes FUT8, MGAT3 and B4GALT in the CG combined group (rs = 0.418, 0.643, 0.534 respectively, p < 0.003) consistent with the shared common N-glycan synthesis pathway.
On account of the known population variation (SNPs) for MGAT3, FUT8 and in the case of possible variation in expression of the ALG9 gene we compared the fold differences in expression between these genes in the Irish and Dutch cohort. There was no statistically significant difference in expression noted.
More than 50% of all proteins are considered to be glycosylated in humans. Glycan oligosaccharide modification affects receptor function, cell signalling, and cell adhesion. Modification of branched N-glycans structures such as bisecting GlcNAc, β-1,6-GlcNAc and core fucose (α-1,6-fucose), the enzymatic products of MGAT3 and FUT6 genes, shown to be abnormal in galactosaemia [9], are highly associated with biological functions involving cell adhesion [34, 36].
Almost all key molecules involved in innate and adaptive immune responses are glycoproteins. IgG is the most abundant circulating glycoprotein (as measured in this study). The addition of different glycans to its Fc regions (region that interacts with cell surface receptors) and changes in core fucosylation can have dramatic effects on its effector function [37, 38].
In addition to N-glycan synthesis genes in this study, we thus sought to examine whether there was also an identifiable anti-inflammatory component to the pathophysiology of galactosaemia. We proposed from our earlier studies that systemic glycosylation abnormalities evidenced in galactosaemia could be associated with abnormal inflammatory responses. We studied two genes as possible anti-inflammatory markers, ANXA1 and ICAM1, the former which we previously reported as dysregulated in galactosaemia [26].
ANXA1 (Annexin A1) is a phospholipid binding protein which responds to a glucocorticoids and carries out an anti-inflammatory response [39]. The expression of ANXA1 was increased 6.2 fold in the CG combined group females vs healthy controls, (p < 0.01), which may reflect upregulated apoptosis pathways, one of the proposed possible mechanisms of primordial follicle depletion.
Another consideration is an anti-inflammatory response to the sub-galactosylation of immunoglobulins resulting in increased immune activation as shown for IgG, whereby reduced galactosylation of the Fc region results in an inflammatory conformation [40].
ICAM1 (Intercellular Adhesion Molecule 1) encodes a cell surface glycoprotein which is typically expressed on endothelial cells and cells of the immune system which binds to integrins. ICAM1’s concentration greatly increases upon cytokine stimulation [41]. ICAM1 was observed to be upregulated 4.79 fold in the CG combined group (p < 0.01). The expression of ANXA1 and ICAM1 expression correlated strongly in galactosaemia patients (rs = 0.624, p < 0.05 × 10− 4), suggesting an inflammatory association.
ANXA1 correlated strongly with expression of the glycan assembly gene ALG9 in the CG combined group (rs = 0.840, p < 0.05 × 10− 12). Also ncreased expression of ICAM1 correlated with the increased expression of ALG9 in the CG combined group (rs = 0.615, p < 0.05 × 10− 4) indicating a possible relationship between inflammation and abnormal glycosylation in these patients.
SEPT4 (the gene that encodes Septin 4) was increased in expression 3.6 fold (p < 0.005). The septin family of proteins are a group of GTP-binding proteins that are essential for biological processes such as cytokinesis and vesical trafficking. SEPT4 has been shown to be involved with sperm terminal differentiation in mice and is a marker of apoptosis, required for the induction of cell death mediated by TGF-beta and by other apoptotic stimuli [42].
In addition to the identification of dysregulation of specific genes in CG, we and others have proposed that abnormal glycosylation leads to systemic signalling abnormalities. We noted significant dysregulation of the leptin receptor (LEPR) in our earlier studies and also reported decreased circulating serum leptin in both CG males and females [27]. We now report dysregulated expression of leptin (LEP) and LEPR in both CG males and females in the present extended study and provide a link between glycosylation (G-ratio) and leptin expression. Both LEP and its receptor (LEPR) were upregulated 8.8 and 6.38 fold respectively (p < 0.005) in the CG combined group (male and females). The fact that we noted upregulation of the leptin gene in CG male and female PBMCs but reduced serum concentration in both CG males and females (not statistically significant in females) requires further investigation.
The finding of normal leptin circulating concentrations in females as previously identified may reflect the fact that the majority of females in the study who manifested POI were on hormone replacement therapy as reviewed earlier in our previous study [27].
Biochemical and endocrine clinical correlates with gene expression
We noted that Gal-1-P levels correlate positively with G-ratios which may be consistent with a recent observation of better clinical outcomes in male and female CG patients who exhibit lower RBC Gal-1-P levels on a galactose restricted diet [17]. Lower Gal-1-P levels with lower G-ratios may be explained by residual GALT activity, accessory pathway metabolism and less endogenous turnover of galactose from sources such as glycoproteins [43].
Testosterone levels have been noted to be in the lower normal range in a small number of galactosaemic males but within reference ranges [33]. Testosterone levels in this patient cohort were within the normal reference range (Table 2). As expected with the normal values for FSH and LH observed in our male cohort, there was no correlation noted between G-ratios and FSH and LH levels in males (data not shown). LH, as an indicator of POI in females, was shown to correlate positively with both abnormal G0/G1 and G0/G2 ratios in females.
Leptin levels (BMI corrected) were shown to correlate negatively with G-ratios in the CG combined group. Leptin levels correlated positively with the hormone FSH in the combined cohort and with testosterone in males.
Leptin is an important hormone predominantly released by adipocytes which has key roles in the regulation of energy balance, body weight, metabolism, neuroendocrine function, reproductive function and bone formation. Leptin exhibits its activity by binding to its fully active receptor (Ob-R, encoded by LEPR) which activates the JAK2 signalling mechanism and activators of transcription (STATs) [44–46] (see Fig. 4).
Reproductive signals integrate at different levels of the hypothalamic-pituitary-gonadal (HPG) axis involving GnRH, the pituitary hormones LH and FSH and gonadal hormones (see Fig. 4). Hypoleptinemia associated with energy deficiency influences several neuroendocrine axes including the thyroid, gonadal, cortisol and growth hormone axes. The identification of humans with mutations of the leptin and leptin receptor gene has illustrated how leptin deficiency influences the onset of puberty [47]. Leptin replacement has been shown to result in resumption in ovulation, increase in LH and oestradiol levels in blood and increase in follicular diameter and number in women with hypothalamic amenorrhea and replacement of leptin in deficient individuals has led to the successful treatment of hypogonadism by gonadotropin secretion and the restoration of puberty and fertility [48].
Although the primary site of leptin is the control of the HPG axis in the brain the actions of leptin have been shown on other reproductive systems including the gonads. Leptin expression has been confirmed in ovarian granulosa and cumulus cells and in mature human oocytes [49]. The leptin receptor also has been shown to be expressed in theca and granulosa cells on the human ovary with a putative function of leptin in the ovary to control folliculogenesis [50]. Expression of leptin receptors has also been reported in the testis [51].
Also leptin has a pro-inflammatory effect stimulating T Lymphocyte proliferation and cytokine production and is proposed to be an important link between nutritional status and the immune system [52]. LEPR activity on astrocytes has been proposed to actively regulate leptin transport across the blood brain barrier, a finding consistent with evidence that central regulatory changes of LepR during obesity and inflammation often occur in astrocytes [53, 54].
In our studies in Galactosaemia, we have identified a primary alteration of the leptin system in CG patients with low circulating leptin levels in males and females which are statistically significant in males with the expected correlation between log leptin and BMI evident in CG females who were mostly on HRT but not in CG males. The corresponding gene expression, the LEP gene and its receptor is significantly overexpressed in galactosaemia vs healthy controls (upregulated 8.88 fold, p < 0.01 and 6.38 fold, p < 0.001, respectively). Both the leptin gene LEP and its receptor gene LEPR are upregulated in CG males (n = 27) and CG females (n = 27).
From our earlier studies, we have suggested that abnormal N-glycosylation could putatively disrupt leptin-HPG signalling resulting from distorted Ob-r, soluble leptin receptor (sOb-R) and GnRH receptor (GnRH-R) function in galactosaemia [53].
Leptin exerts paracrine effects and is predominantly synthesized in adipose tissue. Expression of LEP gene is weight-course dependent and circulating leptin concentrations can be indicative of an adaptive pattern of LEP gene expression in obese subjects undergoing weight reduction [55]. Conversely, expression of LEPR gene, which is abundantly present in adipose tissue specimens from lean subjects, is found reduced in specimens obtained from obese patients [56]. As we could not examine adipose tissue specimens as part of this study it is unclear if the upregulation of expression of LEP and LEPR observed in white blood cells from this Galactosaemia cohort with the existence of decreased circulating leptin levels relates to altered transcriptional regulatory pathways, inflammatory mediators, or adaptive changes directly affecting leptin signalling molecules such as Suppressor of Cytokine Signaling 3 (SOCS3) [56].
The role and context of ANXA1 as an anti-inflammatory and apoptosis marker in females requires further elaboration. Primordial ovarian follicular atresia and dysgenesis can be noted in the first year of life in affected galactosaemia females. It is considered that abnormal signalling for development of this dysgenesis may occur prenatally and during the first years of life linked to an apoptosis pathway. It has recently been demonstrated that human ovarian explants exposed to ibuprofen (anti-inflammatory) showed reduced cell number, less proliferating cells, increased apoptosis and a dramatic loss of germ cell number [57]. Whether ANXA1 is a marker of apoptosis in this pathway remains to be determined.
Limitations of the study: The findings of this study involve a limited number of adult CG patients. As with many rare diseases it can be challenging to recruit large sample sizes, in particular for adult patients when attendance to clinic may be lower than anticipated. Further validation of the significant gene expression abnormalities could be indicated.
In addition, these data relate to a Dutch and Irish population. The population frequency of modifying SNPs for the glycan synthesis genes ALG9, MGAT3, FUT8, and B4GALT1 for the Dutch and Irish populations in comparison to other populations is currently not known. However, we did not note any statistical differences in gene expression differences between the Dutch and Irish study groups for these glycan synthesis genes. Also, as glycosylation is determined not only by genetic control but very significantly by epigenetic effects, variations in environmental influences including changes in dietary substrate (glucose and galactose exposure) would be expected to modify the specific glycan gene expression between subjects on an individual basis dependent on environmental exposures.
This study describes the gene expression in peripheral blood lymphocytes in adults with CG compared with N-glycosylation and endocrine hormonal measurements. The findings cannot describe the ovarian specific tissue expression or prenatal developmental expression or aberrant signalling effects presumed to be in existence prenatally with evidence of profound serum N-glycome intoxication effects (assembly defects) in CG intoxicated neonates with CG (25).
Conclusion
In this study we have demonstrated the validation of specific key N-glycan synthesis genes, and related genes associated with inflammation and fertility in CG patients. These findings were correlated biochemically with IgG N-glycan galactose incorporating phenotypic markers and also with phenotyping endocrine markers of POI in females and fertility in males. As shown in Fig. 4, this study of galactosaemia has illustrated the dysregulation of glycosylation, inflammatory response and leptin metabolism as possible cellular event sequences with apoptosis in CG. These studies further illustrate the complex nature of the CG phenotype, in particular in relation to fertility. We propose also that these studies of a rare inborn error of metabolism involving central N-glycosylation have also illustrated how significant oligosaccharide modification/glycosylation ‘modifier’ epigenetic gene effects involving recently described glycan modifier genes influencing cell signaling converts Galactosaemia and its outcome analysis from a simple mendelian disease to a complex trait, applicable to other related disorders [10, 11].
Methods
Study subjects and characterisation
For this multi-center cross sectional study, RNA samples from 54 adult CG adult patients were included for gene expression analysis. This included 36 Irish patients and 18 Dutch patients (age range 17–51). The genotypes and gender of these subjects are illustrated in Table 1.
The inclusion criterion was a diagnosis of CG (confirmed by the genotypes illustrated in Table 1 or by the presence of absent or virtually absent GALT enzyme activity). All study patients were maintained on a dietary galactose intake of < 1000 mg galactose/day. All patients entered in the study were compliant with regular clinic attendances and adherence to the galactose dietary restriction. Recent biochemical monitoring by RBC Gal-1-P in this adult population was available for only a subset of subjects. 16 apparently healthy adult control subjects aged between 20 and 40 years (9 females and 7 males) provided RNA samples as healthy controls.
Endocrine studies/hormonal assays
FSH, LH, and leptin were analysed in females and males. Oestradiol was measured in females and testosterone in male subjects. FSH, LH and oestradiol were measured by chemiluminescent immunoassay on the Abbott Architect i2000SR (Abbott Diagnostics, Illinois, USA). Between run coefficients of variation (CV) for LH and FSH were < 5%, while the maximum between run CV for oestradiol was 6.3% at 147 pmol/L. The assay limits of quantitation were 0.5 IU/L, 0.5 IU/L and 92 pmol/L for LH, FSH and oestradiol respectively. Testosterone was analysed using liquid chromatography tandem mass spectrometry (LC-MS/MS using a Waters Acquity UPLC coupled to a Xevo TQ tandem quadrupole using positive ion electrospray MS and multiple reaction monitoring (MRM). Serum leptin was measured using a human leptin immunoassay: an in-house immunoassay using DELFIA technology with antibodies and standards purchased from R&D Systems (R&D Systems Europe, Abington UK). CVs were 7.1% at 2.7 ng/mL, 3.9% at 14.9 ng/mL and 5.7% at 54.9 ng/mL (in-house data) and the Lower Limit of Detection (LLOQ) was 0.1 ng/mL. The concentration of sOb-R was determined using a commercially available ligand immunofunctional assay with reagents and standards purchased from R&D Systems. CVs ranged from 5.3 to 8.6% and the Lower Limit of Detection was 0.7 ng/mL.
IgG N-glycan analysis
The method of IgG glycoprofiling was as previously described [22, 23]. Briefly, serum was extracted from whole blood and immediately frozen. IgG was captured and eluted with Protein G spin plates. IgG was then denatured and N-glycans were released with Peptide N-glycosidase F (PNGase F). N-glycans were washed and labelled with anthranilamide (2-AB) and analysed on a Waters Acquity UPLC machine. The ratios of non-galactosylated, mono-galactosylated and di-galactosylated N-glycan species were compared (G-ratios)]. Statistical analysis was performed as described below with SPSS software (IBM).
TaqMan qPCR arrays
Within 3 h of collection, the whole blood samples were spun in BD Vacutainer cell preparation tubes (Fisher Scientific, Loughborough, UK), which allow rapid isolation of PBMCs. Total RNA was extracted from PBMCs using the RNeasy Plus Mini Kit (Qiagen Ltd., Manchester UK). cDNA was produced following reverse transcription of total RNA using the RT2 First Strand Kit (Qiagen Ltd).
Custom format ABI Taqman Array plates (Applied Biosystems, Foster City, CA, USA), were manufactured for 12 selected gene probes; ALG9, ANXA1, B4GALT1, FUT8, ICAM1, LEP, LEPR, MGAT1, MGAT3, SEPT4, UGDH and UGP2 along with three selected controls; ACTB, GAPDH and GUSB, of which ACTB and GUSB were selected as reference genes for normalisation of target genes of interest. cDNA was applied to the customised plates and Quantitative Real-Time PCR analysis was performed on an ABI PRSIM 7900 HT Sequence Detection System with a 96-well Fast thermal cycling block (Applied Biosystems).
Statistical analysis
For the RNA assays: Fold change of gene expression was quantified from raw CT scores with DataAssist software, version 3.01 using the 2ΔΔCT method (Applied Biosystems). Genes with undetermined CT scores were excluded. Two control and three galactosaemia samples with undetermined CTs for the selected normaliser genes were excluded entirely. This was likely due to insufficient PBMC pellet or excessive RNA contamination in these samples. Next, the fold change values were tested for normality of distribution with Shapiro Wilk’s test and checked for homogeneity of variance with Levene’s test.
A t-test was not suitable as most of the genes were not normally distributed. We thus opted for a Mann Whitney U test which evaluates differences between the groups irrespective of normality or variance. Type 1 errors were controlled with application of the Benjamini–Hochberg False Discovery Rate using R-software, version 3.4.0.
Spearman’s rank correlation coefficient (rs) was used to evaluate correlating data. Preparation of boxplots and scatterplots, testing of statistical differences between groups and correlation tests were conducted with SPSS software, version 24 (IBM, New York, USA).
Acknowledgements
The leptin and leptin receptor assays were performed by the NIHR Cambridge Biomedical Research Centre, Core Biochemistry Assay Laboratory. Ms. Marguerite McMahon is thanked for her collaboration for the testosterone assays.
Funding
Funding for these studies was provided by a HRB/HRA award (no. 623 to EPT).
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ALG9
Alpha-1,2-Mannosyltransferase
- AMH
Anti Mullerian Hormone
- ANXA1
Annexin A1
- B4GALT
UDP-Gal:BetaGlcNAc Beta 1,4- Galactosyltransferase
- cDNA
Complimentary Deoxyribonucleic acid
- CV
Coefficient of Variance
- ER
Endoplasmic reticulum
- FSH
Follicle stimulating hormone
- FUT8
- G0
Non-galactosylated
- G1
Monogalactosylated
- G2
Digalactosylated
- Gal-1-P
Galactose-1-phosphate
- GALE
Galactose epimerase
- GALK
Galactokinase
- GALT
Galactose-1-phosphate uridylyltransferase
- GnRH
Gonadotropin Releasing Hormone
- GP
Glycan peak
- HILIC-UPLC
Hydrophilic Interaction Ultra-high Performance Liquid Chromatography
- HRT
Hormone Replacement Therapy
- ICAM1
Intercellular Adhesion Molecule 1
- IMPase1
Inositol monophosphatase 1
- LEP
Leptin
- LEPR
Leptin receptor
- LH
Luteinising hormone
- MGAT1
Alpha-1,3-mannosyl-glycoprotein 2-beta-N-acetylglucosaminyltransferase
- MGAT3
Beta-1,4-mannosyl-glycoprotein 4-beta-N-acetylglucosaminyltransferase
- NS
Not significant
- PNGase
Peptide N-glycosidase F
- POI
Primary ovarian insufficiency
- qRT-PCR
Quantitative real-time polymerase chain reaction
- RBC
Red blood cell
- RNA
Ribonucleic acid
- SD
Standard deviation
- SEPT4
Septin 4
- sOb-R
Soluble leptin receptor
- UDP
Uridine diphosphate
- UGDH
UDP-Glucose 6-Dehydrogenase
- UGP2
UDP-glucose pyrophosphorylase 2
- UPR
Unfolded Protein Response
Authors’ contributions
HOC, EPT and PPD were involved in designing the concept of the study and oversight. HOC and EPT drafted the manuscript. HOC, MERG, AMB, IK, DD, JJB, ROF and KS contributed to the acquisition and interpretation of all data. JJB and MF contributed to the analysis and interpretation of hormonal data. CM contributed to the interpretation and analysis of the gene expression studies. MG and HOC provided the statistical analysis. PB assisted with interpretation of the leptin analysis. All authors contributed to the overall analysis of the results and in writing and reviewing the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Ethical approval for this study was obtained from the Ethics Committees of the Children’s University Hospital, Mater Misericordiae University Hospital, Maastricht University Medical Centre. Ethics committee review was waived at the Academic Medical Center, Amsterdam based on the coordinating site review. All participants provided voluntary informed consent to enter the study.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hugh-Owen Colhoun, Email: hcolhoun@hotmail.com.
Estela M. Rubio Gozalbo, Email: estela.rubio@mumc.nl
Annet M. Bosch, Email: a.m.bosch@amc.uva.nl
Ina Knerr, Email: ina.knerr@cuh.ie.
Charlotte Dawson, Email: charlotte.dawson@uhb.nhs.uk.
Jennifer Brady, Email: jbrady@mater.ie.
Marie Galligan, Email: marie.galligan@ucd.ie.
Karolina Stepien, Email: kstepien@doctors.org.uk.
Roisin O’Flaherty, Email: roisin.oflaherty@nibrt.ie.
C. Catherine Moss, Email: catherine.moss@ucd.ie
P. Peter Barker, Email: peter.barker@addenbrookes.nhs.uk
Maria Fitzgibbon, Email: mfitzgibbon@mater.ie.
Peter P. Doran, Email: peter.doran@ucd.ie
Eileen P. Treacy, Phone: 353 1 8034668, Email: etreacy@mater.ie
References
- 1.Jumbo-Lucioni PP, Garber K, Kiel J, Baric I, Berry GT, Bosch A, et al. Diversity of approaches to classic galactosemia around the world: a comparison of diagnosis, intervention, and outcomes. J Inherit Metab Dis. 2012;35(6):1037–1049. doi: 10.1007/s10545-012-9477-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coss KP, Doran PP, Owoeye C, Codd MB, Hamid N, Mayne PD, et al. Classical Galactosaemia in Ireland: incidence, complications and outcomes of treatment. J Inherit Metab Dis. 2013;36(1):21–27. doi: 10.1007/s10545-012-9507-9. [DOI] [PubMed] [Google Scholar]
- 3.Demirbas D, Coelho AI, Rubio-Gozalbo ME, Berry GT. Hereditary Galactosemia. Metabolism. 2018; 10.1016/j.metabol.2018.01.0125. (Epub ahead of print) [DOI] [PubMed]
- 4.Hughes J, Ryan S, Lambert D, Geoghegan O, Clark A, Rogers Y, et al. Outcomes of siblings with classical galactosemia. J Pediatr. 2009;154(5):721–726. doi: 10.1016/j.jpeds.2008.11.052. [DOI] [PubMed] [Google Scholar]
- 5.Waisbren SE, Potter NL, Gordon CM, Green RC, Greenstein P, Gubbels CS, et al. The adult Galactosemic phenotype. J Inherit Metab Dis. 2012;35(2):279–286. doi: 10.1007/s10545-011-9372-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fridovich-Keil J, Walter J. Galactosaemia. In: Valle D, editor. The Online Metabolic and Molecular Bases of Inherited Disease, OMMBID. The McGraw-Hill Companies, Inc: New York, New York, USA; 2008. [Google Scholar]
- 7.Coelho AI, Rubio-Gozalbo ME, Vicente JB, Rivera I. Sweet and sour: an update on classic galactosemia. J Inherit Metab Dis. 2017;40(3):325–342. doi: 10.1007/s10545-017-0029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Viggiano E, Marabotti A, Politano L, Burlina A. Galactose-1-phosphate uridyltransferase deficiency: a literature review of the putative mechanisms of short and long-term complications and allelic variants. Clin Genet. 2018;93(2):206–215. doi: 10.1111/cge.13030. [DOI] [PubMed] [Google Scholar]
- 9.Maratha A, Stockmann H, Coss KP, Estela Rubio-Gozalbo M, Knerr I, Fitzgibbon M, et al. Classical galactosaemia: novel insights in IgG N-glycosylation and N-glycan biosynthesis. Eur J Hum Genet. 2016;24(7):976–984. doi: 10.1038/ejhg.2015.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dipple KM, McCabe ER. Modifier genes convert "simple" Mendelian disorders to complex traits. Mol Genet Metab. 2000;71(1–2):43–50. doi: 10.1006/mgme.2000.3052. [DOI] [PubMed] [Google Scholar]
- 11.Luac G, Kristic J, Zoldos V. Glycans - the third revolution in evolution. Front Genet. 2014;5:145. doi: 10.3389/fgene.2014.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lai K, Langley SD, Khwaja FW, Schmitt EW, Elsas LJ. GALT deficiency causes UDP-hexose deficit in human galactosemic cells. Glycobiology. 2003;13(4):285–294. doi: 10.1093/glycob/cwg033. [DOI] [PubMed] [Google Scholar]
- 13.Coss KP, Byrne JC, Coman DJ, Adamczyk B, Abrahams JL, Saldova R, et al. IgG N-glycans as potential biomarkers for determining galactose tolerance in classical Galactosaemia. Mol Genet Metab. 2012;105(2):212–220. doi: 10.1016/j.ymgme.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 14.Frederick AB, Cutler DJ, Fridovich-Keil JL. Rigor of non-dairy galactose restriction in early childhood, measured by retrospective survey, does not associate with severity of five long-term outcomes quantified in 231 children and adults with classic galactosemia. J Inherit Metab Dis. 2017;40(6):813–821. doi: 10.1007/s10545-017-0067-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balakrishnan B, Chen W, Tang M, Huang X, Cakici DD, Siddiqi A, et al. Galactose-1 phosphate uridylyltransferase (GalT) gene: a novel positive regulator of the PI3K/Akt signaling pathway in mouse fibroblasts. Biochem Biophys Res Commun. 2016;470(1):205–212. doi: 10.1016/j.bbrc.2016.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krabbi K, Uudelepp ML, Joost K, Zordania R, Ounap K. Long-term complications in Estonian galactosemia patients with a less strict lactose-free diet and metabolic control. Mol Genet Metab. 2011;103(3):249–253. doi: 10.1016/j.ymgme.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 17.Yuzyuk T, Viau K, Andrews A, Pasquali M, Longo N. Biochemical changes and clinical outcomes in 34 patients with classic galactosemia. J Inherit Metab Dis. 2018;41(2):197–208. doi: 10.1007/s10545-018-0136-9. [DOI] [PubMed] [Google Scholar]
- 18.Gubbels CS,Thomas CM, Wodzig WK, Olthaar AJ, Jaeken J, Sweep FC, Rubio-Gozalbo ME FSH isoform pattern in classic galactosaemia. J Inherit Metab Dis. 2011;34(2):387–390. doi: 10.1007/s10545-010-9180-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gubbels CS, Ja L, Evers JL, Bierau J, Menheere PP, Robben SG, Rubio-Gozalbo ME. Primary ovarian insufficiency in classic galactosaemia: role of FSH dysfunction and timing of the lesion. J Inherit Metab Dis. 2013;36(1):29–34. doi: 10.1007/s10545-012-9497-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spencer JB, Badik JR, Ryan EL, Gleason TJ, Broadaway KA, Epstein MP, Fridovich-Keil JL. Modifiers of ovarian function in girls and women with classic galactosaemia. J Clin Endocrinol Metab. 2013;98(7):E1257–E1265. doi: 10.1210/jc.2013-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gudelj I, Lauc G, Pezer M. Immunoglobulin G glycosylation in aging and diseases. Cell Immunol. 2018; [DOI] [PubMed]
- 22.Stockmann H, Coss KP, Rubio-Gozalbo ME, Knerr I, Fitzgibbon M, Maratha A, et al. IgG N-glycosylation galactose incorporation ratios for the monitoring of classical Galactosaemia. JIMD Rep. 2016;27:47–53. doi: 10.1007/8904_2015_490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colhoun Hugh Owen, Treacy Eileen P, MacMahon Marguerite, Rudd Pauline M, Fitzgibbon Maria, O'Flaherty Roisin, Stepien Karolina M. Validation of an automated ultraperformance liquid chromatography IgG N-glycan analytical method applicable to classical galactosaemia. Annals of Clinical Biochemistry: International Journal of Laboratory Medicine. 2018;55(5):593–603. doi: 10.1177/0004563218762957. [DOI] [PubMed] [Google Scholar]
- 24.Wahl A, van den Akker E, Klaric L, Stambuk J, Benedetti E, Plomp R, et al. Genome-wide association study on immunoglobulin G glycosylation patterns. Front Immunol. 2018;9:277. doi: 10.3389/fimmu.2018.00277:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coman DJ, Murray DW, Byrne JC, Rudd PM, Bagaglia PM, Doran PD, et al. Galactosemia, a single gene disorder with epigenetic consequences. Pediatr Res. 2010;67(3):286–292. doi: 10.1203/PDR.0b013e3181cbd542. [DOI] [PubMed] [Google Scholar]
- 26.Coss KP, Treacy EP, Cotter EJ, Knerr I, Murray DW, Shin YS, et al. Systemic gene dysregulation in classical Galactosaemia: is there a central mechanism? Mol Genet Metab. 2014;113(3):177–187. doi: 10.1016/j.ymgme.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 27.Knerr I, Coss KP, Doran PP, Hughes J, Wareham N, Burling K, et al. Leptin levels in children and adults with classic galactosaemia. JIMD Rep. 2013;9:125–131. doi: 10.1007/8904_2012_191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knerr I, Coss KP, Kratzsch J, Crushell E, Clark A, Doran P, et al. Effects of temporary low-dose galactose supplements in children aged 5-12 y with classical galactosemia: a pilot study. Pediatr Res. 2015;78(3):272–279. doi: 10.1038/pr.2015.107. [DOI] [PubMed] [Google Scholar]
- 29.Rubio-Gozalbo ME, Gubbels CS, Bakker JA, Meenheere PPCA, Wodzig WKWH, Land JA. Gonadal function in male and female patients with classic galactosaemia. Hum Reprod Update. 2010;16(2):177–188. doi: 10.1093/humupd/dmp038. [DOI] [PubMed] [Google Scholar]
- 30.Thakur M, Feldman G, Puscheck EE. Primary ovarian insufficiency in classic galactosemia: current understanding and future research opportunities. J Assist Reprod Genet. 2018;35(1):3–16. doi: 10.1007/s10815-017-1039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mamsen Linn Salto, Kelsey Thomas W., Ernst Erik, Macklon Kirsten Tryde, Lund Allan Meldgaard, Andersen Claus Yding. Cryopreservation of ovarian tissue may be considered in young girls with galactosemia. Journal of Assisted Reproduction and Genetics. 2018;35(7):1209–1217. doi: 10.1007/s10815-018-1209-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banerjee S, Chakraborty P, Saha P, Bandyopadhyay SA, Banerjee S, Kabir SN. Ovotoxic effects of galactose involve attenuation of follicle-stimulating hormone bioactivity and up-regulation of granulosa cell p53 expression. PLoS One. 2012;7(2):e30709. doi: 10.1371/journal.pone.0030709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gubbels CS, Welt CK, Dumoulin JC, Robben SG, Gordon CM, Dunselman GA, et al. The male reproductive system in classic galactosemia: cryptorchidism and low semen volume. J Inherit Metab Dis. 2013;36(5):779–786. doi: 10.1007/s10545-012-9539-1. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Y, Sato Y, Isaji T, Fukuda T, Matsumoto A, Miyoshi E, et al. Branched N-glycans regulate the biological functions of integrins and cadherins. FEBS J. 2008;275(9):1939–1948. doi: 10.1111/j.1742-4658.2008.06346.x. [DOI] [PubMed] [Google Scholar]
- 35.Jumbo-Lucioni P, Parkinson W, Broadie K. Overelaborated synaptic architecture and reduced synaptomatrix glycosylation in a Drosophila classic galactosemia disease model. Dis Model Mech. 2014;7(12):1365–1378. doi: 10.1242/dmm.017137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takahashi M, Kuroki Y, Ohtsubo K, Taniguchi N. Core fucose and bisecting GlcNAc, the direct modifiers of the N-glycan core: their functions and target proteins. Carbohydr Res. 2009;344(12):1387–1390. doi: 10.1016/j.carres.2009.04.031. [DOI] [PubMed] [Google Scholar]
- 37.Karsten CM, Pandey MK, Figge J, Kilchenstein R, Taylor PR, Rosas M, et al. Anti-inflammatory activity of IgG1 mediated by fc galactosylation and association of FcgammaRIIB and dectin-1. Nat Med. 2012;18(9):1401–1406. doi: 10.1038/nm.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Novokmet M, Lukić E, Vučković F, Durić Ž, Keser T, Rajšl K, et al. Changes in IgG and total plasma protein glycomes in acute systemic inflammation. Sci Rep. 2014;4:4347. doi: 10.1038/srep04347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perretti M, D'Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. 2009;9(1):62–70. doi: 10.1038/nri2470. [DOI] [PubMed] [Google Scholar]
- 40.de Jong SE, Selman MH, Adegnika AA, Amoah AS, van Riet E, Kruize YC, et al. IgG1 fc N-glycan galactosylation as a biomarker for immune activation. Sci Rep. 2016;6:28207. doi: 10.1038/srep28207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rothlein R, Dustin ML, Marlin SD, Springer TA. A human intercellular adhesion molecule (ICAM-1) distinct from LFA-1. J Immunol. 1986;137(4):1270–1274. [PubMed] [Google Scholar]
- 42.Kissel H, Georgescu MM, Larisch S, Manova K, Hunnicutt GR, Steller H. The Sept4 septin locus is required for sperm terminal differentiation in mice. Dev Cell. 2005;8(3):353–364. doi: 10.1016/j.devcel.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 43.Berry GT, Moate PJ, Reynolds RA, Yager CT, Ning C, Boston RC, et al. The rate of de novo galactose synthesis in patients with galactose-1-phosphate uridyltransferase deficiency. Mol Genet Metab. 2004;81(1):22–30. doi: 10.1016/j.ymgme.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 44.Allison MB, Myers MG. Conecting leptin signaling to biological function. J Endocrinol. 2014;223(1):T25–T35. doi: 10.1530/JOE-14-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chou SH, Mantzoros C. 20 years of leptin: role of leptin in human reproductive disorders. J Endocrinol. 2014;223(1):T49–T62. doi: 10.1530/JOE-14-0245. [DOI] [PubMed] [Google Scholar]
- 46.Polyzos SA, Mantzoros CS. Leptin in health and disease: facts and expectations at its twentieth anniversary. Metabolism. 2015;64(1):5–12. doi: 10.1016/j.metabol.2014.10.017. [DOI] [PubMed] [Google Scholar]
- 47.Farooqi IS, O'Rahilly S. 20 years of leptin: human disorders of leptin action. J Endocrinol. 2014;223(1):T63–T70. doi: 10.1530/JOE-14-0480. [DOI] [PubMed] [Google Scholar]
- 48.Welt CK, Chan JL, Bullen J, Murphy R, Smith P, DePaoli AM, et al. Recombinant human leptin in women with hypothalamic amenorrhea. N Engl J Med. 2004;351(10):987–997. doi: 10.1056/NEJMoa040388. [DOI] [PubMed] [Google Scholar]
- 49.Cioffi J. The expression of leptin and its receptors in pre-ovulatory human follicles. Molecular Human Reproduction. 1997;3(6):467–472. doi: 10.1093/molehr/3.6.467. [DOI] [PubMed] [Google Scholar]
- 50.Karlsson C, Lindell K, Svensson E, Bergh C, Lind P, Billig H, et al. Expression of functional leptin receptors in the human ovary. J Clin Endocrinol Metab. 1997;82(12):4144–4148. doi: 10.1210/jcem.82.12.4446. [DOI] [PubMed] [Google Scholar]
- 51.Tena-Sempere M, Barreiro ML. Leptin in male reproduction: the testis paradigm. Mol Cell Endocrinol. 2002;188(1–2):9–13. doi: 10.1016/S0303-7207(02)00008-4. [DOI] [PubMed] [Google Scholar]
- 52.Gan L, Guo K, Cremona ML, McGraw TE, Leibel RL, Zhang Y. TNF-alpha up-regulates protein level and cell surface expression of the leptin receptor by stimulating its export via a PKC-dependent mechanism. Endocrinology. 2012;153(12):5821–5833. doi: 10.1210/en.2012-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fruhbeck G. Intracellular signalling pathways activated by leptin. Biochem J. 2006;393(Pt 1):7–20. doi: 10.1042/BJ20051578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hsuchou H, Kastin AJ, Tu H, Joan Abbott N, Couraud PO, Pan W. Role of astrocytic leptin receptor subtypes on leptin permeation across hCMEC/D3 human brain endothelial cells. J Neurochem. 2010;115(5):1288–1298. doi: 10.1111/j.1471-4159.2010.07028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Knerr I, Herzog D, Rauh M, Rascher W, Horbach T. Leptin and ghrelin expression in adipose tissues and serum levels in gastric banding patients. Eur J Clin Investig. 2006;36(6):389–394. doi: 10.1111/j.1365-2362.2006.01642.x. [DOI] [PubMed] [Google Scholar]
- 56.Séron K, Corset L, Vasseur F, Boutin P, Gómez-Ambrosi J, Salvador J, Frühbeck G, Froguel P. Distinct impaired regulation of SOCS3 and long and short isoforms of the leptin receptor in visceral and subcutaneous fat of lean and obese women. Biochem Biophys Res Commun. 2006;6; 348(4):1232–1238. doi: 10.1016/j.bbrc.2006.07.068. [DOI] [PubMed] [Google Scholar]
- 57.Leverrier-Penna S, Mitchell R T, Becker E, Lecante L, Ben Maamar M, Homer N, Lavoué V, Kristensen D M, Dejucq-Rainsford N, Jégou B, Mazaud-Guittot S. Ibuprofen is deleterious for the development of first trimester human fetal ovary ex vivo. Human Reproduction. 2018;33(3):482–493. doi: 10.1093/humrep/dex383. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.