

Herpesviruses assemble capsids and encapsidate their genomes by a process that is unlike those of other mammalian viruses but is similar to those of some bacteriophage. Many important aspects of herpesvirus morphogenesis remain enigmatic, including how the capsid shell matures into a stable angularized configuration. Capsid maturation is triggered by activation of a protease that cleaves an internal protein scaffold. We report on the fortuitous discovery that a region of the major capsid protein that is exposed on the outer surface of the capsid also contributes to capsid maturation, demonstrating that the morphogenesis of the capsid shell from its procapsid precursor to the mature angularized form is dependent upon internal and external components of the megastructure.
KEYWORDS: herpesvirus, HSV-1, capsid, procapsid, VP5, maturation, assembly, microscopy, VP5 apical region, axon transport, herpes simplex virus, major capsid protein
ABSTRACT
The herpesvirus capsid assembles in the nucleus as an immature procapsid precursor built around viral scaffold proteins. The event that initiates procapsid maturation is unknown, but it is dependent upon activation of the VP24 internal protease. Scaffold cleavage triggers angularization of the shell and its decoration with the VP26 and pUL25 capsid-surface proteins. In both the procapsid and mature angularized capsid, the apical region of the major capsid protein (VP5) is surface exposed. We investigated whether the VP5 apical region contributes to intracellular transport dynamics following entry into primary sensory neurons and also tested the hypothesis that conserved negatively charged amino acids in the apical region contribute to VP26 acquisition. To our surprise, neither hypothesis proved true. Instead, mutation of glutamic acid residues in the apical region delayed viral propagation and induced focal capsid accumulations in nuclei. Examination of capsid morphogenesis based on epitope unmasking, capsid composition, and ultrastructural analysis indicated that these clusters consisted of procapsids. The results demonstrate that, in addition to established events that occur inside the capsid, the exterior capsid shell promotes capsid morphogenesis and maturation.
IMPORTANCE Herpesviruses assemble capsids and encapsidate their genomes by a process that is unlike those of other mammalian viruses but is similar to those of some bacteriophage. Many important aspects of herpesvirus morphogenesis remain enigmatic, including how the capsid shell matures into a stable angularized configuration. Capsid maturation is triggered by activation of a protease that cleaves an internal protein scaffold. We report on the fortuitous discovery that a region of the major capsid protein that is exposed on the outer surface of the capsid also contributes to capsid maturation, demonstrating that the morphogenesis of the capsid shell from its procapsid precursor to the mature angularized form is dependent upon internal and external components of the megastructure.
INTRODUCTION
The Herpesviridae family of large double-stranded DNA viruses is divided into three subfamilies: the Alpha-, Beta-, and Gammaherpesvirinae. Several members of the Alphaherpesvirinae, including herpes simplex virus type 1 (HSV-1), exhibit robust neuroinvasive ability (1). HSV-1 infection begins in epithelial cells but quickly spreads to nerve terminals and invades the sensory and autonomic ganglia by retrograde axonal transport (2). Within these neuronal cells, HSV-1 establishes the lifelong, recurrent infection that is characteristic of these viruses. All herpesviruses share a virion morphology consisting of three layers: nucleocapsid, tegument, and envelope. Herpesvirus nucleocapsids are approximately 125 nm in diameter, conform to a T=16 icosahedral structure, and consist of 162 capsomeres: 150 hexamers, 11 pentamers, and one portal vertex (3). HSV-1 nucleocapsids are estimated to withstand 18 atmospheres of internal pressure exerted by the tightly packed 152-kbp genome until the capsid docks at a nuclear pore and releases the DNA into the nucleus (4, 5). Delivery of the viral genome initiates a cascade of gene expression leading to the assembly of nascent virions.
Extensive efforts have been undertaken to understand herpesvirus capsid assembly and maturation, which includes analyses of baculovirus and cell-free systems (6–11), temperature-sensitive mutants (12, 13), and three-dimensional reconstructions from cryoelectron microscopy (cryo-EM) (14–20). From this work, an overarching model of HSV-1 capsid morphogenesis has been developed. The major capsid protein (VP5), triplexes (VP19c and VP23), portal vertex (pUL6), and internal scaffold proteins self-assemble into a spherical procapsid within the cell nucleus. There are two types of scaffold proteins, small (pUL26.5) and large (pUL26). Once assembly of the procapsid is complete the catalytic activity of the VP24 protease, housed within the amino terminus of the large scaffold protein, is triggered (21). VP24 cleaves itself from the scaffold and cleaves a release site in the C terminus of the small and large scaffold proteins, disconnecting them from the shell floor (9, 22, 23). These events cause the capsid shell to angularize into a stable form and become a substrate for the binding of two accessory capsid surface proteins: pUL25 and VP26 (22, 24–26). The scaffold fragments internal to the capsid are expelled as the viral genome is packaged, but VP24 is retained (26–32). This process produces three angularized capsid types within infected cell nuclei: capsids that retained the scaffold and lack DNA (B capsids), capsids that expelled the scaffold but failed to stably encapsidate the viral genome (A capsids), and nucleocapsids (C capsids).
The majority of the capsid shell mass is comprised of 955 copies of the 149-kDa VP5 protein (33–36), which is arranged into hexamers and pentamers. Pentamers are found at 11 of the capsid vertices while hexamers occupy the remainder of the icosahedral lattice, with the exception of the unique portal vertex. Hexamers are crowned with VP26 (12 kDa), and pentamers are associated with complexes of pUL25 and pUL17 that radiate outward from the pentamers and serve as tegument binding sites (5, 19, 37–40). The VP5 monomer is a large, tower-like protein divided into three domains: the upper, middle, and floor. The apical region of the upper domain, which consists of a polyproline loop plus an adjacent sequence that serves as an antibody binding epitope (41), is surface exposed in hexamers and pentamers and does not participate in subunit interactions within the capsid shell. Instead, the apical region is proposed to be important for binding VP26 on hexamers and tegument on pentamers (42).
Our studies of herpesvirus neuroinvasion demonstrate that capsid-bound tegument proteins contribute to neuronal transmission and retrograde axonal transport (43–45). To examine if the capsid surface participates in these processes, a collection of HSV-1 variants with mutations in the VP5 apical region was made. Initial characterization of these viruses in cell culture revealed only modest changes in propagation, with the exception of a virus mutated at two conserved acidic residues in the polyproline loop. Neither the polyproline loop mutant nor a mutant lacking the majority of the antibody epitope was impacted for retrograde axonal transport. Further examination of the polyproline loop mutant indicated that VP5 expression and VP26 incorporation into capsids were also unimpaired. Using a combination of fluorescence and ultrastructural imaging, we determined that the polyproline loop mutant offers a unique example of a procapsid maturation defect that maps to the external face of the capsid shell.
(This article was submitted to an online preprint archive [46].)
RESULTS
Mutating VP5 apical region residues impacts herpes simplex virus spread in cell culture.
The majority of the HSV-1 major capsid protein, VP5, is integral to the capsid icosahedral structure and its constituent capsomeres. An exception is a small apical region within the VP5 upper domain that is exposed on the capsid surface (Fig. 1A to C), which consists of a polyproline loop and a binding site for a capsid-specific antibody (41, 42). The accessibility of the apical domain to antibody indicates it could interface with cellular or viral proteins to promote infection. To assess the importance of the apical region during HSV-1 infection, an alignment of eight alphaherpesviruses was used to inform a mutational approach (Fig. 1D). Based on sequence conservation, five VP5 mutants were made by two-step RED-mediated recombination in the F strain of HSV-1 (Fig. 1E). The recombinant viruses exhibited a reduction in cell-cell spread, as evidenced by decreased plaque diameters relative to those of wild-type (WT) virus (Fig. 2A). The most significant decrease in plaque size occurred when two glutamic acid residues in the polyproline loop, E846 and E851, were mutated to alanines (EE>AA). These glutamic acid residues, shown on the VP5 crystal structure in magenta and blue in Fig. 1A to D, are located in the most apical region of the VP5 monomer (Fig. 1C and D). The decrease in plaque size was only observed when both glutamic acid residues were mutated to alanine, and the defect was rescued when both mutations were repaired (Fig. 2B).
FIG 1.

Targeted mutation of the VP5 apical region. (A to D) Representation of the HSV-1 VP5 upper domain crystal structures as arranged in a top view of a peripentonal hexamer (A) and a pentamer (B), and a side view of a peripentonal hexamer (C) and a pentamer (D). Rotations of panels C and D are arbitrary to best display the glutamic acid residues. The apical region is colored green, and side chains of residues E846 (magenta) and E851 (blue) are shown as sticks. (E) Alignment of the apical region of eight alphaherpesvirus major capsid proteins. Amino acid positions for the HSV-1 sequence are indicated below. (F) HSV-1 VP5 apical region mutants used in this study. Predicted amino acid changes are in red.
FIG 2.

Impact of the VP5 mutations on plaque size. Vero cells were infected with VP5 apical region mutants (A) and additional VP5 glutamic acid mutants (B). Plaque diameters were measured 5 days postinfection. Diameters are represented as a percentage of the mean plaque diameter of wild-type HSV-1 strain F (WT). Repaired alleles are listed as E846A>E and E851A>E. Error bars indicate the standard deviations from three independent experiments. Asterisks indicate a statistically significant difference from the WT or VP5 EE>AA, as determined by Tukey's multiple-comparison test (****, P ≤ 0.0001).
The VP5 apical region does not contribute to retrograde viral transport.
The capsid-bound tegument proteins, pUL36 and pUL37, are effectors of intracellular capsid transport (5, 43–45, 47, 48). To examine if the capsid surface-exposed region of VP5 contributes to its transport on microtubules, primary sensory neurons were infected with the EE>AA and Δ6F10 mutant viruses (Fig. 1F). To visualize individual capsids in the process of retrograde axonal transport by time-lapse fluorescence microscopy, the viruses were further modified to encode an mCherry fusion to the N terminus of the maturation protease, VP24 (32). The mCherry-VP24 fusion was chosen because the internal location of VP24 inside the capsid shell predicts that the mCherry tag should not interplay with the mutations introduced into the VP5 apical region. Capsid dynamics were monitored during the first hour postinfection (hpi), and continuous periods of retrograde motion (runs) were analyzed for average velocity and distance traveled. The distribution of run velocities were consistently Gaussian for the wild-type, EE>AA, and Δ6F10 viruses (R2 of ≥0.96 for each), and run distances were decaying exponentials (R2 of ≥0.98 for each), the latter being consistent with processive motion (Fig. 3). These transport dynamics were consistent between the WT and mutant viruses, demonstrating that the VP5 apical region mutations did not impair microtubule-based retrograde axonal transport.
FIG 3.

Mutation of the VP5 upper domain did not impair retrograde axonal transport. Primary sensory neurons were infected with derivatives of the VP5 wild type (WT), glutamic acid mutant (EE>AA), and Δ6F10 mutant that encode an mCherry-VP24 fusion to allow for tracking of individual capsids in axons. Intracellular capsid transport was monitored by time-lapse fluorescence microscopy during the first hour of infection. Run velocity (A) and run distance (B) profiles of individual capsids are representative of three independent experiments.
The VP5 EE>AA mutant forms intranuclear clusters of capsid proteins.
Because the defect underlying the reduction in plaque diameter was not ascribed to defects in retrograde trafficking, de novo capsid production was examined next. Vero cells infected with the mCherry-VP24 variants of the EE>AA and Δ6F10 mutants were examined by live-cell microscopy at 8.5 hpi. As expected, the nuclei of cells infected with either of the mutant viruses filled with diffraction-limited punctae, consistent with high loads of dispersed capsids (32); however, the EE>AA mutant additionally produced large intranuclear structures (Fig. 4A). These structures were also observed in the absence of the mCherry-VP24 tag by immunofluorescence using a VP5 antibody but were not evident with WT HSV-1 or a repair of the EE>AA mutant (Fig. 4B). These results indicated that the EE>AA mutations induced intranuclear clusters of the VP24 and VP5 capsid proteins and that the focal clusters were not an artifact of either the VP24 fluorescent tag or fixation.
FIG 4.
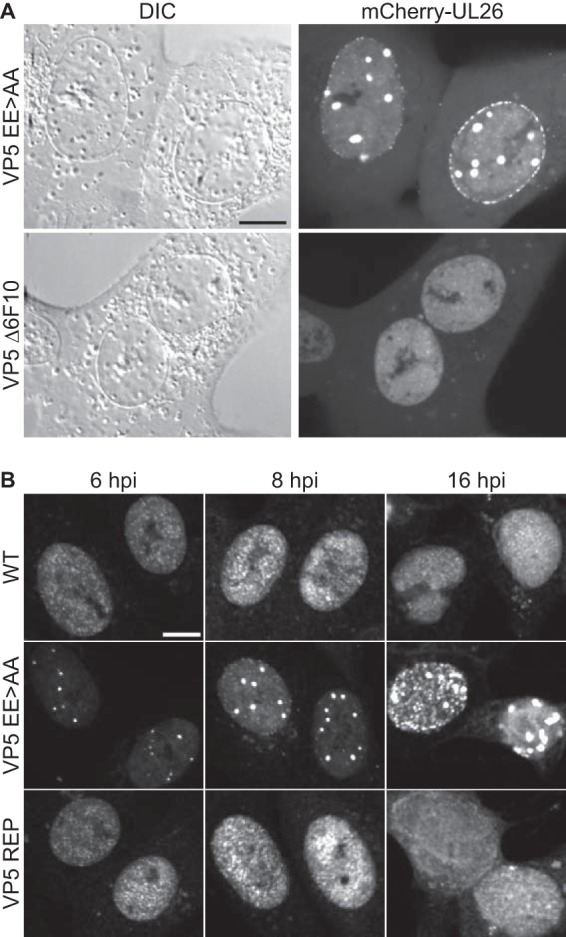
VP5 EE>AA mutant forms intranuclear clusters of capsid proteins. Vero cells were infected at an MOI of 5. (A) Live-cell imaging at 8.5 hpi using derivatives of HSV-1 encoding mCherry-VP24 and either VP5 EE>AA or Δ6F10. (B) Fixed-cell immunofluorescence time course of cells infected with unmodified HSV-1 (WT), EE>AA mutant, and VP5 repair (REP). Cells were fixed at the indicated time postinfection and stained with an αVP5 antibody. Scale bars, 10 μm.
HSV-1 propagation kinetics are reduced by VP5 EE>AA mutation.
Single-step growth kinetics of the EE>AA mutant were attenuated relative to the wild-type and repair viruses (Fig. 5). At 8 and 12 hpi, the EE>AA mutant exhibited a decrease in cell-associated and supernatant PFU, respectively, but attained titers approaching those of the wild type by 30 hpi. The delays in PFU production were confirmed in triplicate: the EE>AA mutant cell-associated titers were consistently 6% ± 1.6% of those of the repaired virus at 8 hpi, and the supernatant titers were 8% ± 3.8% of those of the repaired virus at 12 hpi. We conclude that the aberrant nuclear foci correlated to decreased rates of virus propagation, as well as plaque size (Fig. 2), but had limited impact on the number of PFU per cell (burst size).
FIG 5.
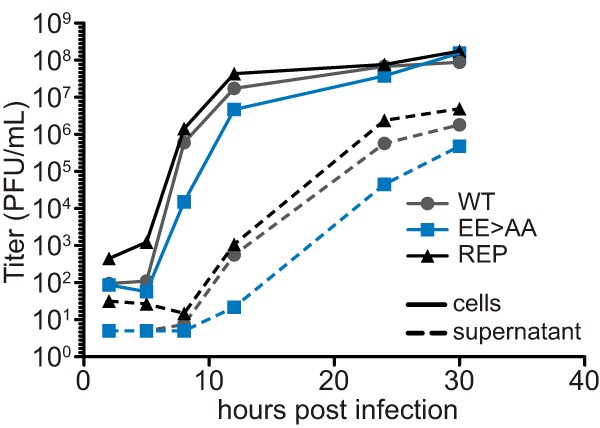
VP5 glutamic acid residues support HSV-1 propagation. Single-step replication kinetics of HSV-1 WT, VP5 glutamic acid mutant (EE>AA), and VP5 repair in Vero cells are shown following infection at an MOI of 10. Cells (solid lines) and supernatants (dashed lines) were collected at the indicated times, and titers were determined by plaque assay on Vero cells. Cell-associated PFU at 8 hpi and supernatant PFU at 12 hpi were confirmed in a separate experiment (see the text).
The conserved glutamic acids of the VP5 polyproline loop are dispensable for VP26 assembly on capsids.
Despite the large foci of VP5 and VP24 proteins in the nuclei of cells infected with the EE>AA mutant, the production of PFU indicated that capsid assembly occurred but is impaired. We were particularly interested in whether acquisition of VP26 to capsids occurred with the EE>AA mutant, because negatively charged residues within the VP5 polyproline loop are predicted to mediate its binding to the surface of capsid hexamers (42). To analyze the protein composition of EE>AA capsids, intranuclear capsids were isolated from Vero cells infected with wild-type HSV-1 (WT) or the glutamic acid mutant (EE>AA). A VP26-null mutant (Δ26) was included as a control. Empty capsids (A capsids), scaffold-containing capsids (B capsids), and genome-containing capsids (C capsids) were separated by density ultracentrifugation and visualized by light scattering. The EE>AA and VP26-null viruses produced each capsid species, similar to the wild type (Fig. 6A). The protein compositions of the A, B, and C capsids produced from the wild-type and EE>AA viruses were indistinguishable from one another; notably, capsids of the EE>AA virus possessed levels of VP26 comparable to those of wild-type capsids (Fig. 6B). These data indicate that the VP5 EE>AA mutant assembles A, B, and C capsids and that E846 and E851 are dispensable for VP26 incorporation.
FIG 6.

VP5 EE>AA capsids acquire VP26. (A) A, B, and C capsids were isolated from nuclei of Vero cells infected with HSV-1 over a 20 to 50% sucrose gradient and visualized by light scattering. (B) Protein profiles of the capsids observed by silver staining of a 4 to 20% gradient denaturing gel. HSV-1 strains used were wild type (WT), the EE>AA mutant (AA), and a VP26-null mutant (Δ26). The latter serves to identify VP26 in the denaturing gel.
The VP5 EE>AA mutant produces wild-type levels of VP5 protein.
The capsid profiles from the previous experiment were consistent with the expression of capsid proteins and their assembly. Nevertheless, the potential impact of the EE>AA mutation on VP5 expression warranted further inspection. Western blot detection of VP5 from cell lysates was performed at 8 hpi, a time point at which EE>AA propagation was attenuated compared to that of wild-type virus (Fig. 5). VP5 was detected as a 149-kDa band in both HSV-1 WT- and EE>AA- but not mock-infected cell lysates (Fig. 7A). Following densitometry analysis, the expression of VP5 was normalized to the pUL37 tegument protein as a loading control. The analysis revealed no significant change in the amount of VP5 produced by the EE>AA mutant (Fig. 7B). Therefore, we next examined the impact of EE>AA mutation on capsid morphogenesis.
FIG 7.

VP5 expression is not impacted by glutamic acid mutations. (A) Western blots of cell lysates prepared from mock-infected or infected cells. Vero cells were infected with HSV-1 encoding the wild-type (WT) or EE>AA VP5 allele at an MOI of 5. Threefold dilutions of cell lysates, prepared at 8 hpi, were probed by Western blotting. The HSV-1 tegument protein, pUL37, and cellular GAPDH served as loading controls. The image shown is representative of three independent experiments. (B) VP5 expression was quantitated by densitometry analysis. Individual measurements are normalized to the pUL37 loading control. The difference was not statistically significant by an unpaired t test. Error bars represent the standard errors of the means.
Nuclear foci formed during infection with the HSV-1 VP5 EE>AA mutant initially lack capsid maturation markers.
To better understand the protein composition of EE>AA foci, Vero cells were infected, fixed at 8 hpi, and processed for fluorescence imaging. Cells infected with HSV-1 encoding wild-type VP5 had capsids dispersed throughout the nuclei, which substantially colocalized with VP24, VP26, and pUL25, with reactivity to the maturation-specific VP5 antibody 8F5 (Fig. 8). In contrast, EE>AA nuclear foci colocalized only with VP24 despite dispersed punctate signals of VP26, pUL25, and VP5 8F5 reactivity in nuclei apart from the foci. Because the VP26 and pUL25 capsid proteins are acquired upon shell angularization (22, 24–26) and the VP5 8F5 epitope (49) is exposed on the tips of hexamers after internal scaffold cleavage by the maturation protease (50, 51), we conclude the EE>AA foci initially consisted of capsids that lacked maturation markers. By 16 hpi, the foci acquired each of these markers (Fig. 9).
FIG 8.

VP5 EE>AA focus composition is consistent with that of procapsids. Vero cells infected with HSV-1 encoding WT or EE>AA VP5 at an MOI of 5 were fixed at 8 hpi and processed for immunofluorescence using αVP5 and αVP26 antibodies. In some instances, the viruses encoded mCherry-VP24 or pUL25/mCherry fusions, as indicated. Scale bar, 10 μm.
FIG 9.

VP5 EE>AA foci acquire capsid maturation markers during late infection. Experiments were carried out as described for Fig. 8 but were examined at 16 hpi. Scale bar, 10 μm.
The VP5 EE>AA foci contain procapsids.
Transmission electron microscopy was used to visualize the EE>AA foci. Vero cells infected with HSV-1 VP5 EE>AA and fixed at 8 hpi contained nuclear foci of electron-dense material surrounding partially and fully formed procapsids (Fig. 10A and B). Consistent with the production of infectious EE>AA virions (Fig. 5), mature capsid species were detected throughout these nuclei but always apart from the foci (Fig. 10C to F). At 16 hpi, the intranuclear foci persisted but now contained mixtures of procapsids and B capsids (Fig. 10G to H). Interestingly, only scaffold-containing capsids were observed within the VP5 EE>AA foci, with C capsids found outside the foci (Fig. 10I). Collectively, the data indicate that mutation of the E846 and E851 residues on the capsid surface delays procapsid maturation.
FIG 10.

VP5 EE>AA foci contain clusters of procapsids. HSV-1 VP5 EE>AA-infected Vero cells (MOI of 5) were fixed at 8 hpi (A to F) or 16 hpi (G to I) and imaged by transmission electron microscopy. (A and B) Focus ultrastructure reveals fully and partially formed capsids containing large-core scaffolds consistent with procapsids. (C, D, E, and F) Infected cells exhibit all capsid species: procapsid (C), A capsid (D), B capsid (E), and C capsid (F). Scale bars are 100 nm (C to F). (G and H) Procapsids persist in foci, and B capsids are now evident as small-core capsids. (I) C capsids (white arrowheads) are present in nuclei but are excluded from foci.
DISCUSSION
Assembly of herpesvirus virions begins with the formation of spherical procapsid shells in cell nuclei. Procapsids are short-lived structures that are unstable when isolated from infected cells but spontaneously angularize into rigid, stable capsids during maturation (13, 32). When shell maturation is accompanied by genome encapsidation, the resulting product is a C capsid, or nucleocapsid. This critical process is a target for antiviral development (52–60), which is bolstered by ongoing refinements of cryo-EM reconstruction techniques that have led to herpesvirus capsid models with near-atomic resolution (19, 61–63).
The surface of the procapsid is decorated by the apical region of the VP5 major capsid protein, which constitutes the tips of the 11 pentamers (accounting for 11 of the icosahedral vertices, with the 12th vertex consisting of 12 copies of pUL6 portal protein) and 150 hexamers that make up the majority of the icosahedral shell. Following maturation, the procapsid acquires the pUL25 and VP26 accessory proteins, the latter of which selectively covers the apical regions of the hexamers. Thus, the procapsid displays 955 copies of the apical region on its surface, which is reduced to 55 exposed copies upon maturation and decoration with VP26. The exposure of the VP5 apical region on the mature capsid led us to investigate whether it contributes to the retrograde axonal transport of capsids that leads to the establishment of latent infections in the nervous system.
Although no functions have formally been ascribed to the apical region, two conserved glutamic acids within the polyproline loop of the apical region (E846 and E851) were proposed as potential sites for recruitment of VP26 upon shell maturation (18, 42). While residues within the VP5 upper domain are undoubtedly important for the VP26-VP5 interaction, our data demonstrated that the two glutamic acid residues are not required for VP26 binding to capsids. Furthermore, they were dispensable for retrograde axonal transport in primary sensory neurons, as was the antibody epitope portion of the apical region (based on analysis of the Δ6F10 mutant). Given that the VP26 protein is also dispensable for axonal transport (64, 65), we conclude that the bulk of exposed outer capsid surface does not promote this critical component of the HSV-1 neuroinvasion mechanism. While the scope of the mutagenesis included in this study does not entirely rule out a role for capsid surface epitopes that contribute to the transport mechanism, it seems likely that the previously identified interaction between pUL25 and the pUL36 tegument protein is sufficient to confer these dynamic properties to the virus particle (66). These results support a model in which the herpesvirus capsid is a dormant cargo that requires the action of attached tegument proteins to produce the dynamics that underlie productive infection.
Despite these findings, the polyproline glutamic acid mutant produced small plaques in cell culture, indicating the apical region was not fully dispensable. Further analysis correlated the propagation defect with the formation of focal structures in infected cell nuclei that consisted of clusters of procapsids. This observation was unexpected because it indicated that capsid maturation, a process dependent on proteolytic processing of the interior scaffold, was delayed. While the VP5 floor domain faces the interior of the capsid and interacts with the scaffold proteins (67–71), the exterior location of the apical region is inconsistent with it promoting scaffold processing. Furthermore, the VP5 apical region does not participate in intercapsomeric interactions (14, 15, 17–19, 63). Importantly, the persistence of procapsids that was initially indicated by the absence of maturation markers (pUL25, VP26, and exposure of the 8F5 epitope) was verified by ultrastructural analysis. The latter revealed the foci to consist of capsids containing large scaffold cores, which are indicative of procapsids (7, 13, 72). While the acquisition of surface markers could be sterically restricted within foci, the large scaffold cores indicate that the VP24 maturation protease was inefficiently activated within these procapsids, an event that does not require any known extrinsic interactions, including with pUL25, VP26, or genome encapsidation (7, 32, 73, 74).
We note that the procapsid foci of the polyproline mutant were associated with electron-dense material when imaged by transmission electron microscopy and in this way resembled procapsids in cell-free assembly systems (9, 75). The procapsid foci were also reminiscent of foci observed when VP24 activity is disrupted through mutation or deletion (12, 13, 76). In contrast, we previously reported a catalytically inactive VP24 mutant that did not produce procapsid foci (32). Thus, the mechanism driving procapsids to cluster in foci is of interest, as the clusters may represent sites of capsid assembly. Of greater interest is why capsid maturation is sensitive to mutations in the polyproline loop of the VP5 apical region. While procapsids in the mutant foci were often seen in a partially assembled state, the defect is inconsistent solely with an assembly defect, as seeing accumulations of procapsids is atypical of wild-type infections. Because angularization of the capsid shell under physiological conditions is dependent solely on VP24 activation, which occurs independently of genome encapsidation (9, 32, 77, 78), we can infer that mutation of the apical region has affected VP24 activation. Although speculative, the results are consistent with VP24 activation being enhanced by a cue external of the capsid. Capsids require ATP for shell maturation (79), and the apical domain could respond to ATP or interface with another factor such as the putative chaperone of preassembled capsids, pUL32 (80). Interestingly, the mutant foci exclusively consisted of procapsids and B capsids, with procapsids being the predominant species at 8 hpi and B capsids accumulating by 16 hpi. While A and C capsids could be found in proximity to the foci, there was no evidence of DNA-packaging occurring within them.
Despite a wealth of knowledge, the details of the molecular mechanisms driving procapsid assembly and maturation remain enigmatic. Viral mutants are a valuable resource to better understanding the complex assembly and maturation of nucleocapsids (13, 79). To the best of our knowledge, this report presents the first attribution of function to the VP5 apical region and documents that the exterior of the capsid contributes to shell maturation.
MATERIALS AND METHODS
Sequences and alignment.
Major capsid protein amino acid sequences obtained from GenBank are listed in order of appearance from top to bottom of the alignment in Fig. 1E: GU734771, Z86099, NC_004812, JF797219, AJ004801, AY665713, NC_002686.2, and NC_001348.1. Protein sequences were aligned using the ClustalW alignment tool in MacVector.
Structural modeling.
Using UCSF chimera (81), the structure of the upper domain of VP5 (PDB entry 1NO7) (42) was fit in both the pentamer and the peripentonal hexamer of the previously published density map of HSV-1 (EMDB entry EMD-6386) (18).
Cells and virus.
Vero (African green monkey kidney epithelial) and Vero cells expressing Cre recombinase (Vero-CRE) were maintained in Dulbecco's modified Eagle medium (DMEM) (11965-118; Invitrogen) supplemented with 10% (vol/vol) bovine growth serum (BGS) (FGR-BBT; Rocky Mountain Biologicals). The latter were generously provided by David Leib (82). Cultured cells were tested for Mycoplasma contamination with the PlasmoTest mycoplasma detection kit (rep-pt1; InvivoGen). Primary sensory neurons were isolated from the dorsal root ganglion (DRG) of embryonic chicks (embryonic days 9 and 10) and cultured as whole explants as previously described (83). Table 1 details all recombinant viruses used in this study. Mutation of UL19 (encodes VP5) or UL35 (encodes VP26) or insertion of coding sequences for fluorescent proteins was achieved by a two-step RED-mediated recombination method (84). Primers used for bacterial artificial chromosome (BAC) mutagenesis are listed in Table 2. Mutations were sequence confirmed at the Northwestern University Genomics Core Facility. HSV-1 recombinant viruses were produced by electroporation of the infectious BAC clone of HSV-1 strain F, pYEbac102, into Vero cells (85). Recombinant viruses were subsequently propagated on Vero-CRE cells to excise the loxP-flanked BAC backbone from the viral genome and produce a working stock of virus. All viral stock titers were determined by plaque assay on Vero cells overlaid with DMEM supplemented with 10 mg/ml methyl cellulose (AA36718-36; VWR), penicillin-streptomycin (Pen-Strep) at 100 U/ml (15140122; Invitrogen), and 2% BGS.
TABLE 1.
Recombinant viruses
| Strain | Fluorescent fusion | Mutant | Titer (PFU/ml) |
|---|---|---|---|
| HSVF-GS2695a | 2.59 × 108 | ||
| HSVF-GS5798 | VP5 E846A/E851A | 4.72 × 107 | |
| HSVF-GS5799 | VP5 N873A/N876A | 9.81 × 107 | |
| HSVF-GS5801 | VP5 H863A/H866A | 1.31 × 108 | |
| HSVF-GS5905 | VP5 H863A/H866A/N869A/L870A/V871A/T874A | 7.56 × 107 | |
| HSVF-GS6276 | VP5 A862-N873>GGSGGS | 1.07 × 108 | |
| HSVF-GS6197 | VP5 E846A | 1.45 × 108 | |
| HSVF-GS6434 | VP5 E851A | 1.93 × 108 | |
| HSVF-GS6462 | VP5 E846A>E/E851A>E | 3.06 × 108 | |
| HSVF-GS5748 | mCherry-5× G-VP24 | 2.95 × 108 | |
| HSVF-GS6460 | mCherry-5× G-VP24 | VP5 E846A/E851A | 8.06 × 107 |
| HSVF-GS6493 | mCherry-5× G-VP24 | VP5 A862-N873>GGSGGS | 1.32 × 107 |
| HSVF-GS4553b | pUL25/mCherry | 7.65 × 107 | |
| HSVF-GS5797 | pUL25/mCherry | VP5 E846A/E851A | 3.70 × 107 |
| HSVF-GS6199 | VP26 null | 1.59 × 108 |
TABLE 2.
Primers used for RED-mediated recombination unique to this study
| Strain | Template | Primer paira |
|---|---|---|
| HSVF-GS5798 | pEP-KanS2 | 5′ TACGCCACCCTGCAGAACATGGTGGTCCCGGCGATCGCCCCCGGCGCGGAGTGCCCCAGCGACAGGATGACGACGATAAGTAGGG 3′ |
| 5′ GTGCAGGGGGTGCGCGGGGTCCGTCACGGGGTCGCTGGGGCACTCCGCGCCGGGGGCGATCGCCAACCAATTAACCAATTCTGATTAG 3′ | ||
| HSVF-GS5799 | pEP-KanS2 | 5′ GTGACGGACCCCGCGCACCCCCTGCACCCGGCCAATCTGGTGGCCGCGACCGTGGCCGCCATGTTTCACAACAGGATGACGACGATAAGTAGGG 3′ |
| 5′ CATGGCGGGCCCGTCCACTACCACGCGCCCGTTGTGAAACATGGCGGCCACGGTCGCGGCCACCAGATTGGCCAACCAATTAACCAATTCTGATTAG 3′ | ||
| HSVF-GS5801 | pEP-KanS2 | 5′ CCCCGGCGAGGAGTGCCCCAGCGACCCCGTGACGGACCCCGCGGCGCCGCTAGCCCCGGCCAATCTGGTGAGGATGACGACGATAAGTAGGG 3′ |
| 5′ GTTGTGAAACATGGCGTTGACCGTGTTGGCCACCAGATTGGCCGGGGCTAGCGGCGCCGCGGGGTCCGTCACCAACCAATTAACCAATTCTGATTAG 3′ | ||
| HSVF-GS5905 | pEP-KanS2 | 5′ CCAGCGACCCCGTGACGGACCCCGCGGCGCCGCTAGCCCCGGCCGCTGCAGCGGCAAACGCGGTCAACGCCATGTTTAGGATGACGACGATAAGTAGGG 3′ |
| 5′ CCGTCCACTACCACGCGCCCGTTGTGAAACATGGCGTTGACCGCGTTTGCCGCTGCAGCGGCCGGGGCTAGCGGCAACCAATTAACCAATTCTGATTAG 3′ | ||
| HSVF-GS6276 | pEP-KanS2 | 5′ CCCCGGCGAGGAGTGCCCCAGCGACCCCGTGACGGACCCCGGAGGCAGCGGAGGATCCACGGTCAACGCCATGAGGATGACGACGATAAGTAGGG 3′ |
| 5′ GTCCACTACCACGCGCCCGTTGTGAAACATGGCGTTGACCGTGGATCCTCCGCTGCCTCCGGGGTCCGTCACGGGCAACCAATTAACCAATTCTGATTAG 3′ | ||
| HSVF-GS6197 | pEP-KanS2 | 5′ CACCCTGCAGAACATGGTGGTCCCGGAGATCGCGCCCGGGGCGGAGTGCCCCAGCGACAGGATGACGACGATAAGTAGGG 3′ |
| 5′ GGGGTGCGCGGGGTCCGTCACGGGGTCGCTGGGGCACTCCGCCCCGGGCGCGATCTCCAACCAATTAACCAATTCTGATTAG 3′ | ||
| HSVF-GS6434 | pEP-KanS2 | 5′ CGCTTCGACCGCGTATACGCCACCCTGCAGAACATGGTGGTCCCGGCGATCGCCCCCGGCGAGAGGATGACGACGATAAGTAGGG 3′ |
| 5′ GGGGTCCGTCACGGGGTCGCTGGGGCACTCCTCGCCGGGGGCGATCGCCGGGACCACCATGTTCAACCAATTAACCAATTCTGATTAG 3′ | ||
| HSVF-GS6462 | pEP-KanS2 | 5′ TACGCCACCCTGCAGAACATGGTGGTCCCGGAGATCGCGCCCGGGGAGGAGTGCCCCAGCGACAGGATGACGACGATAAGTAGGG 3′ |
| 5′ GTGCAGGGGGTGCGCGGGGTCCGTCACGGGGTCGCTGGGGCACTCCTCCCCGGGCGCGATCTCCAACCAATTAACCAATTCTGATTAG 3′ | ||
| HSVF-GS5748 | pEP-mCherry-in | 5′ GACCGTTGCGCCTTTTTTTTTTTTCGTCCACCAAAGTCTCTGTGGGTGCGCGCATGGTGAGCAAGGGCGAGGAG 3′ |
| 5′ CAGGGGCTCCTCCATCCGGTCTCCCGGGGCATCGGCTGCACCACCTCCGCCACCCTTGTACAGCTCGTCCATGC 3′ | ||
| HSVF-GS6460 | pEP-mCherry-in | 5′ GACCGTTGCGCCTTTTTTTTTTTTCGTCCACCAAAGTCTCTGTGGGTGCGCGCATGGTGAGCAAGGGCGAGGAG 3′ |
| 5′ CAGGGGCTCCTCCATCCGGTCTCCCGGGGCATCGGCTGCACCACCTCCGCCACCCTTGTACAGCTCGTCCATGC 3′ | ||
| HSVF-GS6493 | pEP-mCherry-in | 5′ GACCGTTGCGCCTTTTTTTTTTTTCGTCCACCAAAGTCTCTGTGGGTGCGCGCATGGTGAGCAAGGGCGAGGAG 3′ |
| 5′ CAGGGGCTCCTCCATCCGGTCTCCCGGGGCATCGGCTGCACCACCTCCGCCACCCTTGTACAGCTCGTCCATGC 3′ | ||
| HSVF-GS5797 | pEP-KanS2 | 5′ TACGCCACCCTGCAGAACATGGTGGTCCCGGCGATCGCCCCCGGCGCGGAGTGCCCCAGCGACAGGATGACGACGATAAGTAGGG 3′ |
| 5′ GTGCAGGGGGTGCGCGGGGTCCGTCACGGGGTCGCTGGGGCACTCCGCGCCGGGGGCGATCGCCAACCAATTAACCAATTCTGATTAG 3′ | ||
| HSVF-GS6199 | pEP-KanS2 | 5′ CCCCATATCGCTTCCCGACCTCCGGTCCCGATGGCCGTCCCGCAATGAGGCCCAGGGAGTAGGATGACGACGATAAGTAGGG 3′ |
| 5′ TCTTTTATTGATTAAAACACCCCAGAAGGAACTCCCTGGGCCTCATTGCGGGACGGCCATCAACCAATTAACCAATTCTGATTAG 3′ |
Sequences in bold share homology to the template plasmid.
Virus propagation and plaque assays.
Single-step growth curves were determined as previously described (86), with the following changes. In brief, Vero cells seeded in 6-well plates were infected at a multiplicity of infection (MOI) of 10. After 1 h, unabsorbed virus was inactivated with 1 ml of citrate buffer (pH 3.0), and cells were washed and then incubated in 2 ml of DMEM supplemented with 2% BGS at 37°C, 5% CO2. Cell-associated and extracellular virus was harvested at 2, 5, 8, 12, 24, and 30 hpi. Titers were determined in duplicate by thawing and sonicating samples and performing plaque assays on Vero cells.
Plaque assays used for plaque diameter measurements were incubated for 5 days postinfection and then stained with neutral red (sc-281691; Santa Cruz). Cells were gently rinsed with phosphate-buffered saline (PBS) before adding 1 ml of 1:1 neutral red and PBS. After 1 h, stain was replaced with PBS and plaques were imaged on an Epson Perfection V500 photo scanner at 3,200 dots per inch. Plaque diameters were determined by averaging two orthogonal diameter measurements for each individual plaque. Plaque diameters were then expressed as a percentage of wild-type (WT) plaque diameter and plotted in Prism 7 (GraphPad). More than 85 plaques were measured for each virus. Statistically significant differences were determined by a one-way analysis of variance with Tukey's multiple-comparisons posttest.
Live-cell fluorescence microscopy.
To monitor capsid retrograde axonal transport, whole-explant chick DRGs were infected, imaged, and analyzed as previously described (87), with the following changes. Imaging began approximately 30 min after infecting DRGs with 3 × 107 to 5 × 107 PFU. Live-cell images were captured on an inverted wide-field Nikon Eclipse TE2000-U microscope fitted with a 60×/1.40-numerical-aperture (NA) objective and a Cascade II:512 camera. Red fluorescence emissions were captured at 100-ms exposures with the camera's digitizer set at 10 megahertz (MHz) with 4,095 electron-multiplying (EM) gain for 250 frames. Run velocities and run distances traveled were determined by kymograph analysis. Viral particles with a forward (retrograde) motion of >0.5 μm were plotted on a histogram as a fraction of total particles, and run velocities or run distances were fit to a Gaussian or decaying exponential curve with a nonlinear regression, respectively. More than 150 runs were measured for each virus.
For live-cell imaging of infected epithelial cells, Vero cells were seeded on flame-sterilized 22- by 50-mm no. 1.5 cover glass and infected at an MOI of 5. After 1 h, inoculum was replaced with F12 medium supplemented with 2% BGS and Pen-Strep. VALAB chambers were made on 25- by 75- by 1-mm slides as previously described (83) at 8.5 hpi, a time when viral capsids can readily be observed in infected cell nuclei (13, 32). Slides were imaged with a 100× lambda/1.49-NA objective on a Nikon Ti inverted microscope housed in an environmental box set at 37°C (InVivo Scientific) and coupled with a CSU-W1 confocal head (Yokogawa Electric Corporation) and a CascadeII:1024 EM charge-coupled device (Photometrics). Illumination was supplied by a Sapphire 561-nm laser (Coherent) and custom laser launch (Solamere Technology Group, Inc.). Red fluorescence emissions were captured with 4,000-ms exposures with the digitizer set to 1-MHz zero electron-multiplying gain. Differential interference contrast (DIC) images were captured with 200-ms exposures and 1-MHz zero electron-multiplying gain.
Preparation of cell lysates.
Vero cells seeded in a 10-cm dish were infected at an MOI of 5. After 1 h, inoculum was replaced with DMEM supplemented with 2% BGS. At 8 hpi, cells were washed with 10 ml of ice-cold PBS and lysed in 300 μl of radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, pH 8, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate) supplemented with 5 mM dithiothreitol and protease inhibitors (2.5 mM sodium fluoride, 1 mM sodium orthovanadate, 0.5 mM phenylmethylsulfonyl fluoride, 100 μl of protease inhibitor cocktail [P8340; Sigma]). Cells were scraped into lysis buffer, transferred to an Eppendorf tube, and set on ice for 15 min. Lysates were then sonicated for three 1.5-s pulses and set on ice for an additional 15 min. Lysates were spun at 13,201 × g for 20 min, and the supernatants were harvested. Total protein concentration of cell lysates was determined using the Pierce 660-nm protein assay reagent (22660; Thermo Fisher Scientific). Absorbances of lysates were read at 650 nm on a Molecular Devices SpectraMax M5 microplate reader. Sample protein concentration was calculated from the standard curve of Pierce prediluted protein assay standards, bovine serum albumin (BSA) set (23208; Thermo Fisher Scientific). Samples were diluted in 5× final sample buffer (FSB) (60 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 0.01% bromophenol blue) supplemented with 5% β-mercaptoethanol and stored at −20°C.
Western blotting and densitometry analysis.
Cell lysates were thawed, boiled for 5 min, and loaded as three 3-fold dilutions in equal concentrations across samples on a 4 to 20% mini-PROTEAN TGX gel (no. 456-1096). Bio-Rad dual-color standards (1610374; Bio-Rad) were also loaded to mark molecular weights. Proteins were separated and transferred to a PVDF membrane (IPFL00010; Millipore). The membrane was blocked in 5% milk for 80 min and then incubated overnight with primary antibodies diluted in 1% milk in PBST (0.1% Tween 20 in PBS), namely, mouse monoclonal anti-VP5 H1.4 (C05014MA; Meridian Life Science, Inc.) at 1:1,000 and rabbit polyclonal anti-pUL37 (kindly provided by Frank Jenkins) at 1:500. The next day, the membrane was washed and incubated for 1 h at room temperature (RT) with secondary antibodies diluted in 1% milk in PBST, namely, goat anti-mouse IRDye 800CW (926-32210; LI-COR Biosciences) at 1:10,000 and donkey anti-rabbit IRDye 680RD (926-68073; LI-COR Biosciences) at 1:10,000. The membrane was imaged on a LI-COR Odyssey FC imaging system at 700 nm and 800 nm for 3.5 min for each wavelength. The membrane next was blocked again with 5% milk in PBS for 80 min and then incubated for 1 h at RT with primary antibody mouse monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (αGAPDH), clone 6C5 (ab8245; Abcam), diluted 1:500 in 1% milk in PBS. The membrane was washed and then incubated at RT for 1 h with secondary antibody goat anti-mouse IRDye 800CW at 1:10,000 and imaged on the LI-COR Odyssey FC imaging system at 700 nm for 3.5 min.
Densitometry analysis was carried out using Image Studio Lite software. In brief, rectangles were added around each VP5 and pUL37 signal band, with background subtraction set to median and a border width of 3 pixels top and bottom. Boxes were adjusted, if needed, based on the profile shape and intensity tools. The signal values for VP5 bands were calculated as a ratio to their corresponding pUL37 signal value. These ratios were then normalized to wild-type samples.
Immunofluorescence assays.
Vero cells seeded on flame-sterilized cover glass were infected at an MOI of 5 for 1 h and then incubated in DMEM supplemented with 2% BGS at 37°C, 5% CO2. At the indicated time postinfection, infected cells were washed with PBS and then fixed in 4% paraformaldehyde for 15 min. Fixed cells were rinsed three times with PBS and then permeabilized in PBST (0.1% Triton X-100 in PBS) for 10 min. Permeabilized cells were blocked in 5% bovine serum albumin (BSA) (A9647; Sigma) in PBST for 45 min at RT or overnight at 4°C. After blocking, fixed cells were reacted with primary and secondary antibodies each for 1 h at RT, protected from light. Primary antibodies were diluted in 1% BSA in PBST using mouse monoclonal anti-VP5 H1.4 (C05014MA; Meridian Life Science, Inc.) at 1:30, mouse monoclonal anti-VP5 8F5 (kindly provided by Duncan Wilson) at 1:100, and rabbit anti-VP26 (kindly provided by Prashant Desai) at 1:2,000. Secondary antibodies were diluted in 1% BSA in PBST: goat anti-mouse Alexa Fluor 488 (A-11001; Thermo Fisher Scientific) at 1:400 and goat anti-rabbit Alexa Fluor 568 (A-11011; Thermo Fisher Scientific) at 1:400. Cover glass was mounted on slides with ProLong Gold with 4′,6-diamidino-2-phenylindole (DAPI) mounting medium (P36931; Thermo Fisher Scientific). Slides were imaged by confocal microscopy on a Nikon Ti inverted microscope fitted with a 60×/1.49-NA objective as described above. Red and green fluorescence emissions were captured at 1 MHz with zero electron-multiplying gain at the following exposures: DAPI, 2,000 ms; αVP5 (H1.4 and 8F5), 200 ms; αVP26, 200 ms; mCherry-VP24, 4,000 ms; and pUL25/mCherry, 5,000 ms. DIC images were captured with 200-ms exposures with 1-MHz digitizer and zero electron-multiplying gain.
Transmission electron microscopy.
Vero cells seeded in a 10-cm dish were infected at an MOI of 5 for 1 h and then incubated in DMEM supplemented with 2% BGS and Pen-Strep at 37°C and 5% CO2. At the indicated time postinfection, infected cells were rinsed with PBS and fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer for 60 min at RT. After fixation, cells were scraped into fixative and transferred to an Eppendorf tube. Fixed cells were pelleted at 300 × g for 10 min. The cell pellet was enrobed in 5% low-melting point agarose (50101; SeaPlaque Agarose) and delivered to the Northwestern University's Center for Advanced Microscopy to be processed for transmission electron microscopy. In brief, the agarose plug was postfixed with 1% osmium tetroxide, stained with 0.5% uranyl acetate, dehydrated in ascending grades of ethanol from 30 to 100%, infiltrated with propylenoxide, and embedded in hard resin. Blocks were thin sectioned at 50 to 60 nm and stained with uranyl acetate and Reynolds lead citrate. Slices were mounted on 200-mesh copper grids (Electron Microscopy Sciences). Imaging work was performed on an FEI Tecnai Spirit G2 transmission electron microscope at the Northwestern University Center for Advanced Microscopy, generously supported by NCI CCSG P30 CA060553, awarded to the Robert H. Lurie Comprehensive Cancer Center.
Intranuclear capsid isolation and silver staining.
Isolation of intranuclear capsids was performed as previously described (88), with the following changes. Vero cells were seeded in 15-cm dishes and infected at an MOI of 10 with five dishes per virus. Infected cells were harvested between 22 and 24 hpi. A, B, and C capsid bands were pulled from the gradient using a gradient fractionator (BioComp Instruments). Each capsid species was subsequently diluted in a total volume of 2 ml of TNE (20 mM Tris, pH 7.6, 500 mM NaCl, 1 mM EDTA) supplemented with 10 μl of protease inhibitor cocktail (P8340; Sigma) and pelleted in a Beckman SW50.1 rotor at 74,909 × g for 1 h and 4 min at 4°C. Pelleted capsids were resuspended in 50 μl of 5× FSB supplemented with 5% β-mercaptoethanol and stored at −20°C.
Resuspended capsids were thawed, boiled for 5 min, and loaded in equal volumes on a 4 to 20% Mini-PROTEAN TGX gel (no. 456-1096) with Bio-Rad dual-color standards (1610374; Bio-Rad) diluted 1:100 in 5× FSB supplemented with 5% β-mercaptoethanol. Proteins were then detected using the Pierce silver stain kit (24612; Thermo Fisher Scientific). The silver-stained gel was imaged on an Epson Perfection V500 photo scanner at 2,400 dots per inch.
ACKNOWLEDGMENTS
We thank Sarah Antinone for providing embryonic chick DRGs, Duncan Wilson and Prashant Desai for generously providing antibodies, and Ekaterina Heldwein for her assistance with designing the VP5 Δ6F10 mutant. Transmission electron microscopy samples were processed by Northwestern University's Center for Advanced Microscopy and imaged using the facility's FEI Tecnai Spirit G2 transmission electron microscope. Sequencing services were performed at the Northwestern University Genomics Core Facility.
This work was funded by the National Institute of Allergy and Infectious Diseases, including the efforts of Laura L. Ruhge and Gregory A. Smith (R01 AI056346) and the contributions of Alexis G. E. Huet and James F. Conway (R01 AI089803).
REFERENCES
- 1.Steiner I, Kennedy PGE, Pachner AR. 2007. The neurotropic herpes viruses: herpes simplex and varicella-zoster. Lancet Neurol 6:1015–1028. doi: 10.1016/S1474-4422(07)70267-3. [DOI] [PubMed] [Google Scholar]
- 2.Smith G. 2012. Herpesvirus transport to the nervous system and back again. Annu Rev Microbiol 66:153–176. doi: 10.1146/annurev-micro-092611-150051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baines JD. 2011. Herpes simplex virus capsid assembly and DNA packaging: a present and future antiviral drug target. Trends Microbiol 19:606–613. doi: 10.1016/j.tim.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 4.Bauer DW, Huffman JB, Homa FL, Evilevitch A. 2013. Herpes virus genome, the pressure is on. J Am Chem Soc 135:11216–11221. doi: 10.1021/ja404008r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sodeik B, Ebersold MW, Helenius A. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J Cell Biol 136:1007–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newcomb WW, Homa F, Thomsen DR, Ye Z, Brown JC. 1994. Cell-free assembly of the herpes simplex virus capsid. J Virol 68:6059–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tatman JD, Preston VG, Nicholson P, Elliott RM, Rixon FJ. 1994. Assembly of herpes simplex virus type 1 capsids using a panel of recombinant baculoviruses. J Gen Virol 75:1101–1113. doi: 10.1099/0022-1317-75-5-1101. [DOI] [PubMed] [Google Scholar]
- 8.Thomsen DR, Roof LL, Homa FL. 1994. Assembly of herpes simplex virus (HSV) intermediate capsids in insect cells infected with recombinant baculoviruses expressing HSV capsid proteins. J Virol 68:2442–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newcomb WW, Homa F, Thomsen DR, Booy FP, Trus BL, Steven AC, Spencer JV, Brown JC. 1996. Assembly of the herpes simplex virus capsid: characterization of intermediates observed during cell-free capsid formation. J Mol Biol 263:432–446. doi: 10.1006/jmbi.1996.0587. [DOI] [PubMed] [Google Scholar]
- 10.Thomsen DR, Newcomb WW, Brown JC, Homa FL. 1995. Assembly of the herpes simplex virus capsid: requirement for the carboxyl-terminal twenty-five amino acids of the proteins encoded by the UL26 and UL26.5 genes. J Virol 69:3690–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kennard J, Rixon FJ, McDougall IM, Tatman JD, Preston VG. 1995. The 25 amino acid residues at the carboxy terminus of the herpes simplex virus type 1 UL26.5 protein are required for the formation of the capsid shell around the scaffold. J Gen Virol 76:1611–1621. doi: 10.1099/0022-1317-76-7-1611. [DOI] [PubMed] [Google Scholar]
- 12.Preston VG, Coates JAV, Rixon FJ. 1983. Identification and characterization of a herpes simplex virus gene product required for encapsidation of virus DNA. J Virol 45:1056–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Church GA, Wilson DW. 1997. Study of herpes simplex virus maturation during a synchronous wave of assembly. J Virol 71:3603–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trus BL, Booy FP, Newcomb WW, Brown JC, Homa FL, Thomsen DR, Steven AC. 1996. The herpes simplex virus procapsid: structure, conformational changes upon maturation, and roles of the triplex proteins VP19c and VP23 in assembly. J Mol Biol 263:447–462. doi: 10.1016/S0022-2836(96)80018-0. [DOI] [PubMed] [Google Scholar]
- 15.Zhou ZH, Dougherty M, Jakana J, He J, Rixon FJ, Chiu W. 2000. Seeing the herpesvirus capsid at 8.5 A. Science 288:877–880. doi: 10.1126/science.288.5467.877. [DOI] [PubMed] [Google Scholar]
- 16.Heymann JB, Cheng N, Newcomb WW, Trus BL, Brown JC, Steven AC. 2003. Dynamics of herpes simplex virus capsid maturation visualized by time-lapse cryo-electron microscopy. Nat Struct Biol 10:334–341. doi: 10.1038/nsb922. [DOI] [PubMed] [Google Scholar]
- 17.Aksyuk AA, Newcomb WW, Cheng N, Winkler DC, Fontana J, Heymann JB, Steven AC. 2015. Subassemblies and asymmetry in assembly of herpes simplex virus procapsid. mBio 6:e01525-. doi: 10.1128/mBio.01525-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huet A, Makhov AM, Huffman JB, Vos M, Homa FL, Conway JF. 2016. Extensive subunit contacts underpin herpesvirus capsid stability and interior-to-exterior allostery. Nat Struct Mol Biol 23:531–539. doi: 10.1038/nsmb.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dai X, Zhou ZH. 2018. Structure of the herpes simplex virus 1 capsid with associated tegument protein complexes. Science 360:eaao7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu YT, Jiang J, Bohannon KP, Dai X, Gant Luxton GW, Hui WH, Bi GQ, Smith GA, Hong Zhou Z. 2017. A pUL25 dimer interfaces the pseudorabies virus capsid and tegument. J Gen Virol 98:2837–2849. doi: 10.1099/jgv.0.000903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robertson BJ, McCain PJ III, Matusick-Kumar L, Newcomb WW, Brown JC, Colonno RJ, Gao M. 1996. Separate functional domains of the herpes simplex virus type 1 protease: evidence for cleavage inside capsids. J Virol 70:4317–4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newcomb WW, Trus BL, Cheng N, Steven AC, Sheaffer AK, Tenney DJ, Weller SK, Brown JC. 2000. Isolation of herpes simplex virus procapsids from cells infected with a protease-deficient mutant virus. J Virol 74:1663–1673. doi: 10.1128/JVI.74.4.1663-1673.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rixon FJ, McNab D. 1999. Packaging-competent capsids of a herpes simplex virus temperature-sensitive mutant have properties similar to those of in vitro-assembled procapsids. J Virol 73:5714–5721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chi JH, Wilson DW. 2000. ATP-dependent localization of the herpes simplex virus capsid protein VP26 to sites of procapsid maturation. J Virol 74:1468–1476. doi: 10.1128/JVI.74.3.1468-1476.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thurlow JK, Rixon FJ, Murphy M, Targett-Adams P, Hughes M, Preston VG. 2005. The herpes simplex virus type 1 DNA packaging protein UL17 is a virion protein that is present in both the capsid and the tegument compartments. J Virol 79:150–158. doi: 10.1128/JVI.79.1.150-158.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheaffer AK, Newcomb WW, Gao M, Yu D, Weller SK, Brown JC, Tenney DJ. 2001. Herpes simplex virus DNA cleavage and packaging proteins associate with the procapsid prior to its maturation. J Virol 75:687–698. doi: 10.1128/JVI.75.2.687-698.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stevenson AJ, Morrison EE, Chaudhari R, Yang C, Meredith DM. 1997. Processing and intracellular localization of the herpes simplex virus type 1 proteinase. J Gen Virol 78:671–675. doi: 10.1099/0022-1317-78-3-671. [DOI] [PubMed] [Google Scholar]
- 28.Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol 82:8605–8618. doi: 10.1128/JVI.00904-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Person S, Desai P. 1998. Capsids are formed in a mutant virus blocked at the maturation site of the UL26 and UL26.5 open reading frames of herpes simplex virus type 1 but are not formed in a null mutant of UL38 (VP19C). Virology 242:193–203. doi: 10.1006/viro.1997.9005. [DOI] [PubMed] [Google Scholar]
- 30.Kramer T, Greco TM, Enquist LW, Cristea IM. 2011. Proteomic characterization of pseudorabies virus extracellular virions. J Virol 85:6427–6441. doi: 10.1128/JVI.02253-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spear PG, Roizman B. 1972. Proteins specified by herpes simplex virus. V. Purification and structural proteins of the herpesvirion. J Virol 9:143–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maier O, Sollars PJ, Pickard GE, Smith GA. 2016. Visualizing herpesvirus procapsids in living cells. J Virol 90:10182–10192. doi: 10.1128/JVI.01437-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Newcomb WW, Brown JC, Booy FP, Steven AC. 1989. Nucleocapsid mass and capsomer protein stoichiometry in equine herpesvirus 1: scanning transmission electron microscopic study. J Virol 64:3777–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newcomb WW, Trus BL, Booy FP, Steven AC, Wall JS, Brown JC. 1993. Structure of the herpes simplex virus capsid. Molecular composition of the pentons and the triplexes. J Mol Biol 232:499–511. [DOI] [PubMed] [Google Scholar]
- 35.Gibson W, Roizman B. 1972. Proteins specified by herpes simplex virus. VIII. Characterization and composition of multiple capsid forms of subtypes 1 and 2. J Virol 10:1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Newcomb WW, Brown JC. 1991. Structure of the herpes simplex virus capsid: effects of extraction with guanidine hydrochloride and partial reconstitution of extracted capsids. J Virol 65:613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou ZH, He J, Jakana J, Tatman JD, Rixon FJ, Chiu W. 1995. Assembly of VP26 in herpes simplex virus-1 inferred from structures of wild-type and recombinant capsids. Nat Struct Biol 2:1026–1030. doi: 10.1038/nsb1195-1026. [DOI] [PubMed] [Google Scholar]
- 38.Booy FP, Trus BL, Newcomb WW, Brown JC, Conway JF, Steven AC. 1994. Finding a needle in a haystack: detection of a small protein (the 12-kDa VP26) in a large complex (the 200-MDa capsid of herpes simplex virus). Proc Natl Acad Sci U S A 91:5652–5656. doi: 10.1073/pnas.91.12.5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen DH, Jakana J, McNab D, Mitchell J, Zhou ZH, Dougherty M, Chiu W, Rixon FJ. 2001. The pattern of tegument-capsid interaction in the herpes simplex virus type 1 virion is not influenced by the small hexon-associated protein VP26. J Virol 75:11863–11867. doi: 10.1128/JVI.75.23.11863-11867.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou ZH, Chen DH, Jakana J, Rixon FJ, Chiu W. 1999. Visualization of tegument-capsid interactions and DNA in intact herpes simplex virus type 1 virions. J Virol 73:3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spencer JV, Trus BL, Booy FP, Steven AC, Newcomb WW, Brown JC. 1997. Structure of the herpes simplex virus capsid: peptide A862-H880 of the major capsid protein is displayed on the rim of the capsomer protrusions. Virology 228:229–235. doi: 10.1006/viro.1996.8392. [DOI] [PubMed] [Google Scholar]
- 42.Bowman BR, Baker ML, Rixon FJ, Chiu W, Quiocho FA. 2003. Structure of the herpesvirus major capsid protein. EMBO J 22:757–765. doi: 10.1093/emboj/cdg086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zaichick SV, Bohannon KP, Hughes A, Sollars PJ, Pickard GE, Smith GA. 2013. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe 13:193–203. doi: 10.1016/j.chom.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richards AL, Sollars PJ, Pitts JD, Stults AM, Heldwein EE, Pickard GE, Smith GA. 2017. The pUL37 tegument protein guides alpha-herpesvirus retrograde axonal transport to promote neuroinvasion. PLoS Pathog 13:e1006741. doi: 10.1371/journal.ppat.1006741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huffmaster NJ, Sollars PJ, Richards AL, Pickard GE, Smith GA. 2015. Dynamic ubiquitination drives herpesvirus neuroinvasion. Proc Natl Acad Sci U S A 112:12818–12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruhge LL, Huet AGE, Conway JF, Smith GA. 2018. The apical region of the herpes simplex virus major capsid protein promotes capsid maturation. bioRxiv doi: 10.1101/319988. [DOI] [PMC free article] [PubMed]
- 47.Shanda SK, Wilson DW. 2008. UL36p is required for efficient transport of membrane-associated herpes simplex virus type 1 along microtubules. J Virol 82:7388–7394. doi: 10.1128/JVI.00225-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krautwald M, Fuchs W, Klupp BG, Mettenleiter TC. 2009. Translocation of incoming pseudorabies virus capsids to the cell nucleus is delayed in the absence of tegument protein pUL37. J Virol 83:3389–3396. doi: 10.1128/JVI.02090-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trus BL, Newcomb WW, Booy FP, Brown JC, Steven AC. 1992. Distinct monoclonal antibodies separately label the hexons or the pentons of herpes simplex virus capsid. Proc Natl Acad Sci U S A 89:11508–11512. doi: 10.1073/pnas.89.23.11508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matusick-Kumar L, Hurlburt W, Weinheimer SP, Newcomb WW, Brown JC, Gao M. 1994. Phenotype of the herpes simplex virus type 1 protease substrate ICP35 mutant virus. J Virol 68:5384–5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matusick-Kumar L, Newcomb WW, Brown JC, McCann P III, Hurlburt W, Weinheimer SP, Gao M. 1995. The C-terminal 25 amino acids of the protease and its substrate ICP35 of herpes simplex virus type 1 are involved in the formation of sealed capsids. J Virol 69:4347–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Newcomb WW, Brown JC. 2002. Inhibition of herpes simplex virus replication by WAY-150138: assembly of capsids depleted of the portal and terminase proteins involved in DNA encapsidation. J Virol 76:10084–10088. doi: 10.1128/JVI.76.19.10084-10088.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jin F, Li S, Zheng K, Zhuo C, Ma K, Chen M, Wang Q, Zhang P, Fan J, Ren Z, Wang Y. 2014. Silencing herpes simplex virus type 1 capsid protein encoding genes by siRNA: a promising antiviral therapeutic approach. PLoS One 9:e96623. doi: 10.1371/journal.pone.0096623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Inoue N, Matsushita M, Fukui Y, Yamada S, Tsuda M, Higashi C, Kaneko K, Hasegawa H, Yamaguchi T. 2012. Identification of a varicella-zoster virus replication inhibitor that blocks capsid assembly by interacting with the floor domain of the major capsid protein. J Virol 86:12198–12207. doi: 10.1128/JVI.01280-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akanitapichat P, Bastow KF. 2002. The antiviral agent 5-chloro-1,3-dihydroxyacridone interferes with assembly and maturation of herpes simplex virus. Antiviral Res 53:113–126. doi: 10.1016/S0166-3542(01)00203-0. [DOI] [PubMed] [Google Scholar]
- 56.Piret J, Lamontagne J, Bestman-Smith J, Roy S, Gourde P, Desormeaux A, Omar RF, Juhasz J, Bergeron MG. 2000. In vitro and in vivo evaluations of sodium lauryl sulfate and dextran sulfate as microbicides against herpes simplex and human immunodeficiency viruses. J Clin Microbiol 38:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Taylor LF, Yuan Y. 2018. Structure of herpesvirus capsid sheds light on drug discovery. Trends Microbiol 26:391–392. doi: 10.1016/j.tim.2018.03.003. [DOI] [PubMed] [Google Scholar]
- 58.Underwood MR, Harvey RJ, Stanat SC, Hemphill ML, Miller T, Drach JC, Townsend LB, Biron KK. 1998. Inhibition of human cytomegalovirus DNA maturation by a benzimidazole ribonucleoside is mediated through the UL89 gene product. J Virol 72:717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krosky PM, Underwood MR, Turk SR, Feng KWH, Jain RK, Ptak RG, Westerman AC, Biron KK, Townsend LB, Drach JC. 1998. Resistance of human cytomegalovirus to benzimidazole ribonucleosides maps to two open reading frames: UL89 and UL56. J Virol 72:4721–4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goldner T, Hewlett G, Ettischer N, Ruebsamen-Schaeff H, Zimmermann H, Lischka P. 2011. The novel anticytomegalovirus compound AIC246 (Letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J Virol 85:10884–10893. doi: 10.1128/JVI.05265-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dai X, Gong D, Lim H, Jih J, Wu TT, Sun R, Zhou ZH. 2018. Structure and mutagenesis reveal essential capsid protein interactions for KSHV replication. Nature 553:521–525. doi: 10.1038/nature25438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu X, Jih J, Jiang J, Zhou ZH. 2017. Atomic structure of the human cytomegalovirus capsid with its securing tegument layer of pp150. Science 356:eaam6892. doi: 10.1126/science.aam6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yuan S, Wang J, Zhu D, Wang N, Gao Q, Chen W, Tang H, Wang J, Zhang X, Liu H, Rao Z, Wang X. 2018. Cryo-EM structure of a herpesvirus capsid at 3.1 A. Science 360:eaao7283. doi: 10.1126/science.aao7283. [DOI] [PubMed] [Google Scholar]
- 64.Antinone SE, Shubeita GT, Coller KE, Lee JI, Haverlock-Moyns S, Gross SP, Smith GA. 2006. The herpesvirus capsid surface protein, VP26, and the majority of the tegument proteins are dispensable for capsid transport toward the nucleus. J Virol 80:5494–5498. doi: 10.1128/JVI.00026-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dohner K, Radtke K, Schmidt S, Sodeik B. 2006. Eclipse phase of herpes simplex virus type 1 infection: efficient dynein-mediated capsid transport without the small capsid protein VP26. J Virol 80:8211–8224. doi: 10.1128/JVI.02528-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Coller KE, Lee JI, Ueda A, Smith GA. 2007. The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J Virol 81:11790–11797. doi: 10.1128/JVI.01113-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Desai P, Person S. 1999. Second site mutations in the N-terminus of the major capsid protein (VP5) overcome a block at the maturation cleavage site of the capsid scaffold proteins. Virology 261:357–366. doi: 10.1006/viro.1999.9877. [DOI] [PubMed] [Google Scholar]
- 68.Warner SC, Desai P, Person S. 2000. Second-site mutations encoding residues 34 and 78 of the major capsid protein (VP5) of herpes simplex virus type 1 are important for overcoming a blocked maturation cleavage site of the capsid scaffold proteins. Virology 278:217–226. doi: 10.1006/viro.2000.0657. [DOI] [PubMed] [Google Scholar]
- 69.Walters JN, Sexton GL, McCaffery JM, Desai P. 2003. Mutation of single hydrophobic residue I27, L35, F39, L58, L65, L67, or L71 in the N terminus of VP5 abolishes interaction with the scaffold protein and prevents closure of herpes simplex virus type 1 capsid shells. J Virol 77:4043–4059. doi: 10.1128/JVI.77.7.4043-4059.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang E, Perkins EM, Desai P. 2007. Structural features of the scaffold interaction domain at the N terminus of the major capsid protein (VP5) of herpes simplex virus type 1. J Virol 81:9396–9407. doi: 10.1128/JVI.00986-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hong Z, Beaudet-Miller M, Durkin J, Zhang R, Kwong AD. 1996. Identification of a minimal hydrophobic domain in the herpes simplex virus type 1 scaffolding protein which is required for interaction with the major capsid protein. J Virol 70:533–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Addison C, Rixon FJ, Preston VG. 1990. Herpes simplex virus type 1 UL28 gene product is important for the formation of mature capsids. J Gen Virol 71:2377–2384. doi: 10.1099/0022-1317-71-10-2377. [DOI] [PubMed] [Google Scholar]
- 73.McNab AR, Desai P, Person S, Roof LL, Thomsen DR, Newcomb WW, Brown JC, Homa FL. 1998. The product of the herpes simplex virus type 1 UL25 gene is required for encapsidation but not for cleavage of replicated viral DNA. J Virol 72:1060–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Klupp BG, Granzow H, Keil GM, Mettenleiter TC. 2006. The capsid-associated UL25 protein of the alphaherpesvirus pseudorabies virus is nonessential for cleavage and encapsidation of genomic DNA but is required for nuclear egress of capsids. J Virol 80:6235–6246. doi: 10.1128/JVI.02662-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Newcomb WW, Homa FL, Thomsen DR, Trus BL, Cheng N, Steven A, Booy F, Brown JC. 1999. Assembly of the herpes simplex virus procapsid from purified components and identification of small complexes containing the major capsid and scaffolding proteins. J Virol 73:4239–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gao M, Matusick-Kumar L, Hurlburt W, DiTusa SF, Newcomb WW, Brown JC, McCann PJ III, Deckman IC, Colonno RJ. 1994. The protease of herpes simplex virus type 1 is essential for functional capsid formation and viral growth. J Virol 68:3702–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Al-Kobaisi MF, Rixon FJ, McDougall I, Preston VG. 1991. The herpes simplex virus UL33 gene product is required for the assembly of full capsids. Virology 180:380–388. [DOI] [PubMed] [Google Scholar]
- 78.Fuchs W, Klupp BG, Granzow H, Leege T, Mettenleiter TC. 2009. Characterization of pseudorabies virus (PrV) cleavage-encapsidation proteins and functional complementation of PrV pUL32 by the homologous protein of herpes simplex virus type 1. J Virol 83:3930–3943. doi: 10.1128/JVI.02636-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dasgupta A, Wilson DW. 1999. ATP depletion blocks herpes simplex virus DNA packaging and capsid maturation. J Virol 73:2006–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lamberti C, Weller SK. 1998. The herpes simplex virus type 1 cleavage/packaging protein, UL32, is involved in efficient localization of capsids to replication compartments. J Virol 72:2463–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 82.Gierasch WW, Zimmerman DL, Ward SL, Vanheyningen TK, Romine JD, Leib DA. 2006. Construction and characterization of bacterial artificial chromosomes containing HSV-1 strains 17 and KOS. J Virol Methods 135:197–206. doi: 10.1016/j.jviromet.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 83.Smith GA, Gross SP, Enquist LW. 2001. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc Natl Acad Sci U S A 98:3466–3470. doi: 10.1073/pnas.061029798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol 634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- 85.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J Virol 77:1382–1391. doi: 10.1128/JVI.77.2.1382-1391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tirabassi RS, Enquist LW. 1998. Role of envelope protein gE endocytosis in the pseudorabies virus life cycle. J Virol 72:4571–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bohannon KP, Sollars PJ, Pickard GE, Smith GA. 2012. Fusion of a fluorescent protein to the pUL25 minor capsid protein of pseudorabies virus allows live-cell capsid imaging with negligible impact on infection. J Gen Virol 93:124–129. doi: 10.1099/vir.0.036145-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Leelawong M, Guo D, Smith GA. 2011. A physical link between the pseudorabies virus capsid and the nuclear egress complex. J Virol 85:11675–11684. doi: 10.1128/JVI.05614-11. [DOI] [PMC free article] [PubMed] [Google Scholar]