

It is well accepted that herpesviruses suppress the apoptosis of host cells via various strategies to ensure sustained viral replication during infection. However, a recent in vivo study reported the apoptosis of remote uninfected cells during GaHV-1 infection. The mechanism and the biological meaning of this unexpected herpesvirus-host interaction are unclear. This study uncovers the mechanisms of herpesvirus-triggered apoptosis in uninfected cells and may also contribute to a mechanistic illustration of paracrine-regulated apoptosis induced by other viruses in uninfected host cells.
KEYWORDS: alphaherpesviruses, virus-host interactions, paracrine apoptosis, p53
ABSTRACT
Apoptosis is a common innate defense mechanism of host cells against viral infection and is therefore suppressed by many viruses, including herpes simplex virus (HSV), via various strategies. A recent in vivo study reported the apoptosis of remote uninfected cells during Gallid herpesvirus 1 (GaHV-1) infection, yet little is known about this previously unknown aspect of herpesvirus-host interactions. The aim of the present study was to investigate the apoptosis of uninfected host cells during GaHV-1 infection. The present study used in vitro and in ovo models, which avoided potential interference by host antiviral immunity, and demonstrated that this GaHV-1–host interaction is independent of host immune responses and important for both the pathological effect of viral infection and early viral dissemination from the primary infection site to distant tissues. Further, we revealed that GaHV-1 infection triggers this process in a paracrine-regulated manner. Using genome-wide transcriptome analyses in combination with a set of functional studies, we found that this paracrine-regulated effect requires the repression of p53 activity in uninfected cells. In contrast, the activation of p53 not only prevented the apoptosis of remote uninfected cells and subsequent pathological damage induced by GaHV-1 infection but also delayed viral dissemination significantly. Moreover, p53 activation repressed viral replication both in vitro and in ovo, suggesting that dual cell-intrinsic mechanisms underlie the suppression of GaHV-1 infection by p53 activation. This study uncovers the mechanism underlying the herpesvirus-triggered apoptosis of remote host cells and extends our understanding of both herpesvirus-host interactions and the roles of p53 in viral infection.
IMPORTANCE It is well accepted that herpesviruses suppress the apoptosis of host cells via various strategies to ensure sustained viral replication during infection. However, a recent in vivo study reported the apoptosis of remote uninfected cells during GaHV-1 infection. The mechanism and the biological meaning of this unexpected herpesvirus-host interaction are unclear. This study uncovers the mechanisms of herpesvirus-triggered apoptosis in uninfected cells and may also contribute to a mechanistic illustration of paracrine-regulated apoptosis induced by other viruses in uninfected host cells.
INTRODUCTION
The induction of programed host cell death via apoptosis and necrosis is a common response of multicellular organisms to the stress of pathogenic infection and protects the host by directly eliminating infected cells. For example, at the onset of herpes simplex virus (HSV) infection, the function of infected host cells can be induced by viral proteins, such as ICP0 and ICP6, which trigger apoptosis and necrosis, respectively (1–3). To ensure sustained infection, pathogens have evolved many strategies to evade cell death-based host defenses, which have been studied intensively in herpesviruses. Many HSV proteins that manipulate host cell apoptosis have been identified, such as ICP4, ICP27, glycoprotein D, Us3, and ICP6, as reviewed previously (4). However, mounting evidence suggests that HSV-induced apoptosis has different effects on infectious diseases. For example, in contrast to the proposed protective effect, apoptosis induced by HSV-1 infection has been shown to facilitate herpes simplex encephalitis (HSE) in the human brain, which was strongly associated with a high level of soluble Fas in cerebrospinal fluid from consecutive HSE patients (5). However, the exact mechanisms underlying the promotive effect of apoptosis on such HSV infection-associated diseases remain unknown. Studies of human immunodeficiency virus (HIV) infection have revealed that the promotive effect of apoptosis on disease progression is directly associated with the induction of apoptosis in uninfected host cells by HIV infection (6–8). More than 60% and 99% of apoptotic CD4+ T cells are uninfected cells during acute and established HIV infection, respectively (8). Many mechanisms, including activation-induced cell death (9, 10), changes in mitochondrial permeability (11), gp120/160-induced apoptosis (12), and autologous cell-mediated killing (13, 14), have been proposed as possible triggers of HIV-associated apoptosis in uninfected cells. Although the apoptosis of uninfected cells during infection with other viruses is rarely reported, it is possible that the promotive effect of apoptosis on some HSV-associated diseases, such as herpes simplex encephalitis, is also due to the killing of uninfected cells by viral infection.
Gallid herpesvirus 1 (GaHV-1), also known as avian infectious laryngotracheitis virus (ILTV), which belongs to the family Herpesviridae in the subfamily Alphaherpesvirinae, continues to cause substantial economic losses to the poultry industry worldwide (15). Both in vitro and in vivo studies have shown that ILTV infection blocks apoptosis in infected cells, thereby prolonging the life span of infected cells and consequently facilitating viral replication (16, 17). These findings are consistent with previous in vitro observations of reduced apoptosis of cells infected with other alphaherpesviruses, such as HSV-1, HSV-2, and suid herpesvirus 1 (18–20). Interestingly, along with the prosurvival effect of ILTV infection, a recent study by Reddy et al. showed that ILTV infection induces apoptosis in bystander cells in vivo (17). However, the biological significance and underlying mechanisms of this phenomenon remain unclear.
p53, one of the most important tumor suppressors, as evidenced by the malfunction of p53 signaling in most cancers (21), is also an important host antivirus factor. Super-p53 mice (with three copies of the wild-type TP53 gene) are not only resistant to oncogenesis but also have stronger antiviral capabilities than normal wild-type TP53 mice (22, 23), providing the first in vivo evidence of the antiviral function of p53. To date, the antiviral function of p53 has been confirmed in many viruses, such as Marek's disease virus (24), vesicular stomatitis virus (23, 25), poliovirus (26), hepatitis C virus (27), and influenza A virus (28). However, the effect of p53 on ILTV infection has not yet been reported.
Consistent with the findings of Reddy et al. (17), paracrine-regulated apoptosis of uninfected host cells triggered by ILTV in a host immune response-independent manner was observed in the present study. This interaction between ILTV and uninfected host cells is important for the pathological effects of viral infection and for early viral dissemination. By comparing the transcriptional profiles of ILTV-infected cells to those of uninfected apoptotic cells in combination with a set of functional studies, p53 was identified as one of the key determinants of the interaction between ILTV and uninfected host cells.
RESULTS
ILTV infection induces apoptosis in uninfected host cells.
To monitor viral infection, an ILTV strain expressing enhanced green fluorescent protein (EGFP) was generated, as shown in Fig. 1A. This EGFP virus strain was rescued and purified by multiround (>3 rounds) isolations of EGFP-positive plaques. The deletion of the US9 gene was proved by PCR (Fig. 1B). After being confirmed by PCR identification, another round of isolation of EGFP-positive plaques was performed to ensure the purification of the EGFP virus strain. The expression of EGFP in leghorn male hepatoma (LMH) cells infected with ILTV-EGFP was validated by both fluorescence microscopy (Fig. 1C) and flow cytometry (Fig. 1D). Viral replication and the cytopathic effects of infection, the main properties we focused on and investigated throughout the present study, were compared between the EGFP-expressing strain and its parental strain in our in vitro and in ovo models. No significant difference in any characteristic investigated was observed either in vitro (Fig. 1E) or in ovo (Fig. 1F and G).
FIG 1.

Characterization of recombinant ILTV expressing EGFP. (A) Scheme depicting the generation of ILTV-EGFP. (B) PCR validation of the US9 deletion. The vertical dotted line indicates that all lanes are spliced from the same gel. (C and D) Validation of EGFP expression in LMH cells infected with ILTV-EGFP by fluorescence microscopy (C) and flow cytometry (D). Cell nuclei were stained with Hoechst 33342 (blue). The scale bar indicates 400 μm in panel C. (E) The replication of ILTV/ILTV-EGFP in LMH cells was determined with the TCID50 assay (upper, primary y axis), and the cytopathic effect of ILTV/ILTV-EGFP infection on LMH cells was determined with the plaque assay. The spread of CPE was visualized by crystal violet staining (lower) and quantified statistically using ImageJ (upper, secondary y axis). (F) Viral replication in allantoic fluid from 9-day-old specific-pathogen-free (SPF) chicken embryos inoculated with ILTV and ILTV-EGFP was detected by RT-qPCR at 5 days postinfection. Data are presented as the means ± SD (n = 6; P < 0.05). (G) The survival rate of inoculated embryos was calculated at 7 days postinfection. The data are presented as the means ± SD (n = 10; P < 0.05).
Using this strain, the induction of apoptosis was compared between infected and uninfected cells via fluorescence-activated cell sorting (FACS) using annexin V-allophycocyanin (APC) at 36 h postinfection (hpi) at a multiplicity of infection (MOI) of 0.1. Under those conditions, no PI-positive cells were induced by ILTV infection (Fig. 2A). More than 15% of the uninfected cells were positive for annexin V-APC staining, while apoptotic cells were rarely observed among the 35% of cells positive for EGFP (Fig. 2A). The disparity in the induction of apoptosis by ILTV infection between the infected and uninfected cells was also evidenced by immunofluorescence staining, as the majority of annexin V-APC-positive cells and EGFP-positive cells localized differently (Fig. 2B). Specific-pathogen-free (SPF) chicken embryos infected with ILTV next were used as an in ovo model to avoid any potential effect of the host immune system. Notably, the apoptosis of cells uninfected by ILTV was also observed in ovo by the immunofluorescence-based examination of chorioallantoic membranes (CAMs) around plaques and embryo livers using antibodies specific for the gI protein of ILTV and transferase-mediated dUTP-biotin nick end labeling (TUNEL) (Fig. 2C and D).
FIG 2.

ILTV infection induces apoptosis in uninfected host cells. (A) The ILTV infection-induced apoptosis of PI-negative LMH cells was assayed by flow cytometry using annexin V-APC staining. The percentage of apoptotic cells is presented as the means ± SD (n = 3; P < 0.05). (B) ILTV-infected LMH cells and apoptotic cells were detected by fluorescence microscopy via tracing EGFP and annexin V-APC, respectively. (C and D) Immunofluorescence staining of CAMs (C) and embryo livers (D) using a rabbit polyclonal antibody against glycoprotein I of ILTV, followed by an FITC-conjoined anti-rabbit secondary antibody and TUNEL-APC staining in ovo, respectively. Normal rabbit serum was used as a background control. Cell nuclei were stained with Hoechst 33342 (blue). In panels B to D, the scale bars indicate 400 μm.
ILTV-induced apoptosis facilitates the pathological effects and early transmission of ILTV.
To explore the biological significance of ILTV-induced apoptosis, further in ovo studies of the regulation of apoptosis were performed. Two widely used compounds, carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone (Z-VAD-FMK) and N-tosyl-l-phenylalanine chloromethyl ketone (TPCK), were employed to inhibit apoptosis, and a well-known DNA-damaging agent, 5-fluorouracil (5-FU), was used to promote apoptosis. The alterations of ILTV-induced apoptosis by the above-mentioned agents were confirmed by immunofluorescence staining of CAMs via TUNEL-APC staining at 1 day postinfection (dpi) (Fig. 3A). The percentage of apoptotic cells per field was quantified statistically by observing 100 cells per field in 3 fields per slide (Fig. 3B). Upon the addition of these compounds, embryo livers and embryo survival were examined after ILTV infection. Both Z-VAD-FMK and TPCK, which blocked ILTV-induced apoptosis, but not 5-FU, which promoted ILTV-induced apoptosis, reduced the severity of liver damage in ILTV-inoculated embryos at 5 dpi (Fig. 3C). In embryos infected with ILTV alone, embryo death occurred at 5 dpi and reached 30% at 7 dpi (Fig. 3D). Upon 5-FU treatment, ILTV induced embryo death 1 day earlier, and all ILTV-inoculated embryos were dead at 6 dpi. In contrast to the significant reduction of survival rate by 5-FU treatment (P < 0.05), no embryonic death was induced by ILTV infection in embryos administered either Z-VAD-FMK or TPCK. In addition, none of these compounds alone led to embryo death. Thus, the above-described data suggest that ILTV-induced apoptosis is important for the severity of the pathological effect of ILTV infection.
FIG 3.

Biological significance of ILTV-induced apoptosis. (A) TUNEL-APC staining of CAMs in ovo at 24 hpi. Cell nuclei were stained with Hoechst 33342 (blue). The scale bar indicates 50 μm. (B) The percentage of apoptotic cells per field was quantified statistically by observing 100 cells per field in 3 fields per slide. Data are presented as the means ± SD. Asterisks indicate statistical difference (n = 6; P < 0.05). (C) Pathological examinations of livers by H&E staining. The scale bar indicates 50 μm. (D) Survival analysis of embryos inoculated with ILTV (n = 10; P < 0.05).
The effects of apoptosis on ILTV replication and spreading next were investigated using ILTV-specific reverse transcription-quantitative PCR (RT-qPCR) instead of a plaque assay or 50% tissue culture infectious dose (TCID50) assay, since the strain we used is unstable after being released from host cells (29). At 1 dpi, despite the presence of detectable amounts, no difference in viral load in either allantoic fluid or CAMs was observed among the groups (Fig. 4). Compared with the viral load at 1 dpi, a significant increase in ILTV replication at 2 dpi was seen only in CAMs but not allantoic fluid. This increase in the viral load in CAMs at 2 dpi was significantly reduced by all compounds, which demonstrates the repression of ILTV replication in CAMs by all compounds. However, except for 5-FU treatment of allantoic fluid, the viral loads reached similar levels again at 5 dpi in both allantoic fluid and CAMs regardless of the treatment, suggesting that there is little effect of ILTV-induced apoptosis on in ovo ILTV replication in general. The lowest level of viral load in CAMs of 5-FU-treated embryos may be due to the presence of the most severe pathological damage and the highest mortality rate (Fig. 3C and D). Despite having no effect on viral load, both Z-VAD-FMK and TPCK significantly reduced the levels of ILTV transmitted to the brain and liver at 1 dpi (Fig. 4). Especially in the brain, the levels of ILTV in the Z-VAD-FMK- and TPCK-treated groups were substantially lower than those of the control group throughout the observation period, while 5-FU had no effect on ILTV transmission at any time point. These findings demonstrate that ILTV-induced apoptosis helps ILTV to spread from the primary infection site to distant tissues and may be essential for ILTV infection of neurons and subsequent latency.
FIG 4.

Effect of ILTV-induced apoptosis on in ovo viral replication. Viral replication in allantoic fluid, CAMs, brains, and livers at 1, 2, and 5 days postinfection was detected by RT-qPCR. Data are presented in log10 form. Asterisks indicate statistical difference (n = 6; P < 0.05).
Paracrine-regulated apoptosis of uninfected cells by ILTV infection.
Considering the apoptosis of remote uninfected cells induced by ILTV infection (Fig. 2), we hypothesized that physical contact between ILTV-infected cells and uninfected apoptotic cells might not be required for the infection-induced apoptosis of bystander cells. A prediction from this hypothesis is that ILTV induces apoptosis in uninfected cells through a paracrine-regulated mechanism. To address this idea, two strategies were employed. First, a Transwell cell culture system was used to separate the target cells cultured in the lower chamber from the virus-host cell mixture cultured in the upper chamber, as shown in Fig. 5A (left). The success of the separation was evidenced by the absence of EGFP-positive cells from the lower chamber (Fig. 5A, middle). After ILTV infection in the upper chamber, apoptosis was clearly observed in the lower chamber cells, as detected by FACS using annexin V-APC staining (Fig. 5A, middle and right), which demonstrated the induction of paracrine-regulated apoptosis by ILTV infection. This effect was further evidenced in LMH cells pretreated with conditioned medium (CM), as presented in the schematic model, as apoptosis was clearly observed in cells incubated with CM (Fig. 5B). The exclusion of the presence of viable virions was confirmed by the absence of EGFP-positive cells from the lower chamber (Fig. 5B, middle). The induction of apoptosis by CM was also confirmed by immunofluorescence staining with annexin V-APC (Fig. 5C). Taken together, the data obtained using both strategies demonstrate that ILTV infection induces apoptosis in uninfected cells via a paracrine-regulated mechanism.
FIG 5.

Paracrine-regulated apoptosis of uninfected cells during ILTV infection. The apoptosis of uninfected LMH cells was assayed by flow cytometry, as described in the legend to Fig. 1A, using a Transwell system (A) and incubation with conditioned medium (B). The percentage of apoptotic cells is presented as the means ± SD. Asterisks indicate statistical difference (n = 3; P < 0.05). (C) Paracrine-regulated apoptosis of uninfected LMH cells after incubation with conditioned medium was determined via immunofluorescence staining using annexin V-APC, as described in the legend to Fig. 2B. The scale bar indicates 400 μm.
Genome-wide gene expression analyses reveal the repression of p53 signaling in uninfected apoptotic cells upon ILTV infection.
To address the molecular mechanisms regulating the paracrine-regulated apoptosis of uninfected cells triggered by ILTV infection, RNA samples isolated from living cells without any treatment, uninfected apoptotic cells after ILTV inoculation, ILTV-infected cells, and apoptotic cells cultured in CM were submitted for RNA sequencing (Fig. 6A). No significant difference in the total amount of RNA was observed among the groups at the time of detection (Fig. 6B), while the host RNA level of ILTV-infected cells, but not uninfected cells, was greatly repressed by infection, and viral RNA accounted for approximately 20% of the total RNA of infected cells (Fig. 6C). Bioinformatics analysis identified 1,911 genes that were differentially expressed among the groups based on the following criteria: (i) a P value of <0.001, (ii) a q value of <0.001, and (iii) a fold change of >1.5 (see Table S1 in the supplemental material). Hierarchical clustering analysis using these differentially expressed genes demonstrated efficient clustering of biological replicates for each group of cells (Fig. 6D). Compared with control cells, 422 genes were downregulated and 511 were upregulated in infected cells, while 258 genes were downregulated and 360 genes were upregulated in uninfected apoptotic cells after ILTV inoculation; 611 genes were downregulated and 546 genes were upregulated in apoptotic cells following CM incubation. For validation, the transcript levels of 15 genes randomly selected from the 1,911 differentially expressed genes were examined by RT-qPCR analysis. The directions of change determined by RT-qPCR detection and transcriptome sequencing (RNA-seq) analysis are identical in 39 of the 45 conditions tested (∼87%) (Fig. 7), suggesting an acceptable correspondence between these two methods in our study.
FIG 6.

Genome-wide transcriptome analysis. (A) Workflow of the genome-wide transcriptome analysis. (B and C) RNA level per 100 isolated cells (B) and the proportions of viral and host RNA (C) are presented as the means ± SD (n = 3; P < 0.05). FPKM, fragments per kilobase million. (D) Hierarchical clustering analysis of 1,911 genes that were differentially expressed in LMH cells at P < 0.001, q < 0.001, and a fold change of >1.5. Columns indicate arrays, and rows indicate genes. The values are normalized by row. Blue indicates downregulation, and pink indicates upregulation.
FIG 7.

Validation of the genome-wide transcriptome analysis. The transcription of 15 genes selected for the validation of the RNA sequencing data were assayed by RT-qPCR. Both RNA sequencing data and RT-qPCR data are presented as the means ± SD (n = 3).
Among these groups, the genome-wide transcription profiles of uninfected apoptotic cells from the ILTV infection group and apoptotic cells from CM incubation were in the same subcluster regardless of the presence of ILTV in the culture system (Fig. 6D), suggesting that the transcription profiles of these two groups are relatively closer. This could be explained by the finding that 65.5% of the downregulated genes and 63.8% of the upregulated genes altered in the uninfected apoptotic cells cultured in the presence of ILTV were regulated similarly in apoptotic cells cultured with CM (Fig. 8A). However, the general transcription profiles of these two groups of apoptotic cells were very different from each other due to the additional 758 genes that are differentially regulated by CM incubation only (65.5% of the total genes altered by CM). As the CM was produced by collecting culture medium from LMH cells at 24 h postinfection with ILTV, the apoptotic cells isolated from the ILTV infection group and the CM group were under different cellular conditions at the time of collection. The transcriptional profile of uninfected apoptotic cells from the ILTV inoculation group represents the cellular environment for the initiation of paracrine apoptosis, while the additional 758 genes that were altered only by CM may be important for the maintenance of paracrine apoptosis.
FIG 8.

Biological analyses of transcriptome data to identify key modulators of the paracrine-regulated apoptosis of uninfected cells during ILTV infection. (A) A Venn diagram showing the intersections of genes significantly regulated among subgroups in LMH cells (P < 0.05). (B) Combined pathway analysis of significantly expressed genes (P < 0.05). (C) An analysis of functional interactions between the significantly expressed genes reveals p53 as the most promising central modulator of the molecular events that are differentially induced in infected nonapoptotic cells and uninfected apoptotic cells. (Left) The red rectangle in the functional interaction network represents p53, and the blue rectangles represent other proteins. (Right) The top 20 key nodes were arranged in order of their functional connectivity to other proteins in the network.
Pathways enriched with these differently regulated genes were analyzed using DAVID functional annotation with a P value of <0.05. The pathways significantly regulated in each group are listed in Fig. 8B. Among these pathways, p53 pathway and spliceosome were the only two pathways commonly altered in uninfected apoptotic cells but unaffected in infected cells, which may play important roles throughout the initiation and maintenance of the paracrine-regulated apoptosis. However, the other significantly altered pathways might also contribute to the observed paracrine-regulated apoptosis. Thus, it is hard to predict the potential essential mechanisms regulating paracrine-regulated apoptosis based only on an analysis at the pathway level, and further bioinformatic strategies were needed.
Thus, the functional connections between proteins encoded by the genes that were differentially regulated in infected nonapoptotic cells and uninfected apoptotic cells were analyzed using STRING software with the default settings to predict the central modulators of the observed paracrine apoptosis. This analysis revealed a protein-protein interaction network with p53 at the center (Fig. 8C, left, the red rectangle represents p53 and the blue rectangles represent other proteins). The top 20 potential central modulators were arranged in order of their functional connectivity to the other proteins in the network, and p53 was identified as the modulator with the highest connectivity (Fig. 8C, right). Interestingly, most of the above-mentioned differentially regulated pathways, which are mainly involved in metabolism, DNA repair, RNA regulation, and the cell cycle, have been reported to be regulated by p53 (30, 31). Thus, the repression of the p53 pathway was considered a major molecular event inducing paracrine-regulated apoptosis in uninfected cells.
p53 is a key determinant of paracrine-regulated apoptosis in uninfected cells after ILTV infection.
p53 is widely recognized as a transcriptional factor regulating multiple signaling pathways, such as metabolism, the cell cycle, and apoptosis, and its activity can be regulated by different mechanisms, such as posttranslational modifications of p53, isoforms, and cofactors of p53, depending on the specific cellular environment (30). In our model, no alteration of p53 protein levels was observed in either EGFP-positive or EGFP-negative cells upon ILTV infection or CM incubation, as assayed by FACS using an antibody that specifically recognizes chicken p53 (Fig. 9A). However, the repression of p53 basal transcriptional activity (p53 transcriptional activity indicates the ability of p53 in controlling the transcription of its target genes) in uninfected apoptotic cells was evidenced by both RNA-seq (Fig. 8B) and RT-qPCR (Fig. 9B) analyses, indicating that the repression of p53 basal activity in apoptotic cells is achieved via other regulation mechanisms of p53 activity, such as changes in the posttranslational modifications of p53 and alterations of p53 cofactors. Unfortunately, neither mechanism could be detected in chickens in this study.
FIG 9.
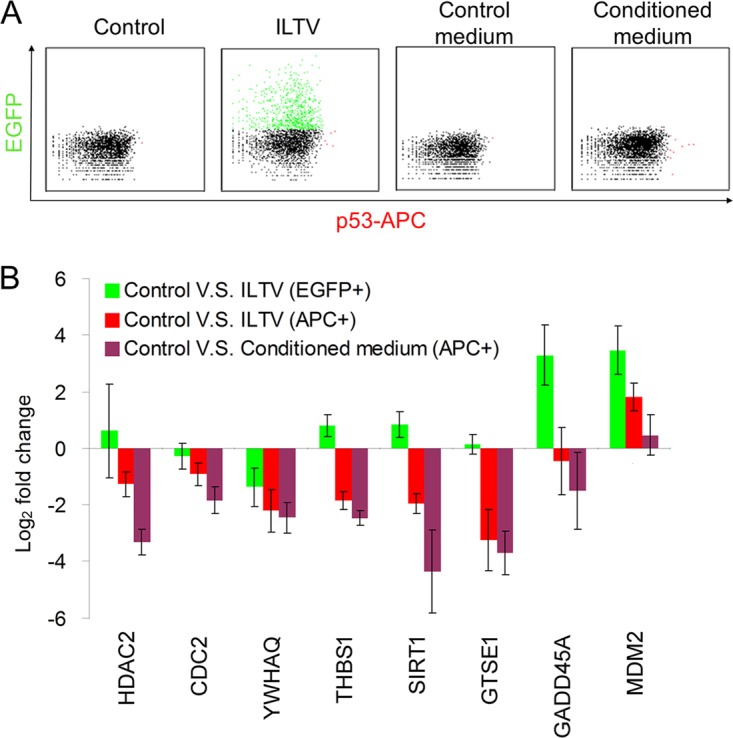
Repression of p53 signaling by ILTV infection in uninfected apoptotic cells. (A) Protein levels of p53 were examined by flow cytometry using an anti-p53 antibody followed by an APC-conjugated anti-mouse secondary antibody in LMH cells after ILTV infection or inoculation with conditioned medium. The mouse IgG was used as an isotype control to determine the level of background. (B) The transcription levels of selected genes involved in p53 signaling were assayed by RT-qPCR. Data are presented as the means ± SD (n = 3; P < 0.05).
To determine the role of p53 repression in the apoptosis of uninfected cells, p53 activity was maintained using a well-known p53 regulator, Nutlin-3a, which has been used in a p53 study of chickens (32). The induction of p53 by Nutlin-3a was confirmed by FACS analysis (Fig. 10A). Paracrine-regulated apoptosis during ILTV infection was greatly reduced by Nutlin-3a in both cells subjected to ILTV inoculation and cells cultured in CM (Fig. 10B).
FIG 10.
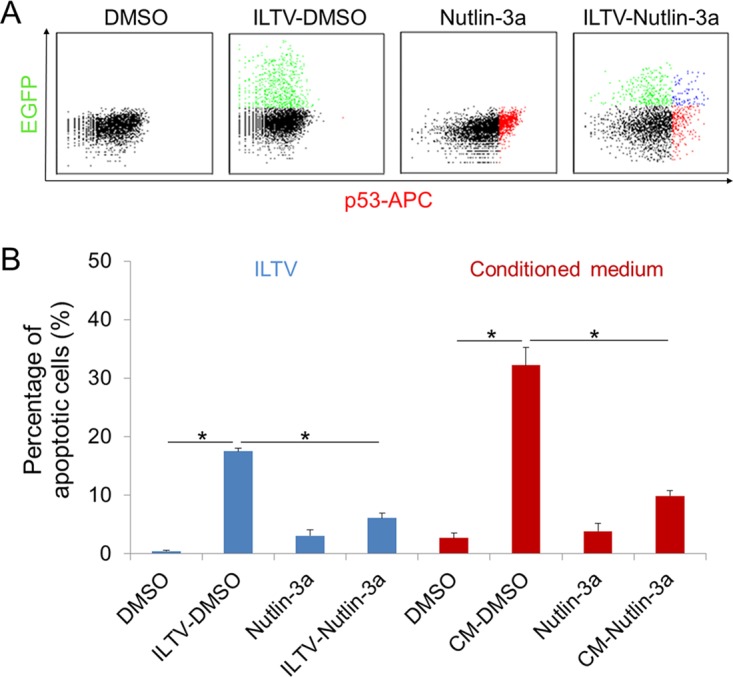
Rescue of LMH cells from paracrine-regulated apoptosis by Nutlin-3a. (A) Protein levels of p53 were examined by flow cytometry in LMH cells after ILTV infection in the presence or absence of Nutlin-3a treatment, as described in the legend to Fig. 9A. (B) Apoptosis of LMH cells upon ILTV infection (blue) or inoculation with conditioned medium (CM; red) in the presence or absence of Nutlin-3a treatment was assayed by flow cytometry, as described in the legend to Fig. 2A. Data are presented as the means ± SD. Asterisks indicate statistical difference (n = 3; P < 0.05).
To test the p53 dependence of the prosurvival effect of Nutlin-3a observed in Fig. 10B, p53 was repressed using a short interfering RNA (siRNA) specifically targeting TP53, the administration of pifithrin-α (PFT-α), a well-known p53 inhibitor, which was shown to inhibit p53 activity in both mammalian and chicken cells (32–34), or the administration of PFT-μ, which inhibits p53 binding to mitochondria by reducing its affinity for the antiapoptotic proteins Bcl-xL and Bcl-2 but has no effect on p53-dependent transactivation, as a control (35). The depletion of p53 by the siRNA was validated with Western blotting (Fig. 11A). The promotive effect of Nutlin-3a on the transcriptional activity of p53 was evidenced by the induction of the p53 target genes p21, GADD45A, and MDM2, which were blocked by the preadministration of PFT-α but unaffected by PFT-μ (Fig. 11B). This prosurvival effect of Nutlin-3a was p53 dependent, as the Nutlin-3a-mediated rescue of uninfected cells from apoptosis was greatly compromised by siRNA-mediated p53 depletion (Fig. 11C). Notably, this p53-mediated rescue of uninfected cells from paracrine-regulated apoptosis was transcription dependent, because it was only halted by PFT-α but was generally unaffected by PFT-μ (Fig. 11D).
FIG 11.
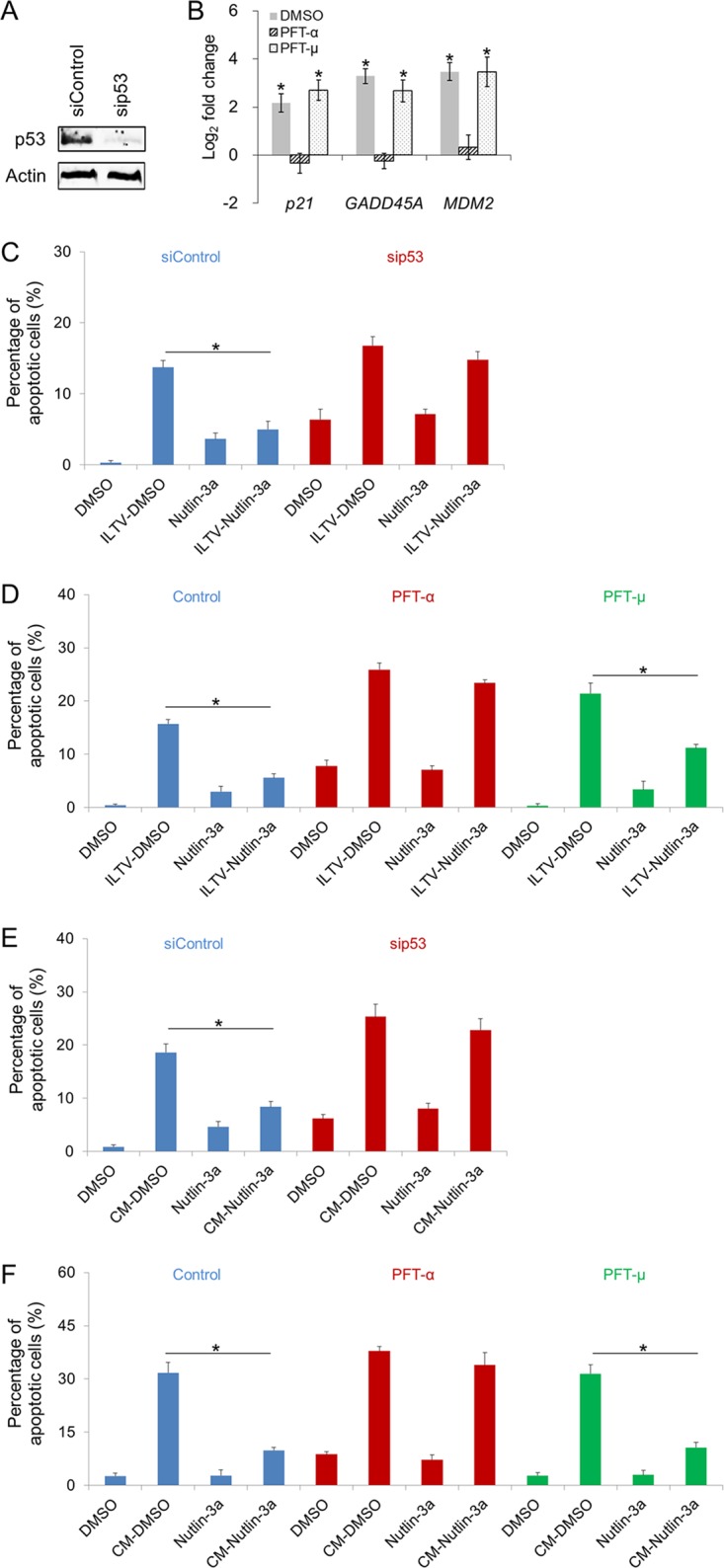
p53 is a key determinant of paracrine-regulated apoptosis in uninfected cells during ILTV infection. (A) The efficiency of p53 depletion by an siRNA specifically targeting p53 and the induction of p53 by Nutlin-3a in LMH cells were evaluated by immunoblotting. Actin was used as a loading control. (B) The transcriptional activity of p53 was determined by assaying the transcription levels of p53 target genes using RT-qPCR. All levels were normalized to the housekeeping gene GAPDH. Data are presented as log2 fold change between Nutlin-3a-treated samples and DMSO mock samples. (C to F) The apoptosis of LMH cells upon ILTV infection (C and D) or inoculation with conditioned medium (CM; E and F) in the presence or absence of Nutlin-3a treatment was assayed by flow cytometry as described in the legend to Fig. 2A. p53 activity was blocked by siRNA-mediated p53 depletion (C and E), PFT-α, an inhibitor of p53 transcriptional activity, or PFT-μ, an inhibitor of p53 posttranslational activity (D and F, respectively). The data in panels B to F are presented as the means ± SD. Asterisks indicate statistical difference (n = 3; P < 0.05).
Considering the induction of p53 by Nutlin-3a in both ILTV-infected and uninfected host cells (Fig. 10A), the prosurvival effect of p53 on the paracrine-regulated apoptosis of uninfected cells was investigated in cells cultured in CM upon siRNA-mediated p53 depletion or PFT-α preadministration. Consistent with our findings in Fig. 11C and E, the reduction of CM-triggered apoptosis by Nutlin-3a treatment was halted by both siRNA-mediated p53 depletion (Fig. 11E) and PFT-α preadministration (Fig. 11F), suggesting that p53 activation in uninfected cells is sufficient to rescue uninfected cells from the paracrine-regulated apoptosis triggered by ILTV infection, while an increase in the p53 protein in infected cells is not required.
Repression of in vitro ILTV replication by p53 activation.
We next investigated the effect of p53 on ILTV replication. Nutlin-3a treatment repressed viral replication significantly at 24 hpi, as evidenced by a TCID50 assay (Fig. 12A to C). This effect of Nutlin-3a was p53 dependent, since the reduction of ILTV replication by Nutlin-3a was compromised by both siRNA-mediated p53 knockdown (Fig. 12A) and pretreatment with PFT-α (Fig. 12B). Neither p53 knockdown using siRNA nor the administration of PFT-α had any effect on ILTV replication at 24 hpi, demonstrating that the basal function of p53, which is essential for the survival of uninfected cells, has no effect of ILTV replication. Furthermore, this p53-mediated repression of ILTV replication was not a transient effect, because the repression of viral replication by Nutlin-3a was also observed at 72 hpi (Fig. 12C). Unexpectedly, PFT-α treatment itself, which had no effect on viral replication at 24 hpi (Fig. 12B), repressed viral replication significantly at 72 hpi (Fig. 12C). This finding was correlated with faster cell death after PFT-α treatment during ILTV infection than with ILTV infection alone (Fig. 12D). In contrast, Nutlin-3a treatment rescued cells from death induced by ILTV infection throughout the observation period after 36 hpi (Fig. 12D). Hence, the repression of viral replication by PFT-α was likely due to rapid cell loss. In addition, both Nutlin-3a and PFT-α were found to induce transient low levels of cell death (less than 10% of cells) at the beginning of infection (Fig. 12D), which was inconsistent with previous findings concerning the induction of low levels of apoptotic cells by the administration of Nutlin-3a or siRNAs targeting p53 and PFT-α (Fig. 11C to F). The transient cell death caused by Nutlin-3a alone was p53 independent, since it could be blocked by neither siRNA-mediated p53 knockdown (Fig. 11C and E) nor pretreatment with PFT-α (Fig. 11D and F), indicating the existence of another target for Nutlin-3a in LMH cells. Given that the LMH chicken hepatoma cell line is the only cell line susceptible to ILTV infection in vitro, in ovo studies were carried out to determine whether the above-mentioned effects of Nutlin-3a and PFT-α were cell line-specific effects.
FIG 12.
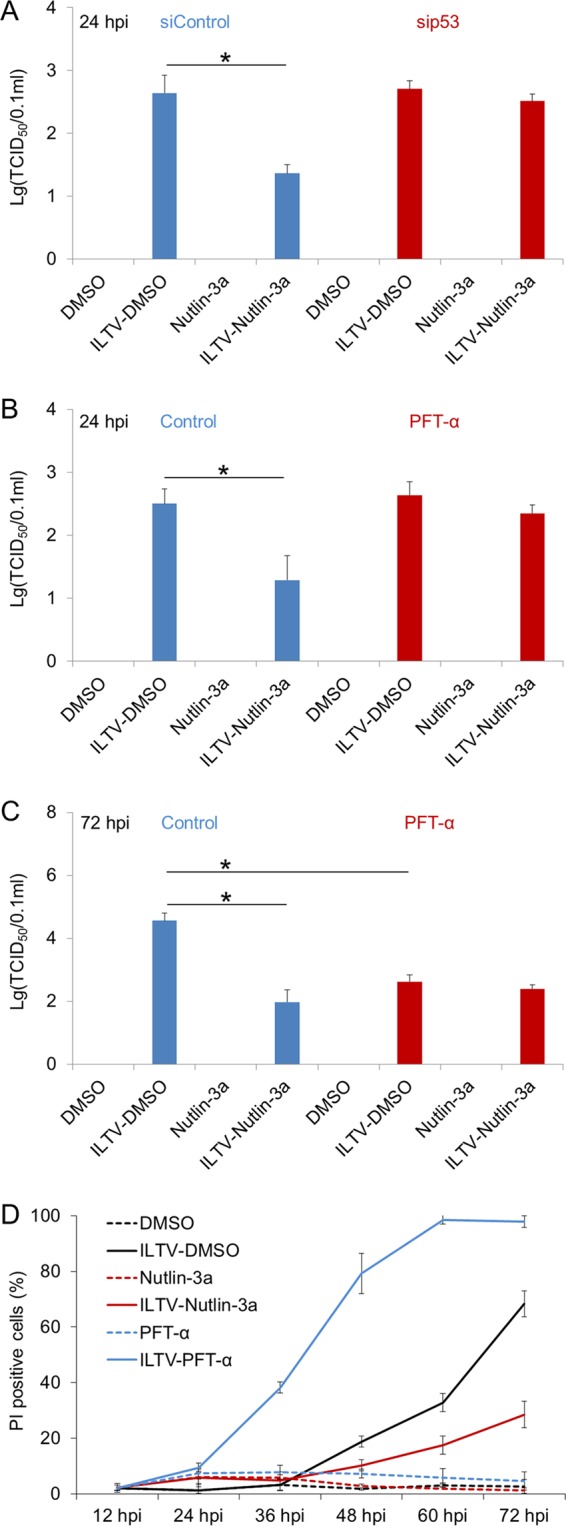
Repression of in vitro ILTV replication by p53 activation. (A to C) The effect of p53 on viral replication in LMH cells upon p53 depletion (A) or pretreatment with PFT-α (B and C) in the presence or absence of Nutlin-3a treatment was determined with the TCID50 assay. (D) The proportions of PI-positive cells were assayed by flow cytometry after pretreatment with Nutlin-3a or PFT-α. The data are presented as the means ± SD. Asterisks indicate statistical difference (n = 3; *, P < 0.05).
p53 is a key determinant of ILTV infection in ovo.
To determine whether the above-mentioned effects of p53 observed in the chicken hepatoma cell line LMH also exist in living organs and tissues, in ovo investigations were conducted using 9-day-old SPF chicken embryos inoculated with ILTV through the allantoic cavity at 1× 105 TCID50 in the presence or absence of 250 μg of PFT-α or 250 μg of Nutlin-3a. Apoptosis was observed in CAMs after ILTV inoculation and was strongly promoted by PFT-α but reduced by Nutlin-3a (Fig. 13A and B). Damage to the hepatic tissue was commonly observed in all embryos inoculated with ILTV, except for embryos coinoculated with Nutlin-3a, in which only the recruitment of lymphocytes around blood vessels occurred (Fig. 13C). The effect of p53 on the survival of inoculated embryos was also investigated. The death of inoculated embryos occurred at 4 dpi, and 40% of inoculated embryos were dead by 7 dpi (Fig. 13D). PFT-α reduced the survival rate of inoculated embryos significantly (P < 0.05), as upon the administration of PFT-α, the death of inoculated embryos occurred 2 days earlier, and all inoculated embryos were dead by 6 dpi (Fig. 13D). In contrast, throughout the observation period, no death of inoculated embryos was observed in the Nutlin-3a-treated group (Fig. 13D). No obvious apoptosis of CAM cells, pathological damage to the hepatic tissue, or embryonic death was found in any uninfected embryos, including embryos treated with Nutlin-3a or PFT-α, suggesting that the transient cell death induced by Nutlin-3a and PFT-α in LMH cells is a cell line-specific effect rather than a complicated effect of p53 (Fig. 13A to C). In addition, Nutlin-3a treatment significantly reduced the viral load in the allantoic fluid, CAMs, and liver at 2 dpi and 5 dpi and reduced the viral load in brain throughout the period of investigation (Fig. 14). Thus, at least two mechanisms, delayed viral dissemination and repressed viral replication, were involved in the reduced viral load caused by Nutlin-3a treatment in the brain and liver.
FIG 13.
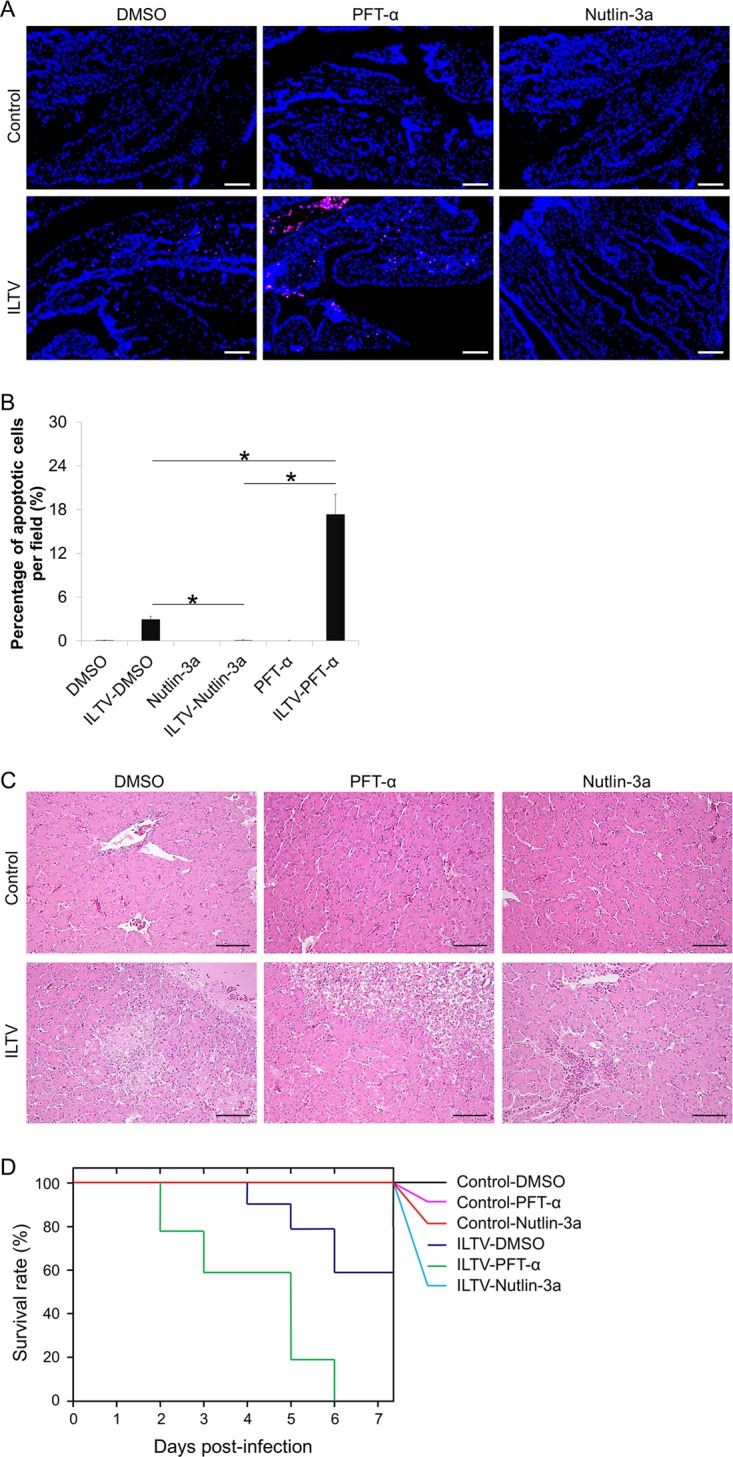
p53 is a key determinant of the pathological effect of ILTV infection in ovo. (A) TUNEL-APC staining of CAMs in ovo at 24 hpi. Cell nuclei were stained with Hoechst 33342 (blue). The scale bar indicates 50 μm. (B) The percentage of apoptotic cells per field was quantified statistically by observing 100 cells per field in 3 fields per slide. Data are presented as the means ± SD. Asterisks indicate statistical difference (n = 6; P < 0.05). (C) Pathological examinations of livers by H&E staining. The scale bar indicates 50 μm. (D) Survival analysis of embryos inoculated with ILTV (n = 10; P < 0.05).
FIG 14.

Effect of p53 on in ovo viral replication. Viral replication in allantoic fluid, CAMs, brains, and livers was detected by RT-qPCR at 1, 2, and 5 days postinfection. Data are presented in log10 form. Asterisks indicate statistical difference (n = 6; P < 0.05).
DISCUSSION
Despite being widely recognized as a common host defense mechanism, apoptosis induced in uninfected cells during viral infection may have the opposite effect. For example, HIV infection induces massive apoptosis in uninfected CD4+ T cells, which is directly associated with disease progression (8). Recently, ILTV was reported to induce the apoptosis of remote uninfected cells in solid tissues (17). However, the biological significance and underlying mechanisms of this phenomenon remain unclear. The results of the present study found that ILTV infection triggers the apoptosis of uninfected cells in a host immune response-independent manner, since the apoptosis of uninfected host cells was observed in both the in vitro and in ovo models without interference by the host immune system during ILTV infection (Fig. 2). This effect of ILTV infection contributed to the pathological effect and early tissue transmission of ILTV (Fig. 3 and 4) and is facilitated by the paracrine repression of p53 activity in uninfected cells (Fig. 5 to 14).
Recent studies have demonstrated that ILTV infection prolongs the survival of infected cells by blocking the apoptosis of infected cells to ensure the greatest ILTV replication while triggering apoptosis in uninfected cells (16, 17). The underlying mechanisms remain largely unknown but are important for developing novel strategies targeting the interactions between ILTV and host cells for better control of diseases, especially when the host immune response is compromised. Our previous studies identified Src-FAK cooperation as an essential determinant of efficient ILTV replication and prolonged survival of infected cells (29). The disruption of Src-FAK cooperation significantly represses ILTV replication but also promotes ILTV-associated cell death and pathological damage, which limits the future clinical application of an Src-FAK-based strategy for ILTV control. Other potential therapeutic targets are required for the development of strategies that can improve ILTV control and reduce side effects during therapy. The results of the present study showed that the pharmacological reactivation of host p53 not only limited ILTV replication and distant tissue dissemination but also alleviated the extent of pathological damage during ILTV infection. The dual cell-intrinsic mechanisms of p53-mediated suppression during ILTV infection suggest that p53 is a promising therapeutic target for combating ILTV infection.
The antiviral function of p53 has been recognized since the generation of super-p53 mice (with three copies of the wild-type TP53 gene) (21). To date, the antiviral function of p53 has been confirmed in many viruses, such as Marek's disease virus (24), vesicular stomatitis virus (23, 25), poliovirus (26), hepatitis C virus (27), and influenza A virus (28). To the best of our knowledge, the present study is the first to provide evidence for the antiviral effect of p53 against ILTV infection. p53 has been shown to control many aspects of host immune responses (36–43) and has been identified as a transcriptional factor for several important antivirus immune ligands (44–47), which provides a link between p53 function and the antiviral immune response. In contrast to these advancements in our mechanistic understanding of the cell-extrinsic antiviral function of p53, the mechanisms that underlie the cell-intrinsic antiviral function of p53 remain to be explored. The present study provides dual cell-intrinsic mechanisms by which p53 suppresses ILTV infection. In uninfected cells, p53 activation protects cells from ILTV-induced paracrine-regulated apoptosis, which may reduce pathological damage and delay viral transmission, while in infected cells, p53 activation suppresses ILTV replication (Fig. 12 and 14).
The establishment of a latent infection in the trigeminal ganglia is a general mechanism by which alphaherpesviruses escape from host immune surveillance. The latent viruses cannot be eliminated in either humans or animals by any currently available therapy and are frequently reactivated once host immunity is suppressed. Here, we observed delayed ILTV transmission from the primary inoculation site to the embryo brain via the repression of apoptosis in host cells (Fig. 4). It is possible that the repression of host apoptosis prevents acute ILTV infection of the trigeminal ganglia. According to the dual mechanisms that underlie the p53-mediated suppression of ILTV infection, as discussed above, we can speculate that p53 reactivation helps to delay disease progression and the establishment of a latent infection, which may in turn provide sufficient time for the activation of host anti-ILTV immune responses and thereby improve the clearance of residual infected cells.
In the present study, Z-VAD-FMK and TPCK were used to block apoptosis of host cells, and we found that both apoptosis inhibitors repressed ILTV infection significantly (Fig. 4). In general, TPCK showed greater effects than Z-VAD-FMK, which is most likely due to the greater reduction of the apoptosis of host cells by TPCK (Fig. 3). However, other mechanisms in terms of the repression of ILTV infection by TPCK should also be taken into account, because TPCK is a nonspecific inhibitor of apoptosis (48). Besides, some p53-regulating compounds we used in the current study seemed to play roles more complicated than those we have revealed in our experimental models. Nutlin-3a treatment itself could induce transient low levels of cell death (less than 10% of cells) at the beginning of infection independent of p53 (Fig. 10B, 11C to F, and 12D), which indicates the existence of another target for Nutlin-3a in LMH cells. Similar to Nutlin-3a, both sip53 and PFT-α trigger cell death in LMH cells (Fig. 11C to F), which suggests a crucial role of p53 basal activity in the survival of LMH cells. Although no apoptosis was induced by PFT-μ, ILTV–PFT-μ interaction promoted apoptosis of LMH cells regardless of Nutlin-3a-mediated p53 activation (Fig. 11D and F). This proapoptotic effect of PFT-μ demonstrates a proapoptotic effect of PFT-μ in ILTV-infected cells in a p53-independent manner, since it was not observed during the induction of apoptosis by conditioned medium (Fig. 11F).
The interaction between the virus and the host is a complex process involving not only the virus and infected cells but also uninfected cells and their microenvironment. Thus, it is difficult to address all of the properties of virus-host interactions using in vitro experimental models with a high MOI. Increasing efforts have been made to monitor virus-host interactions with a low MOI to better mimic in vivo infection, and some previously unexpected findings have been reported. Using an in vitro experimental model with an MOI of 0.001, HSV-1 was found to promote host DNA replication in both infected and uninfected cells via an unknown paracrine-regulated mechanism (49). This finding is consistent with our analysis of genome-wide gene expression data, which demonstrated that ILTV infection elevates the metabolic signaling pathways of both infected and uninfected cells (Fig. 8). Once the abnormal elevation of metabolic activity occurs, apoptosis can be triggered in the cells. During retrovirus and Chlamydia trachomatis infection, host p53 is activated to balance host metabolism and protect host cells from apoptosis (50, 51). It will be interesting to address whether ILTV infection triggers paracrine-regulated apoptosis by promoting metabolism and repressing p53 activity simultaneously in uninfected cells. In addition, although the paracrine-regulated apoptosis of uninfected cells has been observed in both HIV and ILTV infections (6–8) (Fig. 5), several molecules from the host cell and HIV that are essential for the apoptosis of uninfected T cells have been identified (12–14). However, the exact soluble molecules from the host cell or virus that induce this paracrine-regulated mechanism during ILTV infection remain unknown. Given that p53 suppresses ILTV infection through its transcriptional activity (Fig. 11), further investigations of the genome-wide chromatin occupancy of p53 in combination with transcriptional profiling analysis may help to uncover the molecular basis of this paracrine-regulated mechanism.
To the best of our knowledge, this study is the first to uncover the mechanism of ILTV-triggered apoptosis in uninfected host cells, which extends our understanding of both herpesvirus-host interactions and the role of p53 in viral infection and may also contribute to a mechanistic illustration of apoptosis induced by other viruses in uninfected host cells.
MATERIALS AND METHODS
Ethics statement.
The animal experimental protocol was approved and performed in accordance with the ethical guidelines of the Animal Ethics Committee of Harbin Veterinary Research Institute of the Chinese Academy of Agricultural Sciences (approval no. SYXK [Hei] 2011022).
In ovo experiments.
Viral specimens (100 μl) at a median tissue culture infective dose (TCID50) of 1 × 105 cells/ml were inoculated into the allantoic cavities of 9-day-old specific-pathogen-free (SPF) chicken embryos, in addition to 250 μg of the p53 activator Nutlin-3a, 250 μg of the p53 inhibitor pifithrin-α (PFT-α), 250 μg of carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone (Z-VAD-FMK), 1.5 mg of N-tosyl-l-phenylalanine chloromethyl ketone (TPCK), 500 μg of 5-fluorouracil (5-FU), or an equal volume of dimethyl sulfoxide (DMSO). All reagents were purchased from Sigma-Aldrich Corporation (St. Louis, MO, USA). Chicken embryos were incubated at 37°C and examined daily for 7 days. The chorioallantoic membranes (CAMs), allantoic fluid, brains, and livers were then harvested, homogenized, and subjected to three freeze-thaw cycles at the indicated time points.
Viral strain and cell line.
The virulent ILTV strain LJS09 (GenBank accession no. JX458822) is stored at the Harbin Veterinary Research Institute of the Chinese Academy of Agricultural Sciences. This strain can be propagated in a chemically immortalized leghorn male hepatoma (LMH) cell line with clear cytopathic effects (52, 53). LMH cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine. The cell cultures were incubated at 37°C under an atmosphere of 5% CO2. The p53 activator Nutlin-3a, the p53 inhibitor PFT-α, and the p53 inhibitor PFT-μ (Sigma-Aldrich Corporation) were used at 10 μM for in vitro experiments. Control groups were treated with DMSO at the same volumes. The cell medium was prefiltered through a 0.1-μm filter twice and stored at −80°C for use as conditioned medium (CM).
RNA interference and transfection.
A short-interfering RNA (siRNA) that specifically recognized sequences from the TP53 mRNA (NM_205264; sip53, 5′-CGG AGG AGA UGG AAC CAU U-3′) and a control siRNA (siControl, 5′-GCA CUU GAU ACA CGU GUA A-3′) with no specific target site in chickens were used. The transfection of siRNA was conducted using an N-TER nanoparticle siRNA transfection system (Sigma-Aldrich Corporation) according to the manufacturer's instructions.
Viral quantitation.
Levels of viral replication were determined using an ILTV-specific qPCR assay (53) and a TCID50 assay (54), as described previously. LMH cells were infected with LJS09 at a multiplicity of infection (MOI) of 0.1. The indicated MOI was obtained according to the number of cells to be infected and the estimated number of infectious particles, based on PFU detected in LMH cells.
Flow cytometry and immunofluorescence.
Fluorescence-activated cell sorting (FACS) analyses were conducted using a BD FACScan cell sorter and CellQuest software version 4.0.2 (Becton Dickinson-Pharmingen, San Diego, CA, USA). Cell death was assayed by examining cells in the sub-G1 phase of the cell cycle with propidium iodide (PI) staining of permeabilized cells, as described previously (44). Apoptosis was assayed using the annexin V-APC staining kit (KeyGEN BioTECH, Nanjing, China), the annexin V-fluorescein isothiocyanate (FITC)/PI double staining kit (KeyGEN BioTECH), or the TUNEL staining kit (Beyotime BioTECH, Shanghai, China) according to the manufacturer's instructions. For immunofluorescence staining, the samples were washed with phosphate-buffered saline and fixed with 4% paraformaldehyde for 10 min. After quenching excess aldehyde, the samples were permeabilized with 0.1% Triton X-100. Nonspecific antibody binding was blocked with 2% bovine serum albumin for 1 h, and then the samples were incubated with a rabbit polyclonal antibody against ILTV glycoprotein I (29), followed by a secondary FITC-conjugated goat anti-rabbit antibody (The Jackson Laboratory, Bar Harbor, ME, USA). Background was determined by normal rabbit control serum from nonimmunized SPF rabbits. All cell nuclei were stained with Hoechst 33342 (Sigma-Aldrich Corporation).
Histopathological examination.
Embryonic CAMs and livers were collected and fixed in 4% paraformaldehyde. The fixed samples were sent to the Plaques Diagnosis and Technical Service Center of the Harbin Veterinary Research Institute (animal biosafety level 3; accredited by the China National Accreditation Service for Conformity Assessment, or CNAS) for histopathological examination. Briefly, the samples were embedded in paraffin and then sectioned. The sample slides were stained with hematoxylin-eosin (H&E) and observed by light microscopy or used for immunofluorescence staining. Histopathological examination reports were provided by the Plaques Diagnosis and Technical Service Center.
Protein extraction and Western blotting.
Western blotting was performed under reduced denaturing conditions according to previously described procedures (55). Antibodies against p53 (Pab240; Santa Cruz Biotechnology, Dallas, TX, USA) and actin (Sigma-Aldrich Corporation) were used.
RNA sequencing.
Genome-wide gene expression profiling of LMH cells was performed via RNA deep sequencing by Annoroad Gene Technology Co., Ltd. (Beijing, China). Library construction was performed using the Illumina platform (Illumina, Inc., San Diego, CA, USA) according to the manufacturer's instructions. The samples were sequenced on an Illumina HiSeq 2500 instrument.
RT-qPCR.
RNA was isolated from 100 cells using the REPLI-g single-cell kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. RT-qPCR was performed using the SYBR PrimeScript kit (TaKaRa Bio Inc., Tokyo, Japan) as described previously (56). The primer sequences used for RT-qPCR are presented in Table 1. Data were calculated according to the 2−ΔΔCT method, and the results are presented as the log2 fold change.
TABLE 1.
List of RT-qPCR primers
| Gene | Direction | Sequence (5′–3′) |
|---|---|---|
| CCNB2 | F | GATGGGAAGACGATCAATGGAC |
| R | CTGGATGACTTTGTAAGAAGCGG | |
| CDC2 | F | GAATCTTCAGAGCTTTAGGGACAC |
| R | ACAACAGATCGAGTCCATCCT | |
| CNBP | F | GCTTCCAGTTCATGTCTTCATCTC |
| R | GTTATAGCAGGCTTCATCCTCC | |
| DDX47 | F | TACGAGCTCTGAAGTTCCTAGTG |
| R | CAGTATCTTATCCACCTCTGTCTC | |
| DUSP1 | F | AGGCGATTGACTTCATAGATTCTG |
| R | GGCTTCATCCAGCTTGACTC | |
| FARSB | F | AACTTCCACTCAATAAGCTCACAG |
| R | TTCCGTAGAACACAGGGCA | |
| GADD45A | F | CGTCTTGGTCACGAATCCC |
| R | CACCCACTGATCCATATAGCGA | |
| GAPDH | F | GGCACTGTCAAGGCTGAGAA |
| R | TGCATCTGCCCATTTGATGT | |
| GTSE1 | F | GCATGGATGTTTCAGACTTTCCT |
| R | AGACTTCATCTTCACTTCCGCT | |
| HDAC2 | F | CAAACGCATTTCTATTCGAGCA |
| R | GCAACATTTCGCCGTCCA | |
| HSPA14 | F | AGAACGAGGAGGTTGTGGG |
| R | AGGATCTGCTTTACTTTCACCA | |
| HSP70 | F | GTATTCTTGCGTGGGTGTC |
| R | CGCTCTGTATCGGTGAAGG | |
| IFNGR2 | F | AGACTGAGTGCAGTTTCCT |
| R | GGCCCTATGGTAGTGTTTCTC | |
| KIT | F | GGTTAAAGGAAATGCTCGTCTC |
| R | ATTCCATAAGACCAGACATCAC | |
| MDM2 | F | GCCAAATTTCGGCTTGAAAA |
| R | TGTTGTTGGCTGGGAAGTTG | |
| Novel gene | F | TTGCCAAGACTCCGGGT |
| R | CTTCGCTGCTTTGCCCT | |
| p21 | F | CCACCATCAAAGACTTCTACGG |
| R | CTTAGACAGTACAGGGCAGG | |
| RFLB | F | GTGCCGTCATCTTCACCT |
| R | CTTGAGAAGAATCGGCCGC | |
| RPF1 | F | TGTAGATCCCAATGATGAAGAGGT |
| R | TCTCACTGTTCTCCCACGA | |
| SIRT1 | F | GCATTGATTCCAAGTTCCATCC |
| R | TCACAGTCTCCAAGAAGCTCC | |
| THBS1 | F | CCACAGCAGTATGACTATGACAG |
| R | GTTGAGGACACCATCTCCA | |
| TMEM167A | F | TCTTCTGACTGTAATCTTGCTG |
| R | CTCTTTCGTTCACCAATCCTG | |
| TNFRSF21 | F | CGGCATCTCTTCCAAACAC |
| R | AAGAGTTCAGACCTTTGGGA | |
| TUBB6 | F | GAATCATCTTCCCAGAAATACGTG |
| R | CCAGTTGTTTCCTGCACCA | |
| YWHAQ | F | CCAAGACAGCGTTTGATGAG |
| R | CAGCATCACATTCTTCTCCTG |
High-throughput data analysis.
RNA sequencing data were analyzed with the Galaxy web-based tool (57). Pathway analysis was performed with DAVID (gene enrichment analysis using the EASE score, a modified Fisher's exact P value, as the threshold) (58). Functional protein network analysis was performed with STRING (59).
Statistical analysis.
The SPSS software package (SPSS for Windows, version 13.0; SPSS Inc., Chicago, IL, USA) was used for all statistical analyses. Data obtained from several experiments are reported as the means ± standard deviations (SD). The significance of differences between two groups was determined with Student's t test. One-way or two-way analysis of variance with the Bonferroni correction was employed for multigroup comparisons. Kaplan-Meier survival analysis with the log-rank test was performed for survival analysis. For all analyses, a P value of <0.05 was considered statistically significant.
Accession number(s).
Raw RNA sequencing data were uploaded to the Gene Expression Omnibus (GEO) database under accession number GSE103371.
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by the Natural Science Foundation of Heilongjiang Province (no. LC2017016), the Harbin Special Fund for Innovative Talents in Science and Technology (no. 2016RAXYJ072), the China Agriculture Research System (no. CARS-41-K18), and the Elite Youth Program of the Chinese Academy of Agricultural Sciences (CAASQNYC-KYYJ-56).
The publication reflects only the authors' views.
We are greatly indebted to Zhiyong Ma, Yafeng Qiu, Yulong Gao, and our other colleagues who shared valuable reagents with us, and we are grateful to Xijun He and other colleagues from the Plaques Diagnosis and Technical Service Center for providing technical support and valuable suggestions.
We have no conflicts of interest to declare.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00529-18.
REFERENCES
- 1.Samaniego LA, Neiderhiser L, DeLuca NA. 1998. Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J Virol 72:3307–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanfilippo CM, Blaho JA. 2006. ICP0 gene expression is a herpes simplex virus type 1 apoptotic trigger. J Virol 80:6810–6821. doi: 10.1128/JVI.00334-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X, Li Y, Liu S, Yu X, Li L, Shi C, He W, Li J, Xu L, Hu Z, Yu L, Yang Z, Chen Q, Ge L, Zhang Z, Zhou B, Jiang X, Chen S, He S. 2014. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci U S A 111:15438–15443. doi: 10.1073/pnas.1412767111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen ML, Blaho JA. 2007. Apoptosis during herpes simplex virus infection. Adv Virus Res 69:67–97. doi: 10.1016/S0065-3527(06)69002-7. [DOI] [PubMed] [Google Scholar]
- 5.Sabri F, Granath F, Hjalmarsson A, Aurelius E, Sköldenberg B. 2006. Modulation of sFas indicates apoptosis in human herpes simplex encephalitis. J Neuroimmunol 171:171–176. [DOI] [PubMed] [Google Scholar]
- 6.Meyaard L, Otto SA, Jonker RR, Mijnster MJ, Keet RP, Miedema F. 1992. Programmed death of T cells in HIV-1 infection. Science 257:217–219. doi: 10.1126/science.1352911. [DOI] [PubMed] [Google Scholar]
- 7.Badley AD, Pilon AA, Landay A, Lynch DH. 2000. Mechanisms of HIV-associated lymphocyte apoptosis. Blood 96:2951–2964. [PubMed] [Google Scholar]
- 8.Matrajt L, Younan PM, Kiem HP, Schiffer JT. 2014. The majority of CD4+ T-cell depletion during acute simian-human immunodeficiency virus SHIV89.6P infection occurs in uninfected cells. J Virol 88:3202–3212. doi: 10.1128/JVI.03428-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ju ST, Panka DJ, Cui H, Ettinger R, el-Khatib M, Sherr DH, Stanger BZ, Marshak-Rothstein A. 1995. Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature 373:444–448. doi: 10.1038/373444a0. [DOI] [PubMed] [Google Scholar]
- 10.Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. 1995. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature 377:348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]
- 11.Carbonari M, Pesce AM, Cibati M, Modica A, Dell'Anna L, D'Offizi G, Angelici A, Uccini S, Modesti A, Fiorilli M. 1997. Death of bystander cells by a novel pathway involving early mitochondrial damage in human immunodeficiency virus-related lymphadenopathy. Blood 90:209–216. [PubMed] [Google Scholar]
- 12.Aceituno E, Castañón S, Jiménez C, Subirá D, De Górgolas M, Fernández-Guerrero M, Ortíz F, García R. 1997. Circulating immune complexes from HIV-1 patients induces apoptosis on normal lymphocytes. Immunology 92:317–320. doi: 10.1046/j.1365-2567.1997.00361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oyaizu N, Adachi Y, Hashimoto F, McCloskey TW, Hosaka N, Kayagaki N, Yagita H, Pahwa S. 1997. Monocytes express Fas ligand upon CD4 cross-linking and induce CD4 T cells apoptosis. J Immunol 158:2456–2463. [PubMed] [Google Scholar]
- 14.Kojima H, Eshima K, Takayama H, Sitkovsky MV. 1997. Leukocyte function-associated antigen-1 dependent lysis of Fas(CD95/Apo-1) innocent bystanders by antigen-specific CD8 CTL. J Immunol 158:2728–2734. [PubMed] [Google Scholar]
- 15.Ou SC, Giambrone JJ. 2012. Infectious laryngotracheitis virus in chickens. World J Virol 1:142–149. doi: 10.5501/wjv.v1.i5.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burnside J, Morgan R. 2011. Emerging roles of chicken and viral microRNAs in avian disease. BMC Proc Suppl 4:S2. doi: 10.1186/1753-6561-5-S4-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reddy VR, Steukers L, Li Y, Fuchs W, Vanderplasschen A, Nauwynck HJ. 2014. Replication characteristics of infectious laryngotracheitis virus in the respiratory and conjunctival mucosa. Avian Pathol 43:450–457. doi: 10.1080/03079457.2014.956285. [DOI] [PubMed] [Google Scholar]
- 18.Glorieux S, Bachert C, Favoreel HW, Vandekerckhove AP, Steukers L, Rekecki A, Van den Broeck W, Goossens J, Croubels S, Clayton RF, Nauwynck HJ. 2011. Herpes simplex virus type 1 penetrates the basement membrane in human nasal respiratory mucosa. PLoS One 6:e22160. doi: 10.1371/journal.pone.0022160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asano S, Honda T, Goshima F, Watanabe D, Miyake Y, Sugiura Y, Nishiyama Y. 1999. US3 protein kinase of herpes simplex virus type 2 plays a role in protecting corneal epithelial cells from apoptosis in infected mice. J Gen Virol 80(Part 1):51–56. [DOI] [PubMed] [Google Scholar]
- 20.Alemañ N, Quiroga MI, López-Peña M, Vázquez S, Guerrero FH, Nieto JM. 2001. Induction and inhibition of apoptosis by pseudorabies virus in the trigeminal ganglion during acute infection of swine. J Virol 75:469–479. doi: 10.1128/JVI.75.1.469-479.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Menendez D, Inga A, Resnick MA. 2009. The expanding universe of p53 targets. Nat Rev Cancer 9:724–737. doi: 10.1038/nrc2730. [DOI] [PubMed] [Google Scholar]
- 22.García-Cao I, García-Cao M, Martín-Caballero J, Criado LM, Klatt P, Flores JM, Weill JC, Blasco MA, Serrano M. 2002. Super p53′ mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO 21:6225–6235. doi: 10.1093/emboj/cdf595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Munoz-Fontela C, Garcia MA, Garcia-Cao I, Collado M, Arroyo J, Esteban M, Serrano M, Rivas C. 2005. Resistance to viral infection of super p53 mice. Oncogene 24:3059–3062. doi: 10.1038/sj.onc.1208477. [DOI] [PubMed] [Google Scholar]
- 24.Deng X, Li X, Shen Y, Qiu Y, Shi Z, Shao D, Jin Y, Chen H, Ding C, Li L, Chen P, Ma Z. 2010. The Meq oncoprotein of Marek's disease virus interacts with p53 and inhibits its transcriptional and apoptotic activities. Virol J 7:348. doi: 10.1186/1743-422X-7-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garijo R, Hernández-Alonso P, Rivas C, Diallo JS, Sanjuán R. 2014. Experimental evolution of an oncolytic vesicular stomatitis virus with increased selectivity for p53-deficient cells. PLoS One 9:e102365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pampin M, Simonin Y, Blondel B, Percherancier Y, Chelbi-Alix MK. 2006. Cross talk between PML and p53 during poliovirus infection: implications for antiviral defense. J Virol 80:8582–8592. doi: 10.1128/JVI.00031-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dharel N, Kato N, Muroyama R, Taniguchi H, Otsuka M, Wang Y, Jazag A, Shao RX, Chang JH, Adler MK, Kawabe T, Omata M. 2008. Potential contribution of tumor suppressor p53 in the host defense against hepatitis C virus. Hepatology 47:1136–1149. doi: 10.1002/hep.22176. [DOI] [PubMed] [Google Scholar]
- 28.Yan W, Wei J, Deng X, Shi Z, Zhu Z, Shao D, Li B, Wang S, Tong G, Ma Z. 2015. Transcriptional analysis of immune-related gene expression in p53-deficient mice with increased susceptibility to influenza A virus infection. BMC Med Genomics 8:52. doi: 10.1186/s12920-015-0127-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Wang F, Han Z, Gao Q, Li H, Shao Y, Sun N, Liu S. 2016. Genome-wide gene expression analysis identifies the proto-oncogene tyrosine-protein kinase Src as a crucial virulence determinant of infectious laryngotracheitis virus in chicken cells. J Virol 90:9–21. doi: 10.1128/JVI.01817-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meek DW. 2015. Regulation of the p53 response and its relationship to cancer. Biochem J 469:325–346. doi: 10.1042/BJ20150517. [DOI] [PubMed] [Google Scholar]
- 31.Allende-Vega N, Dayal S, Agarwala U, Sparks A, Bourdon JC, Saville MK. 2013. p53 is activated in response to disruption of the pre-mRNA splicing machinery. Oncogene 32:1–14. doi: 10.1038/onc.2012.38. [DOI] [PubMed] [Google Scholar]
- 32.Shirazi Fard S, Thyselius M, All-Ericsson C, Hallböök F. 2014. The terminal basal mitosis of chicken retinal Lim1 horizontal cells is not sensitive to cisplatin-induced cell cycle arrest. Cell Cycle 13:3698–3706. doi: 10.4161/15384101.2014.964985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV. 1999. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285:1733–1737. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 34.Pietrancosta N, Maina F, Dono R, Moumen A, Garino C, Laras Y, Burlet S, Quéléver G, Kraus JL. 2005. Novel cyclized Pifithrin-alpha p53 inactivators: synthesis and biological studies. Bioorg Med Chem Lett 15:1561–1564. doi: 10.1016/j.bmcl.2005.01.075. [DOI] [PubMed] [Google Scholar]
- 35.Strom E, Sathe S, Komarov PG, Chernova OB, Pavlovska I, Shyshynova I, Bosykh DA, Burdelya LG, Macklis RM, Skaliter R, Komarova EA, Gudkov AV. 2006. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat Chem Biol 2:474–479. doi: 10.1038/nchembio809. [DOI] [PubMed] [Google Scholar]
- 36.Albina JE, Cui S, Mateo RB, Reichner JS. 1993. Nitric oxide-mediated apoptosis in murine peritoneal macrophages. J Immunol 150:5080–5085. [PubMed] [Google Scholar]
- 37.Messmer UK, Brüne B. 1996. Nitric oxide-induced apoptosis: p53-dependent and p53-independent signalling pathways. Biochem J 319(Part 1):299–305. doi: 10.1042/bj3190299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwitalla S, Ziegler PK, Horst D, Becker V, Kerle I. 2013. Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell 23:93–106. doi: 10.1016/j.ccr.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 39.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. 2007. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, Lowe SW. 2013. Non-cell-autonomous tumor suppression by p53. Cell 153:449–460. doi: 10.1016/j.cell.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang B, Niu D, Lai L, Ren EC. 2013. p53 increases MHC class I expression by upregulating the endoplasmic reticulum aminopeptidase ERAP1. Nat Commun 4:2359. doi: 10.1038/ncomms3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu K, Wang J, Zhu J, Jiang J, Shou J, Chen X. 1999. p53 induces TAP1 and enhances the transport of MHC class I peptides. Oncogene 18:7740–7747. doi: 10.1038/sj.onc.1203235. [DOI] [PubMed] [Google Scholar]
- 43.Thiery J, Abouzahr S, Dorothee G, Jalil A, Richon C, Vergnon I, Mami-Chouaib F, Chouaib S. 2005. p53 potentiation of tumor cell susceptibility to CTL involves Fas and mitochondrial pathway. Immunology 174:871–878. doi: 10.4049/jimmunol.174.2.871. [DOI] [PubMed] [Google Scholar]
- 44.Li H, Lakshmikanth T, Garofalo C, Enge M, Spinnler C, Anichini A, Szekely L, Kärre K, Carbone E, Selivanova G. 2011. Pharmacological activation of p53 triggers anticancer innate immune response through induction of ULBP2. Cell Cycle 10:3346–3358. doi: 10.4161/cc.10.19.17630. [DOI] [PubMed] [Google Scholar]
- 45.Taura M, Eguma A, Suico MA, Shuto T, Koga T, Komatsu K, Komune T, Sato T, Saya H, Li JD, Kai H. 2008. p53 regulates Toll-like receptor 3 expression and function in human epithelial cell lines. Mol Cell Biol 28:6557–6567. doi: 10.1128/MCB.01202-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shatz M, Menendez D, Resnick MA. 2012. The human TLR innate immune gene family is differentially influenced by DNA stress and p53 status in cancer cells. Cancer Res 72:3948–3957. doi: 10.1158/0008-5472.CAN-11-4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Textor S, Fiegler N, Arnold A, Porgador A, Hofmann TG, Cerwenka A. 2011. Human NK cells are alerted to induction of p53 in cancer cells by upregulation of the NKG2D ligands ULBP1 and ULBP2. Cancer Res 71:5998–6009. doi: 10.1158/0008-5472.CAN-10-3211. [DOI] [PubMed] [Google Scholar]
- 48.Zhu H, Dinsdale D, Alnemri ES, Cohen GM. 1997. Apoptosis in human monocytic THP.1 cells involves several distinct targets of N-tosyl-L-phenylalanyl chloromethyl ketone (TPCK). Cell Death Differ 4:590–599. doi: 10.1038/sj.cdd.4400284. [DOI] [PubMed] [Google Scholar]
- 49.Schmidt N, Hennig T, Serwa RA, Marchetti M, O'Hare P. 2015. Remote Activation of Host Cell DNA Synthesis in Uninfected Cells Signaled by Infected Cells in Advance of Virus Transmission. J Virol 89:11107–11115. doi: 10.1128/JVI.01950-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim SJ, Wong PK. 2015. p53 as a retrovirus-induced oxidative stress modulator. J Gen Virol 96(Part 1):144–149. doi: 10.1099/vir.0.070391-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Siegl C, Prusty BK, Karunakaran K, Wischhusen J, Rudel T. 2014. Tumor suppressor p53 alters host cell metabolism to limit Chlamydia trachomatis infection. Cell Rep 9:918–929. doi: 10.1016/j.celrep.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 52.Kong C, Zhao Y, Cui X, Zhang X, Cui H, Xue M, Wang Y. 2013. Complete genome sequence of the first Chinese virulent infectious laryngotracheitis virus. PLoS One 8:e70154. doi: 10.1371/journal.pone.0070154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao Y, Kong C, Cui X, Cui H, Shi X, Zhang X, Hu S, Hao L, Wang Y. 2013. Detection of infectious laryngotracheitis virus by real-time PCR in naturally and experimentally infected chickens. PLoS One 8:e67598. doi: 10.1371/journal.pone.0067598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wulff NH, Tzatzaris M, Young PJ. 2012. Monte Carlo simulation of the Spearman-Kaerber TCID50. J Clin Bioinforma 2:5. doi: 10.1186/2043-9113-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H, Zhang Y, Ströse A, Tedesco D, Gurova K, Selivanova G. 2014. Integrated high throughput analysis identifies Sp1 as a crucial determinant of p53-mediated apoptosis. Cell Death Differ 21:1493–1502. doi: 10.1038/cdd.2014.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cong F, Liu X, Han Z, Shao Y, Kong X, Liu S. 2013. Transcriptome analysis of chicken kidney tissues following coronavirus avian infectious bronchitis virus infection. BMC Genomics 14:743. doi: 10.1186/1471-2164-14-743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. 2010. Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol. Chapter 19:Unit 19.10.1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang DW, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc 4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 59.Franceschini A, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ. 2013. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 41(Database issue):D808–D815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.