

The innate immune system is crucial for effective responses to viral infection. Type I interferons, central components of innate immunity, induce expression of hundreds of ISGs; however, the mechanisms of action of these antiviral proteins are not well understood. CH25H, encoded by an ISG, represents a significant constituent of these cellular antiviral strategies, as its metabolic product, 25HC, can act in both an autocrine and a paracrine fashion to protect cells from infection and has been shown to limit viral infection in animal models. Further investigation into the mechanism of action of 25HC may inform novel antiviral therapies and influence the use of mammalian reovirus in clinical trials as an oncolytic agent.
KEYWORDS: reovirus, interferon, cholesterol-25-hydroxylase, 25-hydroxycholesterol
ABSTRACT
Following the initial detection of viral infection, innate immune responses trigger the induction of numerous interferon-stimulated genes (ISGs) to inhibit virus replication and dissemination. One such ISG encodes cholesterol-25-hydroxylase (CH25H), an enzyme that catalyzes the oxidation of cholesterol to form a soluble product, 25-hydroxycholesterol (25HC). Recent studies have found that CH25H is broadly antiviral; it inhibits infection by several viruses. For enveloped viruses, 25HC inhibits membrane fusion, likely by altering membrane characteristics such as hydrophobicity or cholesterol aggregation. However, the mechanisms by which 25HC restricts infection of nonenveloped viruses are unknown. We examined whether 25HC restricts infection by mammalian reovirus. Treatment with 25HC restricted infection by reovirus prototype strains type 1 Lang and type 3 Dearing. In contrast to reovirus virions, 25HC did not restrict infection by reovirus infectious subvirion particles (ISVPs), which can penetrate either directly at the cell surface or in early endosomal membranes. Treatment with 25HC altered trafficking of reovirus particles to late endosomes and delayed the kinetics of reovirus uncoating. These results suggest that 25HC inhibits the efficiency of cellular entry of reovirus virions, which may require specific endosomal membrane dynamics for efficient membrane penetration.
IMPORTANCE The innate immune system is crucial for effective responses to viral infection. Type I interferons, central components of innate immunity, induce expression of hundreds of ISGs; however, the mechanisms of action of these antiviral proteins are not well understood. CH25H, encoded by an ISG, represents a significant constituent of these cellular antiviral strategies, as its metabolic product, 25HC, can act in both an autocrine and a paracrine fashion to protect cells from infection and has been shown to limit viral infection in animal models. Further investigation into the mechanism of action of 25HC may inform novel antiviral therapies and influence the use of mammalian reovirus in clinical trials as an oncolytic agent.
INTRODUCTION
A productive innate immune response is dependent on the successful recognition of molecular signatures of infection by infected cells. Detection of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs) results in upregulation of multiple mechanisms that restrict pathogen replication (1). In the case of RNA virus infection, ligation of double-stranded RNA (dsRNA) by Toll-like receptors or cytoplasmic RIG-I-like helicases (RLHs) induces the production of type I interferons (IFNs), a family of major antiviral cytokines (2). IFNs signal through the type I IFN receptor (IFNAR) to elicit an antiviral state characterized by the expression of a broad range of interferon-stimulated genes (ISGs). ISGs inhibit virus replication through a variety of mechanisms, both cell intrinsic and extrinsic. However, despite the identification of over 200 identified ISGs, the molecular mechanism of action has been characterized for only a few of these genes (3).
Cholesterol-25-hydroxylase (CH25H) is an ISG product that acts through both cell-intrinsic and -extrinsic mechanisms. CH25H converts cholesterol to 25-hydroxycholesterol (25HC), one of a large number of endogenous cellular oxysterols that are involved in multiple metabolic pathways (4). 25HC regulates sterol-responsive element binding proteins (SREBPs) to control sterol biosynthesis (5, 6) and can suppress sterol biosynthetic enzymes such as HMG-coenzyme A reductase (7, 8). In addition to these roles in metabolism, CH25H also functions in the immune system, suppressing immunoglobulin A production in B cells (9) and inducing expression of the inflammatory cytokine interleukin-8 (10). More recently, CH25H has been shown to directly restrict target cell entry of enveloped viruses, including herpes simplex virus (HSV), Ebola virus, Lassa virus, Zika virus, porcine reproductive and respiratory syndrome virus (PRRSV), and pseudorabies virus, by inhibiting fusion of virus and cell membranes (11–16). Intriguingly, 25HC acts in both an autocrine and a paracrine manner and may be secreted by macrophages to prevent viral infection in surrounding cells (17). Although an initial report indicated that 25HC was an ineffective antiviral compound for adenovirus (a nonenveloped virus), a more recent report demonstrated significant antiviral activity against other nonenveloped viruses, including human rotavirus, human rhinovirus, and human papillomavirus (HPV) (18). However, neither the stages of virus replication inhibited, nor the mechanism of this activity, are known.
Mammalian orthoreoviruses (reoviruses) are nonenveloped viruses with double-stranded RNA (dsRNA) genomes. Although relatively nonpathogenic in humans, reoviruses have recently been identified as a trigger for celiac disease (19) and are in clinical trials for use as oncolytic agents (20). Reovirus target cell entry involves initial engagement with cell surface carbohydrates prior to ligation of proteinaceous receptors, including junction adhesion molecule A and the Nogo receptor NgR1 (21, 22). Virions are targeted into endosomal compartments via interactions with β1-integrins (23). In late endosomes, acid-dependent proteolysis of outer capsid proteins by cathepsins results in uncoating and cleavage of the membrane penetration protein μ1, which facilitates entry of transcriptionally active viral cores into the cytoplasm (24–28). Reovirus infectivity is sensitive to alterations in the entry process, as correct targeting to late endosomes, as denoted by expression of Rab7, is crucial for establishing productive infection (29). Additionally, alterations to this process, such as expression of the antiviral ISG IFITM3, can limit the capacity of reovirus to infect target cells (30). Given the role of 25HC in restricting entry of both enveloped and nonenveloped viruses, we sought to test whether 25HC inhibits reovirus infection.
In this study, we determined that both CH25H expression and 25HC treatment significantly inhibit reovirus infection and replication. 25HC-mediated restriction occurs after receptor engagement but prior to cytoplasmic entry of the core particle. 25HC treatment altered reovirus targeting to Rab7-containing late endosomes and delayed proteolysis of the reovirus capsid. However, 25HC treatment did not alter the infectivity of in vitro-generated infectious subvirion particles (ISVPs), which do not require entry into acidified compartments for entry, indicating that membrane penetration was not affected. Thus, CH25H and its product, 25HC, likely alter the dynamics of uncoating in the endosomal system, leading to inefficient membrane penetration. CH25H, therefore, represents another member of the growing arsenal of known antiviral ISG products that restrict reovirus infection, which may have implications for the therapeutic use of reoviruses as oncolytic agents.
RESULTS
25HC restricts mammalian reovirus infection.
25-Hydroxycholesterol (25HC), the product of cholesterol-25-hydroxylase (CH25H), restricts the entry of several enveloped viruses by inhibiting fusion between viral and cellular membranes (11–13), though molecular mechanisms for virus restriction are not fully defined. Additionally, although treatment with 25HC restricted infection by some nonenveloped viruses, including human rotavirus, human rhinovirus, and human papillomavirus (18), it did not restrict infection by another nonenveloped virus, adenovirus (12), raising the possibility that 25HC-mediated restriction may be limited to a subset of nonenveloped viruses. Therefore, we tested whether 25HC also restricts mammalian orthoreovirus infection. L929 and HeLa cells were grown in medium conditioned with 25HC at various concentrations, with ethanol as a vehicle control. The cells were pretreated with the conditioned medium for 16 h prior to adsorption with reovirus strains type 1 Lang (T1L) and type 3 Dearing (T3D) at various multiplicities of infection (MOI) (as determined by plaque titer on L929 cells). Cells were infected for 24 h, and the percentage of infected cells was quantified by indirect immunofluorescence using anti-reovirus T1L or T3D antiserum. The presence of 25HC significantly restricted infection by reovirus strain T1L in a dose-dependent manner in both L929 (Fig. 1A) and HeLa (Fig. 1B) (P < 0.05) cells. This effect was serotype independent, as 25HC also significantly restricted infection by reovirus strain T3D (Fig. 1C) (P < 0.05). Importantly, these concentrations of 25HC did not alter cell viability, as determined by trypan blue exclusion (Fig. 1A and B).
FIG 1.
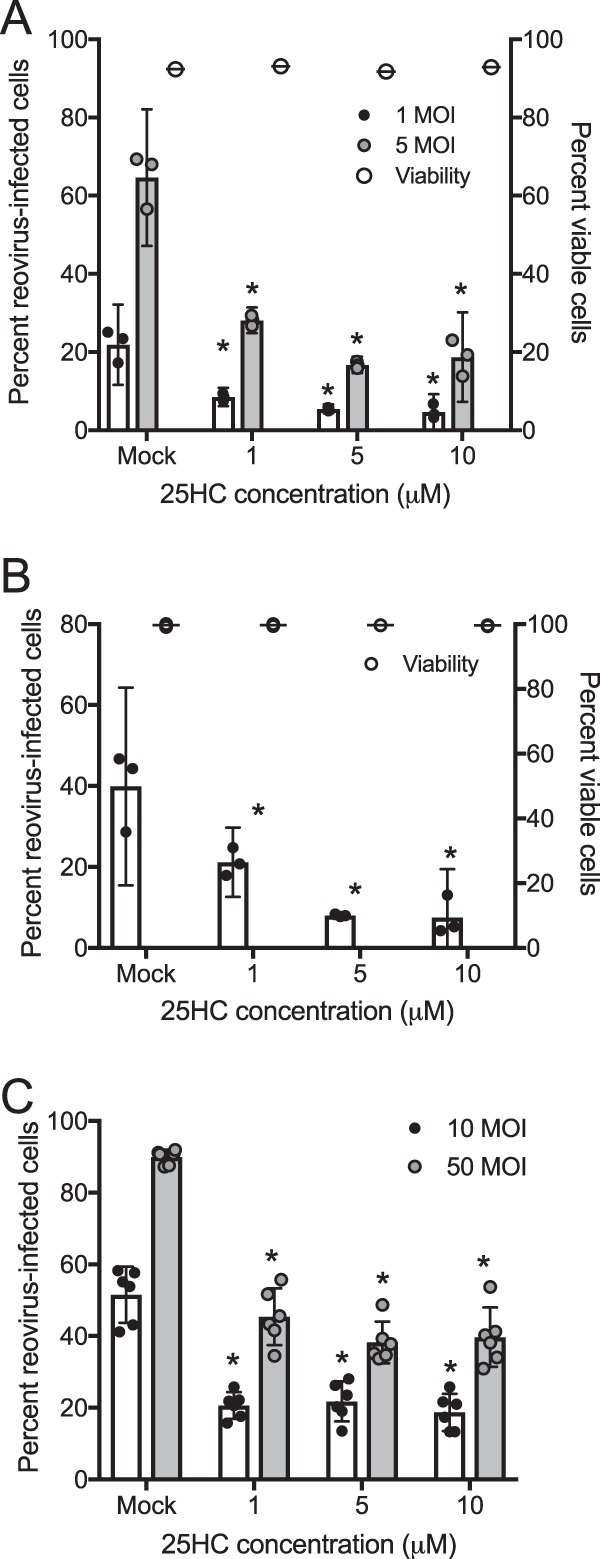
25HC restricts reovirus infection. L929 cells (A) or HeLa cells (B and C) were treated with the ethanol vehicle control or 25HC at the indicated concentrations for 16 h. Cells were infected with T1L (A and B) or T3D (C) at the indicated MOI (MOI of 10 for panel B), fixed at 24 h postinfection, stained with anti-T1L or anti-T3D polyclonal antisera and DAPI, and analyzed by fluorescence microscopy. Cell viability (A and B; right axis) was determined by trypan blue staining. Bars represent the means, and error bars represent 95% confidence intervals (CI) of biological replicates, *, P < 0.05 (versus results for mock-treated cells by Student's t test). Data are representative of three to five independent experiments per panel.
We next tested whether the restriction in the percentage of infected cells induced by the presence of 25HC would result in decreased reovirus replication. L929 cells were treated with 25HC or the vehicle control for 16 h and were then adsorbed with reovirus strain T1L at an MOI of 1 PFU/cell. Medium containing 25HC was replaced and cultures were incubated at 37°C, and viral yield was determined via plaque assay at 24 and 48 h postinfection (hpi). Treatment with 25HC restricted reovirus replication in a dose-dependent manner, with titers in cultures treated with 10 μM 25HC reduced by >10-fold in comparison to mock-treated samples at both 24 and 48 h postinfection (Fig. 2) (P < 0.05). These results suggest that 25HC-mediated restriction limits the replicative potential of reovirus in cell culture.
FIG 2.
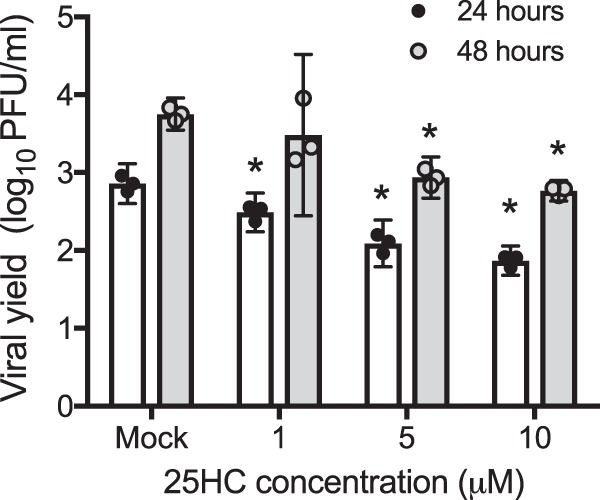
25HC restricts reovirus replication. L929 cells treated with the vehicle control or 25HC at the indicated concentrations for 16 h were infected with reovirus strain T1L at an MOI of 1 PFU/cell, at which time the 25HC was replaced. Viral titers at 24 hpi and 48 hpi were determined by plaque assay. The results indicate mean viral yields, calculated by dividing the titer at the indicated time points by the titer at 0 hpi. Bars represent the means, and error bars represent 95% CI of biological replicates. *, P < 0.05 by analysis of variance (ANOVA; compared to results for control-treated cells). Data are representative of three independent experiments.
CH25H expression is induced by, and restricts, reovirus infection.
Expression of CH25H is induced in cells following stimulation by type I interferons (31). Mammalian reoviruses are known inducers of type I IFNs, so we sought to determine whether CH25H is induced by reovirus infection. To ensure uniform kinetics of cellular gene expression, HeLa cells were mock infected or infected with a high MOI of T3D, 100 PFU/cell, and RNA was extracted from cells at various times postinfection. Levels of CH25H mRNA were quantified using quantitative reverse transcriptase PCR (qPCR). Expression of CH25H mRNA was upregulated by >15-fold at 8 h postinfection, with levels decreasing at 16 h and 24 h postinfection (Fig. 3). This pattern is consistent with previously observed kinetics of IFN production and ISG stimulation following reovirus infection (30, 32, 33).
FIG 3.
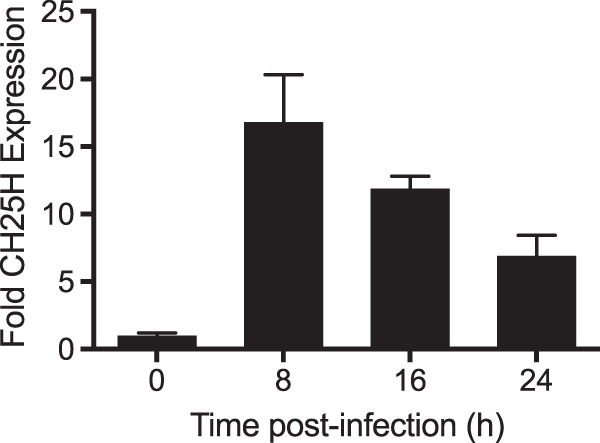
Reovirus infection induces CH25H expression. HeLa cells were mock infected or infected with reovirus T3D at an MOI of 100 PFU/cell for 24 h. RNA was extracted from cultures at the indicated time points, converted to cDNA, and used for qPCR analysis using primers for human CH25H. The level of CH25H expression was normalized to GAPDH in each culture. Data represent the mean and standard deviation (SD) of the results of triplicate biological samples.
To determine whether CH25H expression also restricts infection, we exogenously expressed green fluorescent protein (GFP)-tagged CH25H from a plasmid following transient transfection. HeLa cells were transfected overnight with CH25H-GFP and then infected with reovirus T1L at an MOI of 10 PFU/cell. After 18 h, the cells were fixed and stained using an anti-T1L antisera, and the percentages of GFP-positive and reovirus-positive cells were determined by indirect immunofluorescence microscopy. Exogenous expression of CH25H diminished the percentage of cells infected with reovirus (Fig. 4) from ∼40% to around ∼25% of the cells in the culture. This effect was even more pronounced in the GFP-positive cells expressing CH25H, as only around 13% of GFP-positive cells were also positive for reovirus antigen. Nevertheless, since reovirus staining was also diminished in GFP-negative cells, this indicates that the product of CH25H, 25HC, could be secreted from cells to restrict infection in cells that do not express CH25H, similar to previous reports (12). Of note, in examining the subcellular location of GFP, we observed GFP fluorescence primarily in small puncta throughout the cytoplasm. These were determined to be lipid droplets, as confirmed by staining with Nile red (data not shown).
FIG 4.
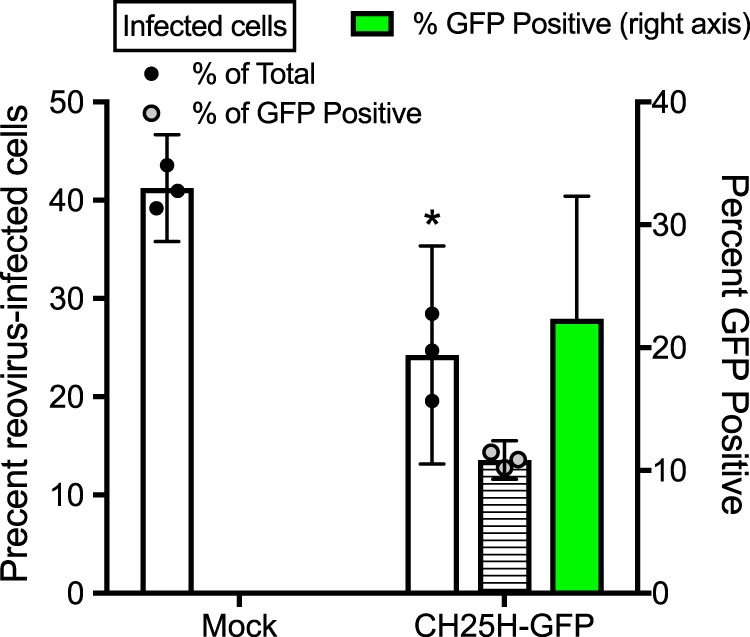
CH25H expression restricts reovirus infection. HeLa cells were mock transfected or transfected with a construct expressing GFP-tagged CH25H. At 18 h posttransfection, the cells were infected with T1L at an MOI of 10 PFU/cell for 18 h. The cells were fixed and stained with anti-T1L polyclonal antisera and DAPI, and the percentage of reovirus-infected cells, GFP-positive cells, and reovirus/GFP double-positive cells was determined by fluorescence microscopy. Error bars indicate 95% CI of biological replicates. A minimum of 30 GFP-positive cells were counted in each field. *, P < 0.05 by Student's t test (compared to results for mock-transfected cells). Data are representative of three independent experiments.
25HC pretreatment restricts reovirus infection at the entry stage.
We next sought to understand the kinetics of the restriction mediated by 25HC using a time-of-addition experiment, pretreating cells with 25HC for different intervals prior to or after infection with reovirus. Addition of 25HC between 1 h prior to infection and 1 h postinfection did not affect reovirus infectivity (Fig. 5). However, pretreatment at or before 2 h prior to infection significantly inhibited infection, with maximal inhibition occurring with pretreatment 8 h or more prior to infection. These results indicate that 25HC does not inhibit reovirus during the process of infection or after infection has taken place but likely establishes a refractory state prior to infection. As 25HC influences several metabolic pathways, including sterol biosynthesis and protein prenylation, it is possible that the refractory state may be induced by alterations in the levels of cellular metabolites regulated by 25HC. Sterol biosynthesis is sensitive to the key intermediate mevalonate, which is produced by HMG-coenzyme A (HMG-CoA) reductase; 25HC suppresses HMG-CoA reductase activity (8). 25HC also inhibits production of another key biochemical intermediate, isopentyl-pyrophosphate, which is utilized by farnesyltransferase and geranylgeranyltransferase for protein prenylation (17). Neither the addition of exogenous mevalonate (30 to 300 μM) nor inhibitors of farnesyltransferase (FTI-276; 5 to 20 μM) or geranylgeranyltransferase (GGTI-298; 5 to 20 μM) altered the infectivity of reovirus (not shown). Additionally, staining with filipin, a marker of free cellular cholesterol, showed no significant differences between vehicle- and 25HC-treated cells (not shown). Together, these data suggest that 25HC does not exert its refractory effects on reovirus infection via alteration of cellular biosynthetic pathways.
FIG 5.
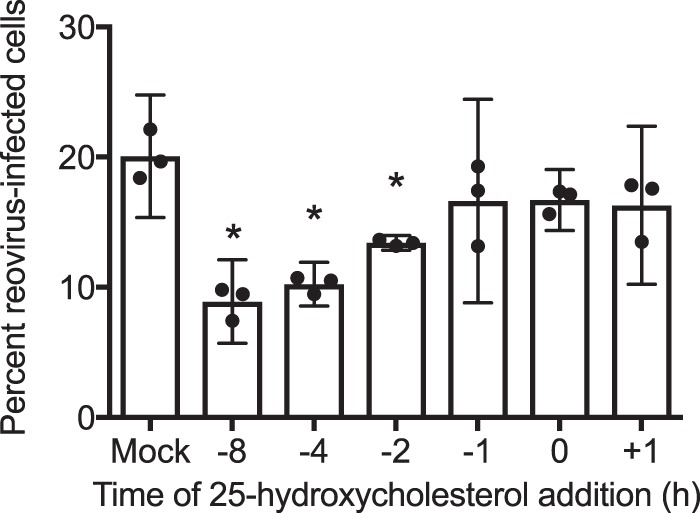
25HC restricts reovirus infection as early as 2 h posttreatment. HeLa cells were treated with the ethanol vehicle control or 10 μM 25HC at the indicated times prior to infection. Cells were infected with T1L at an MOI of 10 PFU/cell and were fixed at 24 h postinfection, stained with anti-T1L antisera and DAPI, and analyzed by fluorescence microscopy. Error bars indicate 95% CI of the results of biological replicates. *, P < 0.05 by Student's t test. Data are representative of two independent experiments.
We next sought to determine the stage of the reovirus replication cycle at which 25HC exerts its restriction. Since 25HC was shown to restrict the entry of enveloped viruses, we hypothesized that it might also restrict the entry of reovirus. We then tested to see whether the presence of 25HC restricted infection caused by delivery of reovirus core particles into cells via an alternative mechanism, transient transfection. HeLa cells were pretreated with 10 μM 25HC or the vehicle control for 12 h and then transfected with purified in vitro-generated reovirus cores. After 18 h, the percentage of infected cells was determined via indirect immunofluorescence. The presence of 25HC did not alter the percentage of cells infected following this delivery mechanism (Fig. 6A), suggesting that 25HC acts prior to membrane penetration and not during the subsequent stages of the reovirus replication cycle. Importantly, no infection was observed in cultures incubated with reovirus cores in the absence of transfection reagent, indicating that the infection observed was not due to the incomplete processing of virions in the preparation of cores.
FIG 6.
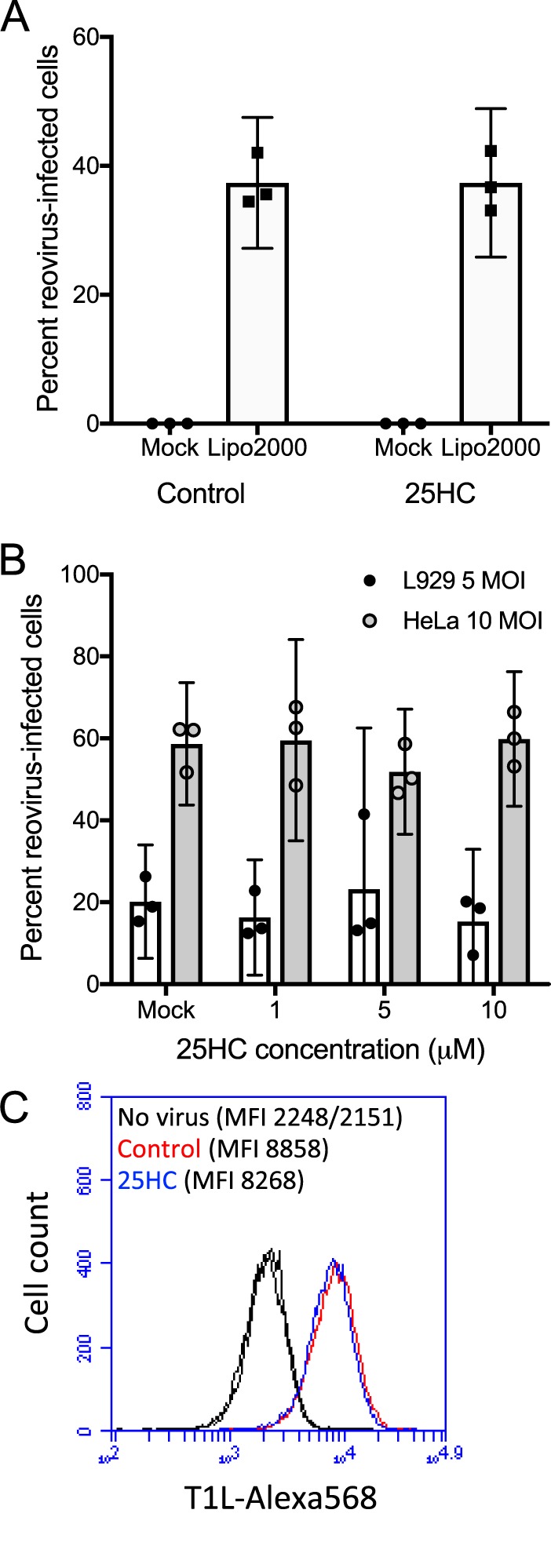
25HC does not restrict infection by reovirus ISVPs or transfected cores and does not alter virion binding to target cells. HeLa (A, B, and C) or L929 (B) cells were treated with the ethanol vehicle control or 10 μM 25HC at 12 h prior to inoculation. (A) Cells were mock transfected or transfected with in vitro-generated T1L cores using Lipofectamine 2000. (B) Cells were infected with in vitro-generated T1L ISVPs at the indicated MOI and were fixed at 24 h postinfection, stained with anti-T1L antisera and DAPI, and analyzed by fluorescence microscopy. Error bars represent 95% CI of results for biological replicates. Data are representative of three independent experiments per panel. (C) Cells were incubated at 4°C for 1 h, mock treated or adsorbed with 10,000 particles/cell of Alexa 568-labeled T1L virions, washed in cold PBS, fixed, and analyzed by flow cytometry. The mean fluorescence intensities (MFI) of the resulting histograms are indicated.
We then tested whether 25HC restricted infection by reovirus infectious subvirion particles (ISVPs), which do not require endosomal protease activity for membrane penetration and infectivity (34). HeLa cells were incubated with 10 μM 25HC or the vehicle control for 16 h, followed by incubation with either T1L virions or in vitro-generated ISVPs at an MOI of 10 PFU/cell, and the percentage of infected cells was determined at 16 hpi by indirect immunofluorescence microscopy. In contrast to virions, 25HC did not alter the capacity of ISVPs to infect HeLa cells (Fig. 6B). Additionally, no differences were observed in the initial binding of fluorescently tagged T1L virions to the surface of cells in the presence or absence of 25HC, as assessed by flow cytometry (Fig. 6C). These results indicate that 25HC restricts infection at a phase after receptor binding on target cells, likely during endosomal uptake or trafficking.
25HC alters reovirus endocytic trafficking, resulting in delayed outer capsid proteolysis and uncoating.
Next, we sought to determine whether 25HC affects the kinetics of reovirus particles entering into endosomal compartments necessary for acid-dependent proteolysis of the outer capsid during the uncoating process. Productive reovirus entry occurs in late endosomes that contain specific Rab GTPases, such as Rab7 and Rab-interacting lysosomal protein (RILP) (29). HeLa cells were transfected with Rab7-enhanced GFP (EGFP) and preincubated for 12 h in the presence or absence of 10 μM 25HC. The cells were then chilled at 4°C for 1 h, adsorbed with Alexa 568-labeled reovirus particles at 4°C for 1 h, and then incubated at 37°C for 0, 60, 90, or 120 min. At these times, the cells were fixed with 2% paraformaldehyde and visualized by confocal microscopy (Fig. 7A). Quantification of colocalization between reovirus particles and Rab7-EGFP-labeled endosomes showed an increase over background levels in the absence of 25HC (Fig. 7B). In contrast, in the presence of 25HC, colocalization between particles and Rab7-containing endosomes was significantly reduced at 90 and 120 min postadsorption. These data suggest that 25HC may alter endosomal trafficking dynamics. Additionally, 25HC treatment did not overtly affect the localization, morphology, or number of Rab7-EGFP-containing compartments, suggesting that 25HC does not cause global alterations in cellular endocytic pathways (Fig. 7). As a control, the cells were adsorbed with EGFP-labeled transferrin in the presence or absence of 25HC pretreatment, and the uptake of transferrin was monitored by confocal microscopy. No difference in the kinetics of transferrin uptake was observed in the presence of 25HC, indicating that 25HC does not globally alter clathrin-dependent endocytic pathways (Fig. 8).
FIG 7.

25HC decreases reovirus particle trafficking to late endosomes. HeLa cells were transfected with Rab7-EGFP 18 h prior to infection and pretreated with the vehicle control or 10 μM 25HC for 12 h prior to infection. The cells were chilled at 4°C for 1 h and then adsorbed with 10,000 particles/cell of reovirus-Alexa 568 at 4°C for 1 h. The inoculum was removed, and the cells were washed to remove unbound virus and then either fixed with 2% paraformaldehyde or supplemented with complete medium and incubated at 37°C for the indicated times (in minutes) before fixing. (A) Cells were imaged by confocal microscopy. Insets depict enlarged images of boxed regions. (B) The percentage of reovirus-Alexa 568 particles colocalized with Rab7-EGFP was determined. Data are presented as the percentage of virus particles exhibiting spectral overlap with EGFP expression (n = 45 to 94 cells per time point; average of 2,348 particles per time point). Error bars represent SD. *, P < 0.05 by Student's t test (in comparison to results for control treatment at the same time point).
FIG 8.

25HC does not affect transferrin uptake. HeLa cells were treated with the ethanol vehicle control or 10 μM 25HC for 12 h. Cells were serum starved for 30 min at 37°C and pulse-labeled with Alexa 594-labeled human transferrin for 10 min, washed, and incubated in serum-containing medium for the indicated times, at which point the cells were fixed, stained with DAPI, and imaged by confocal microscopy.
Finally, we examined the kinetics of outer capsid proteolysis in the presence or absence of 25HC. HeLa cells were preincubated with 10 μM 25HC for 12 h, adsorbed with T1L virions for 1 h at 4°C, and then incubated at 37°C for 0, 60, 90, or 120 min. At these times postinfection, cell lysates were analyzed by SDS-PAGE, followed by immunoblotting using a T1L-specific antiserum. Cells treated with 25HC exhibited delayed cleavage of μ1 into μ1C and δ in comparison to mock-treated cells, with initial cleavage products detected in mock-treated cells by 60 min postinfection and in 25HC-treated cells by 120 min postinfection (Fig. 9A). Reovirus uncoating is mediated by acid-dependent cathepsin proteases, including cathepsin L (24). To determine whether 25HC directly altered cathepsin L activity, cells were treated with 25HC or the vehicle control for 12 h, at which point the cathepsin L activity of culture lysates was determined using a luminescence-based proteolysis assay (Fig. 9B). No differences were observed between mock- and 25HC-treated cultures. Together with data regarding colocalization with Rab7-containing endosomes, these data indicate that 25HC alters the endocytic trafficking patterns of reovirus particles, leading to delayed outer capsid proteolysis and subsequent membrane penetration and thereby decreasing the efficiency of infection.
FIG 9.
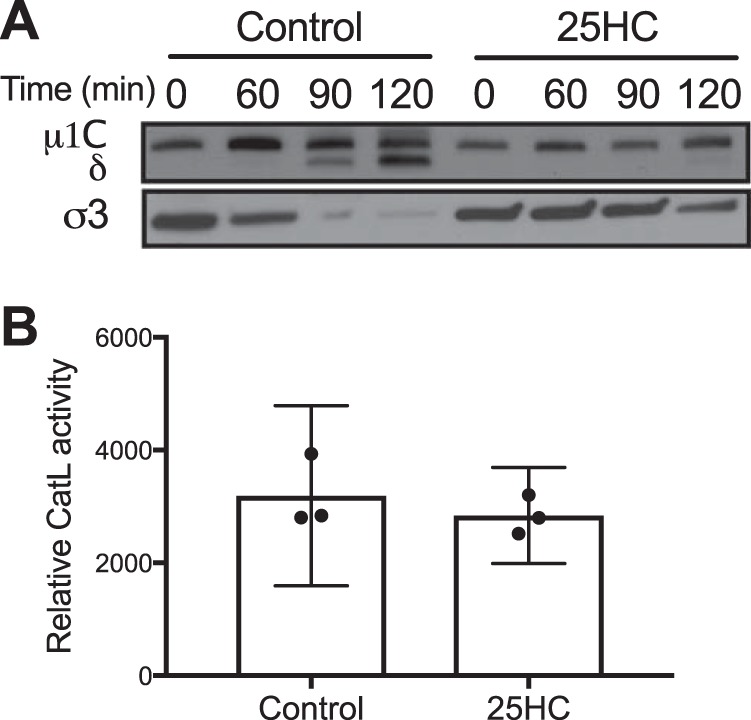
25HC delays reovirus outer capsid cleavage. (A) HeLa cells were pretreated with the vehicle control or 10 μM 25HC for 12 h prior to infection. Cells were then adsorbed with reovirus T1L at 4°C for 1 h. The inoculum was removed, and cells were washed two times with PBS to remove unbound virus and incubated with prewarmed medium at 37°C. At the indicated times after adsorption, the cells were scraped into PBS, pelleted at 3,000 × g, and resuspended in radioimmunoprecipitation buffer. Lysates were clarified by centrifugation at 13,000 × g, resolved in 4 to 12% polyacrylamide gels, and transferred to PVDF membranes. The membranes were probed with a rabbit polyclonal anti-T1L antiserum and an anti-rabbit horseradish peroxidase-conjugated secondary antibody and visualized using chemiluminescence. Data are representative of five independent experiments. (B) HeLa cells were pretreated with the vehicle control or 10 μM 25HC for 12 h prior to determination of cathepsin L (CatL) activity in cell lysates. Error bars represent 95% CI of the results of biological replicates.
DISCUSSION
The type I IFN system is pivotal in responding to viral infection. The activities of hundreds of ISGs elicit an antiviral state to curtail nascent infections prior to the induction of a specific adaptive immune response. Collectively, ISGs are known to act at multiple stages of viral replication cycles, from attachment and entry, to RNA synthesis, to particle exit (3). However, many ISGs target cell entry processes as a crucial step in all viral replication cycles. Preventing entry, and therefore subsequent viral protein production, negates the activity of virally encoded innate immune antagonists. The CH25H system, through the production of the secreted metabolite 25HC, protects cells from infection by multiple viruses, including mammalian reoviruses, by restricting viral entry mechanisms.
CH25H is a 233-amino-acid protein that catalyzes the oxidation of cholesterol to 25-hydroxycholesterol (4). CH25H mRNA expression is induced following infection by reovirus, consistent with the prior identification of the CH25H gene as an interferon-stimulated gene (31). While we did not determine whether reovirus induces a concomitant increase in endogenous CH25H protein level or 25HC production, prior reports have observed increases in both CH25H protein expression and 25HC secretion following upregulation of mRNA levels (17, 35), suggesting that reovirus infection likely induces expression of CH25H at both the mRNA and protein level, resulting in the production of 25HC. Previous data suggest that CH25H localizes to the endoplasmic reticulum, consistent with its function in oxysterol synthesis (5). However, our observations indicate that GFP-tagged CH25H also associates with lipid droplets, as determined by Nile red staining, indicating that its localization may vary with expression level and cellular phenotype. Regardless of the subcellular location of CH25H, it is widely understood that 25HC is soluble and membrane penetrable, suggesting that it can exert its antiviral effects in both an autocrine and a paracrine fashion. 25HC administration limits viral infection in vivo, suggesting an important physiologic relevance for these antiviral effects (12, 13).
The mechanism of action for 25HC is not completely understood. For enveloped viruses, 25HC is thought to alter the properties of cellular membranes, making them refractory to fusion with viral membranes. 25HC alters cholesterol dynamics by directly mobilizing cholesterol from membranes and thereby altering membrane fluidity (36, 37). 25HC may also alter the exposure of phospholipid head groups and lead to membrane expansion (38). As viral fusion mechanisms are dependent on the properties of the target membrane, including cholesterol content, spacing of phospholipids, and membrane curvature (39, 40), it is plausible that 25HC disrupts the normal membrane physiology to prevent fusion. The mechanism of action of 25HC for nonenveloped viruses, however, is less clear. 25HC has been shown to inhibit infection by HPV, human rotavirus, human rhinovirus, and now mammalian reoviruses (18). These viruses share a requirement for receptor-mediated endocytosis and pH-dependent uncoating in late endosomes (41–43). Our data suggest that 25HC alters the kinetics of trafficking of viral particles to late endosomes, which results in altered uncoating dynamics and decreased membrane penetration efficiency. Previous studies suggest that reovirus particles that do not successfully uncoat in late endosomes traffic to lysosomes (23, 25) or are shunted to other nonproductive endocytic routes that do not facilitate membrane penetration (29). Therefore, slight delays in uncoating mediated by 25HC can significantly decrease the percentage of cells that become infected and the subsequent production of viral progeny. Similar results were observed following expression of another ISG product, IFITM3, which also acts at the entry phase to delay uncoating and inhibit reovirus infection (30). Together, these studies highlight the reovirus entry process as being particularly sensitive to inhibition via multiple cellular mechanisms. In contrast to reovirus, adenovirus, a nonenveloped virus that is not inhibited by 25HC, also undergoes a pH-dependent uncoating step but is thought to have a more disruptive membrane penetration process, mediated by the membrane lytic protein VI (44, 45). This more disruptive process, which is capable of activating the NLRP3 inflammasome, may be sufficient to overcome the restriction mediated by 25HC.
25HC treatment did not alter the infectivity of in vitro-generated reovirus ISVPs. ISVPs require endocytosis for cell entry but do not require endosomal acidification for uncoating and membrane penetration (34, 46). Additionally, entry of virions, but not ISVPs, is sensitive to cholesterol extraction by methyl-β-cyclodextrin (46). Therefore, it is possible that 25HC exerts its effects against reovirus by mobilizing cholesterol from endosomal membranes and thereby decreasing the efficiency of endosomal trafficking or altering the cholesterol-dependent membrane penetration of reovirus virions. However, HPV infection is cholesterol independent (47), whereas adenovirus type 2 infection is cholesterol dependent (48). Additionally, filipin staining showed no gross changes in cellular free cholesterol following 25HC treatment. Therefore, alterations to cellular cholesterol are likely not the primary mechanism of action of 25HC against all nonenveloped viruses.
Similar to results with enveloped viruses, 25HC-mediated restriction of reovirus requires pretreatment of cells for at least 2 h prior to virus adsorption (12) (Fig. 5). This suggests that 25HC does not exert its effects by directly modifying virus particles but by altering cellular conditions to become refractory to virus entry. The timing of this preincubation is consistent with the effects of 25HC in regulating sterol biosynthesis, via suppression of HMG-CoA reductase (8) or the production of isopentenyl-pyrophosphate (17). However, also similar to results with enveloped viruses, the addition of the metabolic intermediate mevalonate did not rescue the effect of 25HC, suggesting that the restrictive mechanism of 25HC does not act via suppression of sterol biosynthesis. Additionally, inhibitors of protein prenylation enzymes, including farnesyltransferase and geranylgeranyltransferase, did not alter reovirus infectivity, suggesting that 25HC does not act to restrict infection via alterations in cellular metabolic pathways. Interestingly, the effect of exogenous 25HC on reovirus infection was not fully dose dependent. This may indicate that a threshold level of 25HC is sufficient to alter endocytic trafficking patterns enough to disrupt normal viral uncoating processes, which would be beneficial in limiting virus dissemination from infected regions following interferon-mediated upregulation of CH25H.
CH25H has emerged as an important component of both cellular metabolism and antiviral mechanisms. The production of 25HC as a soluble factor that can elicit an antiviral state in not just the producing cell but in surrounding cells as well allows for a much broader antiviral response than many other ISG-mediated effects. Indeed, in animal models, administration of 25HC is protective against HIV-1 and Zika virus infection (12, 13), indicating an important physiologic role for this antiviral defense. Therefore, further investigation of the CH25H system is necessary to inform novel therapeutic strategies for a wide range of viral pathogens, as well as its implications for the use of viruses, such as mammalian reoviruses, as oncolytic agents.
MATERIALS AND METHODS
Cells, viruses, and reagents.
HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented to contain 10% fetal bovine serum, 2 mM l-glutamine, 100 U/ml of penicillin, 100 μg/ml streptomycin, and 25 ng/ml of amphotericin B (Sigma). L929 cells were maintained in Joklik's minimum essential medium supplemented to contain 5% fetal bovine serum, 2 mM l-glutamine, 100 U/ml of penicillin, 100 μg/ml streptomycin, and 25 ng/ml of amphotericin B.
Reovirus strains type 1 Lang (T1L) and type 3 Dearing (T3D) are laboratory stocks. Purified reovirus virions were generated using second- or third-passage L-cell lysate stocks of twice-plaque-purified reovirus as described previously (49). Viral particles were Freon extracted from infected cell lysates, layered onto 1.2- to 1.4-g/cm3 CsCl gradients, and centrifuged at 62,000 × g for 18 h. Bands corresponding to virions (1.36 g/cm3) were collected and dialyzed in virion storage buffer (150 mM NaCl, 15 mM MgCl2, 10 mM Tris-HCl, pH 7.4). Concentrations of reovirus virions in purified preparations were determined from an equivalence of 1 optical density (OD) unit at 260 nm equal to 2.1 × 1012 virions (50). Viral titer was determined by plaque assay using murine L929 cells (51). ISVPs were generated as described previously (52) and confirmed by SDS-PAGE and Coomassie brilliant blue staining. Rabbit anti-T3D and anti-T1L antisera were generously provided by Terence S. Dermody (University of Pittsburgh). Reovirus cores were generated and transfected as described previously (53). Reovirus particles were labeled with Alexa 568 by succinimidyl ester labeling as previously described (30).
Plasmids encoding human GFP-tagged CH25H were purchased from OriGene Technologies and GeneCopoeia. Plasmids encoding Rab7-EGFP and Rab-interacting lysosomal protein (RILP)-EGFP were obtained from Terence S. Dermody (University of Pittsburgh). R-mevalonic acid, GGTI-298, and FTI-276 were purchased from Sigma-Aldrich.
25HC treatment.
25-Hydroxycholesterol (25HC; Sigma) was dissolved in 95% ethanol (EtOH) at a concentration of 5 mM and stored at room temperature. At the time of treatment, the 25HC was added to complete medium at concentrations of 1, 5, and 10 μM. Vehicle control cells were treated with the corresponding volume of 95% EtOH.
Cell viability assay.
Cell viability was assessed using the trypan blue dye exclusion method. L cells (5 × 104 cells) were seeded onto a 24-well plate and incubated with the EtOH vehicle control or 1, 5, or 10 μM 25HC-conditioned medium for 24 h at 37°C. At 24 h, the cell were trypsinized and treated with 2× trypan blue (Sigma-Aldrich) for 10 min, and the numbers of viable and nonviable cells were quantified using a hemocytometer.
Fluorescent focus assay.
HeLa cells (4 × 104) were grown in 24-well tissue culture plates, pretreated with 25HC at various times and concentrations, and adsorbed with reovirus strains at various MOIs for 1 h at 4°C. After adsorption, 0.5 ml of fresh medium was added and the cells were incubated at 37°C for 18 h. Cells were fixed with 2% paraformaldehyde for 30 min, washed twice with phosphate-buffered saline (PBS), and permeabilized and blocked in PBS–2% bovine serum albumin (BSA)–0.1% Triton X-100 (PBS-T) for at least 1 h at 4°C. The cells were then incubated with rabbit anti-reovirus T1L or T3D antiserum (1:1,000) in PBS-T for at least 1 h at 4°C, washed three times with PBS-T, and incubated with an anti-rabbit Alexa 568-conjugated antibody (1:2,000) in PBS-T for at least 1 h at 4°C. The cells were washed three times with PBS and visualized using fluorescence microscopy. Reovirus antigen-positive cells were quantified by counting fluorescent cells in at least two random fields of view per well at a magnification of ×113. The total cell number was quantified using background fluorescence or by DAPI (4′,6-diamidino-2-phenylindole) staining of cell nuclei. The average percent infectivity was calculated for at least three independently infected wells per experiment, and each experiment was repeated at least three times. Due to small fluctuations in the percentage of cells infected in mock-treated cells in each experiment, data across multiple experiments were not combined; one representative experiment is shown, selected to illustrate levels of infection in mock-treated cells between ∼20 to 80% to ensure that potentially both positive and negative effects of treatment could be observed. All effects of treatment are compared within a given experiment. Note that MOI reflect virus stocks for which titers were determined on L929 cells; relative MOI on other cell lines may be different based on differences in infectivity and in the relative sensitivity of focus-forming unit (FFU) assays versus plaque assays. All cell lines used have been well characterized for their capacity to support productive reovirus infection (54, 55).
CH25H expression.
HeLa cells (4 × 104) were grown in 24-well tissue culture plates and transfected overnight with constructs expressing CH25H-EGFP using FuGene6 (Promega), as indicated by the manufacturer. The cells were then infected with reovirus T1L at an MOI of 50 PFU/cell for 18 h, whereupon cells were analyzed by fluorescent focus assay.
Assessment of virus replication by plaque assay.
Cells (4 × 104) grown in 24-well tissue culture plates were adsorbed with the reovirus strains T1L and T3D at an MOI of 1 PFU/cell for 1 h at 4°C. After adsorption, the cells were washed two times with PBS and incubated in 0.5 ml of fresh medium at 37°C. After different time intervals, the cells were freeze-thawed twice and the viral titer was determined by plaque assay. The viral yields were calculated by dividing the viral titers at the indicated times by the viral titer at 0 h.
Analysis of viral disassembly.
Cells (5 × 105) grown in 60-mm dishes were pretreated with 25HC at various times and concentrations and then inoculated with T3D at an MOI of 100 PFU/cell for 1 h at 4°C. The inoculum was removed and the cells were washed two times with PBS and incubated in fresh prewarmed medium at 37°C. At various times after adsorption, the cells were scraped into 1 ml of ice-cold PBS and pelleted at 3,000 × g for 5 min at 4°C. The supernatant was aspirated, and the pellet was resuspended in 100 μl of ice-cold modified radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% sodium deoxycholate, 1% IGEPAL CA-630, 1 mM phenylmethylsulfonyl fluoride [PMSF]) supplemented with protease inhibitor cocktail (Roche). The lysate was clarified by centrifugation at 13,000 × g for 10 min at 4°C, and the supernatant was removed to a fresh tube and frozen at −20°C. Extracts (10 μg of total protein) were resolved by electrophoresis in 4 to 12% Bis-Tris gels and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked overnight at room temperature in PBS–1% Tween 20 (PBS-Tween) containing 5% milk and incubated with a rabbit anti-T3D antiserum (1:500) in PBS-Tween plus milk at room temperature for 3 h. The membranes were washed three times for 10 min with PBS-Tween and incubated with an alkaline phosphatase-conjugated goat anti-rabbit antibody (Bio-Rad) diluted 1:2,000 for 3 h. Following three washes with PBS-Tween, the membranes were incubated for 5 min with chemiluminescent alkaline phosphate substrate (Bio-Rad) and visualized using a ChemiDock XRS+ molecular imager (Bio-Rad).
qPCR.
Cells (5 × 105) grown in 60-mm dishes were adsorbed with T3D in PBS at an MOI of 100 PFU/cell at 4°C for 1 h. The cells were incubated in medium at 37°C for various intervals, removed from the plates with a scraper, washed once with PBS, and centrifuged at 500 × g for 5 min. The supernatant was removed, and the cell pellet was frozen at −20°C. RNA was extracted by using an RNeasy Plus RNA extraction minikit (Qiagen) according to the manufacturer's instructions. RNA was converted to cDNA by using an Omniscript RT cDNA synthesis kit (Qiagen) with an oligo(dT) primer, a reovirus L1 minus-strand-specific primer (5′-GGGCTCTATGCTGTGCTTTC-3′), or a reovirus L1 plus-strand-specific primer (5′-GGGCGTATCAAGCTAATCCA-3′) according to the manufacturer's instructions. Quantitative reverse transcriptase PCR (qPCR) was performed by using the Express SYBR GreenER system (Invitrogen). Primers specific for human GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (F primer, 5′-GATCATCAGCAATGCCTCCT-3′, and R primer, 5′-TGTGGTCATGAGTCCTTCCA-3′) or human CH25H (F primer, 5′-GCTGGCAACGCAGTATATGA-3′, and R primer, 5′-ACGGAAAGCCAGATGTTGAC-3′) were used at a final concentration of 0.2 μM. Quantification and melt curve analyses were performed according to the manufacturer's protocol. For each sample, the CT (threshold cycle) for the RNA of interest was normalized to that for GAPDH. Fold induction was calculated by comparing normalized CT values (ΔΔCT) of duplicate cDNA synthesis reactions to those of samples taken at the time of infection (T = 0) for two independent experiments. For the quantification of reovirus L1 gene segments, samples were normalized to GAPDH expression levels in matched cDNA synthesized by using an oligo(dT) primer.
Virion binding assay.
Cells (106) were plated in 6-well plates at 37°C and treated with 10 μM 25HC or the vehicle control for 12 h, at which point the cells were removed from the plates using CellStripper solution (Corning-CellGro), rinsed with PBS, and chilled at 4°C for 1 h. The cells were then adsorbed with 10,000 Alexa 568-labeled reovirus particles/cell at 4°C for 1 h. The inoculum was removed, and the cells were washed three times with cold PBS to remove unbound virus. The cells were fixed briefly with 2% paraformaldehyde for 5 min, washed with cold PBS, and analyzed with a BD Accuri C6 flow cytometer. Cell fluorescence was quantified using BD Accuri C6 software.
Confocal microscopy of reovirus internalization.
Cells were plated on glass coverslips (no. 1.5; Thermo Scientific) in 24-well plates at 37°C overnight. Cells were transfected with plasmids using Lipofectamine 2000 (Life Sciences) according to the manufacturer's instructions. After incubation at 37°C for 16 h, the cells were chilled at 4°C for 1 h. The cells were then adsorbed with 10,000 Alexa 568-labeled reovirus particles/cell at 4°C for 1 h. The inoculum was removed, and the cells were washed three times with PBS and either fixed with 2% paraformaldehyde or supplemented with complete medium and incubated at 37°C for various intervals. Cells were washed once with PBS and fixed for 20 min with 2% paraformaldehyde, quenched with 0.1 M glycine, and washed three times with PBS. Coverslips were removed from the wells and placed on slides using ProLong Gold mounting medium (Life Sciences). Images were captured using a Zeiss LSM710 laser-scanning confocal microscope using a 63× Plan-Apochromat objective lens. Images were thresholded for pixel intensity, and the pinhole size used was the same for all fluorophores. All images represent single sections and were adjusted for brightness and contrast to the same extent. Colocalization analysis was performed using ImageJ, taking into consideration endosomal vesicle size to try to isolate individual endosomes. Virions within the boundary of single cells were quantified.
Measurement of transferrin uptake.
Cells (5 × 104) were plated on coverslips at 37°C and treated with 10 μM 25HC or the vehicle control for 12 h, at which point they were serum starved in serum-free DMEM supplemented with 25 mM HEPES and 0.5% BSA for 30 min at 37°C. The cells were then incubated with 50 μg/ml of Alexa 594-labeled human transferrin (Thermo Fisher) for 5 min at 37°C, at which point they were washed two times with PBS and either fixed in 2% paraformaldehyde or incubated at 37°C for various intervals prior to fixing, followed by mounting on slides using ProLong Gold and imaging using a Zeiss LSM710 laser-scanning confocal microscope with a 63× Plan-Apochromat objective lens.
Measurement of cathepsin L activity.
Cells (106) were plated in 6-well plates at 37°C and treated with 10 μM 25HC or the vehicle control for 12 h, at which point the cells were removed from the plates using CellStripper solution (Corning-CellGro) and rinsed with PBS, and the cathepsin L activity was determined fluorometrically using a cathepsin L activity assay kit (Abcam) according to the manufacturer's protocol.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (NIH) Public Health Service Award R15 AI094440 (to G.H.H.) and by the Intramural Research Program of the NHLBI, NIH (to N.A.-B.). Additional support was provided by the Colgate University Department of Biology.
We thank members of our laboratories for helpful discussions and Karl Boehme, Pranav Danthi, Terence Dermody, Tanner Gill, and John Parker for technical assistance, reagents, and advice.
REFERENCES
- 1.Mogensen TH. 2009. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22:240–273. doi: 10.1128/CMR.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jensen S, Thomsen AR. 2012. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol 86:2900–2910. doi: 10.1128/JVI.05738-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borden EC, Williams BR. 2011. Interferon-stimulated genes and their protein products: what and how? J Interferon Cytokine Res 31:1–4. doi: 10.1089/jir.2010.0129. [DOI] [PubMed] [Google Scholar]
- 4.Holmes RS, VandeBerg JL, Cox LA. 2011. Genomics and proteomics of vertebrate cholesterol ester lipase (LIPA) and cholesterol 25-hydroxylase (CH25H). 3 Biotech 1:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lund EG, Kerr TA, Sakai J, Li W-P, Russell DW. 1998. cDNA cloning of mouse and human cholesterol 25-hydroxylases, polytopic membrane proteins that synthesize a potent oxysterol regulator of lipid metabolism. J Biol Chem 273:34316–34327. doi: 10.1074/jbc.273.51.34316. [DOI] [PubMed] [Google Scholar]
- 6.Adams CM, Reitz J, De Brabander JK, Feramisco JD, Li L, Brown MS, Goldstein JL. 2004. Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J Biol Chem 279:52772–52780. doi: 10.1074/jbc.M410302200. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J, Dricu A, Sjövall J. 1997. Studies on the relationships between 7α-hydroxylation and the ability of 25- and 27-hydroxycholesterol to suppress the activity of HMG-CoA reductase. Biochim Biophys Acta 1344:241–249. doi: 10.1016/S0005-2760(96)00148-8. [DOI] [PubMed] [Google Scholar]
- 8.Smith LL, Johnson BH. 1989. Biological activities of oxysterols. Free Radic Biol Med 7:285–332. doi: 10.1016/0891-5849(89)90136-6. [DOI] [PubMed] [Google Scholar]
- 9.Bauman DR, Bitmansour AD, McDonald JG, Thompson BM, Liang G, Russell DW. 2009. 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc Natl Acad Sci U S A 106:16764–16769. doi: 10.1073/pnas.0909142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang F, Ma Y, Barrett JW, Gao X, Loh J, Barton E, Virgin HW, McFadden G. 2004. Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nat Immunol 5:1266–1274. doi: 10.1038/ni1132. [DOI] [PubMed] [Google Scholar]
- 11.Shrivastava-Ranjan P, Bergeron É, Chakrabarti AK, Albariño CG, Flint M, Nichol ST, Spiropoulou CF. 2016. 25-Hydroxycholesterol inhibition of Lassa virus infection through aberrant GP1 glycosylation. mBio 7:e01808-16. doi: 10.1128/mBio.01808-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu S-Y, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, Pernet O, Guo H, Nusbaum R, Zack JA, Freiberg AN, Su L, Lee B, Cheng G. 2013. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 38:92–105. doi: 10.1016/j.immuni.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li C, Deng Y-Q, Wang S, Ma F, Aliyari R, Huang X-Y, Zhang N-N, Watanabe M, Dong H-L, Liu P, Li X-F, Ye Q, Tian M, Hong S, Fan J, Zhao H, Li L, Vishlaghi N, Buth JE, Au C, Liu Y, Lu N, Du P, Qin FX-F, Zhang B, Gong D, Dai X, Sun R, Novitch BG, Xu Z, Qin C-F, Cheng G. 2017. 25-Hydroxycholesterol protects host against Zika virus infection and its associated microcephaly in a mouse model. Immunity 46:446–456. doi: 10.1016/j.immuni.2017.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ke W, Fang L, Jing H, Tao R, Wang T, Li Y, Long S, Wang D, Xiao S. 2017. Cholesterol 25-hydroxylase inhibits porcine reproductive and respiratory syndrome virus replication through enzyme activity-dependent and -independent mechanisms. J Virol 91:e00827-17. doi: 10.1128/JVI.00827-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song Z, Zhang Q, Liu X, Bai J, Zhao Y, Wang X, Jiang P. 2017. Cholesterol 25-hydroxylase is an interferon-inducible factor that protects against porcine reproductive and respiratory syndrome virus infection. Vet Microbiol 210:153–161. doi: 10.1016/j.vetmic.2017.09.011. [DOI] [PubMed] [Google Scholar]
- 16.Wang J, Zeng L, Zhang L, Guo Z-Z, Lu S-F, Ming S-L, Li G-L, Wan B, Tian K-G, Yang G-Y, Chu B-B. 2017. Cholesterol 25-hydroxylase acts as a host restriction factor on pseudorabies virus replication. J Gen Virol 98:1467–1476. doi: 10.1099/jgv.0.000797. [DOI] [PubMed] [Google Scholar]
- 17.Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, Lacaze P, Watterson S, Griffiths SJ, Spann NJ, Meljon A, Talbot S, Krishnan K, Covey DF, Wenk MR, Craigon M, Ruzsics Z, Haas J, Angulo A, Griffiths WJ, Glass CK, Wang Y, Ghazal P. 2013. The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity 38:106–118. doi: 10.1016/j.immuni.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Civra A, Cagno V, Donalisio M, Biasi F, Leonarduzzi G, Poli G, Lembo D. 2014. Inhibition of pathogenic non-enveloped viruses by 25-hydroxycholesterol and 27-hydroxycholesterol. Sci Rep 4:7487. doi: 10.1038/srep07487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bouziat R, Hinterleitner R, Brown JJ, Stencel-Baerenwald JE, Ikizler M, Mayassi T, Meisel M, Kim SM, Discepolo V, Pruijssers AJ, Ernest JD, Iskarpatyoti JA, Costes LMM, Lawrence I, Palanski BA, Varma M, Zurenski MA, Khomandiak S, McAllister N, Aravamudhan P, Boehme KW, Hu F, Samsom JN, Reinecker H-C, Kupfer SS, Guandalini S, Semrad CE, Abadie V, Khosla C, Barreiro LB, Xavier RJ, Ng A, Dermody TS, Jabri B. 2017. Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science 356:44–50. doi: 10.1126/science.aah5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gong J, Sachdev E, Mita AC, Mita MM. 2016. Clinical development of reovirus for cancer therapy: an oncolytic virus with immune-mediated antitumor activity. World J Methodol 6:25–42. doi: 10.5662/wjm.v6.i1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Danthi P, Holm GH, Stehle T, Dermody TS. 2013. Reovirus receptors, cell entry, and proapoptotic signaling. Adv Exp Med Biol 790:42–71. doi: 10.1007/978-1-4614-7651-1_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konopka-Anstadt JL, Mainou BA, Sutherland DM, Sekine Y, Strittmatter SM, Dermody TS. 2014. The Nogo receptor NgR1 mediates infection by mammalian reovirus. Cell Host Microbe 15:681–691. doi: 10.1016/j.chom.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maginnis MS, Mainou BA, Derdowski A, Johnson EM, Zent R, Dermody TS. 2008. NPXY motifs in the beta1 integrin cytoplasmic tail are required for functional reovirus entry. J Virol 82:3181–3191. doi: 10.1128/JVI.01612-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ebert DH, Deussing J, Peters C, Dermody TS. 2002. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J Biol Chem 277:24609–24617. doi: 10.1074/jbc.M201107200. [DOI] [PubMed] [Google Scholar]
- 25.Chandran K, Parker JS, Ehrlich M, Kirchhausen T, Nibert ML. 2003. The delta region of outer-capsid protein m1 undergoes conformational change and release from reovirus particles during cell entry. J Virol 77:13361–13375. doi: 10.1128/JVI.77.24.13361-13375.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nibert ML, Fields BN. 1992. A carboxy-terminal fragment of protein m1/m1C is present in infectious subvirion particles of mammalian reoviruses and is proposed to have a role in penetration. J Virol 66:6408–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nibert ML, Odegard AL, Agosto MA, Chandran K, Schiff LA. 2005. Putative autocleavage of reovirus m1 protein in concert with outer-capsid disassembly and activation for membrane permeabilization. J Mol Biol 345:461–474. doi: 10.1016/j.jmb.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 28.Odegard AL, Chandran K, Zhang X, Parker JS, Baker TS, Nibert ML. 2004. Putative autocleavage of outer capsid protein m1, allowing release of myristoylated peptide m1N during particle uncoating, is critical for cell entry by reovirus. J Virol 78:8732–8745. doi: 10.1128/JVI.78.16.8732-8745.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mainou BA, Dermody TS. 2012. Transport to late endosomes is required for efficient reovirus. J Virol 86:8346–8358. doi: 10.1128/JVI.00100-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anafu AA, Bowen CH, Chin CR, Brass AL, Holm GH. 2013. Interferon-inducible transmembrane protein 3 (IFITM3) restricts reovirus cell entry. J Biol Chem 288:17261–17271. doi: 10.1074/jbc.M112.438515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park K, Scott AL. 2010. Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J Leukoc Biol 88:1081–1087. doi: 10.1189/jlb.0610318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stewart MJ, Smoak K, Blum MA, Sherry B. 2005. Basal and reovirus-induced beta interferon (IFN-β) and IFN-β-stimulated gene expression are cell type specific in the cardiac protective response. J Virol 79:2979–2987. doi: 10.1128/JVI.79.5.2979-2987.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Donnell SM, Holm GH, Pierce JM, Tian B, Watson MJ, Chari RS, Ballard DW, Brasier AR, Dermody TS. 2006. Identification of an NF-k B-dependent gene network in cells infected by mammalian reovirus. J Virol 80:1077–1086. doi: 10.1128/JVI.80.3.1077-1086.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sturzenbecker LJ, Nibert ML, Furlong DB, Fields BN. 1987. Intracellular digestion of reovirus particles requires a low pH and is an essential step in the viral infectious cycle. J Virol 61:2351–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eibinger G, Fauler G, Bernhart E, Frank S, Hammer A, Wintersperger A, Eder H, Heinemann A, Mischel PS, Malle E, Sattler W. 2013. On the role of 25-hydroxycholesterol synthesis by glioblastoma cell lines. Implications for chemotactic monocyte recruitment. Exp Cell Res 319:1828–1838. doi: 10.1016/j.yexcr.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lange Y, Ye J, Strebel F. 1995. Movement of 25-hydroxycholesterol from the plasma membrane to the rough endoplasmic reticulum in cultured hepatoma cells. J Lipid Res 36:1092–1097. [PubMed] [Google Scholar]
- 37.Olsen BN, Schlesinger PH, Ory DS, Baker NA. 2011. 25-Hydroxycholesterol increases the availability of cholesterol in phospholipid membranes. Biophys J 100:948–956. doi: 10.1016/j.bpj.2010.12.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gale SE, Westover EJ, Dudley N, Krishnan K, Merlin S, Scherrer DE, Han X, Zhai X, Brockman HL, Brown RE, Covey DF, Schaffer JE, Schlesinger P, Ory DS. 2009. Side chain oxygenated cholesterol regulates cellular cholesterol homeostasis through direct sterol-membrane interactions. J Biol Chem 284:1755–1764. doi: 10.1074/jbc.M807210200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pécheur E-I, Sainte-Marie J, Bienvenüe A, Hoekstra D. 1999. Lipid headgroup spacing and peptide penetration, but not peptide oligomerization, modulate peptide-induced fusion. Biochemistry 38:364–373. doi: 10.1021/bi981389u. [DOI] [PubMed] [Google Scholar]
- 40.Teissier É, Pécheur E-I. 2007. Lipids as modulators of membrane fusion mediated by viral fusion proteins. Eur Biophys J 36:887–899. doi: 10.1007/s00249-007-0201-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arias CF, Silva-Ayala D, López S. 2015. Rotavirus entry: a deep journey into the cell with several exits. J Virol 89:890–893. doi: 10.1128/JVI.01787-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DiGiuseppe S, Bienkowska-Haba M, Sapp M. 2016. Human papillomavirus entry: hiding in a bubble. J Virol 90:8032–8035. doi: 10.1128/JVI.01065-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blaas D, Fuchs R. 2016. Mechanism of human rhinovirus infections. Mol Cell Pediatr 3:21. doi: 10.1186/s40348-016-0049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wiethoff CM, Nemerow GR. 2015. Adenovirus membrane penetration: tickling the tail of a sleeping dragon. Virology 0:591–599. doi: 10.1016/j.virol.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barlan AU, Griffin TM, Mcguire KA, Wiethoff CM. 2011. Adenovirus membrane penetration activates the NLRP3 inflammasome. J Virol 85:146–155. doi: 10.1128/JVI.01265-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schulz WL, Haj AK, Schiff LA. 2012. Reovirus uses multiple endocytic pathways for cell entry. J Virol 86:12665–12675. doi: 10.1128/JVI.01861-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schelhaas M, Shah B, Holzer M, Blattmann P, Kühling L, Day PM, Schiller JT, Helenius A. 2012. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog 8:e1002657. doi: 10.1371/journal.ppat.1002657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Imelli N, Meier O, Boucke K, Hemmi S, Greber UF. 2004. Cholesterol is required for endocytosis and endosomal escape of adenovirus type 2. J Virol 78:3089–3098. doi: 10.1128/JVI.78.6.3089-3098.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Furlong DB, Nibert ML, Fields BN. 1988. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol 62:246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith RE, Zweerink HJ, Joklik WK. 1969. Polypeptide components of virions, top component and cores of reovirus type 3. Virology 39:791–810. doi: 10.1016/0042-6822(69)90017-8. [DOI] [PubMed] [Google Scholar]
- 51.Virgin HW IV, Bassel-Duby R, Fields BN, Tyler KL. 1988. Antibody protects against lethal infection with the neurally spreading reovirus type 3 (Dearing). J Virol 62:4594–4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Connolly JL, Dermody TS. 2002. Virion disassembly is required for apoptosis induced by reovirus. J Virol 76:1632–1641. doi: 10.1128/JVI.76.4.1632-1641.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thete D, Danthi P. 2015. Conformational changes required for reovirus cell entry are sensitive to pH. Virology 483:291–301. doi: 10.1016/j.virol.2015.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gomatos PJ. 1967. RNA synthesis in reovirus-infected L929 mouse fibroblasts. Proc Natl Acad Sci U S A 58:1798–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tyler KL, Squier MK, Rodgers SE, Schneider SE, Oberhaus SM, Grdina TA, Cohen JJ, Dermody TS. 1995. Differences in the capacity of reovirus strains to induce apoptosis are determined by the viral attachment protein s1. J Virol 69:6972–6979. [DOI] [PMC free article] [PubMed] [Google Scholar]