

The accumulation of alpha-synuclein (αS) inclusions is a hallmark feature of Parkinson's disease (PD) and PD-related diseases. Recently, a number of studies have demonstrated similarities between the prion protein and αS, including its ability to spread along neuroanatomical tracts throughout the central nervous system (CNS). However, there are caveats in each of these studies in which the injection routes used had the potential to result in a widespread dissemination of the αS-containing inocula, making it difficult to precisely define the mechanisms of spread. In this study, we assessed the spread of pathology following a localized induction of αS inclusions in the lumbar spinal cord following a unilateral injection in the sciatic nerve. Using this paradigm, we demonstrated the ability for αS inclusion spread and/or induction along neuroanatomical tracts within the CNS of two αS-overexpressing mouse models.
KEYWORDS: α-synuclein, prion, sciatic nerve, axonal transport, propagation, alpha-synuclein, prions
ABSTRACT
Misfolded alpha-synuclein (αS) may exhibit a number of characteristics similar to those of the prion protein, including the apparent ability to spread along neuroanatomical connections. The demonstration for this mechanism of spread is largely based on the intracerebral injections of preaggregated αS seeds in mice, in which it cannot be excluded that diffuse, surgical perturbations and hematogenous spread also contribute to the propagation of pathology. For this reason, we have utilized the sciatic nerve as a route of injection to force the inoculum into the lumbar spinal cord and induce a localized site for the onset of αS inclusion pathology. Our results demonstrate that mouse αS fibrils (fibs) injected unilaterally in the sciatic nerve are efficient in inducing pathology and the onset of paralytic symptoms in both the M83 and M20 lines of αS transgenic mice. In addition, a spatiotemporal study of these injections revealed a predictable spread of pathology to brain regions whose axons synapse directly on ventral motor neurons in the spinal cord, strongly supporting axonal transport as a mechanism of spread of the αS inducing, or seeding, factor. We also revealed a relatively decreased efficiency for human αS fibs containing the E46K mutation to induce disease via this injection paradigm, supportive of recent studies demonstrating a diminished ability of this mutant αS to undergo aggregate induction. These results further demonstrate prion-like properties for αS by the ability for a progression and spread of αS inclusion pathology along neuroanatomical connections.
IMPORTANCE The accumulation of alpha-synuclein (αS) inclusions is a hallmark feature of Parkinson's disease (PD) and PD-related diseases. Recently, a number of studies have demonstrated similarities between the prion protein and αS, including its ability to spread along neuroanatomical tracts throughout the central nervous system (CNS). However, there are caveats in each of these studies in which the injection routes used had the potential to result in a widespread dissemination of the αS-containing inocula, making it difficult to precisely define the mechanisms of spread. In this study, we assessed the spread of pathology following a localized induction of αS inclusions in the lumbar spinal cord following a unilateral injection in the sciatic nerve. Using this paradigm, we demonstrated the ability for αS inclusion spread and/or induction along neuroanatomical tracts within the CNS of two αS-overexpressing mouse models.
INTRODUCTION
Alpha-synuclein (αS) is a natively unfolded cytosolic protein but is observed to form amyloidogenic inclusions in a spectrum of neurodegenerative diseases referred to as α-synucleinopathies (1–6). In Parkinson's disease (PD) αS inclusions, termed Lewy bodies or Lewy neurites, are believed to have a toxic role as directly supported by mutations in the αS gene, SNCA, resulting in autosomal-dominant PD (1, 7–13); however, the exact mechanisms these inclusions carry out in the neurodegenerative cascade of PD are still debated (1, 3, 4, 14). In recent years, findings have demonstrated the prion-like nature of the αS protein, with some investigators going so far as to refer to them as αS prions (15–17). The basis for this came about by the observed progression of αS pathology into different regions of the brain and the demonstration that soluble αS can undergo conformational templating leading to its aggregation and the formation of inclusions (18–22). To better understand the spread of αS pathology, several in vivo studies have utilized intracerebral injection in nontransgenic and αS transgenic mouse models and describe a propagation of αS inclusions away from the injection site (23–27). Additionally, investigators have peripherally administered human or mouse αS inclusions into mice and demonstrated the ability for these to induce central nervous system (CNS) αS pathology, suggesting a neuroinvasive nature of αS and supporting the theory that αS pathology begins in the peripheral nervous system (PNS) and spreads to the CNS (18, 19, 28–33). Although these data are analogous to the propagation of prions along neuroanatomically connected neuronal populations, there are caveats in each of these studies in which the injection routes had the potential to result in a widespread dissemination of the αS-containing inocula. For instance, for both the intracerebral and intramuscular (i.m.) routes of injections, there is a strong potential for the inoculum to get into the bloodstream or the needle to alter the cellular homeostasis, thereby allowing the αS seeds to access the CNS via multiple points of entry (32, 33). Due to these scenarios, there still exists a lack of understanding as to the mechanism(s) of spread and/or induction of αS pathology.
To better assess the potential for αS pathology to undergo propagation along axonal projections, we utilized a sciatic nerve injection paradigm which has been used to study the contribution of neuroanatomical transport for other neurodegenerative proteins (34–36). We studied the temporal and spatial propagation of αS pathology in two lines of αS-overexpressing mouse lines and discovered that following a unilateral injection in the sciatic nerve with mouse αS fibrils (fibs), pathology began in the lumbar spinal cord and progressed rostrally to several brain nuclei, strongly supporting disease spread along direct neuroanatomical connections.
RESULTS
Efficiency of disease induction via sciatic nerve injection in M83+/− mice.
We previously reported that injection of mouse αS fibrils (fibs) via peripheral routes, including the intramuscular (i.m.), intraperitoneal (i.p.), and intravenous (i.v.) routes, are capable of inducing a robust αS pathology in the spinal cords and brains of hemizygous M83 αS transgenic mice (M83+/−) and, to a lesser extent, in hemizygous M20 αS transgenic mice (M20+/−), indicating an efficient mechanism for neuroinvasion (29, 32). To better understand the role of axonal transport in the spread of αS into and throughout the CNS, we used a previously published sciatic nerve injection paradigm that targets the inoculum to a defined group of motor neurons in the lumbar spinal cord (34–36). We injected various forms of recombinant αS fibs and controls unilaterally into the sciatic nerve in M83+/− mice and let them age to determine the efficiency of this route of injection to induce disease. Importantly, naive M83+/− mice do not begin to accumulate αS inclusion pathology prior to 21 months of age (37). All 14 M83+/− mice injected in the sciatic nerve with 4 μg of mouse wild-type (WT) αS fibs developed hind-limb motor impairment (i.e., foot drop followed by paralysis), which first appeared in the ipsilateral injected limb. This progressed to bilateral hind-limb paralysis by 3.9 ± 0.1 months postinjection (p.i.), at which point the mice had to be euthanized (Fig. 1). Injections of E46K human αS fibs intramuscularly into M83+/− mice have previously revealed a decreased attack rate and delayed incubation period compared to injections with human WT αS fibs or fibs containing the H50Q, G51D, and A53E mutations (33). We found that injection of M83+/− mice unilaterally in the sciatic nerve with 4 μg of E46K human αS fibs resulted in a similar phenomenon. Of the 7 mice injected with these fibs, 5 developed hind-limb paralysis at an average age of 7.0 ± 1.3 months p.i. (Fig. 1). The 2 unaffected mice were aged to 12 months p.i. prior to harvesting. Controls included M83+/− mice injected unilaterally in the sciatic nerve with phosphate-buffered saline (PBS), 4 μg of soluble human αS containing a deletion of amino acids 71 to 82 (Δ71–82), and 1.6 μg of recombinant WT superoxide dismutase 1 (SOD1) fibs. Human αS protein lacking amino acids 71 to 82 is nonamyloidogenic and is unable to seed αS in vitro and in cultured cells (20, 22, 38, 39). WT SOD1 fibs have been demonstrated to induce disease in mice overexpressing SOD1 following exogenous administration (40) and were used in this study to ensure that disease induction in M83+/− mice could not be achieved with other disease-specific proteins. Mice injected with these control inocula were all aged to 6 months p.i. without developing any phenotype (Fig. 1).
FIG 1.
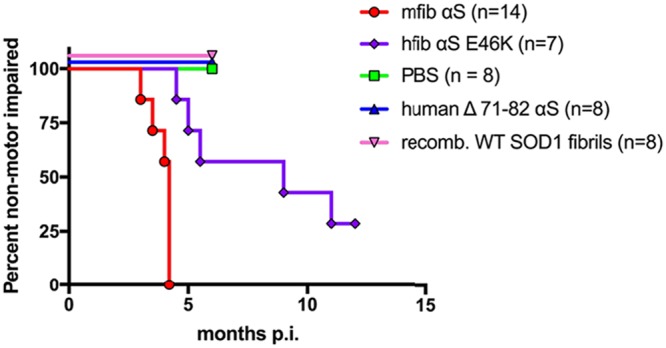
Induction of disease in the M83+/− αS transgenic mouse model following unilateral sciatic nerve injections. Kaplan-Meier curves reveal the transmissibility of the indicated recombinant proteins following unilateral sciatic nerve injection in the M83+/− line of αS transgenic mice. Mice were injected with mouse WT αS fibs (mfib αS), E46K human αS fibs (hfib αS E46K), human Δ71–82 αS, recombinant WT SOD1 fibrils, or PBS.
Disease progression in M83+/− mice following sciatic nerve injection of mouse WT αS fibs.
To better determine the mechanism(s) of pathological spread in M83+/− mice, we performed a longitudinal study following sciatic nerve injection with mouse αS fibs. At 1 month p.i., αS inclusions were observed in a few neurons in the ipsilateral dorsal root ganglia (DRG) and within the ventral horn of the lumbar spinal cord (Fig. 2a). No αS deposition was found in the thoracic or cervical sections of the spinal cord at this time point. At 2 months p.i., αS deposits were still only observed in the ipsilateral DRG, with no immunoreactivity detected in the contralateral DRG (Fig. 2b). Within the spinal cord, deposition had increased in abundance in the lumbar spinal but had not progressed up to the level of cervical regions. Interestingly, αS deposition was found in several neurons located in the reticular formation and periaqueductal gray area (PAG), both of which have direct synaptic connections to the ventral motor neurons in the lumbar spinal cord. At the end stage of disease (3.9 ± 0.1 months p.i.), αS deposition had increased dramatically throughout the spinal cord and brainstem (Fig. 2c; Table 1). Deposition was at that point observed in both the ipsilateral and contralateral DRG and in all levels of the spinal cord. αS pathology was also abundant in the reticular formation, lateral vestibular nucleus, PAG, the thalamus, and hypothalamus. The presence and distribution of αS inclusion pathology were confirmed by staining with antibody Syn 506, which selectively recognizes αS inclusions, and an antibody to p62/sequestrome, another established marker of inclusion pathology (Fig. 3). In motor-impaired M83+/− mice that had been injected with E46K human αS fibs, αS pathology was observed in the same neuroanatomical locations as observed following mouse WT αS fibril injections, including deposition in the DRG (Fig. 2d). No pathology was observed in the 2 M83+/− mice injected with E46K human αS fibs that did not develop motor symptoms up to 12 months p.i. Mice injected unilaterally in the sciatic nerve with PBS, soluble human αS containing Δ71–82, and recombinant WT SOD1 fibs revealed no αS immunoreactivity in the spinal cord or brain at 6 months p.i. (Fig. 4). These data support a spread of the A53T αS seeding component along neuroanatomical pathways following the initial deposition in the lumbar spinal cord. In support of this finding, we also detected αS inclusion pathology in the white matter of spinal cords in animals in which disease was induced by sciatic nerve injection of mouse αS fibs, whereas no inclusions were detected in control injected animals (Fig. 5).
FIG 2.

Induction and spread of αS pathology in the M83+/− mouse model following injection with mouse αS fibs. Mice were injected unilaterally in the sciatic nerve with mouse WT αS fibs (mfib αS) and harvested at 1 month (a) or 2 months (b) p.i. or when the mice reached the end stage of disease (c). (d) Mice were also injected with human αS fibs containing the E46K mutation (hfib αS E46K) and harvested at the end stage of disease or, for those with no symptoms, at 12 months p.i. Representative images show αS pathology in both the ipsilateral (ipsi-) and contralateral (contra-) DRG and several CNS regions, depicted by cartoons with black dots representing the specific locations the images were taken (n ≥ 8 animals per cohort). Tissue sections were stained with an antibody to αS phosphorylated at Ser129 (2G12) and counterstained with hematoxylin. Scale bars = 50 μm. Sp.C., spinal cord.
TABLE 1.
Spatiotemporal distribution and abundance of αS pathology in M83+/− mice following sciatic nerve inoculation with mouse WT αS fibs
| Body site | Relative abundance of αS inclusion pathologya |
||
|---|---|---|---|
| 1 mo p.i. | 2 mo p.i. | Clinical (3.9 ± 0.1 mo p.i.) | |
| DRG | |||
| Ipsilateral | + | ++ | ++ |
| Contralateral | − | − | + |
| Spinal cord | |||
| Lumbar | + | ++ | +++ |
| Thoracic | − | + | +++ |
| Cervical | − | − | +++ |
| Brain | − | ||
| Reticular formation | − | ++ | +++ |
| Lateral vestibular nucleus | − | − | ++ |
| Red nucleus | − | + | +++ |
| PAG | − | + | +++ |
| Motor cortex | − | − | + |
−, none; +, rare; ++, numerous; +++, abundant and widespread.
FIG 3.
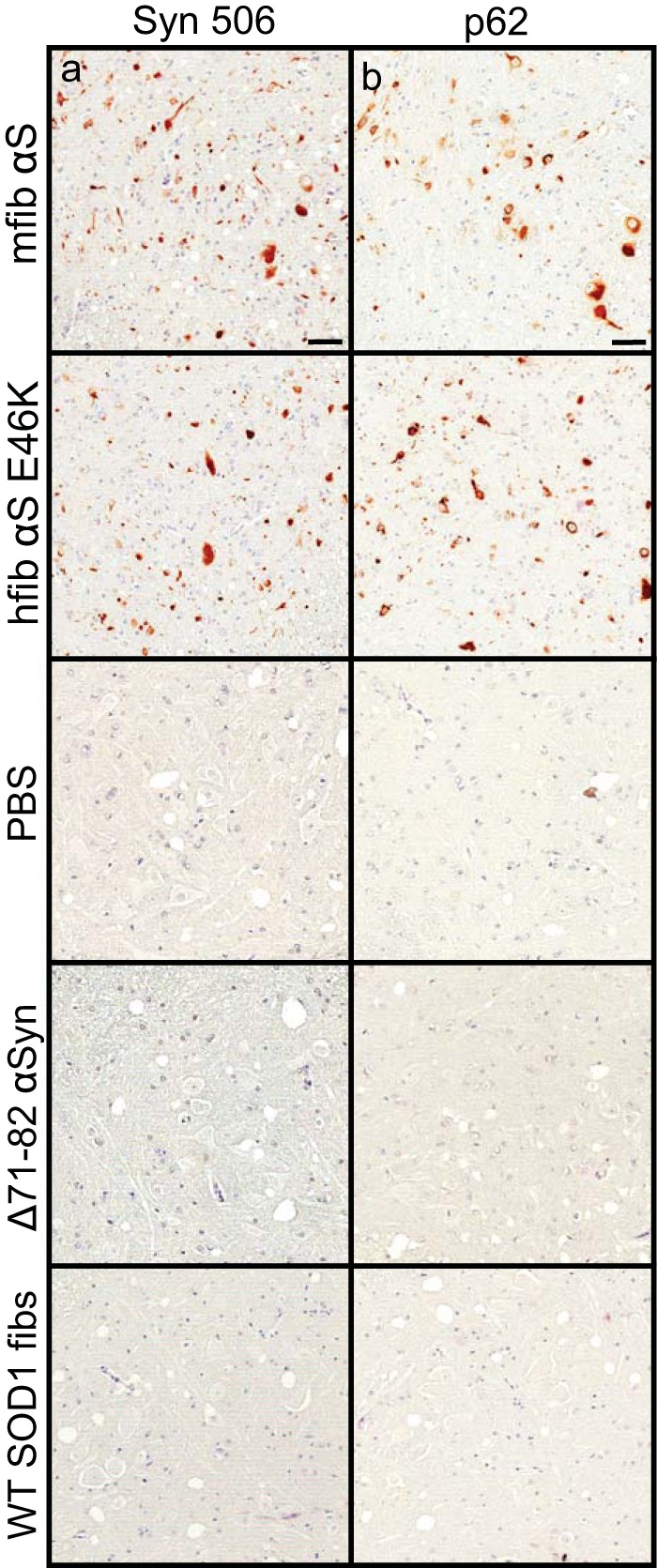
αS inclusion pathology in M83+/− injected mice. M83+/− mice were injected unilaterally in the sciatic nerve with the indicated homogenates and harvested at the end stage of disease (mfib αS and hfib αS E46K) or at 6 months p.i. (PBS, human Δ71–82 αS, and WT SOD1 fibs). Spinal cords were stained with a conformation-specific antibody against αS, Syn 506 (a), and an antibody specific for cytoplasmic Lewy body inclusions, p62/Sqstm1 (b).
FIG 4.
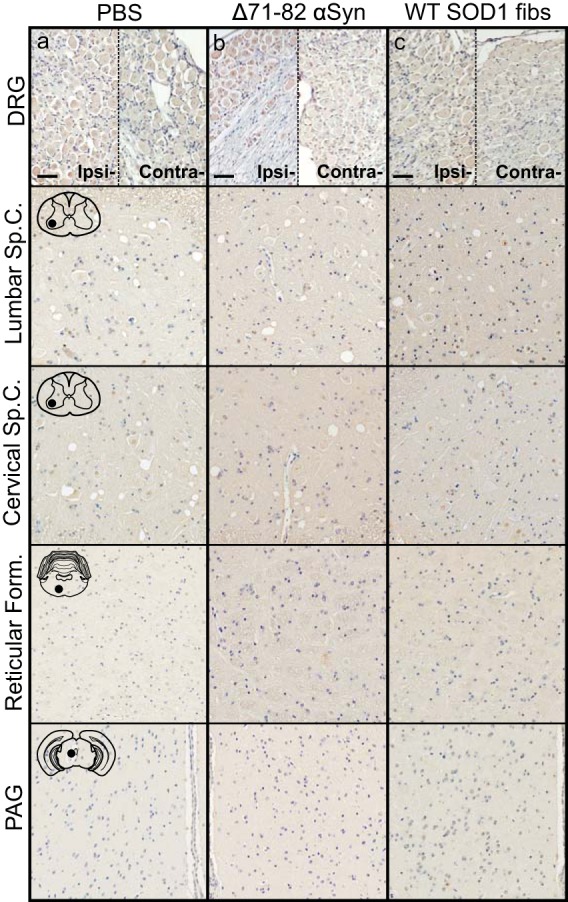
Absence of αS inclusion pathology in M83+/− mice injected with control inocula. Mice were injected unilaterally in the sciatic nerve with PBS (a), soluble human αS containing Δ71–82 (b), or WT SOD1 fibs (c) and aged to 6 months p.i. prior to harvesting tissue (n = 6 animals per cohort). Representative images show αS pathology in both the ipsilateral and contralateral DRG and several CNS regions, depicted by cartoons with black dots representing the specific locations the images were taken. Tissue sections were stained with an antibody to αS phosphorylated at Ser129 (2G12) and counterstained with hematoxylin. Scale bars = 50 μm.
FIG 5.
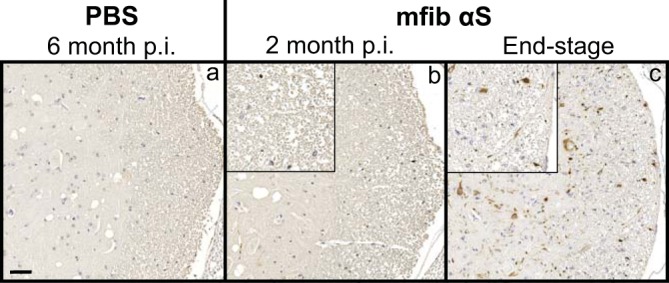
Detection of αS inclusion pathology in the white matter of the spinal cord. Representative images show αS pathology in the white matter surrounding the ventral horn of the spinal cord in mice injected unilaterally in the sciatic nerve with PBS (a) or mfib αS (b and c) and harvested at the indicated time points p.i. Tissue sections were stained with an antibody to αS phosphorylated at Ser129 (2G12) and counterstained with hematoxylin. Scale bars = 50 μm.
We also investigated the accumulation and progression of glial reactivity during the course of disease transmission in the sciatic nerve-injected M83+/− mice. Astrocytic glial fibrillary acidic protein (GFAP) and microglial cd11b immunoreactivity displayed a remarkably similar spatiotemporal accumulation in the spinal cord compared to the accumulation of αS inclusions (Fig. 6). We observed almost no glial pathology at 1 month following injection with mouse αS fibs, while a slight increase was observed by 2 months p.i. and a widespread, robust accumulation was observed at the end stage of disease (Fig. 6). Interestingly, there was a significant increase in the GFAP immunoreactivity in the lumbar spinal cord compared to the cervical spinal cord at the end stage of disease, strongly supporting a spread of disease pathology by injecting αS fibs in the sciatic nerve (Fig. 6b).
FIG 6.

Accumulation of gliosis in M83+/− injected mice. M83+/− mice were injected unilaterally in the sciatic nerve with control inocula (PBS and WT SOD1 fibs) and harvested at 6 months p.i. or with mouse WT αS fibs (mfib αS) and harvested at 1 month or 2 months p.i. or when the mice reached the end stage of disease. (a) Spinal cords were immunostained to visualize astrocytes using an antibody to GFAP and representative images were captured (n = 3 animals per cohort). (b) GFAP immunoreactivity in the cervical and lumbar segments of the spinal cord was quantified for each cohort (n = 3 animals per cohort; mean ± SD). ***, P ≤ 0.001. (c) Spinal cords were also immunostained for microglia using an antibody to cd11b, and representative images were captured (n = 3 animals per cohort). Scale bars = 100 μm.
Efficiency of disease induction via sciatic nerve injection in M20+/− mice.
We also conducted sciatic nerve transmission studies in the M20+/− mouse model that overexpresses human WT αS but never intrinsically develops motor impairments or αS inclusion pathology (37, 41). Similar to the M83+/− model, the expression of human αS in this model is driven by the mouse prion protein promoter and displays a neuroanatomical distribution of αS similar to that observed in the M83+/− model (37, 41, 42). However, unlike with the M83+/− mice, injection within the hind-limb muscle with human αS fibs in the M20+/− mice was unable to induce a motor phenotype when aged to 12 months p.i. (32). When we injected 4 μg of mouse WT αS fibs unilaterally within the sciatic nerve of M20+/− mice, 7 of the 9 mice developed a motor phenotype at 8.3 ± 0.6 months p.i. (Fig. 7), while the 2 other mice had to be euthanized at 8.0 months p.i. for non-disease-related complications. All M20+/− mice injected with controls, including PBS, recombinant WT SOD1 fibs, and soluble human αS containing Δ71–82, were aged to 12 months p.i. without any observable motor impairment (Fig. 7).
FIG 7.
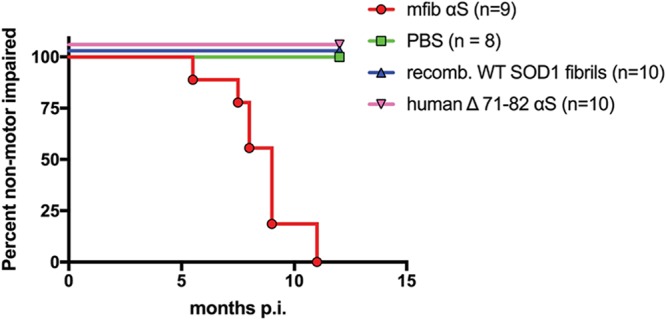
Induction of disease in the M20+/− αS transgenic mouse model following unilatereal sciatic nerve injections. Kaplan-Meier curves reveal the transmissibility of the indicated recombinant proteins following unilateral sciatic nerve injection in M20+/− line of αS transgenic mice. Mice were injected with mouse WT αS fibs (mfib αS), human Δ71–82 αS, recombinant WT SOD1 fibrils, or PBS.
Disease progression in M20+/− mice following sciatic nerve injection of mouse WT αS fibs.
To determine whether the spread of pathology induced in the M20+/− mice following sciatic nerve injection was similar to that observed in the M83+/− mice, we harvested mice at 2 and 4 months p.i. of mouse αS fibs. At 2 months p.i., αS pathology was only sparsely observed in the neuropil of the lumbar spinal cord (Fig. 8a). At 4 months p.i., pathology had increased in abundance in the lumbar spinal cord but had not progressed up to the level in the cervical spinal cord (Fig. 8b). Similar to the spread of pathology observed in the M83+/− mice, fibrillar αS deposits were observed in the in the reticular formation and in the PAG at 4 months p.i. (Fig. 8b). By the end stage of disease, when mice were displaying hind-limb motor impairments, αS pathology was infrequently detected in the ipsilateral DRG but was found in abundance in all levels of the spinal cord along with several brain and brain stem nuclei, including the reticular formation, lateral vestibular nucleus, thalamus, and hypothalamus (Fig. 8c; Table 2). At the end stage of disease, there was also abundant αS inclusion deposition in the white matter of spinal cords, similar to that observed in the M83+/− mice, while no immunoreactivity was observed in the control injected animals (data not shown). The presence and distribution of αS inclusion pathology in these mice were confirmed by staining with antibody Syn 506, which selectively recognizes αS inclusions, and an antibody to p62/sequestrome, another established marker of inclusion pathology (Fig. 9). M20+/− mice injected unilaterally in the sciatic nerve with PBS, recombinant WT SOD1 fibs, and soluble human αS containing Δ71–82 had no αS pathology in the CNS when aged to 12 months p.i. (Fig. 10).
FIG 8.
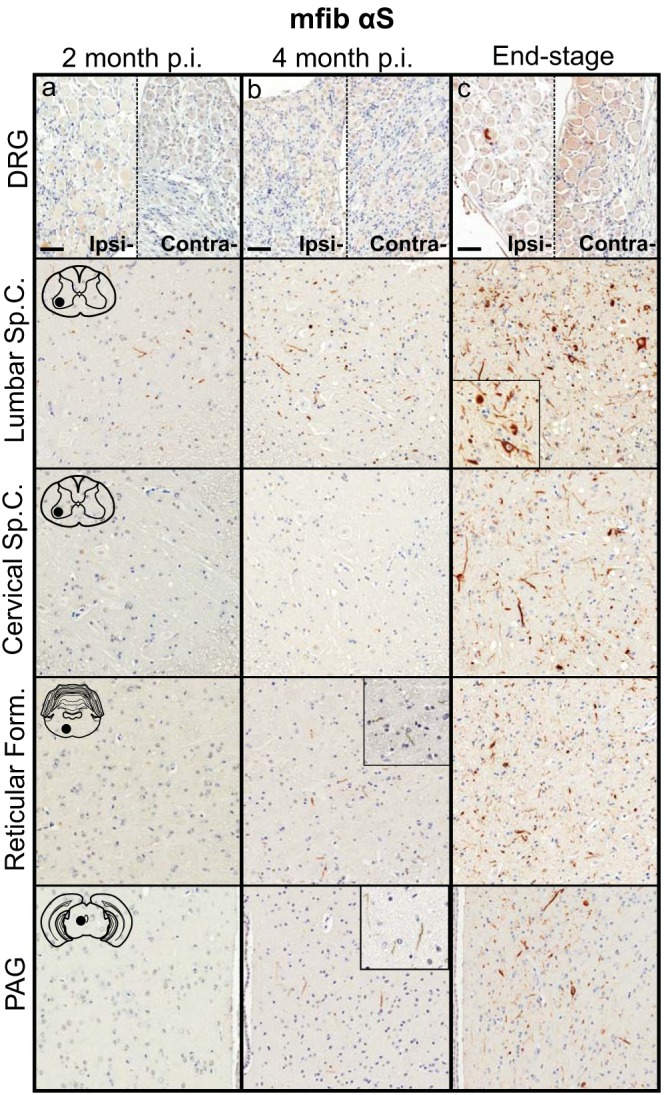
Induction and spread of αS pathology in the M20+/− mouse model following injection with mouse αS fibs. Mice were injected unilaterally in the sciatic nerve with mouse WT αS fibs (mfib αS) and harvested at 2 months (a) or 4 months (b) p.i. or when the mice reached the end stage of disease (c) (n ≥ 8 animals per cohort). Representative images show αS pathology in both the ipsilateral and contralateral DRG and several CNS regions, depicted by cartoons with black dots representing the specific locations the images were taken. Tissue sections were stained with an antibody to αS phosphorylated at Ser129 (2G12) and counterstained with hematoxylin. Scale bars = 50 μm.
TABLE 2.
Spatiotemporal distribution and abundance of αS pathology in M20+/− mice following sciatic nerve inoculation with mouse WT αS fibs
| Body site | Relative abundance of αS inclusion pathologya |
||
|---|---|---|---|
| 2 mo p.i. | 4 mo p.i. | Clinical (8.3 ± 0.6 mo p.i.) | |
| DRG | |||
| Ipsilateral | − | − | + |
| Contralateral | − | − | − |
| Spinal cord | |||
| Lumbar | + | ++ | +++ |
| Thoracic | − | + | +++ |
| Cervical | − | − | +++ |
| Brain | |||
| Reticular formation | − | + | +++ |
| Lateral vestibular nucleus | − | − | ++ |
| Red nucleus | − | + | +++ |
| PAG | − | + | +++ |
| Motor cortex | − | − | − |
−, none; +, rare; ++, numerous; +++, abundant and widespread.
FIG 9.
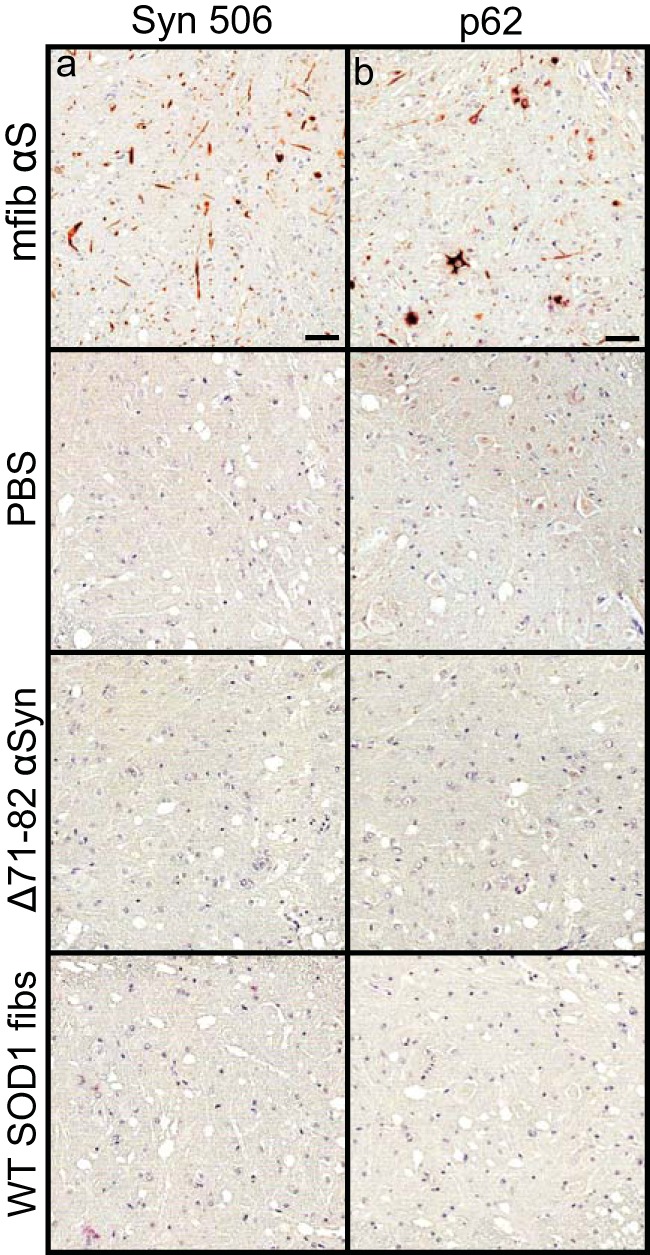
αS inclusion pathology in M20+/− injected mice. M20+/− mice were injected unilaterally in the sciatic nerve with the indicated homogenates and harvested at the end stage of disease (mfib αS) or at 12 months p.i. (PBS, Δ71–82 αS, and WT SOD1 fibs). Spinal cords were stained with a conformation-specific antibody against αS, Syn 506 (a), and an antibody specific for cytoplasmic Lewy body inclusions, p62/Sqstm1 (b).
FIG 10.
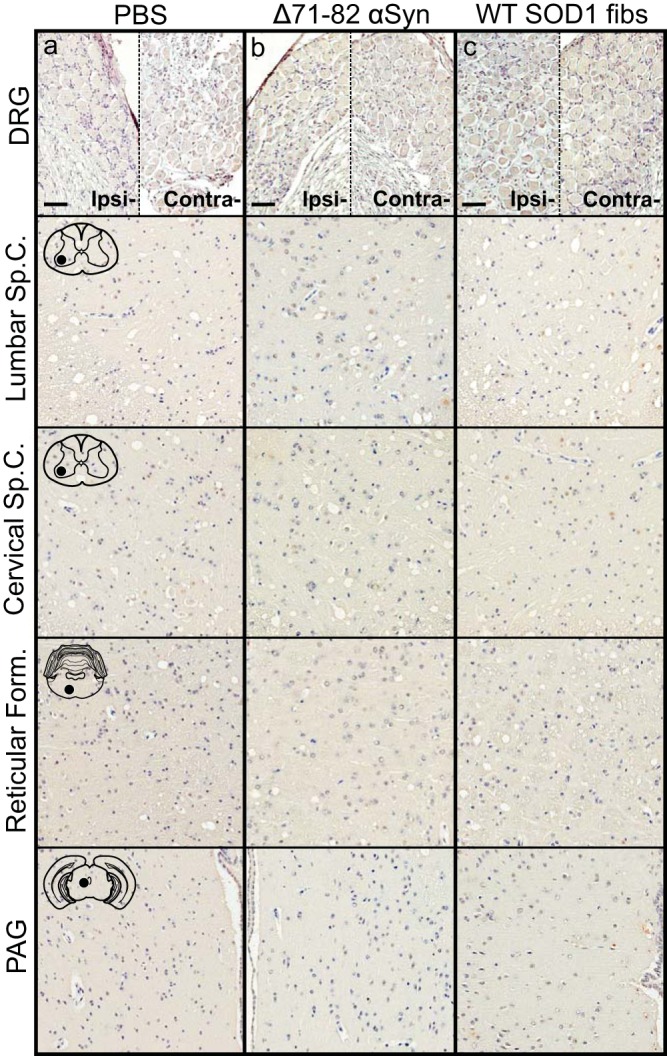
Absence of αS pathology in M20+/− mice injected with control inocula. Mice were injected unilaterally in the sciatic nerve with PBS (a), soluble human αS containing Δ71–82 (b), or WT SOD1 fibs (c) and aged to 12 months p.i. prior to harvesting tissue (n = 6 animals per cohort). Representative images show αS pathology in both the ipsilateral and contralateral DRG and several CNS regions, depicted by cartoons with black dots representing the specific locations the images were taken. Tissue sections were stained with an antibody to αS phosphorylated at Ser129 (2G12) and counterstained with hematoxylin. Scale bars = 50 μm.
We also investigated the progression and accumulation of glial pathology in the injected M20+/− mice and obtained a finding similar to that in the M83+/− injected mice: the glial pathology closely mirrored the spatiotemporal accumulation of αS pathology in the spinal cord (Fig. 11). Following sciatic nerve injection with mouse αS fibs in M20+/− mice, we observed no astrocytic pathology at 2 months p.i., a significant increase in the lumbar spinal cord by 4 months p.i., and a widespread and robust accumulation throughout the spinal cord by the end stage of disease (Fig. 11). We also observed a higher accumulation of GFAP immunoreactivity at end stage in the lumbar spinal cord than in the cervical spinal cord (Fig. 11b).
FIG 11.

Accumulation of gliosis in M20+/− injected mice. M20+/− mice were injected unilaterally in the sciatic nerve with control inocula (PBS and WT SOD1 fibs) and harvested at 12 months p.i. or with mouse WT αS fibs (mfib αS) and harvested at 2 months or 4 months p.i. or when the mice reached the end stage of disease. (a) Spinal cords were immunostained to visualize astrocytes using an antibody to GFAP and representative images were captured (n = 3 animals per cohort). (b) GFAP immunoreactivity in the cervical and lumbar segments of the spinal cord was quantified for each cohort (n = 3 animals per cohort; mean ± SD). *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001. (c) Spinal cords were also immunostained for microglia using an antibody to cd11b and representative images were captured (n = 3 animals per cohort). Scale bars = 100 μm.
Transmissibility in nontransgenic mice following sciatic nerve injection of mouse WT αS fibs.
Recent studies have indicated that intracerebral injection of mouse WT αS fibs efficiently induces αS pathology in the brains of nontransgenic mice, while human αS fibs appear to be much less efficient (24, 43). Based on these data, we sought to determine whether we could achieve a spread of αS pathology following a single unilateral injection of 4 μg of mouse WT αS fibs in the sciatic nerve of WT mice. The mice were aged to 12 months p.i., with no observable motor impairment or any detectable αS inclusions detected in the DRG or CNS (Fig. 12). Similarly, no pathology was observed in WT mice injected in the sciatic nerve with PBS and aged to 12 months p.i. (Fig. 12b).
FIG 12.
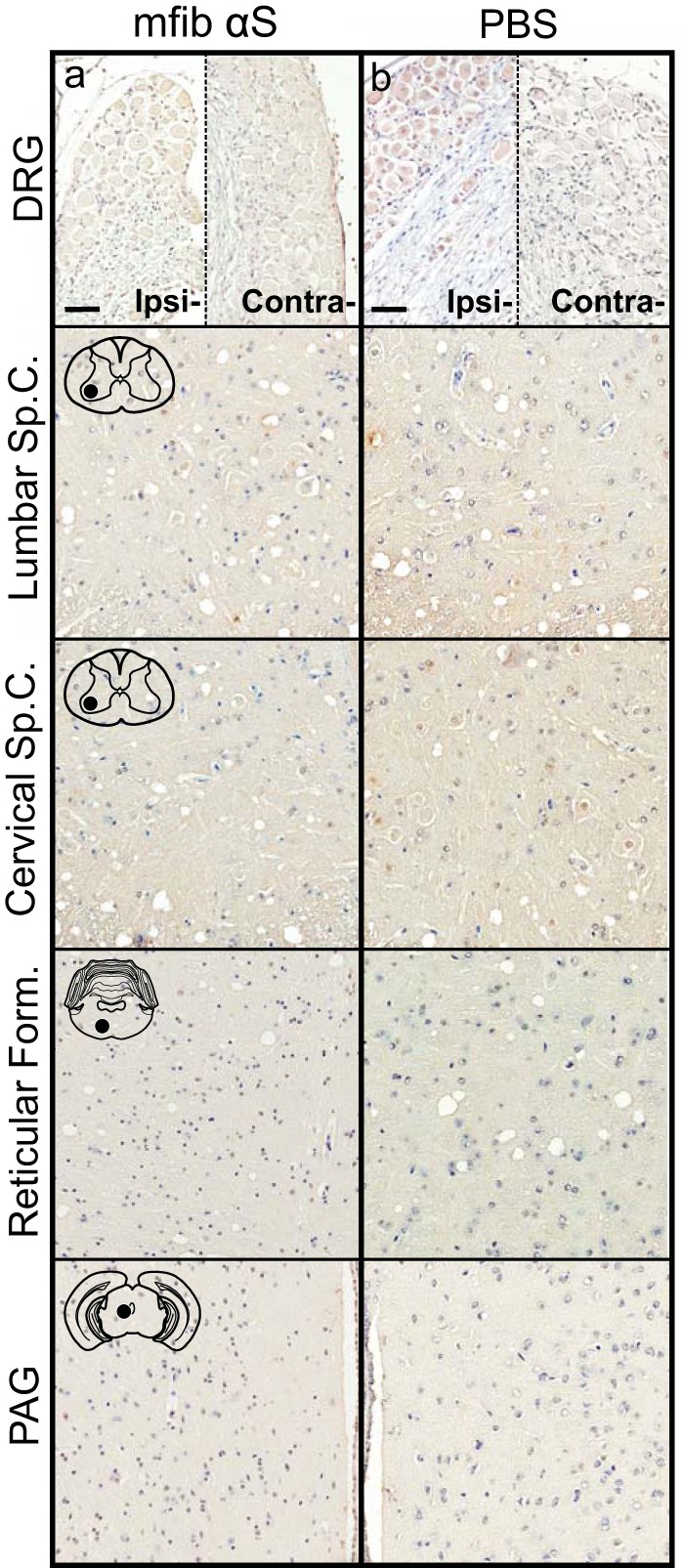
Inability of mouse αS fibs to induce αS pathology in WT mice. WT mice were injected unilaterally in the sciatic nerve with mouse WT αS fibs (mfib αS) (a) or PBS (b) and harvested at 12 months p.i. (n ≥ 6 animals per cohort). Representative images show the lack of αS pathology in both the ipsilateral and contralateral DRG and several CNS regions, depicted by cartoons with black dots representing the specific locations the images were taken. Tissue sections were stained with an antibody to αS phosphorylated at Ser129 (2G12) and counterstained with hematoxylin. Scale bars = 50 μm.
DISCUSSION
The results presented here demonstrate the efficiency of αS induction and its subsequent pathological spread via a single unilateral injection in the sciatic nerve in αS transgenic mouse models. Recombinant mouse WT αS fibs were capable of inducing robust αS pathology, which was closely mirrored by the accumulation of glial pathology, and disease symptoms in both the M83+/− mouse model, overexpressing the A53T mutation of human αS, and the M20+/− mouse model, overexpressing human WT αS. Importantly, when analyzing the spread of αS pathology over the course of disease, we found data to suggest the neuroanatomical spread of αS seeds, or inducing factor in both mouse models to connected populations of neurons. Although this has been reported previously, the presence of αS deposition in areas distant from the site of inoculation, as observed in these studies, could have occurred through widespread dissemination of the αS seeds due to the route of inoculation. This is resolved via the sciatic nerve injection, which forces the inoculum to a defined group of neurons in the lumbar spinal cord, thereby creating an initial point of αS accumulation.
Following sciatic nerve injection, the initial deposition of αS was observed in lamina IX of the lumbar spinal cord, around the ventral motor neurons whose axons give rise to the sciatic nerve. Although a large proportion of axons that comprise the sciatic nerve are myelinated and unmyelinated sensory axons (∼70%) that originate from the DRG, we found very little αS deposition in any of the analyzed DRG around the level of the lumbar spinal cord at any time point p.i. in both the M83+/− and M20+/− mouse models, in contrast to the abundance of pathology observed in the spinal cord. This may suggest a potential preference for transport via myelinated motor axons, which comprise only ∼6% of the sciatic nerve (44), or the inability of αS to accumulate in the DRG neurons. Deposition of αS inclusion pathology in alpha-synucleinopathies has not been well documented for DRG neurons, but in studies in which the tissue was investigated, Braak et al. (45) reported no αS deposition in the DRG, while Sumikura et al. (46) demonstrated Lewy bodies in the soma and processes of DRG neurons in only those cases in which αS deposition was also detected in dorsal horn and dorsal root.
The progression of αS deposition following sciatic nerve injection with mouse WT αS fibs in both the M83+/− and M20+/− mice followed a predictable accumulation of pathology in brain nuclei suggestive of spread via neuroanatomical connections. This spatiotemporal pattern of αS accumulation is identical to both the spread of prions and superoxide dismutase 1 (SOD1) inclusion pathology when induced via the same route of injections (34–36). Following the initial induction of pathology in lamina IX of the lumbar spinal cord, pathology was observed in several brain nuclei whose axons synapse directly on the ventral motor neurons in the spinal cord via descending motor pathways. Pathology was first observed in the reticular formation, red nucleus, and PAG in both the M83+/− and M20+/− mice at 2 months p.i. and 4 months p.i., respectively. An important finding was that the deposition in these nuclei preceded any αS inclusions in the cervical spinal cord, strongly supporting the idea that deposition was induced in these areas through retrograde axonal transport of a seeding factor rather than a cell-to-cell propagative mechanism. Indeed, all three of these nuclei have direct connections to the ventral motor neurons in the lumbar spinal cord: the vestibular nucleus via the vestibulospinal tract, the red nucleus via the rubrospinal tract, and the PAG via neurons that have projections to both cervical and lumbar regions of the spinal cord (47). By the end stage of disease, the pathology in both the M83+/− and M20+/− mice had significantly increased in abundance in all of the aforementioned nuclei and had also progressed rostrally to envelop the entire spinal cord. In addition, αS pathology was detected in several other brain regions, including the lateral vestibular nucleus, thalamus, and hypothalamus. Although layer V of the motor cortex projects axons that synapse directly onto motor neurons in the spinal cord via the corticospinal tract, we never detected abundant αS pathology in either αS mouse model as was seen following sciatic nerve injection with the prion agent in a model of transmissible mink encephalopathy (35, 36). One potential explanation for the lack of pathology in this region could be that it is more distant from the lumbar spinal cord than other brain regions and there was insufficient time for αS pathology to accumulate due to the rapid disease course. Nevertheless, the spatiotemporal nature of this study revealed the induction and accumulation of αS inclusions in locations that support the retrograde axonal transport of an αS inducing factor.
Injection of E46K human αS fibs unilaterally in the sciatic nerve of M83+/− mice was much less efficient than the transmissibility of mouse WT αS fibs in the M83+/− mice. Only 5 of the mice injected with the mutant αS developed motor symptoms at an average age of 7.0 ± 1.3 months p.i., compared with all 14 of the mouse WT αS fib-injected M83+/− mice developing disease at 3.9 ± 0.1 months p.i. (Fig. 1). The E46K mutation has been studied previously and has been shown to be both relatively inefficient at seeding αS inclusion pathology in the M83+/− line of mice and resistant to forming αS inclusions when brain lysates from multiple system atrophy (MSA) patients are incubated in a cell line expressing this particular mutation (33, 48). Although it is tempting to suggest that the distinct characteristics of this particular αS mutant arise due to the distinct structural conformer the E46K point mutation produces, as suggested by numerous in vitro findings (49–54), in our studies these differences may have been due to variations in the mouse versus human αS protein or in differences in the amount of the αS seeding factor in each sample. However, our previous αS seeding studies have revealed that WT human and mouse αS fibs have similar efficiencies in inducing pathology and disease in the M83+/− mouse model (32). In addition, as different prion conformers can be differentiated due to the localization of pathology in the CNS (55–57), we observed no differences in the location of αS deposition in diseased mice following injection with either E46K or WT fibs.
Based on previous reports of the efficiency for mouse αS fibs to induce αS inclusion pathology of the endogenous protein in nontransgenic mice (24, 43), we attempted to test the fibs' effectiveness for seeding mouse αS via sciatic nerve inoculation. Following a 12-month incubation period, no pathology was observed in the ipsilateral DRG, spinal cord, or brain in any of the injected animals. Thus, at equivalent doses of injected seed, induction of αS pathology in nontransgenic mice is much less efficient. It is possible that injection of larger amounts of αS fibs could produce pathology; recent studies demonstrated the induction of endogenous αS pathology by injection of 10 μg and 5 μg directly into the brain (24, 43). Nevertheless, the lack of induction of CNS αS pathology following the direct sciatic nerve injection of mouse αS fibs in nontransgenic mice is consistent with the normally low level of expression of endogenous αS in peripheral nerves and the spinal cord in mice (32, 37, 58). Indeed, the intrinsic low expression of αS in the PNS could constitute another natural barrier to transmission of αS inclusion pathology.
Using human αS transgenic mice and the direct sciatic nerve injection of mouse αS fibs, we have established a robust model of progressive αS inclusion pathology spread along neuroanatomical connections. The sciatic nerve model enables experimental induction of CNS pathology without any direct surgical alteration of the CNS. The direct injection of mouse αS fibs in the sciatic nerve was demonstrated to be more efficient and reliable at inducing αS CNS inclusion pathology in M20 αS transgenic mice than hind-leg muscle injection (32). Further, we observed overt motor symptoms for the first time in M20 mice injected with αS fibs. Our comparison between αS transgenic mice and nontransgenic mice suggests that αS inclusion pathology that has been observed in some human studies might reflect aberrant expression in some individuals that heightens the risk for transmission. In studies to determine whether the spread of αS pathology in CNS can be attenuated by antibody treatments, the sciatic nerve paradigm offers a uniquely useful preclinical model.
MATERIALS AND METHODS
αS transgenic mice and husbandry.
All procedures were performed according to the Guide for the Care and Use of Laboratory Animals (59) and were approved by the University of Florida Institutional Animal Care. M83 transgenic mice expressing human αS with the A53T mutation or M20 transgenic mice expressing WT human αS driven by the mouse prion protein promoter were previously described (37, 41). M83 and M20 mice also both express endogenous mouse αS. M83+/− mice overexpress approximately 3-fold human αS in the brain cortex and 19-fold human αS in the spinal cord relative to endogenous mouse αS (37). M20+/− mice overexpress approximate 6-fold human αS in the brain cortex and 28-fold human αS in the spinal cord relative to endogenous mouse αS (37).
Histology.
Mice were sacrificed by CO2 euthanization and perfused with phosphate-buffered saline (PBS)-heparin. The brain, spinal cord, and DRG were then removed and fixed with 70% ethanol–150 mM NaCl for at least 48 h. As previously described, tissues were dehydrated at room temperature through a series of ethanol solutions, followed by xylene, and then were infiltrated with paraffin at 60°C (60). The tissues were then embedded into paraffin blocks, which were cut into 5- to 6-μm sections. Immunostaining was performed using an established method (60), with the modification that antigen retrieval for the cd11b antibody was performed in a steam bath for 30 min in citrate buffer (target retrieval solution and citrate, pH 6; Agilent, Santa Clara, CA). An avidin-biotin complex (ABC) system (Vectastain ABC Elite kit; Vector Laboratories, Burlingame, CA) was used to enhance detection of the immunocomplexes, which were visualized with the chromogen 3,3′-diaminobenzidine (DAB kit; KPL, Gaithersburg, MD). Sections were counterstained with hematoxylin. All slides were scanned using an Aperio ScanScope CS (×40 magnification; Aperio Technologies Inc., Vista, CA), and images of representative areas of αS pathology were taken using ImageScope software (Aperio Technologies Inc.). Quantitation of GFAP positivity was performed on both cervical and lumbar spinal gray matter sections (n = 3 mice per cohort). For each mouse, multiple spinal sections were analyzed using the positive pixel count algorithm (Aperio) with the same intensity threshold values for all sections. The mean area ± the standard deviation (SD) of the spinal cord that the GFAP immunoreactivity covered was calculated and for statistical comparison, a two-way analysis of variance (ANOVA) with Tukey's multiple comparison's test was performed.
The antibodies used consisted of a mouse monoclonal antibody, termed LS4-2G12 (2G12), that detects pSer129 αS (61), Syn 506, which is a conformational anti-αS mouse monoclonal antibody that preferentially detects αS in pathological inclusions (62, 63), a rabbit polyclonal antibody to p62, which is a general inclusion marker (SQSTM1; Proteintech), rabbit anti-glial fibrillary acidic protein (anti-GFAP; Wako), and rabbit anti-cd11b (Abcam).
Expression and purification of recombinant proteins.
Recombinant full-length mouse αS, human αS with a deletion of amino acids 71 to 82 (Δ71–82), and E46K full-length human αS were expressed and purified to homogeneity as previously described (39, 51). Δ71–82 αS has a deletion in the middle of the hydrophobic region of αS that is required for amyloid formation and therefore lacks the ability to form or seed αS amyloid in vitro and in vivo under physiological conditions (20, 22, 39). Recombinant human SOD1 (hSOD1) proteins were expressed and purified as previously described (64). All protein concentrations were determined using the bicinchoninic acid protein assay (Pierce, Rockford, IL), with bovine serum albumin as a standard.
Fibril preparation of recombinant αS and SOD1 for injection.
Mouse αS and human E46K αS were assembled into fibrils by incubation at 37°C and 5 mg/ml in sterile PBS (Invitrogen) with continuous shaking at 1,050 rpm (Thermomixer R, Eppendorf, Westbury, NY). αS amyloid fibril assembly was monitored as previously described with K114 fluorometry (39, 65). αS fibrils were diluted to 2 mg/ml in sterile PBS and treated by mild water bath sonication for 1 h at room temperature. These fibrils were tested for induction of intracellular amyloid inclusion formation as previously described (21).
SOD1 was fibrillized as previously described (40). Briefly, recombinant SOD1 was fibrillated using 50 μM protein in 20 mM potassium phosphate, pH 7.2, with the addition of 10 mM Tris(2-carboxyethyl)phosphine (TCEP); for those samples used for screening fibril formation, 4 μM thioflavin T was also added. Protein solutions were incubated in a 96-well plate with the addition of a Teflon ball (1/8-in. diameter) at 37°C with constant agitation in a Synergy HT plate reader (BIO-TEK, Winooski, VT). Fluorescence measurements were recorded every 15 min using a 440/30-nm excitation wavelength (λex) filter to excite and a 485/20-nm emission wavelength (λem) filter to detect emission using the Gen5 software (v1.10.8).
Animal inoculations.
Sciatic nerve injections were performed as previously described (34). Prior to the injection, mice were injected with meloxicam (2 mg/kg of body weight) (Norbrook, Overland Park, KS), to relieve pain, and the injection site was shaved and sterilized. The mice were then deeply anesthetized with isoflurane using a precision vaporizer machine with gas scavenging system attached, a small incision was then made in the skin of the hind limb, and the sciatic nerve was exposed at the popliteal fossa. A 33-gauge needle containing the inoculum was inserted in the sciatic nerve and reciprocated 10 times, which has been shown to greatly enhance the efficiency of prion transport to the spinal cord (35). Two microliters was injected under the perineurium of the sciatic nerve and the incision was then closed with stainless steel clips and cleaned. Following surgery, 2 mg/kg of meloxicam was administered at 24 and 48 h postsurgery.
ACKNOWLEDGMENT
This work was supported by a grant from the National Institute of Neurological Disorders and Stroke (NS089622).
REFERENCES
- 1.Waxman EA, Giasson BI. 2009. Molecular mechanisms of alpha-synuclein neurodegeneration. Biochim Biophys Acta 1792:616–624. doi: 10.1016/j.bbadis.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goedert M. 1997. Familial Parkinson's disease. The awakening of alpha-synuclein. Nature 388:232–233. [DOI] [PubMed] [Google Scholar]
- 3.Goedert M. 2001. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci 2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- 4.Cookson MR. 2005. The biochemistry of Parkinson's disease. Annu Rev Biochem 74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- 5.Goedert M, Spillantini MG, del Tredici K, Braak H. 2013. 100 years of Lewy pathology. Nat Rev Neurol 9:13–24. doi: 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- 6.Uchihara T, Giasson BI. 2016. Propagation of alpha-synuclein pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol 131:49–73. doi: 10.1007/s00401-015-1485-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. 1997. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 8.Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schols L, Riess O. 1998. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet 18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 9.Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG. 2004. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 10.Farrer M, Kachergus J, Forno L, Lincoln S, Wang D-S, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW. 2004. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol 55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- 11.Kiely AP, Asi YT, Kara E, Limousin P, Ling H, Lewis P, Proukakis C, Quinn N, Lees AJ, Hardy J, Revesz T, Houlden H, Holton JL. 2013. α-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson's disease and multiple system atrophy? Acta Neuropathol 125:753–769. doi: 10.1007/s00401-013-1096-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. 2003. α-Synuclein locus triplication causes Parkinson's disease. Science 302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 13.Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, Houlden H, Schapira AH. 2013. A novel α-synuclein missense mutation in Parkinson disease. Neurology 80:1062–1064. doi: 10.1212/WNL.0b013e31828727ba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dawson T, Mandir A, Lee M. 2002. Animal models of PD: pieces of the same puzzle? Neuron 35:219–222. doi: 10.1016/S0896-6273(02)00780-8. [DOI] [PubMed] [Google Scholar]
- 15.Woerman AL, Stöhr J, Aoyagi A, Rampersaud R, Krejciova Z, Watts JC, Ohyama T, Patel S, Widjaja K, Oehler A, Sanders DW, Diamond MI, Seeley WW, Middleton LT, Gentleman SM, Mordes DA, Südhof TC, Giles K, Prusiner SB. 2015. Propagation of prions causing synucleinopathies in cultured cells. Proc Natl Acad Sci U S A 112:E4949–E4958. doi: 10.1073/pnas.1513426112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prusiner SB, Woerman AL, Mordes DA, Watts JC, Rampersaud R, Berry DB, Patel S, Oehler A, Lowe JK, Kravitz SN, Geschwind DH, Glidden DV, Halliday GM, Middleton LT, Gentleman SM, Grinberg LT, Giles K. 2015. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A 112:E5308–E5317. doi: 10.1073/pnas.1514475112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sorrentino ZA, Brooks MMT, Hudson V, Rutherford NJ, Golde TE, Giasson BI, Chakrabarty P. 2017. Intrastriatal injection of α-synuclein can lead to widespread synucleinopathy independent of neuroanatomic connectivity. Mol Neurodegener 12:40. doi: 10.1186/s13024-017-0182-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Braak H, del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. 2003. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211. doi: 10.1016/S0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 19.Braak H, Rüb U, Gai WP, Del Tredici K. 2003. Idiopathic Parkinson's disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm 110:517–536. doi: 10.1007/s00702-002-0808-2. [DOI] [PubMed] [Google Scholar]
- 20.Sacino AN, Thomas MA, Ceballos-Diaz C, Cruz PE, Rosario AM, Lewis J, Giasson BI, Golde TE. 2013. Conformational templating of α-synuclein aggregates in neuronal-glial cultures. Mol Neurodegener 8:17. doi: 10.1186/1750-1326-8-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waxman EA, Giasson BI. 2010. A novel, high-efficiency cellular model of fibrillar alpha-synuclein inclusions and the examination of mutations that inhibit amyloid formation. J Neurochem 113:374–388. doi: 10.1111/j.1471-4159.2010.06592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luk KC, Song C, O'Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM-Y. 2009. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A 106:20051–20056. doi: 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM-Y. 2012. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J Exp Med 209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM-Y. 2012. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watts JC, Giles K, Oehler A, Middleton L, Dexter DT, Gentleman SM, Dearmond SJ, Prusiner SB. 2013. Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci U S A 110:19555–19560. doi: 10.1073/pnas.1318268110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mougenot A-L, Nicot S, Bencsik A, Morignat E, Verchere J, Lakhdar L, Legastelois S, Baron T. 2012. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol Aging 33:2225–2228. doi: 10.1016/j.neurobiolaging.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 27.Recasens A, Dehay B, Bové J, Carballo-Carbajal I, Dovero S, Pérez-Villalba A, Fernagut P-O, Blesa J, Parent A, Perier C, Fariñas I, Obeso JA, Bezard E, Vila M. 2014. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 75:351–362. doi: 10.1002/ana.24066. [DOI] [PubMed] [Google Scholar]
- 28.Breid S, Bernis ME, Babila JT, Garca MC, Wille H, Tamgüney G. 2016. Neuroinvasion of α-synuclein prionoids after intraperitoneal and intraglossal inoculation. J Virol 90:9182–9193. doi: 10.1128/JVI.01399-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ayers J, Brooks MM, Rutherford NJ, Howard JK, Sorrentino ZA, Riffe CJ, Giasson BI. 2017. Robust central nervous system pathology in transgenic mice following peripheral injection of α-synuclein fibrils. J Virol 91:e02095-16. doi: 10.1128/JVI.02095-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Braak H, Bohl JR, Müller CM, Rüb U, de Vos RAI, del Tredici K. 2006. Stanley Fahn Lecture 2005: the staging procedure for the inclusion body pathology associated with sporadic Parkinson's disease reconsidered. Mov Disord 21:2042–2051. doi: 10.1002/mds.21065. [DOI] [PubMed] [Google Scholar]
- 31.Ulusoy A, Phillips RJ, Helwig M, Klinkenberg M, Powley TL, Di Monte DA. 2017. Brain-to-stomach transfer of α-synuclein via vagal preganglionic projections. Acta Neuropathol 133:381–393. doi: 10.1007/s00401-016-1661-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sacino AN, Brooks M, Thomas MA, McKinney AB, Lee S, Regenhardt RW, McGarvey NH, Ayers J, Notterpek L, Borchelt DR, Golde TE, Giasson BI. 2014. Intramuscular injection of α-synuclein induces CNS α-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc Natl Acad Sci U S A 111:10732–10737. doi: 10.1073/pnas.1321785111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rutherford NJ, Dhillon J-KS, Riffe CJ, Howard JK, Brooks M, Giasson BI. 2017. Comparison of the in vivo induction and transmission of α-synuclein pathology by mutant α-synuclein fibril seeds in transgenic mice. Hum Mol Genet 26:4906–4915. doi: 10.1093/hmg/ddx371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ayers J, Fromholt SE, O'Neal VM, Diamond JH, Borchelt DR. 2016. Prion-like propagation of mutant SOD1 misfolding and motor neuron disease spread along neuroanatomical pathways. Acta Neuropathol 131:103–114. doi: 10.1007/s00401-015-1514-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bartz JC, Kincaid AE, Bessen RA. 2002. Retrograde transport of transmissible mink encephalopathy within descending motor tracts. J Virol 76:5759–5768. doi: 10.1128/JVI.76.11.5759-5768.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ayers J, Kincaid AE, Bartz JC. 2009. Prion strain targeting independent of strain-specific neuronal tropism. J Virol 83:81–87. doi: 10.1128/JVI.01745-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM-Y. 2002. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 34:521–533. doi: 10.1016/S0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- 38.Giasson BI, Murray IV, Trojanowski JQ, Lee VM. 2001. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem 276:2380–2386. doi: 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
- 39.Waxman EA, Mazzulli JR, Giasson BI. 2009. Characterization of hydrophobic residue requirements for alpha-synuclein fibrillization. Biochemistry 48:9427–9436. doi: 10.1021/bi900539p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ayers J, Diamond J, Sari A, Fromholt S, Galaleldeen A, Ostrow LW, Glass JD, Hart PJ, Borchelt DR. 2016. Distinct conformers of transmissible misfolded SOD1 distinguish human SOD1-FALS from other forms of familial and sporadic ALS. Acta Neuropathol 132:827–840. doi: 10.1007/s00401-016-1623-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emmer KL, Waxman EA, Covy JP, Giasson BI. 2011. E46K human alpha-synuclein transgenic mice develop Lewy-like and tau pathology associated with age-dependent, detrimental motor impairment. J Biol Chem 286:35104–35118. doi: 10.1074/jbc.M111.247965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sacino AN, Brooks M, McKinney AB, Thomas MA, Shaw G, Golde TE, Giasson BI. 2014. Brain injection of α-synuclein induces multiple proteinopathies, gliosis, and a neuronal injury marker. J Neurosci 34:12368–12378. doi: 10.1523/JNEUROSCI.2102-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DMA, Hasegawa M. 2013. Prion-like spreading of pathological α-synuclein in brain. Brain 136:1128–1138. doi: 10.1093/brain/awt037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmalbruch H. 1986. Fiber composition of the rat sciatic nerve. Anat Rec 215:71–81. doi: 10.1002/ar.1092150111. [DOI] [PubMed] [Google Scholar]
- 45.Braak H, Sastre M, Bohl JRE, de Vos RAI, del Tredici K. 2007. Parkinson's disease: lesions in dorsal horn layer I, involvement of parasympathetic and sympathetic pre- and postganglionic neurons. Acta Neuropathol 113:421–429. doi: 10.1007/s00401-007-0193-x. [DOI] [PubMed] [Google Scholar]
- 46.Sumikura H, Takao M, Hatsuta H, Ito S, Nakano Y, Uchino A, Nogami A, Saito Y, Mochizuki H, Murayama S. 2015. Distribution of α-synuclein in the spinal cord and dorsal root ganglia in an autopsy cohort of elderly persons. Acta Neuropathol Commun 3:57. doi: 10.1186/s40478-015-0236-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mantyh PW, Peschanski M. 1982. Spinal projections from the periaqueductal grey and dorsal raphe in the rat, cat and monkey. Neuroscience 7:2769–2776. doi: 10.1016/0306-4522(82)90099-9. [DOI] [PubMed] [Google Scholar]
- 48.Woerman AL, Kazmi SA, Patel S, Aoyagi A, Oehler A, Widjaja K, Mordes DA, Olson SH, Prusiner SB. 2018. Familial Parkinson's point mutation abolishes multiple system atrophy prion replication. Proc Natl Acad Sci U S A 115:409–414. doi: 10.1073/pnas.1719369115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi W, Zibaee S, Jakes R, Serpell LC, Davletov B, Crowther RA, Goedert M. 2004. Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS Lett 576:363–368. [DOI] [PubMed] [Google Scholar]
- 50.Fredenburg RA, Rospigliosi C, Meray RK, Kessler JC, Lashuel HA, Eliezer D, Lansbury PT. 2007. The impact of the E46K mutation on the properties of alpha-synuclein in its monomeric and oligomeric states. Biochemistry 46:7107–7118. doi: 10.1021/bi7000246. [DOI] [PubMed] [Google Scholar]
- 51.Greenbaum EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, Axelsen PH, Giasson BI. 2005. The E46K mutation in alpha-synuclein increases amyloid fibril formation. J Biol Chem 280:7800–7807. doi: 10.1074/jbc.M411638200. [DOI] [PubMed] [Google Scholar]
- 52.Ono K, Ikeda T, Takasaki J-I, Yamada M. 2011. Familial Parkinson disease mutations influence α-synuclein assembly. Neurobiol Dis 43:715–724. doi: 10.1016/j.nbd.2011.05.025. [DOI] [PubMed] [Google Scholar]
- 53.Rospigliosi CC, McClendon S, Schmid AW, Ramlall TF, Barré P, Lashuel HA, Eliezer D. 2009. E46K Parkinson's-linked mutation enhances C-terminal-to-N-terminal contacts in alpha-synuclein. J Mol Biol 388:1022–1032. doi: 10.1016/j.jmb.2009.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brucale M, Sandal M, Di Maio S, Rampioni A, Tessari I, Tosatto L, Bisaglia M, Bubacco L, Samorì B. 2009. Pathogenic mutations shift the equilibria of alpha-synuclein single molecules towards structured conformers. Chembiochem 10:176–183. doi: 10.1002/cbic.200800581. [DOI] [PubMed] [Google Scholar]
- 55.Bessen RA, Marsh RF. 1994. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 68:7859–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fraser H, Dickinson AG. 1968. The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol 78:301–311. doi: 10.1016/0021-9975(68)90006-6. [DOI] [PubMed] [Google Scholar]
- 57.Hecker R, Taraboulos A, Scott M, Pan KM, Yang SL, Torchia M, Jendroska K, Dearmond SJ, Prusiner SB. 1992. Replication of distinct scrapie prion isolates is region specific in brains of transgenic mice and hamsters. Genes Dev 6:1213–1228. doi: 10.1101/gad.6.7.1213. [DOI] [PubMed] [Google Scholar]
- 58.Giasson BI, Duda JE, Forman MS, Lee VM, Trojanowski JQ. 2001. Prominent perikaryal expression of alpha- and beta-synuclein in neurons of dorsal root ganglion and in medullary neurons. Exp Neurol 172:354–362. doi: 10.1006/exnr.2001.7805. [DOI] [PubMed] [Google Scholar]
- 59.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 60.Duda JE, Giasson BI, Gur TL, Montine TJ, Robertson D, Biaggioni I, Hurtig HI, Stern MB, Gollomp SM, Grossman M, Lee VM, Trojanowski JQ. 2000. Immunohistochemical and biochemical studies demonstrate a distinct profile of alpha-synuclein permutations in multiple system atrophy. J Neuropathol Exp Neurol 59:830–841. doi: 10.1093/jnen/59.9.830. [DOI] [PubMed] [Google Scholar]
- 61.Rutherford NJ, Brooks M, Giasson BI. 2016. Novel antibodies to phosphorylated α-synuclein serine 129 and NFL serine 473 demonstrate the close molecular homology of these epitopes. Acta Neuropathol Commun 4:80. doi: 10.1186/s40478-016-0357-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Duda JE, Giasson BI, Mabon ME, Lee VM-Y, Trojanowski JQ. 2002. Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann Neurol 52:205–210. doi: 10.1002/ana.10279. [DOI] [PubMed] [Google Scholar]
- 63.Waxman EA, Duda JE, Giasson BI. 2008. Characterization of antibodies that selectively detect alpha-synuclein in pathological inclusions. Acta Neuropathol 116:37–46. doi: 10.1007/s00401-008-0375-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Seetharaman SV, Taylor AB, Holloway S, Hart PJ. 2010. Structures of mouse SOD1 and human/mouse SOD1 chimeras. Arch Biochem Biophys 503:183–190. doi: 10.1016/j.abb.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Crystal AS, Giasson BI, Crowe A, Kung M-P, Zhuang Z-P, Trojanowski JQ, Lee VM-Y. 2003. A comparison of amyloid fibrillogenesis using the novel fluorescent compound K114. J Neurochem 86:1359–1368. doi: 10.1046/j.1471-4159.2003.01949.x. [DOI] [PubMed] [Google Scholar]