

Abstract
Background
Colorectal cancer is one of the leading causes of death in China, and the development of effective drugs is urgently needed. Here, we report on Paeoniflorin (PF), a product isolated from the roots of the peony plant, as a possible candidate because of its anti-tumor effects on epithelial-to-mesenchymal transition (EMT) of PF in human colorectal cancer (CRC).
Material/Methods
Cell proliferation, wound healing, and Transwell assays were used to analyze the effects of PF on in vitro cell migration and invasion of HCT116 and SW480, 2 colorectal cancer cell lines. The tumor xenograft model was used to verify the anti-metastasis effects of PF in vivo. The RNA and protein levels of epithelia-cadherin (E-cadherin), Vimentin, and histone deacetylase2 (HDAC2) were measured by qPCR and Western blot analysis to explore the mechanism involved.
Results
Our results showed that PF inhibited colorectal cancer cell migration and invasion and suppressed the metastatic potential of the cancer cells in vivo. Moreover, PF significantly decreased the expression of HDAC2 and Vimentin, while increasing the expression of E-cadherin.
Conclusions
These results suggest that PF inhibits colorectal cancer cell migration and invasion ability and reverses the EMT process, through inhibiting the expression of HDAC2, and then affects the expression level of E-cadherin and Vimentin at the cell level. Our results were also verified in the tumor xenograft model. This indicates that PF may be a candidate for colorectal cancer treatment.
MeSH Keywords: Colorectal Neoplasms; Epithelial-Mesenchymal Transition; Medicine, Chinese Traditional; Neoplasm Metastasis
Background
Colorectal cancer (CRC) is one of the most common malignant tumors. Its morbidity and mortality are 5th among all malignant tumors, and show an increasing trend year by year [1]. About 60% of cancer deaths in CRC patients are due to liver metastasis [2]. Therefore, it is very important to carry out in-depth research and improve the treatment of colorectal liver metastases (CLM).
In the process of tumor cell growth and metastasis, the epithelial cells undergo a phenotypic switch to form mesenchymal cells. The change in cell type results in the loss of polarity and the loss of tight intracellular adhesions maintained by epithelial cells via adherences junctions. Tumor cells gain increasing migration and invasion abilities, resistance to apoptosis, and the ability to degrade the extracellular matrix, so the cancer cells turn into exfoliated cells and migrate. This transformation is known as the epithelial-to-mesenchymal transition (EMT). Studies have shown that the aberrant activation of EMT in adult epithelia can promote tumor metastasis by repressing cell adhesion molecules, including epithelial (E)-cadherin. Reduced intracellular adhesion may allow tumor cells to disseminate and spread throughout the body. This change is EMT [3]. Research has confirmed that EMT plays an important role in bladder cancer, primary liver cancer, CRC, melanoma, and infiltration and metastasis in other tumor cells [4–6]. In recent years, many kinds of Chinese medicine been proved to have good anti-tumor effects. With the increase of tumor incidence, the anti-tumor effect of traditional Chinese medicine is getting more and more attention. Traditional Chinese medicines such as ginkgolide, resveratrol, salvia miltiorrhizae, and total paeony glycoside can invigorate the circulation of blood and have been proven to have anti-inflammatory, anti-tumor, and other effects.
Paeoniflorin (PF), the main active ingredient of the total paeony glycosides, can activate blood circulation and has anti-inflammation and anti-tumor effects. It was proved that PF has an inhibitory effect on cell growth, invasion, and metastasis of gastric, hepatocellular, pancreatic, and breast cancers [7–10]. However, there are few studies on the effect of PF on CRC metastasis. In this study, we explored the anti-tumor effects of PF on EMT in human CRC cells by in vitro and in vivo experiments.
Material and Methods
Cell culture and reagents
PF (purity >98%) was purchased from Kailai Bio-Engineering Co. (Xi’an, China). The CRC cell lines HCT116 and SW480 were purchased from the Chinese Academy of Sciences Cell Bank (Shanghai, China). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Hyclone, Utah, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, MA, USA) at 37ºC and 5% CO2 in a humidified incubator. PF (Kailai Biol-Tech, Xi’an, China) was dissolved in Dulbecco’s modified Eagle’s medium. The cell counting kit-8 (CCK8), Bicinchoninic Acid (BCA) protein assay kit, antibody, and crystal violet were purchased from Beyotime Biotech (Nanjing, China). Matrigel glue was purchased from BD Biosciences (N023J, USA), and other reagents were purchased from Hyclone Biotech (Utah, USA). The primers for the genes of interest (E-cadherin, Vimentin, HDAC2) were synthesized by Sangon Biotech Co. Ltd (Shanghai, China), as follows: 5′-ACAACTTTGGTATCGTGGAAGG-3′ (forward) and 5′-GCCATCACGCCACAGTTTC-3′ (reverse) for GAPDH; 5′-CGAGAGCTACACGTTCACGG-3′ (forward) and 5′-GGGTGTCGAGGGAAAAATAGG-3′ (reverse) for E-cadherin; 5′-AGTCCACTGAGTACCGGAGAC-3′ (forward) and 5′-CATTTCACGCATCTGGCGTTC-3′ (reverse) for Vimentin; 5′-ATGGCGTACAGTCAAGGAGG-3′ (forward) and 5′-TGCGGATTCTATGAGGCTTCA (reverse) for HDAC2. The primary antibody against β-actin, E-cadherin, Vimentin, and HDAC2 were purchased from Cell Signaling Technology (MA, USA).
Cell proliferation assays of PF
Cell proliferation was determined by Cell Counting Kit-8 assay. CRC cells of HCT116 and SW480 were seeded in a 96-well plate at a density of 3×103/well in a humidified incubator with 5% CO2 and 95% air at 37ºC. Then, cells were treated with 0, 2.5, 5.0, 10.0, 20.0, and 40.0 mM PF for 48 h. After incubation with 10 μl CCK8 reagent and 90 μl DMEM each well for 2 h at 37ºC in the dark, the optical density (OD) of each well at 450/620 nm was measured. Results are presented as means ± standard deviation (SD) of 3 independent experiments.
Wound healing assay
Methods of colorectal cancer cell lines HCT116 and SW480 culture are described above. When the degree of fusion was up to 80% or 90%, HCT116 and SW480 cells were resuspended and inoculated into 6-pore plates. The drawn monolayer was scraped at a constant width when the cells adhered to the wall. After that, the cells were rinsed slowly with PBS and exposed to the indicated concentrations of PF (0, 2.5, 5.0, and 10.0 mM). We observed the distances of monolayer scraped and photographed it at 0, 12, 24, and 48 h after treatment with PF. Cell motility rate was calculated as (distance at 12, 24, or 48 h – distance at 0 h)/distance at 0 h. Results are represented as means ±SD of 3 independent experiments.
Transwell-migration/invasion assay
The Transwell permeable support system containing 24-well Transwell (unit 0.8-μm pore size polyvinylidene fluoride) filters were used to analyze the migration ability of HCT116 and SW480 cells. HCT116 and SW480 cells were pretreated with 2.5 mM, 5.0 mM, and 10.0 mM PF for 48 h and then a total of 5×104 cells was seeded into the upper insert in 100 μl of serum-free DMEM. The lower chamber was filled with 600 μl DMEM containing 10% FBS as a chemoattractant. After culturing for 48 h, the non-invading cells were removed from the upper surface of the membrane. The migrated cells on the lower surface were fixed with 4% formaldehyde for 15 min at room temperature then stained with crystal violet for 25 min, and their numbers in 5 fields of each triplicate filter were counted. The cell invasion assays were performed in a similar manner except that 1.0×105 cells were seeded into the upper inserts with serum-free DMEM supplemented with Matrigel. Results are presented as means ±SD of 3 independent experiments.
Quantitative real-time polymerase chain reaction (qPCR) assay
After centrifugation, the HCT116 and SW480 cells were removed and seeded onto 12-well plates, and treated with 0, 2.5, 5.0, and 10.0 mM PF for 48 h. We collected cell RNA, and before reverse transcription, we discarded the genomic DNA from the RNA, and then used 2 μg of total RNA for first-stand DNA synthesis. For mRNA detection, 500 ng of total RNA was used for complementary DNA synthesis with a PrimeScript RT reagent kit. Real-time PCR was performed with SYBR Premix Ex Taq II (Tli RNaseH Plus). The mRNA level of targets was calculated using the −ΔCt method and expressed as 2(−ΔCt) values based on threshold cycle (Ct) values, which were obtained by normalizing to the endogenous reference and relative to a control (GAPDH). Results are presented as means ±SD of 3 independent experiments.
Western blot analysis
HCT116 and SW480 cells (1×106 cells/well) were seeded into 6-well plates, cultivated as previously described, and treated with 0, 2.5, 5.0, and 10.0 mM PF for 48 h. In accordance with the manufacturer’s protocol, cells were lysed using RIPA and centrifuged at 15 000×g for 10 min at 4ºC. Then, protein concentrations were determined by BCA assay. Twenty μg/lane of samples were electrophoresed by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) on 6–15% gels and transferred onto PVDF membranes. After blocking with 5% nonfat dry milk in TBST buffer, the membranes were incubated with anti-E-cadherin, anti-Vimentin, and anti-HDAC2 overnight at 4°C. After 3 washes with TBST, the membranes were incubated with HRP-conjugated secondary antibody for 1 h at room temperature. Protein bands were developed using the ECL kit and imaged by the Tanon imaging system (Shanghai, China). β-actin was used as a control for normalization.
Tumor xenograft study
We established a CRC liver metastasis model to explore tumor invasion and metastasis in vivo. A specific pathogen-free (SPF) environment was used to breed the BALB/c nude mice (male, average weight 18–20 g). HCT116 cells (5×105) was mixed under the spleen envelop of each nude mouse and all nude mice were assigned randomly to 2 groups (n=3 mice per group). The negative control group was gavaged with saline and the experimental group was gavaged with PF (1 g/kg/d) for 6 days/week for 4 weeks. The nude mouse body weights were monitored weekly during the entire experiment. At the end of the experiment, all mice were sacrificed. The livers were collected whole, then we assessed liver metastases and stored them in formalin for future analysis with hematoxylin-eosin (HE) staining and immunohistochemistry (ICH) assays. All procedures were approved by the Animal Ethics Committee of Zhongshan Hospital; Fudan University, China (Permit number: 2017-049).
Statistical analysis
The data were analyzed with GraphPad Prism 6 software and are presented as means ± standard deviation (SD). Analysis of variance was performed to compare multiple data. Probability value (P) <0.05 was considered statistically significant.
Results
Effect of PF on CRC cell proliferation
CCK8 was carried out to investigate the cell proliferation of PF on CRC. As shown in Figure 1, PF inhibited cell proliferation of HCT116 and SW480 cells in a dose-dependent manner. The viabilities of HCT116 and SW480 cells were mildly suppressed at doses of 2.5, 5.0, and 10.0 mM PF and were strongly suppressed at doses of 20.0 and 40.0 mM PF as compared with non-treated cells at 48 h (P<0.05). The 50% inhibition concentrations (IC50) of PF on HCT116 and SW480 cells were 13.34 and 12.60 mM, respectively. The findings indicate that PF strongly suppresses CRC cell proliferation.
Figure 1.

Effect of paeoniflorin on HC116 and SW480 proliferation at 48 h. HCT116 (A) and SW480 cells (B) were treated with increasing concentrations of PF (2.5–40 mM) for 48 h. Data are expressed as means and SD of 3 different experiments. ** p<0.01 (one-way ANOVA).
Effect of PF on CRC cell migration/invasion
We used the wound healing and Transwell-migration assays to examine the effects of PF on migration in the CRC cell lines. All wound healing images showed that PF suppressed the cell migration ability and this effect was obviously concentration- and time-dependent. Results showed that the area of wound healing for HCT116 and SW480 cells were approximately 25.9±3.6% and 24.3±0.6%, respectively, at 10 mM at 48 h. The cell motility for HCT116 and SW480 cells was reduced by PF compared with the control group (Figure 2). The Transwell migration assays revealed that PF effectively inhibited the numbers of HCT116 and SW480 cells in the lower chamber (Figure 3). We used the Transwell-invasion assay to determine the invasive activity of CRC cells. As illustrated in Figure 4, after HCT116 and SW480 cells were treated with 2.5–10.0 mM PF for 48 h, PF inhibited the invasive potential of CRC cells in a dose-dependent manner (Figure 4).
Figure 2.

Effect of paeoniflorin on wound healing ability of HCT116 and SW480. HCT116 (A, C) and SW480 (B, D) cells were treated with increasing concentrations of PF (2.5–10 mM) for 48 h. Data are expressed as means and SD of 3 different experiments. * P<0.05; ** P<0.01. (One-Way ANOVA).
Figure 3.

Effect of paeoniflorin on HCT116 and SW480 migration. Cell migration of HCT116 (A) and SW480 (B) cells were evaluated by Transwell-migration assay. With the increase of paeoniflorin concentration, the invasion ability of HCT116 (C) and SW480 (D) cells was decreased (n=3). * P<0.05; ** P<0.01.
Figure 4.

Effect of paeoniflorin on HCT116 and SW480 cell invasion. Invasion of HCT116 (A) and SW480 (B) cells was evaluated by Transwell invasion assay. With the increase of paeoniflorin concentration, the invasive ability of HCT116 (C) and SW480 (D) cells was decreased (n=3). * P<0.05; ** P<0.01.
Effect of PF on the expression of E-cadherin, Vimentin, and HDAC2 at RNA and protein levels
The role of EMT in the progression of tumor cell migration and invasion is very important. To further elucidate the underlying mechanism of the effect of PF on CRC, several putative EMT-related markers, such as E-cadherin and Vimentin, were evaluated by real-time PCR. After treatment with 0, 2.5, 5.0, or 10.0 mM PF for 48 h, the results revealed that the expression of mRNA of E-cadherin was significantly increased and Vimentin was decreased in HCT116 and SW480 cells (Figure 5), while the expressions of Vimentin and HDAC2 were downregulated in a dose-dependent manner. These protein changes of these genes were further confirmed by Western blot analysis (Figure 6). These observations indicate that PF can suppress the EMT of CRC.
Figure 5.
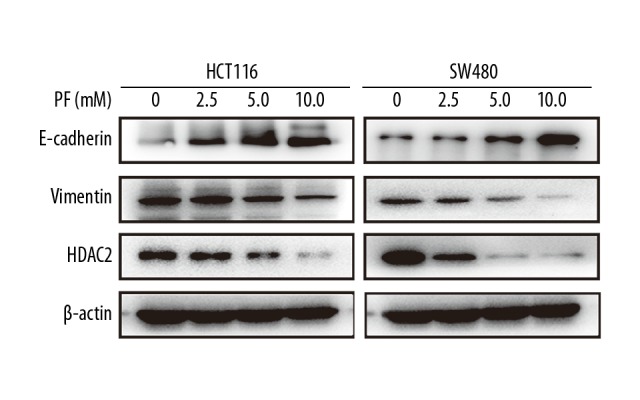
Effect of paeoniflorin on E-cadherin, Vimentin, and HDAC2 expression at the RNA level in HCT116 and SW480 cells. PCR was used to detect E-cadherin, Vimentin, and HDAC2 expression of RNA levels in HCT116 and SW480 cells after paeoniflorin treatment. With the increase of paeoniflorin concentration, E-cadherin was upregulated, while Vimentin and HDAC2 were downregulated in a dose-dependent manner (n=3). * P<0.05; ** P<0.01.
Figure 6.

Effect of paeoniflorin on E-cadherin, Vimentin, and HDAC2 expression in HCT116 (A) and SW480 (B) cells. The protein expression was analyzed by Western blot after paeoniflorin treatment. The results are consistent with PCR results.
Effect of PF on CRC HCT116 tumor xenograft metastases
PF suppresses the invasion and migration of cultured CRC cells, as indicated by the preceding results. Thus, the liver metastasis model of CRC was used to explore whether PF affects CRC metastasis in vivo. On day 30 after transplantation, PF-treated xenograft tumors were significantly smaller and less numerous compared with saline-treated xenograft tumors (Figure 7A, 7B). As shown in HE of tumor issue, PF significantly reduced hepatic metastasis compared to the control group (Figure 7C). Immunohistochemistry analysis of target molecules of EMT was performed on tumor tissue. The results revealed that the E-cadherin was increased in tumors of nude mice gavaged with PF, while the Vimentin and HDAC2 levels were decreased (Figure 7D). Collectively, our results show that PF negatively regulates tumor metastasis of CRC.
Figure 7.

Effect of paeoniflorin on colorectal cancer metastasis in vivo. When gavaged with paeoniflorin, the experimental group mice had significantly fewer and smaller liver metastatic tumors (A–C). The expression levels of E-cadherin, Vimentin, and HDAC2 in liver tissues were measured by immunohistochemical analysis (D), and the results are in accord with the in vitro test results.
Discussion
The traditional Chinese medicine PF has also been shown to inhibit proliferation and metastasis and induces cell apoptosis in gastric cancer, pancreatic cancer, hepatocellular carcinoma, and breast cancer through various mechanisms, such as the modulation of the PI3K/Akt or STAT3 signaling pathway, Bcl-2 and Bax expression, ERK signaling pathway, or the Notch-1 expression [7–10]. Few studies have described the effect of PF on CRC. Our results for the first time indicate that PF has different effects on CRC. At a high dose (40 mM), which is greater than IC50 (13.34 mM of HCT116 and 12.60 mM of SW480), the cell proliferation was inhibited. At a low dose (<10 mM), cell migration and invasion were inhibited and this effect may be related to EMT.
Research has shown that the EMT initiates cancer cell dissemination, inducing non-cancer stem cells to enter into a cancer stem cell-like state [11], and promotes metastatic seeding accompanying the downregulation of E-cadherin and upregulation of Vimentin [12,13]. Gene mutation and transcriptional and post-transcriptional modification play an important part in the process of EMT [12]. The absence of E-cadherin is the basis of EMT occurrence [14].
Many studies have revealed that histone acetylation plays an important role in the transcriptional modifications of EMT-related markers [15]. The acetylation involves histone acetyltransferase (HAT) and histone deacetylase (HDAC). In general, the acetylation level of histones in the nucleus is in a state of dynamic equilibrium. Histone was acetylated by HAT and prompted histone to dissociate from DNA, which makes the nucleosome structure relax. As a result, it can increase the DNA binding sites and regulate the specific binding of transcription factors and the cofactor, as well as activating gene transcription. However, HDAC can reduce the acetylation level of histone and promotes its binding on the charged DNA, thus inhibiting the transcription of target genes [16]. Previous studies revealed that HDAC2, which is one of the most important HDACs, is overexpressed in many tumors and is closely related to tumor metastasis [17–22]. HDAC2 can reduce E-cadherin expression with EZH2, ZEB1, and SNAIL [23,24]. Limiting HDAC2 can suppress E-cadherin transcription in the process of EMT and then inhibit the invasion and metastasis of tumor cells [20,21,25]. Our study verifies this conclusion.
Conclusions
We found that with the increased PF concentration and longer treatment period, the migration and invasion abilities of both CRC cell lines were remarkably inhibited. Moreover, the therapeutic effect of PF was confirmed once more in the HCT116 xenograft mouse model. All the above evidence clearly shows that PF has potential in treatment of CRC, probably by targeting HDAC2 and negative modulation of the EMT in CRC, although the detailed mechanisms and its clinical implication need further study.
Footnotes
Source of support: This project was jointly supported by the Three-Year Action Plan for the Development of Traditional Chinese Medicine (TCM) in Shanghai (grant number ZY3-CCCX-3-3036), the TCM Superiority Disease Species Breeding Program (grant number Zybz-2017027), the Shanghai Science and Technology Commission Project (grant number 2014JP009A), and the National Science Foundation of China (grant number 81472675)
References
- 1.Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. Cancer J Clin. 2016;66:115–32. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 2.Devetzi M, Kosmidou V, Vlassi M, et al. Death receptor 5 (DR5) and a 5-gene apoptotic biomarker panel with significant differential diagnostic potential in colorectal cancer. Sci Rep. 2016;6 doi: 10.1038/srep36532. 36532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Song J. EMT or apoptosis: A decision for TGF-beta. Cell Res. 2007;17:289–90. doi: 10.1038/cr.2007.25. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Wu Z, Hu L. Epithelial-mesenchymal transition phenotype, metformin, and survival for colorectal cancer patients with diabetes mellitus II. Gastroenterol Res Pract. 2017;2017 doi: 10.1155/2017/2520581. 2520581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu W, Wang NR, Wang HF, et al. Analysis of epithelial-mesenchymal transition markers in the histogenesis of hepatic progenitor cell in HBV-related liver diseases. Diagn Pathol. 2016;11:136. doi: 10.1186/s13000-016-0587-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Franzen CA, Blackwell RH, Todorovic V, et al. Urothelial cells undergo epithelial-to-mesenchymal transition after exposure to muscle invasive bladder cancer exosomes. Oncogenesis. 2015;4:e163. doi: 10.1038/oncsis.2015.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang N, Cui H, Han F, et al. Paeoniflorin inhibits human pancreatic cancer cell apoptosis via suppression of MMP-9 and ERK signaling. Oncol Lett. 2016;12:1471–76. doi: 10.3892/ol.2016.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Q, Yuan Y, Cui J, et al. Paeoniflorin inhibits proliferation and invasion of breast cancer cells through suppressing Notch-1 signaling pathway. Biomed Pharmacother. 2016;78:197–203. doi: 10.1016/j.biopha.2016.01.019. [DOI] [PubMed] [Google Scholar]
- 9.Zheng YB, Xiao GC, Tong SL, et al. Paeoniflorin inhibits human gastric carcinoma cell proliferation through up-regulation of microRNA-124 and suppression of PI3K/Akt and STAT3 signaling. World J Gastroenterol. 2015;21:7197–207. doi: 10.3748/wjg.v21.i23.7197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu JT, He W, Song SS, Wei W. Paeoniflorin inhibited the tumor invasion and metastasis in human hepatocellular carcinoma cells. Bratisl Lek Listy. 2014;115(7):427–33. doi: 10.4149/bll_2014_084. [DOI] [PubMed] [Google Scholar]
- 11.Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wells A, Yates C, Shepard CR. E-cadherin as an indicator of mesenchymal to epithelial reverting transitions during the metastatic seeding of disseminated carcinomas. Clin Exp Metastasis. 2008;25:621–28. doi: 10.1007/s10585-008-9167-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 14.Qi P, Xu MD, Ni SJ, et al. Low expression of LOC285194 is associated with poor prognosis in colorectal cancer. J Transl Med. 2013;11:122. doi: 10.1186/1479-5876-11-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang F, Zhang H, Mei Y, Wu M. Reciprocal regulation of HIF-1alpha and lincRNA-p21 modulates the Warburg effect. Mol Cell. 2014;53:88–100. doi: 10.1016/j.molcel.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 16.Krämer OH. HDAC2: A critical factor in health and disease. Trends Pharmacol Sci. 2009;30:647–55. doi: 10.1016/j.tips.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 17.Bieliauskas AV, Pflum MK. Isoform-selective histone deacetylase inhibitors. Chem Soc Rev. 2008;37:1402–13. doi: 10.1039/b703830p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009;280:168–76. doi: 10.1016/j.canlet.2008.10.047. [DOI] [PubMed] [Google Scholar]
- 19.Chang HH, Chiang CP, Hung HC, et al. Histone deacetylase 2 expression predicts poorer prognosis in oral cancer patients. Oral Oncol. 2009;45:610–14. doi: 10.1016/j.oraloncology.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 20.Zhu P, Martin E, Mengwasser J, et al. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell. 2004;5:455–63. doi: 10.1016/s1535-6108(04)00114-x. [DOI] [PubMed] [Google Scholar]
- 21.Huang BH, Laban M, Leung CH, et al. Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ. 2005;12:395–404. doi: 10.1038/sj.cdd.4401567. [DOI] [PubMed] [Google Scholar]
- 22.Ye F, Chen Y, Hoang T, et al. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat Neurosci. 2009;12:829–38. doi: 10.1038/nn.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tong Z, Cai M, Wang X, et al. EZH2 supports nasopharyngeal carcinoma cell aggressiveness by forming a co-repressor complex with HDAC1/HDAC2 and Snail to inhibit E-cadherin. Oncogene. 2012;31:583–94. doi: 10.1038/onc.2011.254. [DOI] [PubMed] [Google Scholar]
- 24.Aghdassi A, Sendler M, Guenther A, et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut. 2012;61:439–48. doi: 10.1136/gutjnl-2011-300060. [DOI] [PubMed] [Google Scholar]
- 25.Fritsche P, Seidler B, Schuler S, et al. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut. 2009;58:1399–409. doi: 10.1136/gut.2009.180711. [DOI] [PubMed] [Google Scholar]