

This study highlights the multiple modes of action of ribavirin, including depletion of intracellular GTP and increased hepatitis C virus mutagenesis. In cell culture, reduced ITP pyrophosphatase (ITPase) enzyme activity affected the intracellular concentrations of ribavirin triphosphate (RTP) and augmented the impact of ribavirin on the mutation rate and virus production. Additionally, our results imply that RTP, similar to ITP, a naturally occurring substrate of ITPase, is dephosphorylated in vitro by ITPase.
KEYWORDS: hepatitis C virus, ITPA, ITP pyrophosphatase, ITPase, ribavirin, mutagenesis, inosine triphosphate pyrophosphatase, mutagenesis
ABSTRACT
A third of humans carry genetic variants of the ITP pyrophosphatase (ITPase) gene (ITPA) that lead to reduced enzyme activity. Reduced ITPase activity was earlier reported to protect against ribavirin-induced hemolytic anemia and to diminish relapse following ribavirin and interferon therapy for hepatitis C virus (HCV) genotype 2 or 3 infections. While several hypotheses have been put forward to explain the antiviral actions of ribavirin, details regarding the mechanisms of interaction between reduced ITPase activity and ribavirin remain unclear. The in vitro effect of reduced ITPase activity was assessed by means of transfection of hepatocytes (Huh7.5 cells) with a small interfering RNA (siRNA) directed against ITPA or a negative-control siRNA in the presence or absence of ribavirin in an HCV culture system. Low ribavirin concentrations strikingly depleted intracellular GTP levels in HCV-infected hepatocytes whereas higher ribavirin concentrations induced G-to-A and C-to-U single nucleotide substitutions in the HCV genome, with an ensuing reduction of HCV RNA expression and HCV core antigen production. Ribavirin triphosphate (RTP) was dephosphorylated in vitro by recombinant ITPase to a similar extent as ITP, a naturally occurring substrate of ITPase, and reducing ITPA expression in Huh 7.5 cells by siRNA increased intracellular levels of RTP in addition to increasing HCV mutagenesis and reducing progeny virus production. Our results extend the understanding of the biological impact of reduced ITPase activity, demonstrate that RTP is a substrate of ITPase, and may point to personalized ribavirin dosage according to ITPA genotype in addition to novel antiviral strategies.
IMPORTANCE This study highlights the multiple modes of action of ribavirin, including depletion of intracellular GTP and increased hepatitis C virus mutagenesis. In cell culture, reduced ITP pyrophosphatase (ITPase) enzyme activity affected the intracellular concentrations of ribavirin triphosphate (RTP) and augmented the impact of ribavirin on the mutation rate and virus production. Additionally, our results imply that RTP, similar to ITP, a naturally occurring substrate of ITPase, is dephosphorylated in vitro by ITPase.
INTRODUCTION
Two variants of the ITP pyrophosphatase (ITPase) gene (ITPA), a missense variant in exon 2 (rs1127354, P32T) and a splice-altering single nucleotide polymorphism (SNP) in intron 2 (rs7270101, IVS2), have been demonstrated in a genome-wide association study (GWAS) to protect against ribavirin-induced hemolytic anemia during therapy with peginterferon in combination with ribavirin for hepatitis C virus (HCV) genotype 1 infection (1). In patients with chronic HCV genotype 2 or 3 infection receiving peginterferon and ribavirin, ITPA variants encoding reduced ITPase activity were found in approximately one-third of patients and were associated with reduced relapse risk following antiviral therapy despite lower plasma ribavirin concentrations (2).
The evolutionarily conserved ITPase is a cytosolic enzyme that recycles purines by the pyrophosphohydrolysis of ITP to IMP and thus protects against integration of noncanonical nucleotides such as ITP and XTP, as well as their deoxy forms, into RNA or DNA (Fig. 1). These nucleotides may otherwise be incorrectly incorporated to produce genetic instability, anomalous proteins, and altered ATP-dependent signaling (3–6). ITPase deficiency has been associated with augmented toxicity in patients receiving purine analogues such as 6-mercaptopurine and azathioprine (7, 8). Studies of human cell lines have ascribed ITPase a role in protection against DNA damage (9), and in genetic knockout mouse models, the absence of ITPase generates growth retardation with cardiac myofiber disarray and death in utero or within 2 weeks of birth (10).
FIG 1.
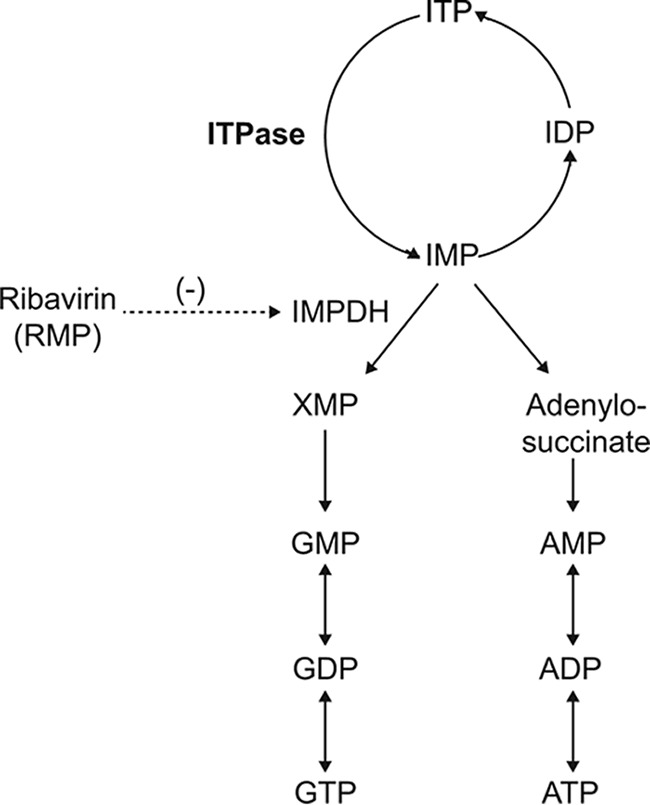
ITP to GTP and ATP and the nucleotide salvage pathway.
Ribavirin originally was synthesized as a guanosine analogue (11) and was soon noted to exert broad-spectrum in vitro activity against many RNA and DNA viruses (12). In addition to its utility in HCV therapy, treatment with ribavirin is of benefit in severe respiratory syncytial virus infections (13), viral hemorrhagic fevers such as Lassa and Crimean-Congo (14, 15), and hepatitis E virus infections (16). Unaided, ribavirin is insufficient to achieve clearance of HCV infection (17, 18) and has a modest effect on systemic levels of HCV RNA, with mean reductions of approximately 0.5 log10 IU/ml (19, 20). In the previously commonly used interferon-based therapy for chronic HCV infection, the addition of ribavirin markedly increased the likelihood of achieving sustained virological responses (SVR; defined as undetectable HCV RNA 24 weeks after the completion of treatment) by means of reduced relapse risk (21, 22). Presently, the use of ribavirin in HCV therapy is diminishing, but it continues to be recommended for some interferon-free, direct-acting antiviral (DAA) treatments (23), especially for HCV genotype 3 infection (24). Among HCV patients with decompensated cirrhosis receiving the recently approved pan-genotypic combination of sofosbuvir and velpatasvir in the ASTRAL-4 trial, the highest SVR rate was observed among patients additionally receiving ribavirin although the difference among rates was not statistically significant (25). Thus, in the setting of decompensated cirrhosis (Child-Pugh B or C), ribavirin likely will remain a component of HCV therapy as protease inhibitors should not be used in these patients (24).
Ribavirin has a large distribution volume, an elimination that is dependent on renal function, and a long half-life requiring in excess of 4 weeks to reach steady state (26–28). Ribavirin is subsequently activated by intracellular phosphorylation into mono-, di-, and triphosphates and upon phosphorylation becomes irreversibly entrapped in erythrocytes. Ribavirin therapy is hampered by adverse effects, most importantly dose-dependent hemolytic anemia, possibly secondary to oxidative stress caused by posterior depletion of ATP in erythrocytes (29, 30).
The detailed molecular mechanism underlying the action of ribavirin on HCV infection remains unclear, but several mechanisms of action have been proposed, including inhibition of IMP dehydrogenase (IMPDH) by ribavirin monophosphate (RMP) with ensuing GTP depletion (31), inhibition of capping of viral RNA (32), direct inhibition of the viral RNA polymerase (33, 34), and modulation of host immune responses (35). Additionally, ribavirin was shown to trigger viral mutagenesis leading to error catastrophe by means of incorporation of ribavirin triphosphate (RTP) (34, 36, 37) although this mechanism has been disputed by other investigators (38). In vivo (39, 40) and in vitro (41, 42) studies also imply that ribavirin downregulates the expression of interferon-stimulated genes (ISGs) in addition to reducing systemic concentrations of liver enzymes (19) and IP-10 (also known as CXCL10) (20).
The aim of the present study was to elucidate the mechanisms of action of the previously reported in vivo impact of reduced ITPase activity on the outcome of HCV therapy by means of assessing the in vitro effect of reduced enzymatic activity in the presence or absence of ribavirin in an HCV culture system.
RESULTS
To mimic the previously reported in vivo impact of reduced ITPase activity, ranging from 60% to <5% of normal full activity (2), a small interfering RNA (siRNA) was utilized to decrease ITPA RNA expression in vitro. Huh7.5 cells were transfected with a pool consisting of four siRNAs directed against ITPA or a scrambled RNA (negative-control siRNA) for 48 h. Real-time PCR quantification of ITPA mRNA demonstrated that expression levels were significantly reduced to around 20% of the level observed in negative-control siRNA-transfected cells (Fig. 2C). Immunoblot detection of ITPase validated the effect of ITPA siRNA also on protein expression (Fig. 2D).
FIG 2.

The effect of ITPA siRNA transfection and ribavirin treatment on HCV replication (A and B) and efficiency of ITPA siRNA transfection (C and D). (A) HCV RNA expression in HCV-infected Huh7.5 cells transfected with ITPA siRNA or a negative-control siRNA and ribavirin treatment as determined by real-time PCR and normalized to RPL4 expression. (B) HCV core antigen production in the supernatant of HCV-infected ITPA siRNA- or negative-control siRNA-transfected and ribavirin-treated Huh7.5 cells. (C) Efficiency of ITPA siRNA transfection on ITPA RNA expression in Huh7.5 cells transfected with ITPA siRNA or negative-control siRNA as determined by real-time PCR and normalized to RPL4 expression. (D) ITPA protein expression in Huh7.5 cells transfected with ITPA siRNA or negative-control siRNA and normalized to beta-actin protein levels as determined by immunoblotting. (E) Toxicity of ribavirin and siRNA treatment was determined by a proliferation assay of Huh7.5 cells treated with ribavirin and ITPA siRNA or a negative-control siRNA. Statistical significance was determined by a t test on logarithmic values (**, P < 0.01; ***, P < 0.001).
The HCV cell culture system was utilized to investigate effects of ITPA siRNA transfection and ribavirin on HCV replication. ITPA siRNA- or negative-control siRNA-transfected cells were incubated with graded concentrations of ribavirin (1 to 200 μM) and infected with the J6/JFH-1 HCV strain. Higher concentrations of ribavirin (100 to 200 μM) did not exert any effect on cell proliferation, and no difference was seen between ITPA siRNA-treated cells and those treated with a negative-control siRNA (Fig. 2E). Ribavirin exposure, as expected, decreased HCV RNA (normalized to expression of ribosomal protein L4, RPL4) and HCV core protein concentration levels in the infected cells transfected with a negative-control siRNA. Significant reduction in core antigen levels was seen in the 100 and 200 μM ribavirin-treated cells, and an HCV RNA reduction was seen in 200 μM ribavirin-treated cells compared to the level in untreated cells (P < 0.0005) (Fig. 2A and B). More interestingly, ITPA siRNA transfection in cells treated with 100 or 200 μM ribavirin significantly further reduced HCV RNA (P < 0.005) and HCV antigen (P < 0.0001) concentrations compared to levels with a negative-control siRNA (Fig. 2A and B). Decreased ITPA expression in HCV-infected cells thus promoted the ribavirin-induced reduction of HCV production, as measured by HCV RNA and HCV core antigen quantification.
As both ITPase and IMPDH are involved in the nucleotide salvage pathway, the relative concentrations of all nucleoside triphosphates (NTPs) were measured in ITPA siRNA- or negative-control siRNA-transfected hepatocytes across a range of ribavirin concentrations. Quantification of cellular nucleotides at 260 nm showed that even a low ribavirin concentration (10 μM) profoundly decreased relative GTP levels (from 6.3% to 2.5% of the nucleotide pool; P = 0.0001). Higher concentrations of ribavirin (100 and 200 μM) resulted in a severe depletion of the GTP pool, combined with a modest decrease in relative levels of ATP and an increase in CTP and UTP. No significant differences were observed between ITPA siRNA- and negative-control siRNA-transfected cells with regard to relative GTP, ATP, CTP, or UTP concentrations (Fig. 3B). To also allow for the quantification of RTP, measurement of the samples was repeated at 215 nm (Fig. 3A). As expected, RTP concentrations increased with increasing ribavirin concentrations. However, ITPA siRNA transfection unexpectedly further increased RTP levels, resulting in approximately 19% of the total nucleotide pool consisting of RTP at 200 μM ribavirin in contrast to 10% for negative-control siRNA-transfected cells (P = 0.02) (Fig. 3A), suggesting that RTP is a substrate of ITPase, converting it into ribavirin monophosphate (RMP). To further explore this hypothesis, an in vitro phosphate detection assay using recombinant ITPase was used with RTP, ITP, and GTP as substrates. This assay confirmed the reactivity of ITPase for RTP, similar to that noted for ITP, a naturally occurring substrate of ITPase, whereas no reactivity was seen for GTP (Fig. 4). Together these data demonstrate that ITPase dephosphorylates RTP.
FIG 3.

Intracellular CTP, UTP, ATP, GTP, and RTP amounts in ITPA siRNA- or negative-control (NC) siRNA-transfected and ribavirin-treated Huh7.5 cells detected by HPLC at 215 nm (A) and (excluding RTP) at 260 nm (B). Nucleoside triphosphate levels are quantified relative to the total amounts of nucleoside triphosphates. Statistical significance was determined by t test (*, P < 0.05; **, P < 0.01). Please note that RTP can be detected at 215 nm but not at 260 nm.
FIG 4.

In vitro ITPase activity as measured by the dephosphorylation of RTP in comparison to that of ITP and GTP (positive and negative controls, respectively).
Next-generation sequencing (NGS) was used to determine mutations in the HCV RNA sequences, which were mainly C-to-U or G-to-A substitutions (Fig. 5A to C). Ribavirin at 200 μM dramatically increased HCV mutagenesis compared to levels with the negative-control siRNA-transfected cells (P < 0.001). Likewise, ITPA siRNA transfection resulted in increased mutagenesis already at 100 μM ribavirin and a prominent increase compared with the level for negative-control siRNA-transfected cells treated with 200 μM ribavirin, representing a significant difference for all C-to-U and G-to-A mutations (P < 0.005) (Fig. 5A). Interestingly, ITPA siRNA transfection led to a mutation pattern similar to that in negative-control siRNA-transfected cells treated with ribavirin but at a higher frequency. It should be noted, however, that while all the NGS analyses were sequenced at similar depths, the HCV RNA levels decreased when cells were treated with higher concentrations of ribavirin and transfected with ITPA siRNA (Fig. 2A). Indeed, NGS analyses of PCR amplimers may give rise to false perceptions of depth, especially if the original copy numbers are low.
FIG 5.
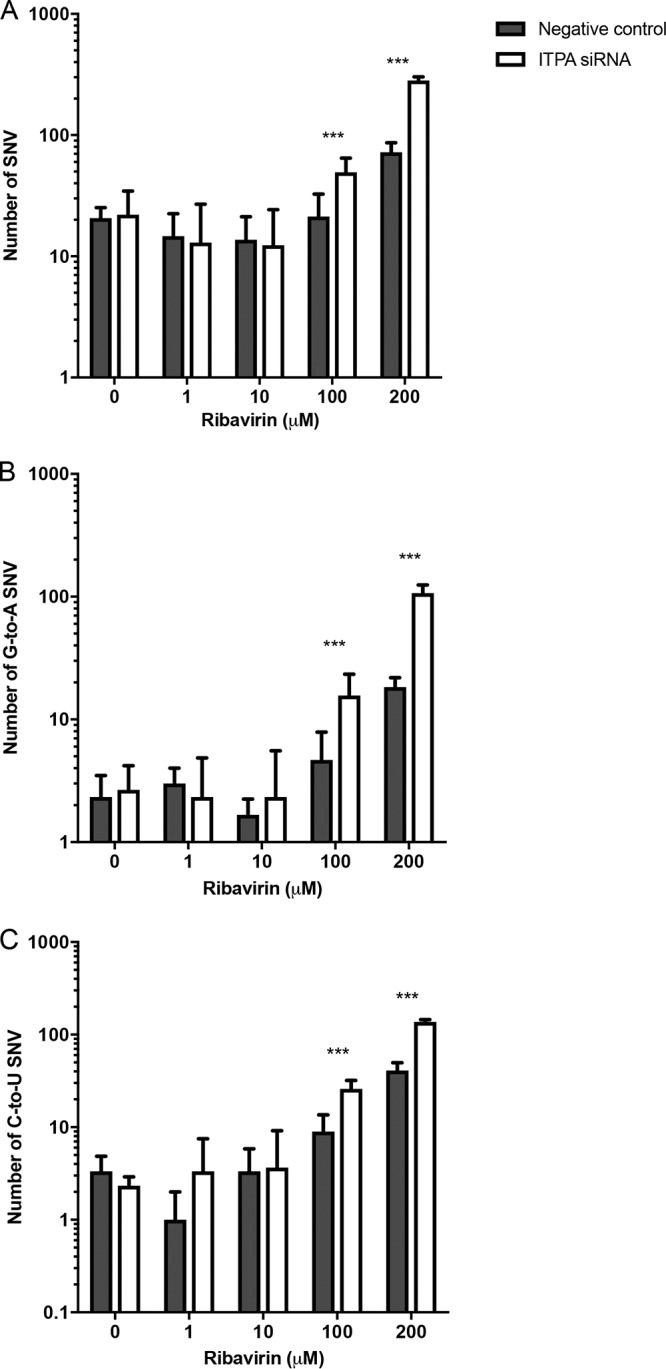
Single nucleotide variations (SNVs) (A) and G-to-A (B) and C-to-U (C) mutations across the HCV genome in HCV-infected Huh7.5 cells treated with ribavirin and transfected with ITPA siRNA or a negative-control siRNA. Statistical significance was determined by Poisson distribution (***, P < 0.001).
To determine the impact of GTP depletion on the antiviral efficacy of ribavirin in ITPA siRNA- and negative-control siRNA-transfected cells, a guanosine salvage experiment was performed (Fig. 6). Guanosine supplementation significantly reduced the combined inhibitory effect of ribavirin and ITPA siRNA transfection as measured by HCV RNA expression (P < 0.01) and HCV core antigen quantification (P < 0.001). Of note, 200 μM guanosine supplementation significantly reduced HCV core antigen and HCV RNA when ribavirin was not added. Thus, GTP depletion may play a role in the combined antiviral activity of ribavirin and reduced ITPase activity.
FIG 6.

HCV RNA (A and B) and HCV core antigen (C and D) in Huh7.5 cells treated with ribavirin or ribavirin and guanosine and transfected with ITPA siRNA (A and C) or a negative-control siRNA (B and D). Statistical significance was determined by a t test on logarithmic values (**, P < 0.01; ***, P < 0.001).
DISCUSSION
HCV-infected patients with reduced ITPase activity experience (i) reduced toxicity and (ii) improved efficacy of treatment with ribavirin, but the detailed mechanisms of ITPase and ribavirin interaction have remained unknown. This study was designed to clarify the impact of reduced ITPase activity on the actions of ribavirin in HCV-infected human liver cells. For this purpose, we introduced ITPA siRNA in HCV-infected Huh7.5 cells and achieved ITPA expression levels (20%) comparable to the level of ITPase activity reported in patients carrying genetic variants of ITPA encoding ITPase deficiency, i.e., activity of <5 to 60% of normal, wild-type ITPase activity. It was observed that the silencing of ITPA (i) promoted ribavirin-induced reduction in HCV RNA and HCV core antigen, (ii) potentiated the mutagenesis noted in the presence of higher concentrations of ribavirin, and (iii) increased the intracellular levels of RTP. Additionally, for the first time to our knowledge, ITPase was demonstrated to accept RTP as a substrate for dephosphorylation to RMP.
The effects of ribavirin on viral replication and mutagenesis were observed only at high doses of ribavirin (≥100 μM) that did not affect cell proliferation or viability, which is in line with a previous study showing a 50% inhibitory concentration (IC50) for ribavirin on J6/JFH-1 recombinant virus of 214 μM and an LC50 for ribavirin on Huh7.5 cells of 123 mM, with 90% live cells at 1 mM (43). Additionally, ≥10 μM ribavirin resulted in markedly reduced GTP levels, unrelated to ITPA siRNA, as well as an increase in RTP concentration when ITPA expression was reduced. Ribavirin at 10 μM is comparable to the steady-state concentrations achieved in patients during standard ribavirin weight-based dosing for HCV infection (20), whereas considerably higher ribavirin concentrations likely are achieved following dosing for Lassa virus infection (44). It should be noted that while patients generally achieve lower ribavirin plasma concentrations than those used in the in vitro HCV cell culture system in this study, patients are exposed to ribavirin for considerably longer times than the cells in the 72-h culture, which was the maximum time that the cells could be exposed without passage. Additionally, the Huh7.5 cell line is a hepatoma cell line and may differ from primary hepatocytes with regard to ribavirin metabolism.
Our finding that ITPase dephosphorylates RTP warrants further investigation. Interestingly, it has previously been reported that reduced ITPase activity is associated with lower plasma concentrations of unphosphorylated ribavirin in patients (2, 45), in spite of also being associated with improved outcomes and reduced toxicity. These clinical observations thus may be secondary to the association between reduced ITPase activity and increased intracellular RTP concentrations as the phosphorylation of ribavirin which occurs within cells leads to intracellular entrapment.
This study supports the previously suggested notion of multiple mechanisms of action of ribavirin (46); i.e., at low concentrations ribavirin markedly depleted intracellular GTP levels while inducing HCV mutagenesis at higher concentrations. The finding that low concentrations of ribavirin reduce GTP levels thus corroborates the previously reported competitive inhibition of IMP dehydrogenase (IMPDH) by RMP (47). Similarly, the observed increase in single nucleotide variations (SNVs) as ribavirin concentrations increased lends credence to the hypothesis that viral mutagenesis is caused by the incorporation of ribavirin triphosphate (RTP) (34, 36, 37) into the viral genome. Human polymerases, having proofreading capacity, are less likely to substitute RTP or noncanonical nucleotides for GTP. However, the profound reduction in intracellular GTP concentrations noted at relatively low ribavirin levels likely might have an additional impact on the levels of cellular gene transcription, as previously proposed (35, 39–42).
The majority of the mutations observed in this study using next-generation sequencing were G-to-A or C-to-U SNVs. This likely results from the incorrect substitution of RTP or ITP for GTP during the replication of viral RNA as intracellular GTP concentrations decline. Unlike GTP, RTP can form two hydrogen bonds with UTP or CTP with equal efficiencies (36), and ITP reportedly binds thermodynamically to CTP > ATP > UTP ≈ GTP (48). Either of these incorrect incorporations by the HCV RNA-dependent RNA polymerase, which lacks proofreading, would cause the observed mutation pattern. Although we did not document incorporation of RTP or ITP into the HCV genome, incorrect incorporation of RTP into the viral genome previously has been demonstrated for poliovirus and HCV (34, 36, 37). Additionally, recent in vivo evidence supports ribavirin-induced mutagenesis of the hepatitis E virus (HEV) genome (49). It should be noted that we did not detect quantifiable intracellular levels of ITP in this study despite repeated efforts, implying that the majority of the mutagenesis is driven by RTP rather than ITP incorporation, where increasing intracellular concentrations of RTP are observed in parallel with increasing numbers of SNVs.
The presence of intracellular ITP is a prerequisite for normal cellular function. It was long recognized that if only canonical pairing were allowed in nature, 64 species of tRNA would be required. However, empirically it was noted that most organisms have fewer than 45 species of tRNA, and thus some tRNA species must pair with more than one codon. To account for these observations, the wobble hypothesis postulated that the 5′ base on the anticodon in tRNA, which binds to the 3′ base on the mRNA, could have nonstandard base pairing (50). Similar to these in vivo observations in tRNA, replacement of canonical nucleotides by ITP often is utilized in vitro to induce wobbling, e.g., deoxy-ITP (dITP) may be incorporated into hybridization probes (51) or primers for PCR (52) at ambiguous codon positions to improve PCR efficiency. Additionally dITP may be incorporated in vitro to promote random mutagenesis (53) or directed evolution (54). In our study very few SNVs were noted in the absence of ribavirin. This may be an effect of ITPA siRNA not being able to completely silence expression of ITPA RNA or a result of the current experimental cell culture infections being standardized to 72 h. Greater reduction in ITPase activity or extending the duration of infection may have been required to document SNVs in the absence of ribavirin. Moreover, it is possible that relatively few substitutions in the viral genome, i.e., below the detection threshold in this study, could be sufficient to impact viral propagation, as previously noted for poliovirus, where low numbers of incorrect substitutions led to error catastrophe for the virus (34, 36, 37). The previous observation that the Cassava brown streak virus genome encodes an ITPase homologue (Maf/HAM1) (55) suggests that reduction of ITPase activity in infected cells in order to increase the mutagenesis of viral RNA may occur in some species and that some viruses might attempt to overcome this by increasing ITPase activity to maintain their genomic stability.
Thus, stable or transient reduction of ITPase activity potentially may constitute a novel form of innate immunity in some organisms. This notion may be supported even in humans by observations among HCV-infected patients treated with ribavirin-free direct-acting antivirals (DAAs). Among 25 HCV genotype 1-infected, noncirrhotic patients treated with the interferon- and ribavirin-free combination of daclatasvir, asunaprevir, and beclabuvir for 12 weeks in the UNITY-1 trial (56) and having resistance-associated substitutions (RASs; also known as resistance-associated variants or RAVs) directed against the HCV protein NS5A at baseline prior to therapy, a nonsignificant trend toward improved outcome was observed in the seven patients having genetic variants encoding reduced ITPase activity (Fig. 7). A similar trend was observed among 17 HCV genotype 3-infected cirrhotic and noncirrhotic patients with baseline NS5A RASs/RAVs who were treated with daclatasvir and sofosbuvir without ribavirin for 12 weeks in the ALLY-3 study (57). Together, these studies demonstrate that in the presence of baseline NS5A RASs/RAVs, HCV-infected patients with reduced ITPase activity are significantly more likely to achieve SVR when treated with DAA therapy without ribavirin (P = 0.03 Fisher's exact test) (Fig. 7). Thus, even in the absence of ribavirin, reduced ITPase activity may beneficially impact the outcome of HCV infection. It should be noted, however, that the low number of subjects with RASs/RAVs limits the ability to make robust conclusions about the association of ITPase activity with the response in these subpopulations as ITPase activity was not associated with response on its own in these clinical trials.
FIG 7.
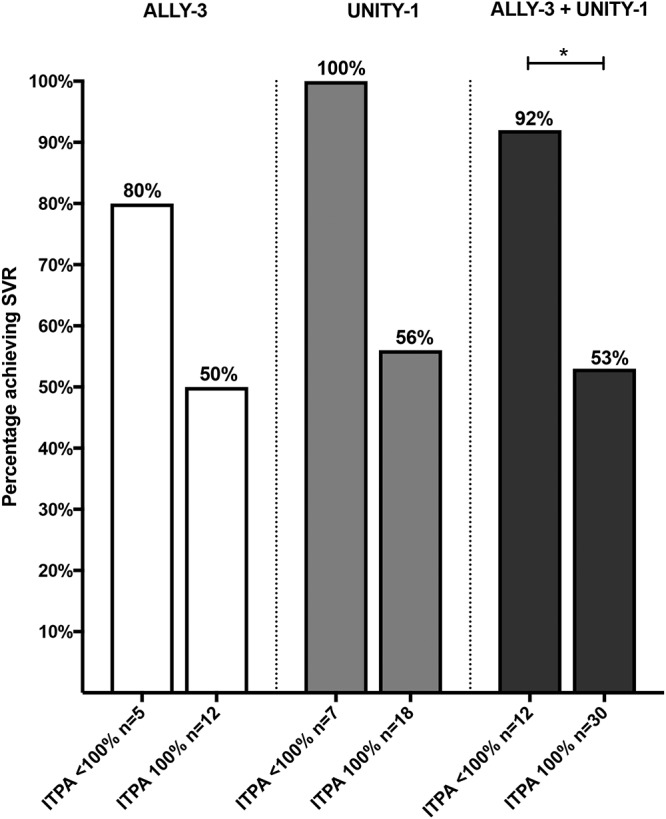
Percentage of patients with normal (100%) or reduced (<100%) ITPase activity having baseline, pretreatment NS5A resistance-associated substitutions (RASs; also known as resistance-associated variants, or RAVs) that achieved SVR following treatment with daclatasvir and sofosbuvir without ribavirin for 12 weeks in the ALLY-3 study (HCV genotype 3-infected patients with noncirrhosis or cirrhosis) and with daclatasvir, asunaprevir, and beclabuvir without ribavirin for 12 weeks in the UNITY-1 trial (HCV genotype 1-infected noncirrhotic patients). Statistical significance was determined using Fisher's exact test (*, P < 0.05).
The protection against ribavirin-induced anemia in patients with reduced predicted ITPase activity has been suggested to occur by means of avoidance of ATP reduction with ensuing diminution of oxidative stress and hemolysis (58). Interestingly, no major impact on ATP concentrations was detected in the presence or absence of ribavirin, and, likewise, ATP was not affected by the presence or absence of ITPA siRNA. This observation likely is secondary to differences in enzymatic activities between the hepatocyte-derived cells used in this study and erythrocytes. Also, in the present study guanosine supplementation partially reversed the effect of ITPA siRNA transfection on HCV propagation in vitro. This finding is coherent with previous reports on attempts to reverse the effects of ribavirin by guanosine supplementation where full reversion was not achieved (34, 36, 37).
The main clinical impact of the present study is further support for the notion that dosing of ribavirin likely may be individualized based on predicted ITPase activity rather than merely on weight and renal function, as is the current norm (59), although this hypothesis warrants confirmation in a prospective randomized trial. The findings from the NORDynamIC study, enrolling 382 HCV genotype 2- or 3-infected patients treated with standard 800-mg dosing of ribavirin in combination with interferon, demonstrated that patients carrying ITPA variants encoding reduced ITPase activity showed increased treatment efficacy mediated by reduced relapse risk and less anemia (2). Thus, development of a partial inhibitor of ITPase conceivably may improve the antiviral efficacy of ribavirin and attenuate its adverse effects. Such an inhibitor of ITPase could also be a potential antiviral agent in the absence of ribavirin.
In conclusion, this study demonstrated that the introduction of ITPA siRNA in an HCV cell culture system aiming at mimicking the reduced ITPase activity previously reported among one-third of humans (2) (i) reduced progeny virus production as measured by HCV core antigen and HCV RNA expression, (ii) increased ribavirin-induced mutagenesis, and (iii) increased the intracellular concentrations of ribavirin triphosphate. Our results demonstrate that RTP is a substrate of ITPase, extend the understanding of the biological impact of reduced ITPase activity, and may point to personalized ribavirin dosage according to ITPA genotype in addition to novel antiviral strategies based on modulation of ITPase activity.
MATERIALS AND METHODS
Virus and cells.
Huh7.5 cells (Apath, New York, NY) were cultured in cell culture medium consisting of Dulbecco's minimum essential medium (DMEM; Gibco, Carlsbad, CA) supplemented with 10% fetal calf serum (FCS), 1% nonessential amino acids (NEAA; Sigma, St. Louis, MO), and 1% penicillin-streptomycin. Huh7.5 cells are of wild-type ITPA genotype. An HCV J6/JFH-1 plasmid (kindly provided by Charles M. Rice, The Rockefeller University, New York) was used for production of infectious HCV.
Production of HCV.
MEGAscript T7 RNA polymerase (Ambion, Carlsbad, CA) was used for in vitro transcription of a J6/JFH-1 plasmid according to the manufacturer's instructions. Briefly, 1 ng of plasmid was linearized with Xbal (Fermentas, Waltham, MA); the reaction was terminated, and the DNA was precipitated using 1/20 volume of 0.5 M EDTA, 1/10 volume of 5 M ammonium acetate, and 2 volumes of ethanol and incubated at −20°C for at least 15 min. The linearized plasmid was pelleted and resuspended in double-distilled H2O (ddH2O), and RNA was transcribed with T7 RNA polymerase at 37°C for 10 h, according to the manufacturer's protocol, with the addition of 1 μl of RiboLock RNase inhibitor (ThermoFisher Scientific, Carlsbad, CA). RNA was purified with an RNeasy RNA purification kit (Qiagen, Hilden, Germany) according to a standard protocol. The RNA was quantified using a NanoDrop 2000 instrument (Thermo Scientific, Carlsbad, CA). RNA quality was analyzed on a 1% agarose gel with SYBR green II RNA gel stain (ThermoFisher Scientific, Carlsbad, CA). Four million Huh7.5 cells were washed in buffered NaCl and resuspended in 400 μl of CytoMix (10 mM K2HPO4/KH2PO4 with 120 mM KCl, 0.15 mM CaCl2, 2 mM EGTA, 25 mM HEPES, 5 mM MgCl2, pH 7.6, and 2 mM ATP). Ten micrograms of RNA was added to the cells in a 0.4-cm cuvette and electroporated (960 μF, 260V) (Gene Pulser II; Bio-Rad, Hercules, CA). Twenty milliliters of cell culture medium was added to the cells, which were cultivated in six-well plates for 96 h. The cell culture medium was collected, and cell debris was removed by centrifugation at 350 × g for 7 min.
HCV culture system.
During HCV infection, Huh7.5 cells were pretreated with ribavirin (Sigma, St. Louis, MO) at 2 h prior to infection. When cells were treated with guanosine (Sigma, St. Louis, MO), 200 μM guanosine was added simultaneously with ribavirin. Cells were infected with 15 pmol/liter HCV from cell culture medium, as quantified by HCV core antigen measurement, in 300 μl in 12-well plates. After 4 h, cells were washed three times with buffered NaCl and medium, or medium supplemented with ribavirin was added. Cells were analyzed at 72 h postinfection.
siRNA treatment.
Huh7.5 cells were seeded at 1 × 105 cells per well (12-well plate) in DMEM, 10% FCS, and 1% NEAA. Lipofectamine (ThermoFisher Scientific, Carlsbad, CA) and Opti-Mem (Gibco, Carlsbad, CA) were used for transfection of siRNA according to the manufacturer's instructions. The cells were transfected with four ITPA siRNAs (ITPA4, -5, -6, and -7) (Qiagen, Hilden, Germany) and a negative-control siRNA (scrambled RNA; Qiagen, Hilden, Germany) and incubated for 48 h.
Antigen measurement.
Supernatant from each HCV-infected well was analyzed in duplicate with an Architect HCV Ag assay (Abbott, Chicago, IL) for automated detection of HCV core antigen (n = 12 for the experiments shown in Fig. 2B, n = 8 to 14 for the experiment shown in Fig. 6 C, and n = 8 to 14 for the experiment shown in Fig. 6D in each group).
HCV RNA isolation.
Infected Huh7.5 cells were harvested by adding RLT lysis buffer (Qiagen, Hilden, Germany), and RNA was isolated using an RNeasy RNA isolation kit (Qiagen, Hilden Germany) according to the manufacturer's instructions. The purified RNA was treated with Turbo DNase (Ambion, Carlsbad, CA) at 37°C for 30 min for removal of cellular DNA.
Real-time PCR.
The RNA (n = 6 in each group) was quantified with a NanoDrop 2000 instrument (ThermoFisher Scientific, Carlsbad, CA), and all samples were diluted to 20 ng/μl. Real-time PCR was performed using a Superscript III Platinum One-Step quantitative reverse transcription-PCR (qRT-PCR) kit with 6-carboxy-X-rhodamine (ROX) (Invitrogen, Carlsbad, CA) and with 40 ng of RNA, 500 nM primer, and 300 nM probe to detect ribosomal protein L4 (RPL4), ITPA, and HCV RNA. Primers and probes detecting HCV were used as previously published (60), and primers and probes detecting RPL4 and ITPA were purchased from ThermoFisher Scientific. Expression was calculated using the ΔCT (where CT is threshold cycle) method, with RPL4 used as a reference gene (61).
Immunoblotting.
SDS-PAGE and immunoblotting were performed as previously described (62). Briefly, ITPA siRNA- and negative-control siRNA-transfected Huh7.5 cells were harvested in SDS sample buffer under reducing conditions and separated on a gradient polyacrylamide gel (Invitrogen, Carlsbad, CA). The proteins were transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA) and detected using rabbit antibodies against ITPA (1/2,000; Abcam, Cambridge, United Kingdom) with beta-actin (1/6,000; Abcam) as the control. Secondary anti-rabbit IgG peroxidase-conjugated antibodies (1/5,000; Abcam, Cambridge, United Kingdom) were used with SuperSignal West Dura Extended Duration Substrate (ThermoFisher Scientific, Carlsbad, CA). The detection of bands was viewed using a ChemiDoc MP System (Bio-Rad, Hercules, CA), and band intensities were calculated using ChemiDoc MP software, with ITPA protein expression normalized to beta-actin levels.
Proliferation assay.
Huh7.5 cells of low confluence were cultured in medium supplemented with ribavirin and/or following ITPA siRNA transfection by trypsinization, and quantification of the number of cells present after treatment in comparison to the number of cells seeded at the start of treatment was performed.
HPLC.
Huh7.5 cells were cultured in 10-cm dishes and transfected with ITPA siRNA or a negative-control siRNA (n = 4 in each group), as described above. Cells were harvested after 72 h and washed with cold-buffered NaCl. The cell material was collected using a cell scraper in 15% trichloroacetic acid (TCA) and 30 mM MgCl2 and snap-frozen in liquid nitrogen. Standard NTPs and intracellular ribavirin triphosphate (RTP) were extracted and measured as previously described (63). Briefly, NTPs were neutralized with two rounds of Freon-trioctylamine extraction, adjusted to pH 3.4, and analyzed on a LaChrom Elite high-performance liquid chromatography (HPLC) system (Hitachi, Tokyo, Japan) with a Partisphere SAX column (Hicrom, Berkshire, United Kingdom). To enable measurement of RTP, detection was carried out at 215 nm. The level of each nucleotide is expressed as a percentage of the total sample NTPs.
Next-generation sequencing (NGS).
Isolated RNA (n = 4) was reverse transcribed using Superscript III (Invitrogen, Carlsbad, CA) with HCV-specific primers in NS4A (GCATTCCTCCATCTCATCAAAAGC) and NS5B (CTACCGAGCGGGGAGTAGGAAGAG). The cDNA was amplified in a nested PCR using Platinum HiFi Taq polymerase (Invitrogen, Carlsbad, CA) along with primer pairs for the 5′ (forward, ATGAGCACAAATCCTAAACCTCAAAGA, and reverse, GCATTCCTCCATCTCATCAAAAGC) and 3′ regions (forward, CCTCACACACATAGACGCCCACTT, and reverse, GTCCTGGGACGCCATGTGGAA) of the genome. The PCR was nested with forward primer TCAAAGAAAAACCAAAAGAAACACCAACCG along with reverse primer GAAACGCATCCAGTCGCCAGGCAA or GTCCTGGGACGCCATGTGGAA and GAAAAGTAGGAGTAGGCCGAAGAGTAATGA, respectively. The PCR product was visualized on a 1% agarose gel, and concentrations were measured with a NanoDrop 2000 instrument. The 5′ and 3′ PCR products were pooled, barcoded, and size selected to 200 bp using an Ion Xpress Plus fragment library kit (Thermo Scientific, Carlsbad, CA) together with an AB Library Builder System (ThermoFisher, Carlsbad, CA). The generated libraries were then amplified and purified according to protocol before quantification, and quality was assessed using a High Sensitivity D1000 screen tape kit (Agilent Technologies, Santa Clara, CA) together with an Agilent 2200 TapeStation system (Agilent Technologies, Santa Clara, CA). Six barcoded libraries were pooled before being amplified, conjugated to sphere particles, and loaded on an Ion 314 chip using Ion Chef (ThermoFisher, Carlsbad, CA). The samples were sequenced on an Ion PGM System (ThermoFisher, Carlsbad, CA). The results were analyzed with CLCbio (Qiagen, Hercules, CA), where each barcoded sample was quality checked. The quality trim threshold was set at 0.01 (PHRED score of >25), and sequences above 30 nucleotides in length were aligned to the J6/JFH-1 strain. All sequences had a read depth of at least 1,000×. The aligned sequences were analyzed using low-frequency variation. To validate reproducibility and to check for PCR errors, the first PCR and the nested PCR were sequenced for five samples, resulting in a comparable number of mutations, insertions, and deletions in the two PCRs run from the same sample (data not shown). The total number of detected mutations and the specific nucleotide substitutions were summarized for each sample.
ITPase enzymatic assay.
Dephosphorylation of ITP, RTP, and GTP (n = 6) was analyzed using a PiColorLock Gold Phosphate detection system (Inova Biosciences, Cambridge, United Kingdom), according to the manufacturer's instructions. Nucleotides (0.1 mM) and 15 nM human ITPase (BioSite, San Diego, CA) in 50 mM Tris-HCl, pH 8, with 10 mM MgCl2 and 1 mM dithiothreitol (DTT) together with 0.5 U of inorganic pyrophosphatase (Roche Diagnostics, Risch, Switzerland), were incubated at 37°C for 10 min. Gold Mix was added to each well, followed by a stabilizer, and incubated in room temperature for 30 min. The plates were measured at 620 nm on a Multiskan FC (Thermo Scientific, Carlsbad, CA).
Statistics.
One-way analysis of variance (ANOVA) with Dunnett's test was used to analyze differences in HCV-RNA, HCV antigen levels, and nucleotide concentrations between different treatments. A t test on logarithmic (when applicable) values was used to compare differences between results for ITPA siRNA-treated cells or guanosine-treated cells and those for controls. Differences in numbers of mutation were analyzed by Poisson distribution. Fisher's exact test was used to evaluate differences in cure rates of the ALLY-3 and UNITY-1 studies. Statistics were done in Prism (version 6.0c; GraphPad Software, La Jolla, CA) or SPSS (version 20.0.0; IBM Corp., Armonk, NY, USA) software. All reported P values are two sided, and P values of <0.05 were considered significant.
ACKNOWLEDGMENTS
We thank Gustav Pettersson and Giorgia Ortolani for expert technical assistance and Sushma Sharma for helpful discussions.
Megan Wind-Rotolo is an employee and stockholder of Bristol-Myers Squibb. None of the other authors have competing financial interests or an association that might pose a conflict of interest.
The Swedish Medical Research Council, ALF Foundation at Sahlgrenska University Hospital, the Swedish Society for Medical Research, and the Swedish Cancer Society supported this study.
REFERENCES
- 1.Fellay J, Thompson AJ, Ge D, Gumbs CE, Urban TJ, Shianna KV, Little LD, Qiu P, Bertelsen AH, Watson M, Warner A, Muir AJ, Brass C, Albrecht J, Sulkowski M, McHutchison JG, Goldstein DB. 2010. ITPA gene variants protect against anaemia in patients treated for chronic hepatitis C. Nature 464:405–408. doi: 10.1038/nature08825. [DOI] [PubMed] [Google Scholar]
- 2.Rembeck K, Waldenstrom J, Hellstrand K, Nilsson S, Nystrom K, Martner A, Lindh M, Norkrans G, Westin J, Pedersen C, Farkkila M, Langeland N, Buhl MR, Morch K, Christensen PB, Lagging M. 2014. Variants of the inosine triphosphate pyrophosphatase gene are associated with reduced relapse risk following treatment for HCV genotype 2/3. Hepatology 59:2131–2139. doi: 10.1002/hep.27009. [DOI] [PubMed] [Google Scholar]
- 3.Sumi S, Marinaki AM, Arenas M, Fairbanks L, Shobowale-Bakre M, Rees DC, Thein SL, Ansari A, Sanderson J, De Abreu RA, Simmonds HA, Duley JA. 2002. Genetic basis of inosine triphosphate pyrophosphohydrolase deficiency. Hum Genet 111:360–367. doi: 10.1007/s00439-002-0798-z. [DOI] [PubMed] [Google Scholar]
- 4.Shipkova M, Lorenz K, Oellerich M, Wieland E, von Ahsen N. 2006. Measurement of erythrocyte inosine triphosphate pyrophosphohydrolase (ITPA) activity by HPLC and correlation of ITPA genotype-phenotype in a Caucasian population. Clin Chem 52:240–247. doi: 10.1373/clinchem.2005.059501. [DOI] [PubMed] [Google Scholar]
- 5.Maeda T, Sumi S, Ueta A, Ohkubo Y, Ito T, Marinaki AM, Kurono Y, Hasegawa S, Togari H. 2005. Genetic basis of inosine triphosphate pyrophosphohydrolase deficiency in the Japanese population. Mol Genet Metab 85:271–279. doi: 10.1016/j.ymgme.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 6.Simone PD, Pavlov YI, Borgstahl GE. 2013. ITPA (inosine triphosphate pyrophosphatase): from surveillance of nucleotide pools to human disease and pharmacogenetics. Mutat Res 753:131–146. doi: 10.1016/j.mrrev.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stocco G, Cheok MH, Crews KR, Dervieux T, French D, Pei D, Yang W, Cheng C, Pui CH, Relling MV, Evans WE. 2009. Genetic polymorphism of inosine triphosphate pyrophosphatase is a determinant of mercaptopurine metabolism and toxicity during treatment for acute lymphoblastic leukemia. Clin Pharmacol Ther 85:164–172. doi: 10.1038/clpt.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bierau J, Lindhout M, Bakker JA. 2007. Pharmacogenetic significance of inosine triphosphatase. Pharmacogenomics 8:1221–1228. doi: 10.2217/14622416.8.9.1221. [DOI] [PubMed] [Google Scholar]
- 9.Waisertreiger IS, Menezes MR, Randazzo J, Pavlov YI. 2010. Elevated levels of DNA strand breaks induced by a base analog in the human cell line with the P32T ITPA variant. J Nucleic Acids 2010:872180. doi: 10.4061/2010/872180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Behmanesh M, Sakumi K, Abolhassani N, Toyokuni S, Oka S, Ohnishi YN, Tsuchimoto D, Nakabeppu Y. 2009. ITPase-deficient mice show growth retardation and die before weaning. Cell Death Differ 16:1315–1322. doi: 10.1038/cdd.2009.53. [DOI] [PubMed] [Google Scholar]
- 11.Revankar GR, Robins RK. 1975. Synthesis and biological activity of some nucleosides resembling guanosine: imidazo(1,2-alpha)pyrimidine nucleosides. Ann N Y Acad Sci 255:166–176. doi: 10.1111/j.1749-6632.1975.tb29221.x. [DOI] [PubMed] [Google Scholar]
- 12.Sidwell RW, Huffman JH, Khare GP, Allen LB, Witkowski JT, Robins RK. 1972. Broad-spectrum antiviral activity of Virazole: 1-beta-d-ribofuranosyl-1,2,4-triazole-3-carboxamide. Science 177:705–706. doi: 10.1126/science.177.4050.705. [DOI] [PubMed] [Google Scholar]
- 13.Wang EE, Randolph AG. 2000. Ribavirin for respiratory syncytial virus infection of the lower respiratory tract. Cochrane Database Syst Rev 2:CD000181. [DOI] [PubMed] [Google Scholar]
- 14.Ascioglu S, Leblebicioglu H, Vahaboglu H, Chan KA. 2011. Ribavirin for patients with Crimean-Congo haemorrhagic fever: a systematic review and meta-analysis. J Antimicrob Chemother 66:1215–1222. doi: 10.1093/jac/dkr136. [DOI] [PubMed] [Google Scholar]
- 15.Gowen BB, Bray M. 2011. Progress in the experimental therapy of severe arenaviral infections. Future Microbiol 6:1429–1441. doi: 10.2217/fmb.11.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee KY, Hung CC. 2014. Ribavirin for chronic hepatitis E virus infection. N Engl J Med 370:2447–2448. doi: 10.1056/NEJMoa1310476. [DOI] [PubMed] [Google Scholar]
- 17.Dusheiko G, Main J, Thomas H, Reichard O, Lee C, Dhillon A, Rassam S, Fryden A, Reesink H, Bassendine M, Norkrans G, Cuypers T, Lelie N, Telfer P, Watson J, Weegink C, Sillikens P, Weiland O. 1996. Ribavirin treatment for patients with chronic hepatitis C: results of a placebo-controlled study. J Hepatol 25:591–598. doi: 10.1016/S0168-8278(96)80225-X. [DOI] [PubMed] [Google Scholar]
- 18.Di Bisceglie AM, Conjeevaram HS, Fried MW, Sallie R, Park Y, Yurdaydin C, Swain M, Kleiner DE, Mahaney K, Hoofnagle JH. 1995. Ribavirin as therapy for chronic hepatitis C. A randomized, double-blind, placebo-controlled trial. Ann Intern Med 123:897–903. [DOI] [PubMed] [Google Scholar]
- 19.Pawlotsky JM, Dahari H, Neumann AU, Hezode C, Germanidis G, Lonjon I, Castera L, Dhumeaux D. 2004. Antiviral action of ribavirin in chronic hepatitis C. Gastroenterology 126:703–714. doi: 10.1053/j.gastro.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Waldenstrom J, Westin J, Nystrom K, Christensen P, Dalgard O, Farkkila M, Lindahl K, Nilsson S, Norkrans G, Krarup H, Norrgren H, Rauning Buhl M, Stenmark S, Lagging M. 2016. Randomized trial evaluating the impact of ribavirin mono-therapy and double dosing on viral kinetics, ribavirin pharmacokinetics and anemia in hepatitis C virus genotype 1 infection. PLoS One 11:e0155142. doi: 10.1371/journal.pone.0155142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL Jr, Haussinger D, Diago M, Carosi G, Dhumeaux D, Craxi A, Lin A, Hoffman J, Yu J. 2002. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 22.Schvarcz R, Ando Y, Sonnerborg A, Weiland O. 1995. Combination treatment with interferon alfa-2b and ribavirin for chronic hepatitis C in patients who have failed to achieve sustained response to interferon alone: Swedish experience. J Hepatol 23(Suppl 2):S17–S21. [PubMed] [Google Scholar]
- 23.Feld JJ, Jacobson IM, Sulkowski MS, Poordad F, Tatsch F, Pawlotsky JM. 2017. Ribavirin revisited in the era of direct-acting antiviral therapy for hepatitis C virus infection. Liver Int 37:5–18. doi: 10.1111/liv.13212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.European Association for the Study of the Liver. 2017. EASL recommendations on treatment of hepatitis C 2016. J Hepatol 66:153–194. doi: 10.1016/j.jhep.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 25.Curry MP, O'Leary JG, Bzowej N, Muir AJ, Korenblat KM, Fenkel JM, Reddy KR, Lawitz E, Flamm SL, Schiano T, Teperman L, Fontana R, Schiff E, Fried M, Doehle B, An D, McNally J, Osinusi A, Brainard DM, McHutchison JG, Brown RS Jr, Charlton M. 2015. Sofosbuvir and velpatasvir for HCV in patients with decompensated cirrhosis. N Engl J Med 373:2618–2628. doi: 10.1056/NEJMoa1512614. [DOI] [PubMed] [Google Scholar]
- 26.Morello J, Rodriguez-Novoa S, Jimenez-Nacher I, Soriano V. 2008. Usefulness of monitoring ribavirin plasma concentrations to improve treatment response in patients with chronic hepatitis C. J Antimicrob Chemother 62:1174–1180. doi: 10.1093/jac/dkn421. [DOI] [PubMed] [Google Scholar]
- 27.Bruchfeld A, Lindahl K, Schvarcz R, Stahle L. 2002. Dosage of ribavirin in patients with hepatitis C should be based on renal function: a population pharmacokinetic analysis. Ther Drug Monit 24:701–708. doi: 10.1097/00007691-200212000-00004. [DOI] [PubMed] [Google Scholar]
- 28.Paroni R, Del Puppo M, Borghi C, Sirtori CR, Galli Kienle M. 1989. Pharmacokinetics of ribavirin and urinary excretion of the major metabolite 1,2,4-triazole-3-carboxamide in normal volunteers. Int J Clin Pharmacol Ther Toxicol 27:302–307. [PubMed] [Google Scholar]
- 29.Brochot E, Castelain S, Duverlie G, Capron D, Nguyen-Khac E, Francois C. 2010. Ribavirin monitoring in chronic hepatitis C therapy: anaemia versus efficacy. Antivir Ther 15:687–695. doi: 10.3851/IMP1609. [DOI] [PubMed] [Google Scholar]
- 30.De Franceschi L, Fattovich G, Turrini F, Ayi K, Brugnara C, Manzato F, Noventa F, Stanzial AM, Solero P, Corrocher R. 2000. Hemolytic anemia induced by ribavirin therapy in patients with chronic hepatitis C virus infection: role of membrane oxidative damage. Hepatology 31:997–1004. doi: 10.1053/he.2000.5789. [DOI] [PubMed] [Google Scholar]
- 31.Streeter DG, Witkowski JT, Khare GP, Sidwell RW, Bauer RJ, Robins RK, Simon LN. 1973. Mechanism of action of 1–d-ribofuranosyl-1,2,4-triazole-3-carboxamide (Virazole), a new broad-spectrum antiviral agent. Proc Natl Acad Sci U S A 70:1174–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scheidel LM, Stollar V. 1991. Mutations that confer resistance to mycophenolic acid and ribavirin on Sindbis virus map to the nonstructural protein nsP1. Virology 181:490–499. doi: 10.1016/0042-6822(91)90881-B. [DOI] [PubMed] [Google Scholar]
- 33.Eriksson B, Helgstrand E, Johansson NG, Larsson A, Misiorny A, Noren JO, Philipson L, Stenberg K, Stening G, Stridh S, Oberg B. 1977. Inhibition of influenza virus ribonucleic acid polymerase by ribavirin triphosphate. Antimicrob Agents Chemother 11:946–951. doi: 10.1128/AAC.11.6.946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maag D, Castro C, Hong Z, Cameron CE. 2001. Hepatitis C virus RNA-dependent RNA polymerase (NS5B) as a mediator of the antiviral activity of ribavirin. J Biol Chem 276:46094–46098. doi: 10.1074/jbc.C100349200. [DOI] [PubMed] [Google Scholar]
- 35.Fang SH, Hwang LH, Chen DS, Chiang BL. 2000. Ribavirin enhancement of hepatitis C virus core antigen-specific type 1 T helper cell response correlates with the increased IL-12 level. J Hepatol 33:791–798. doi: 10.1016/S0168-8278(00)80312-8. [DOI] [PubMed] [Google Scholar]
- 36.Crotty S, Maag D, Arnold JJ, Zhong W, Lau JY, Hong Z, Andino R, Cameron CE. 2000. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat Med 6:1375–1379. doi: 10.1038/82191. [DOI] [PubMed] [Google Scholar]
- 37.Crotty S, Cameron CE, Andino R. 2001. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc Natl Acad Sci U S A 98:6895–6900. doi: 10.1073/pnas.111085598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chevaliez S, Brillet R, Lazaro E, Hezode C, Pawlotsky JM. 2007. Analysis of ribavirin mutagenicity in human hepatitis C virus infection. J Virol 81:7732–7741. doi: 10.1128/JVI.00382-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waldenstrom J, Farkkila M, Rembeck K, Norkrans G, Langeland N, Morch K, Pedersen C, Rauning Buhl M, Nieminen U, Nuutinen H, Alsio A, Holmstrom L, Jungnelius R, Lund K, Rubensson A, Torell E, Westin J, Lagging M. 2016. Short interferon and ribavirin treatment for HCV genotype 2 or 3 infection: NORDynamIC trial and real-life experience. Scand J Gastroenterol 51:337–343. doi: 10.3109/00365521.2015.1087588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feld JJ, Nanda S, Huang Y, Chen W, Cam M, Pusek SN, Schweigler LM, Theodore D, Zacks SL, Liang TJ, Fried MW. 2007. Hepatic gene expression during treatment with peginterferon and ribavirin: Identifying molecular pathways for treatment response. Hepatology 46:1548–1563. doi: 10.1002/hep.21853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas E, Feld JJ, Li Q, Hu Z, Fried MW, Liang TJ. 2011. Ribavirin potentiates interferon action by augmenting interferon-stimulated gene induction in hepatitis C virus cell culture models. Hepatology 53:32–41. doi: 10.1002/hep.23985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rotman Y, Noureddin M, Feld JJ, Guedj J, Witthaus M, Han H, Park YJ, Park SH, Heller T, Ghany MG, Doo E, Koh C, Abdalla A, Gara N, Sarkar S, Thomas E, Ahlenstiel G, Edlich B, Titerence R, Hogdal L, Rehermann B, Dahari H, Perelson AS, Hoofnagle JH, Liang TJ. 2014. Effect of ribavirin on viral kinetics and liver gene expression in chronic hepatitis C. Gut 63:161–169. doi: 10.1136/gutjnl-2012-303852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galli A, Mens H, Gottwein JM, Gerstoft J, Bukh J. 2018. Antiviral effect of ribavirin against HCV associated with increased frequency of G-to-A and C-to-U transitions in infectious cell culture model. Sci Rep 8:4619. doi: 10.1038/s41598-018-22620-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bausch DG, Hadi CM, Khan SH, Lertora JJ. 2010. Review of the literature and proposed guidelines for the use of oral ribavirin as postexposure prophylaxis for Lassa fever. Clin Infect Dis 51:1435–1441. doi: 10.1086/657315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vasanthakumar A, Davis JW, Abunimeh M, Soderholm J, Zha J, Dumas EO, Cohen DE, Waring JF, Lagging M. 2018. Reduced ITPase activity and favorable IL28B genetic variant protect against ribavirin-induced anemia in interferon-free regimens. PLoS One 13:e0198296. doi: 10.1371/journal.pone.0198296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feld JJ, Hoofnagle JH. 2005. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 436:967–972. doi: 10.1038/nature04082. [DOI] [PubMed] [Google Scholar]
- 47.Scheidel LM, Durbin RK, Stollar V. 1987. Sindbis virus mutants resistant to mycophenolic acid and ribavirin. Virology 158:1–7. doi: 10.1016/0042-6822(87)90230-3. [DOI] [PubMed] [Google Scholar]
- 48.Watkins NE Jr, SantaLucia J Jr. 2005. Nearest-neighbor thermodynamics of deoxyinosine pairs in DNA duplexes. Nucleic Acids Res 33:6258–6267. doi: 10.1093/nar/gki918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Todt D, Gisa A, Radonic A, Nitsche A, Behrendt P, Suneetha PV, Pischke S, Bremer B, Brown RJ, Manns MP, Cornberg M, Bock CT, Steinmann E, Wedemeyer H. 2016. In vivo evidence for ribavirin-induced mutagenesis of the hepatitis E virus genome. Gut 65:1733–1743. doi: 10.1136/gutjnl-2015-311000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crick FH. 1966. Codon–anticodon pairing: the wobble hypothesis. J Mol Biol 19:548–555. doi: 10.1016/S0022-2836(66)80022-0. [DOI] [PubMed] [Google Scholar]
- 51.Ohtsuka E, Matsuki S, Ikehara M, Takahashi Y, Matsubara K. 1985. An alternative approach to deoxyoligonucleotides as hybridization probes by insertion of deoxyinosine at ambiguous codon positions. J Biol Chem 260:2605–2608. [PubMed] [Google Scholar]
- 52.Loakes D. 2001. Survey and summary: the applications of universal DNA base analogues. Nucleic Acids Res 29:2437–2447. doi: 10.1093/nar/29.12.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Spee JH, de Vos WM, Kuipers OP. 1993. Efficient random mutagenesis method with adjustable mutation frequency by use of PCR and dITP. Nucleic Acids Res 21:777–778. doi: 10.1093/nar/21.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tee KL, Wong TS. 2013. Polishing the craft of genetic diversity creation in directed evolution. Biotechnol Adv 31:1707–1721. doi: 10.1016/j.biotechadv.2013.08.021. [DOI] [PubMed] [Google Scholar]
- 55.Mbanzibwa DR, Tian Y, Mukasa SB, Valkonen JP. 2009. Cassava brown streak virus (Potyviridae) encodes a putative Maf/HAM1 pyrophosphatase implicated in reduction of mutations and a P1 proteinase that suppresses RNA silencing but contains no HC-Pro. J Virol 83:6934–6940. doi: 10.1128/JVI.00537-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Poordad F, Sievert W, Mollison L, Bennett M, Tse E, Brau N, Levin J, Sepe T, Lee SS, Angus P, Conway B, Pol S, Boyer N, Bronowicki JP, Jacobson I, Muir AJ, Reddy KR, Tam E, Ortiz-Lasanta G, de Ledinghen V, Sulkowski M, Boparai N, McPhee F, Hughes E, Swenson ES, Yin PD. 2015. Fixed-dose combination therapy with daclatasvir, asunaprevir, and beclabuvir for noncirrhotic patients with HCV genotype 1 infection. JAMA 313:1728–1735. doi: 10.1001/jama.2015.3860. [DOI] [PubMed] [Google Scholar]
- 57.Nelson DR, Cooper JN, Lalezari JP, Lawitz E, Pockros PJ, Gitlin N, Freilich BF, Younes ZH, Harlan W, Ghalib R, Oguchi G, Thuluvath PJ, Ortiz-Lasanta G, Rabinovitz M, Bernstein D, Bennett M, Hawkins T, Ravendhran N, Sheikh AM, Varunok P, Kowdley KV, Hennicken D, McPhee F, Rana K, Hughes EA. 2015. All-oral 12-week treatment with daclatasvir plus sofosbuvir in patients with hepatitis C virus genotype 3 infection: ALLY-3 phase III study. Hepatology 61:1127–1135. doi: 10.1002/hep.27726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hitomi Y, Cirulli ET, Fellay J, McHutchison JG, Thompson AJ, Gumbs CE, Shianna KV, Urban TJ, Goldstein DB. 2011. Inosine triphosphate protects against ribavirin-induced adenosine triphosphate loss by adenylosuccinate synthase function. Gastroenterology 140:1314–1321. doi: 10.1053/j.gastro.2010.12.038. [DOI] [PubMed] [Google Scholar]
- 59.Lagging M, Wejstal R, Norkrans G, Karlstrom O, Aleman S, Weiland O, Castedal M, Josephson F, Swedish Consensus Group. 2016. Treatment of hepatitis C virus infection for adults and children: updated Swedish consensus recommendations. Infect Dis (Lond) 48:251–261. doi: 10.3109/23744235.2015.1113438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lindh M, Hannoun C. 2005. Genotyping of hepatitis C virus by Taqman real-time PCR. J Clin Virol 34:108–114. doi: 10.1016/j.jcv.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 61.Nystrom K, Biller M, Grahn A, Lindh M, Larson G, Olofsson S. 2004. Real time PCR for monitoring regulation of host gene expression in herpes simplex virus type 1-infected human diploid cells. J Virol Methods 118:83–94. doi: 10.1016/j.jviromet.2004.01.019. [DOI] [PubMed] [Google Scholar]
- 62.Sjoblom I, Glorioso JC, Sjogren-Jansson E, Olofsson S. 1992. Antigenic structure of the herpes simplex virus type 1 glycoprotein C: demonstration of a linear epitope situated in an environment of highly conformation-dependent epitopes. APMIS 100:229–236. doi: 10.1111/j.1699-0463.1992.tb00865.x. [DOI] [PubMed] [Google Scholar]
- 63.Jia S, Marjavaara L, Buckland R, Sharma S, Chabes A. 2015. Determination of deoxyribonucleoside triphosphate concentrations in yeast cells by strong anion-exchange high-performance liquid chromatography coupled with ultraviolet detection. Methods Mol Biol 1300:113–121. doi: 10.1007/978-1-4939-2596-4_8. [DOI] [PubMed] [Google Scholar]