

Identification of viral mutations that compromise HIV's ability to replicate may aid rational vaccine design. However, while certain escape mutations in Gag have been shown to reduce HIV replication and influence clinical progression, less is known about the consequences of mutations that naturally arise in other HIV proteins. Pol is a highly conserved protein, but the impact of Pol function on HIV disease progression is not well defined. Here, we generated recombinant viruses using the RT-integrase region of Pol derived from HIV-1C-infected individuals with recent and chronic infection and measured their ability to replicate in vitro. We demonstrate that RT-integrase-driven replication ability significantly impacts HIV disease progression. We further show evidence of immune-mediated attenuation in RT-integrase and identify specific polymorphisms in RT-integrase that significantly decrease HIV-1 replication ability, suggesting which Pol epitopes could be explored in vaccine development.
KEYWORDS: replication capacity, HIV-1 subtype C, HLA polymorphisms
ABSTRACT
CD8+ T cell-mediated escape mutations in Gag can reduce HIV-1 replication capacity (RC) and alter disease progression, but less is known about immune-mediated attenuation in other HIV-1 proteins. We generated 487 recombinant viruses encoding RT-integrase from individuals with chronic (n = 406) and recent (n = 81) HIV-1 subtype C infection and measured their in vitro RC using a green fluorescent protein (GFP) reporter T cell assay. In recently infected individuals, reverse transcriptase (RT)-integrase-driven RC correlated significantly with viral load set point (r = 0.25; P = 0.03) and CD4+ T cell decline (P = 0.013). Moreover, significant associations between RT integrase-driven RC and viral load (r = 0.28; P < 0.0001) and CD4+ T cell count (r = −0.29; P < 0.0001) remained in chronic infection. In early HIV infection, host expression of the protective HLA-B*81 allele was associated with lower RC (P = 0.05), as was expression of HLA-B*07 (P = 0.02), suggesting early immune-driven attenuation of RT-integrase by these alleles. In chronic infection, HLA-A*30:09 (in linkage disequilibrium with HLA-B*81) was significantly associated with lower RC (P = 0.05), and all 6 HLA-B alleles with the lowest RC measurements represented protective alleles, consistent with long-term effects of host immune pressures on lowering RT-integrase RC. The polymorphisms V241I, I257V, P272K, and E297K in reverse transcriptase and I201V in integrase, all relatively uncommon polymorphisms occurring in or adjacent to optimally described HLA-restricted cytotoxic T-lymphocyte epitopes, were associated with reduced RC. Together, our data suggest that RT-integrase-driven RC is clinically relevant and provide evidence that immune-driven selection of mutations in RT-integrase can compromise RC.
IMPORTANCE Identification of viral mutations that compromise HIV's ability to replicate may aid rational vaccine design. However, while certain escape mutations in Gag have been shown to reduce HIV replication and influence clinical progression, less is known about the consequences of mutations that naturally arise in other HIV proteins. Pol is a highly conserved protein, but the impact of Pol function on HIV disease progression is not well defined. Here, we generated recombinant viruses using the RT-integrase region of Pol derived from HIV-1C-infected individuals with recent and chronic infection and measured their ability to replicate in vitro. We demonstrate that RT-integrase-driven replication ability significantly impacts HIV disease progression. We further show evidence of immune-mediated attenuation in RT-integrase and identify specific polymorphisms in RT-integrase that significantly decrease HIV-1 replication ability, suggesting which Pol epitopes could be explored in vaccine development.
INTRODUCTION
CD8+ cytotoxic T lymphocytes (CTL), which recognize infected cells through the surface presentation of various HIV-1 epitopes by different HLA class I molecules, play an important role in controlling HIV-1 replication (1, 2). However, mutations may arise in or adjacent to epitopes which allow HIV-1-infected cells to evade detection by CD8+ T cells (1). Some of these escape mutations, particularly those in conserved regions, confer substantial costs to HIV-1 replication (3). While the ideal CTL-based vaccine would target CD8+ T cell responses to multiple vulnerable regions of HIV-1 where escape is impossible, the highly mutable nature of HIV-1 renders this strategy not feasible in practice. Another approach is therefore to focus CTL responses to regions where immune escape can only occur at major cost to viral replication, thereby delaying the ultimate time to escape and, when it occurs, reducing its net immune evasion benefit to HIV by lowering viral load set point, slowing clinical progression, and reducing onward transmission of the virus (4, 5).
While substantial replication costs of several immune escape mutations in HIV-1 Gag have been demonstrated (3, 6–9), less is known about functional consequences of immune-driven mutations in other HIV-1 proteins. Gag has been the focus of such studies, since there is much evidence that Gag CD8+ T cell responses are key to mediating viral control (10, 11) and that human leukocyte antigen (HLA) alleles associated with slower disease progression mediate their protective effects through restricting strong CD8+ T cell responses to key conserved epitopes in Gag, where escape mutations occur at a cost to viral replicative fitness (12, 13). However, Pol (comprised of protease, reverse transcriptase [RT], and integrase) is another conserved protein that is essential to viral replication and contains many CD8+ T cell epitopes (14 and http://www.hiv.lanl.gov/content/immunology/). Indeed, protease-RT replication capacity contributes significantly to whole isolate replication capacity (r2 = 0.53 and P = 0.007) and correlates with plasma viral load (r2 = 0.44 and P = 0.019), supporting that this Pol region contributes significantly to HIV disease progression (15). Interestingly, although Pol CD8+ T cell responses are generally subdominant in natural infection (16), in a phase I clinical trial of a conserved-elements vaccine, the Pol-specific CD8+ T cells induced correlated most strongly with the ability of vaccine-induced CD8+ T cells to suppress viral growth in vitro, indicating that induction of Pol responses that are normally subdominant in natural infection represents a favorable vaccine strategy (17, 18). Furthermore, in a study identifying peptides to which CD8+ T cell responses are associated with significantly lower viral loads, more beneficial Pol peptides (n = 12) than Gag peptides (n = 10) were identified (19, 20), suggesting that there are vulnerable regions in Pol suitable for vaccine inclusion. Finally, the observation that mutations in Pol can significantly affect HIV replicative fitness is supported by studies of drug resistance mutations (21–23) as well as studies of natural Pol sequences in persons infected with HIV-1 subtype B (24, 25).
Here, we aimed to comprehensively investigate the effect of immune-driven mutations in Pol, specifically RT-integrase, on HIV-1 replication capacity (RC) and its impact on the clinical course of HIV infection. To do this, we constructed a large (n = 487) panel of recombinant viruses expressing RT-integrase sequences from individuals recently (n = 81) and chronically (n = 406) infected with HIV-1 subtype C, the most prevalent subtype globally. Our results indicate that RT-integrase-mediated RC is clinically relevant and further suggest that immune-driven mutations in RT-integrase can significantly attenuate HIV.
RESULTS
Construction and replicative assessment of recombinant viruses derived from early and chronic HIV-1C infection.
HIV-1 recombinant viruses expressing bulk plasma HIV RNA-derived RT-integrase sequences from 81 recently and 406 chronically subtype C-infected antiretroviral-naive individuals (Table 1) were constructed via homologous recombination in an HIV-1 subtype B (NL4-3) backbone. The mean time from cotransfection to recombinant virus harvest was 15 days (interquartile range [IQR], 14 to 16 days). Resequencing and phylogenetic comparison of the RT-integrase region of 52 randomly selected recombinant viruses from recently (n = 9) and chronically (n = 42) infected individuals with their original plasma HIV-1 RNA sequences confirmed participant origin in all cases (Fig. 1A) and also confirmed that recombinant virus sequences were highly representative of the original plasma sequences. The median number of full nucleotide differences between recombinant virus and bulk plasma sequences was 3.5 (IQR, 2 to 7.75) out of 2,547 nucleotides (99.92% nucleotide similarity), while the median number of full amino acid differences was 1 (IQR, 0 to 2) of 849 codons (99.97% amino acid similarity). Recombinant viruses also retained a substantial amount of the original within-host plasma HIV diversity in both recent and chronic infection. Consistent with minimal viral diversity in early infection, recombinant viruses generated from recently infected persons contained a mean of 2.9 (standard deviation [SD], 2.33) nucleotide mixtures compared to 2 (SD, 2.05) in the original plasma, while recombinant viruses generated from individuals in chronic infection contained means of 6 (SD, 6) compared to 17 (SD, 13) in the original plasma sequences (Fig. 1B).
TABLE 1.
Clinical characteristics
| Parameter | Median value (IQR) or no. of individuals from each cohorte |
|
|---|---|---|
| Recent infection (n = 81) | Chronic (n = 406) | |
| Plasma viral load (log10 HIV RNA copies/ml) | 5.0 (4.2–5.3) | 4.8b (4.2–5.3) |
| Baseline CD4+ T cell count (cells/mm3) | 419 (230–482) | 338c (230–479) |
| Baseline age (yr) | 26 (23–30) | 31d (26–36) |
| Gender, no. (%) | ||
| Female | 56 (69) | 320 (79) |
| Male | 25 (31) | 86 (21) |
| Estimated time postinfection (days) | 49a (34–69) | ND |
Days postinfection could be estimated for 76 individuals; the remaining 5 were identified in Fiebig stage VI.
Plasma viral load was available for 400 individuals.
Baseline CD4+ T cell count was available for 405 individuals.
Age was available for 402 individuals.
All values, except for those for gender, are indicated as median and IQR. ND, not determined.
FIG 1.
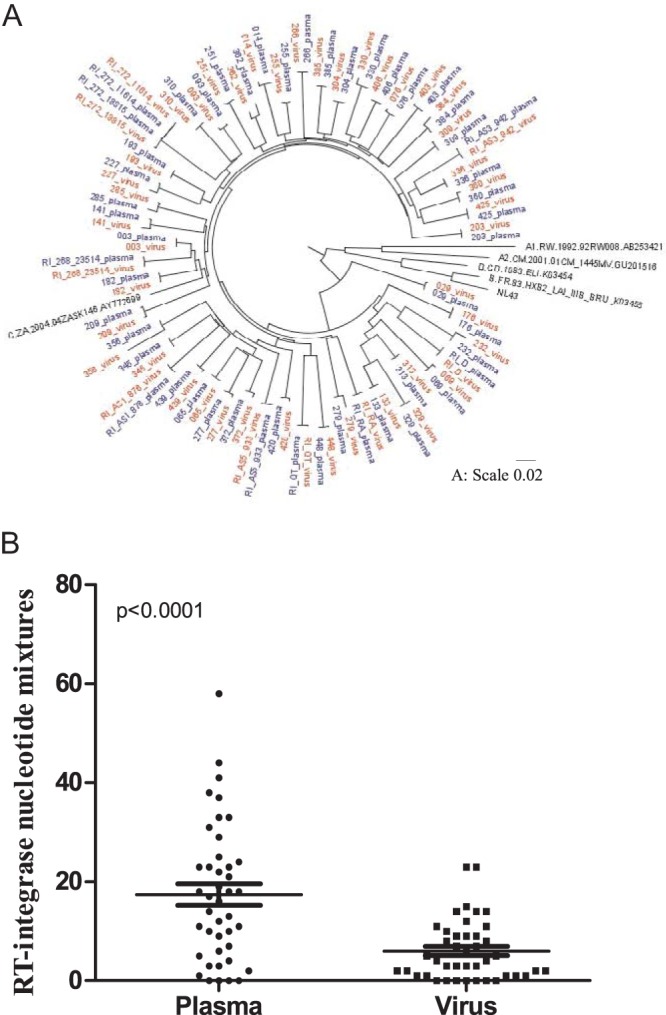
Phylogenetic relatedness and diversity of the RT-integrase sequences from bulk plasma and matched recombinant viruses. (A) A maximum likelihood tree showing the phylogenetic relatedness of HIV-1 subtype C RT-integrase sequences from the plasma and respective recombinant viruses at a scale of 0.02. The prefix RI indicates sequences derived from recently infected individuals, while the rest of the sequences were derived from chronically infected individuals. Blue indicates the bulk plasma sequences, while red indicates the recombinant virus sequences. (B) The mean number of nucleotide mixtures in plasma sequences derived from chronic infection (mean, 17; standard deviation, 13) compared to their respective recombinant virus sequences (mean, 6; standard deviation, 6) is shown. This indicates reduced diversity in the recombinant virus sequences (paired t test). For example, for participant SK313, there were 6 full nucleotide differences between the plasma and virus sequences and the number of mixtures in the plasma and virus sequences was 17 and 6, respectively.
In vitro RC of each participant-derived RT-integrase recombinant viral stock, where RC was expressed as the slope of viral spread from days 3 to 6 postinfection relative to that of the control NL4-3 strain (26), was assessed in duplicate in independent experiments (replicate measurements were highly concordant: r = 0.85 by Pearson's correlation and P < 0.0001; data not shown) and subsequently were reported as the averages from these two measurements. Overall, the RC of the recombinant viruses derived from recent infection (median RC, 0.92; IQR, 0.85 to 0.96) were comparable to those derived from the chronic infection (median RC, 0.92; IQR, 0.87 to 0.98) (P = 0.2 by Student's t test) (Fig. 2A) and approximated a Gaussian distribution. Gag-protease-mediated RC was previously measured using the same cohorts described here (13, 27). A weak yet statistically significant correlation was observed between Gag-protease- and RT-integrase-mediated replication capacity in the subset of 65 recently and 388 chronically infected individuals for whom both measurements were available (r = 0.18 by Pearson's correlation and P = 0.0002) (Fig. 2B).
FIG 2.
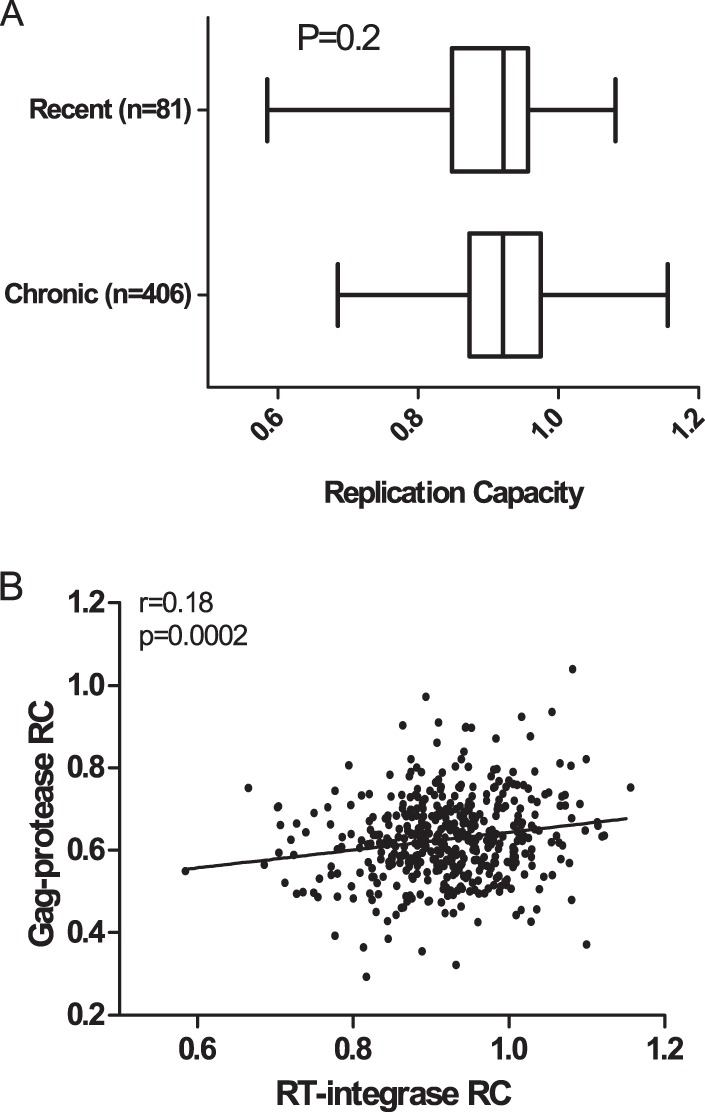
Relationship between Gag-protease- and RT-integrase-mediated RC. (A) The distributions of RT-integrase-driven RC for HIV-1C recent and chronic infection are shown. (B) The RC of NL4-3 recombinant viruses encoding participant-derived RT-integrase and those encoding Gag-protease from the same individuals were significantly correlated (P = 0.0002 by Pearson's correlation).
Longer RT-integrase sequences are associated with reduced RC.
A substantial minority of integrase sequences from both the chronic (n = 89 [22%]) and recent infection (n = 19 [23.5%]) cohorts had an extra 17 amino acids at their 3′ ends where the stop codon at the end of integrase was mutated, leading to the usage of a stop codon 17 residues downstream. Full HIV genome sequences were available for a subset (n = 114) of chronically infected participants (28). RT-integrase sequences from the present study clustered with their previously amplified full-genome counterparts, and the extended RT-integrase sequences were also featured in the full-genome counterparts (in 34 of 114; 30%) (data not shown). Of note, recombinant viruses with extended integrase sequences displayed lower RC than sequences of the usual length (P = 0.0047 by Student's t test) (Fig. 3). To confirm whether this is a subtype C-specific phenomenon or common in other HIV-1 subtypes, we downloaded 8,776 (one sequence per individual) RT-integrase sequences (5′-3′ HXB2 positions 2,550 to 5,147) from the LANL database (https://www.hiv.lanl.gov/content/sequence/HIV/mainpage.html) and checked for mutation at the 849 codon, which would indicate use of the stop codon at position 866. There were 686 (7.8%) sequences mutated at the first stop codon, of which 498 (72.6%) were subtype C or C-containing recombinants, while the remaining 27.4% comprised other non-B subtypes.
FIG 3.
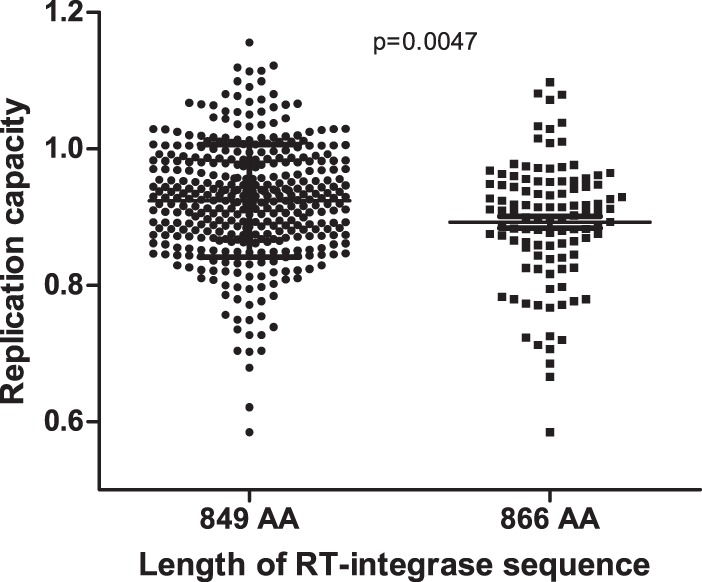
Length of RT-integrase sequence impacts RC. NL4-3 recombinant viruses encoding participant-derived RT-integrase sequences of 866 amino acids (AA) had a lower RC than those encoding participant-derived RT-integrase sequences of the same length as HXB2 (849 AA) (P = 0.0047 by Student's t test).
RT-integrase-mediated RC is clinically relevant.
To address whether RT-integrase function influences HIV-1 subtype C disease progression, we correlated the RC of the recombinant viruses for the recently infected individuals with subsequent viral load set point and rate of CD4+ T cell decline. Only 72 and 78 (of 81 total) individuals had sufficient treatment-free follow-up data available to calculate viral load set point (median of 4.7 [IQR, 4.1 to 5.1] log10 copies/ml) and rate of CD4+ T cell decline (median of 4 [IQR, −11.7 to 0.6] cells/mm3 per month), respectively. The RT-integrase RC correlated positively with subsequent viral load set point (r = 0.25 by Spearman's correlation and P = 0.03) (Fig. 4A). These results remained consistent (r = 0.28 by Spearman's correlation and P = 0.03) when the participants from the Females Rising through Education, Support and Health (FRESH), cohort were excluded from the analysis to minimize possible confounding effects of recruitment stage (FRESH participants were identified in the hyperacute phase, a median of 16 [IQR, 15 to 17] days postinfection, while the participants in the Botswana Tshedimoso and HIV Pathogenesis Programme [HPP] cohorts were recruited a median of 66 [IQR, 32 to 79] and 49 [IQR, 44 to 62] days postinfection, respectively.
FIG 4.
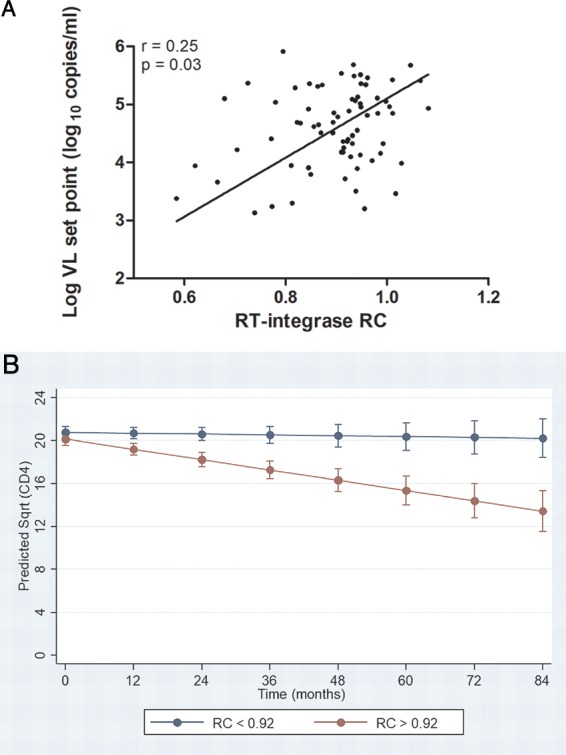
Relationship between RT-integrase-mediated RC in early infection and markers of disease progression. (A) The RC of the recombinant viruses encoding RT-integrase derived from individuals with early HIV-1 C infection correlated negatively with subsequent viral load set point (P = 0.03 by Spearman's correlation). (B) Higher RC corresponds to a higher rate of CD4 decline. The regression lines show prediction of CD4 decline when RT-integrase RC is stratified by the median as RC of >0.92 (red) and RC of <0.92 (blue) (P < 0.001 by GEE model). The bars represent 95% confidence intervals. Sqrt, square root.
Analyses of rates of CD4+ T cell decline were restricted to the Tshedimoso and HPP participants only (as FRESH participants had substantially higher baseline CD4+ T cell counts owing to their recruitment during hyperacute infection): in total, 68 participants were included in the generalized estimating equation (GEE) model, with RT-integrase and time × RT-integrase RC interaction terms as predictors. The mean number of CD4+ T cell count measurements per individual was 7 (range, 2 to 20). As evidenced by the interaction term in the model, there was a significant association of higher RT-integrase-driven RC and higher rate of CD4+ T cell decline for the recently infected individuals (P = 0.013). To facilitate graphical depiction of these results, a similar model was fitted that dichotomized RC at the population median, yielding high (RC of >0.92) and low (RC of <0.92) groups; high RC was consistently associated with faster CD4+ T cell decline (P < 0.001) (Fig. 4B). In contrast, similar analysis of the relationship between Gag-protease RC values and rates of CD4+ T cell decline in the subset of 58 participants for whom these values were available revealed no statistically significant relationship (P = 0.124; n = 58) (data not shown).
This finding suggests that RT-integrase-driven RC is a stronger correlate of CD4+ T cell decline than Gag-protease RC, at least in this cohort subset, but it is important to acknowledge the reduced power in the latter analysis due to a smaller sample size. Indeed, given that Gag-protease and RT-integrase RC are significantly associated (Fig. 2B) and that Gag-driven replication capacity has been previously associated with CD4+ T cell decline in larger cohorts (27, 29), we wished to further explore whether RT-integrase-driven RC represents the primary correlate of CD4+ T cell decline or whether there are additive effects of RT-integrase and Gag-protease RC on clinical outcome. To do this, we categorized participants according to whether their RT-integrase and Gag-protease RC values were above or below the population medians for these measurements. Notably, we observed that the rate of CD4+ T cell decline was significantly higher in those participants with RT-integrase and Gag-protease RC values both above the population medians than in those participants with RT-integrase RC values above the population median but Gag-protease RC values below the population median (P = 0.002). Taken together, the models indicate that RT-integrase RC and Gag RC have an additive effect on the rate of CD4+ T cell decline.
To confirm whether the relationship between RT-integrase function and HIV-1 subtype C disease progression persists long term, for the chronically infected individuals we correlated RT-integrase-mediated RC with contemporaneous viral loads and CD4+ T cell counts. There was a significant correlation between RC and viral load (r = 0.28 by Pearson's correlation and P < 0.0001) and between RC and CD4+ T cell count (r = −0.29 by Spearman's correlation and P < 0.0001) (Fig. 5A and B, respectively), suggesting that RT-integrase function continues to influence HIV clinical parameters into chronic infection.
FIG 5.
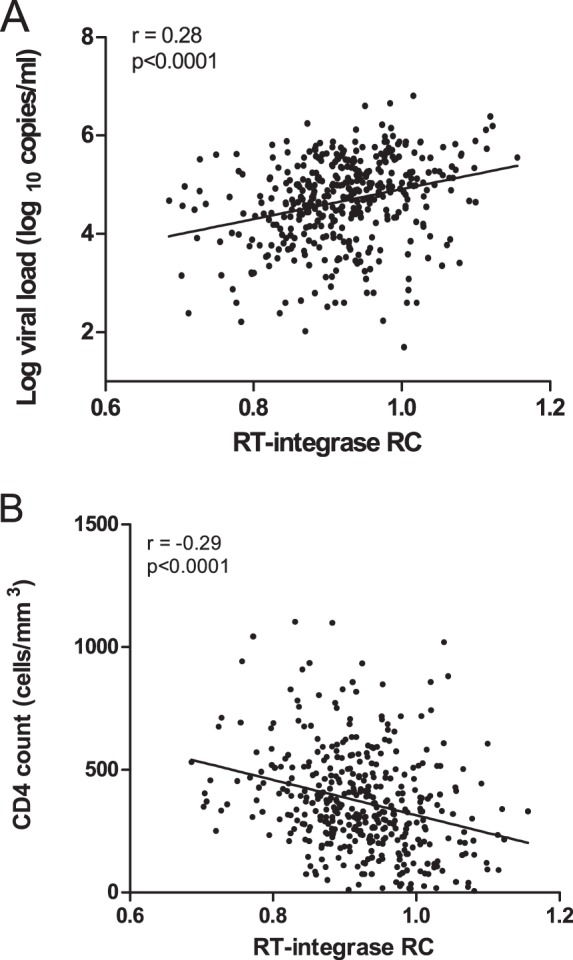
Relationship between RT-integrase-mediated RC in chronic infection and markers of disease progression. The RC of NL4-3 recombinant viruses encoding RT-integrase derived from chronically HIV-1C-infected individuals correlated positively with the plasma log viral load (A) and negatively with the CD4 count (B). Pearson's (P < 0.0001) and Spearman's (P < 0.0001) correlations were used, respectively.
Taken together, our observations of a significant relationship between RT-integrase RC and markers of HIV progression in both early and chronic infection strongly support RT-integrase-mediated RC as a clinically relevant viral attribute whose effects are evident immediately following infection and persist into chronic infection. This relationship of RC and disease progression in early infection could be attributed to RC of the transmitted virus being an inherent property that influences clinical progression (30). In addition, this could be associated with the selection of early immune-driven mutations that compromise viral RC (14, 31).
Relationship between RT-integrase-driven RC and host HLA-I allele expression.
If fitness-costly RT-integrase escape mutations were selected as a result of potent HLA-restricted host immune responses in early HIV infection, then one would expect to see a modulatory effect of host HLA allele carriage on RT-integrase RC, even in early infection. For this reason, we stratified RT-integrase RC data by host HLA-I allele carriage and compared RC distributions in participants expressing versus not expressing that allele using Student's t test (Fig. 6). For the recent infection data set, FRESH cohort participants (n = 9) were excluded, as it is unlikely that HLA alleles would have impacted HIV evolution measurably at this very early stage (hyperacute infection). The remaining recent infection samples ranged from 26 to 209 days postinfection; therefore, there is potential for HLA-mediated selection of these viruses. Of note, the known protective alleles HLA-B*81 (P = 0.05) (9, 32, 33) and -B*07 (P = 0.02) were associated with lower RT-integrase-mediated RC, while HLA-C*03 (P = 0.04) was associated with higher RC. However, these associations did not remain significant when corrected for multiple comparisons (q > 0.2) due to the small sample size of the recent infection data set.
FIG 6.

Associations between HLA class I allele expression and RT-integrase RC. The RC of recent (A to C) and chronic (D to F) viruses were stratified by expression of the host HLA-A (A and D), HLA-B (B and E), and HLA-C (C and F) alleles. The box plots display RC results arranged by lowest mean RC at the bottom and the highest mean RC at the top. Dots indicate the means, and vertical lines indicate the medians. Boundaries of the boxes indicate the interquartile ranges, while the whiskers display the maximum and minimum viral RC values. The continuous vertical line on each graph indicates the mean RC for each cohort. HLA-I alleles with a minimum of n = 5 are shown. Asterisks indicate HLA alleles that are significantly associated with either higher or lower RC (Student's t test).
Studies of HIV-1 Gag-protease RC in subtype B infection have indicated that fitness-costly immune escape mutations selected in early infection are largely compensated for via secondary mutations by chronic infection, largely obscuring HLA relationships with RC at this disease stage (6, 14). Nevertheless, relationships between HLA alleles and Gag-protease RC have been shown to persist into chronic infection in subtype C (13). We investigated this for RT-integrase in the chronic cohort and observed that HLA-A*30:09 (P = 0.048) was associated with lower RC while HLA-B*18:01 (P = 0.02) and B*49:01 (P = 0.03) were associated with higher RC, and these associations remained significant after correction for multiple comparisons (q < 0.07) (Fig. 6D to F). Consistent with the idea that RC influences clinical outcomes, 4 out of the 5 individuals expressing HLA-A*30:09 also coexpressed the protective HLA-B*81 allele (9, 32, 33), while HLA-B*18:01 is a reported detrimental allele in subtype C infection (33). It was notable that previously described protective alleles in HIV-1 subtype C infection (B*39:10 [34, 35], B*13 [35], B*57 [5, 6, 12], B*81 [9, 32, 33], and B*1516 [36]) ranked in the bottom third for RC data, and all of these had a lower average RC than the mean for the cohort (Fig. 6E). In addition, HLA B*14 and B*1516 have been reported as favorable alleles in HIV-1 subtype B infection (36–38), and they displayed low average RC. Indeed, RT-integrase RC values for individuals expressing at least one of these alleles were significantly lower than those for individuals lacking them (average of 0.91 versus 0.93; P = 0.05; data not shown).
The linkage of HLA-B*81 with lower RC was consistent across recent and chronic infection and achieved statistical significance in the former (despite the small sample size of n = 5), supporting long-term effects of this allele on reducing viral RC. Moreover, 3 of the 5 HLA-B*81-expressing individuals with early infection harbored mutations in the HLA-B*81-restricted epitope SL10 (RT codons 3 to 12), which is otherwise relatively conserved in the rest of the cohort (P = 0.0022 by Fisher's exact test), identifying these variants as candidate fitness-costly mutations. Moreover, the observation that, on average, individuals expressing known protective HLA class I alleles harbor RT-integrase sequences with the lowest RC values more broadly supports the notion of long-lasting immune-mediated attenuation by protective HLA alleles.
Relationship between HLA-associated polymorphisms and RT-integrase RC.
To estimate the possible contributions of HLA-associated polymorphisms on RT-integrase function, we used a previously reported list of HLA-associated polymorphisms obtained from statistical analysis of 1,866 antiretroviral-naive HIV-1 subtype C chronically infected individuals from southern Africa, which included the Sinikithemba participants studied here (39). In this analysis, we considered all polymorphisms irrespective of host HLA alleles (which provides a combined estimate of transmitted and de novo selected immune escape mutations).
In early infection, we observed a weak positive correlation between numbers of polymorphisms in RT and RC (r = 0.22 by Spearman correlation and P = 0.05) (Fig. 7A), which was strengthened when we further restricted the analysis to HLA-associated polymorphisms in or adjacent to optimally described CTL epitopes (http://www.hiv.lanl.gov/content/immunology/) restricted by the same HLA allele (P = 0.02 by Student's t test) (Fig. 7B). Conversely, in integrase, the burden of HLA-associated polymorphisms correlated inversely with RC (r = −0.25 by Spearman's correlation and P = 0.02) (Fig. 7C), a relationship that was again strengthened when we restricted analysis to optimally described CTL epitopes (P < 0.0001 by analysis of variance [ANOVA]) (Fig. 7D). These findings raise the intriguing hypothesis that HLA-associated polymorphisms in RT generally increase RC while those in integrase generally reduce RC, an observation which is reminiscent of our previous finding that the majority of Gag p24 mutations lower RC while accumulation of mutations in p17 correlates with increased RC (13). We did not observe this in chronic infection, although there was a weak positive trend for RT (r = 0.08 by Spearman's correlation and P = 0.09) and a weak negative trend for integrase (r = −0.08 by Spearman's correlation and P = 0.11) (data not shown). This could be due to compensation that occurs in chronic infection (6, 25).
FIG 7.

Relationships between HLA-associated polymorphisms in RT or integrase and RT-integrase-mediated RC in early infection. The number of RT HLA-associated polymorphisms correlated positively with RC (P = 0.05 by Spearman's correlation) (A), and the association was stronger when considering only those polymorphisms present in or adjacent to A list epitopes (P = 0.02 by Student's t test) (B). The number of integrase polymorphisms correlated negatively with RC (P = 0.024 by Spearman's correlation) (C), and the association was strengthened when limited to those polymorphisms present in or adjacent to A list epitopes (P < 0.0001 by ANOVA) (D). Counts were performed irrespective of the host HLA alleles.
Amino acids associated with altered RT-integrase RC.
In order to identify specific RT-integrase amino acid variants that are significantly associated with altered viral RC, we performed a codon-by-codon genotype/phenotype analysis in the combined chronic and recent infection data sets (total of 487 sequences) (Table 2). Of note, 18 out of 20 (90%) polymorphisms identified to be associated with altered RC were inside or within 5 amino acids of optimal A-list epitopes (http://www.hiv.lanl.gov/content/immunology/) represented published HLA-associated polymorphisms specific to HIV subtype C (39) and/or represented published experimentally confirmed CTL escape mutations (Table 2). The polymorphisms associated with the most pronounced decreases in RC (average RC of ≤0.84) were V241I (RT), I257V (RT), P272K (RT), E297K (RT), and I201V (integrase). These are all relatively rare polymorphisms (all <1.5% frequency in our data set) and are in, or adjacent to, optimal A-list CTL epitopes (Table 2), identifying them as potential novel fitness-costly immune-driven escape mutations in Pol. Other more common polymorphisms, namely, E6K (RT), A158S (RT), V112I (integrase), Q136K (integrase), and K211R (integrase), were associated with more moderate decreases in RC, while the remainder of the polymorphisms were associated with either a marginal decrease in RC (yet were sufficiently common to yield statistically significant results) or with increased RC. E6K (RT) is a documented CTL escape mutation (40) and Q136K (integrase) is an HLA-associated polymorphism, while A158S (RT), V112I (integrase), and K211R (integrase) are in/adjacent to optimal list CTL epitopes (Table 2). Taken together, these results support a role for immune-driven mutations in Pol to modulate HIV replication capacity.
TABLE 2.
Codon-by-codon analysis of Pol (RT and integrase) polymorphisms and viral replication capacity for the combined (chronic and recent)h
| Pol codon | aa | Median replication |
No. of viruses |
P value | q value | A-list epitopea | HLA-associated polymorphismb | Documented CTL escapec | ||
|---|---|---|---|---|---|---|---|---|---|---|
| With aa | Without aa | With aa | Without aa | |||||||
| E6(RT) | K | 0.87 | 0.92 | 19 | 382 | 0.017 | 0.34 | 6K (C*18) | 6K, SL10 (B*8101) (41) | |
| V8 | V | 0.92 | 0.98 | 468 | 14 | 0.006 | 0.19 | IL-8 (B*40:01)d | ||
| V8 | I | 0.98 | 0.92 | 14 | 468 | 0.006 | 0.19 | |||
| K22 | K | 0.92 | 1.01 | 473 | 6 | 0.005 | 0.19 | GL9 (B*08) | ||
| K22 | R | 1.01 | 0.92 | 6 | 473 | 0.005 | 0.19 | |||
| A36 | E | 0.94 | 0.92 | 123 | 336 | 0.003 | 0.19 | AK11 (A*02:01/A*03:01) | 36A (B*42:01) | |
| T 48 | T | 0.92 | 0.95 | 458 | 22 | 0.021 | 0.37 | Flank AK11 (A*02:01/A*03:01) | 48T (A*74:01); 48S (B*15:16) | |
| A158 | A | 0.92 | 0.88 | 453 | 25 | 0.001 | 0.19 | LA10 (B*54:01); SM9 (B*07); AK9 (A*03:01/A*11:01)e | ||
| A158 | S | 0.88 | 0.92 | 25 | 453 | 0.001 | 0.19 | |||
| A200 | E | 0.98 | 0.92 | 9 | 467 | 0.021 | 0.37 | Flank IL-9 (B*40:01) | 200A (A*29:02) | |
| V241 | V | 0.92 | 0.82 | 479 | 5 | 0.005 | 0.19 | Flank IW9 (B*57:01) | ||
| V241 | I | 0.82 | 0.92 | 5 | 479 | 0.005 | 0.19 | |||
| I257 | I | 0.92 | 0.82 | 475 | 7 | 0.018 | 0.35 | Flank IW9 (B*57:01) and LY12 (B*15:01) | ||
| I257 | V | 0.80 | 0.92 | 5 | 477 | 0.008 | 0.22 | |||
| P272 | P | 0.93 | 0.91 | 350 | 128 | 0.002 | 0.19 | QR9 (A*03:01); YL9 (B*42:01)f | 272A,S,Q (B*42/42:01) | 272A,S,Q, YL9 (B*4201)(50) |
| P272 | K | 0.84 | 0.92 | 5 | 473 | 0.013 | 0.28 | |||
| E297 | K | 0.80 | 0.92 | 7 | 386 | 0.003 | 0.21 | IL-9 (B*3501,/B*5101) | 297A,G,Q,V, IL-9 (B*7) (51) | |
| K311 | K | 0.92 | 0.89 | 458 | 17 | 0.012 | 0.28 | IY10 (A*02:01, B*15:01, C*12:02) | K311R (52) K311E (51) | |
| M357 | R | 0.93 | 0.92 | 171 | 299 | 0.014 | 0.30 | RK11 (A*30:02, A*03:01) | 357R (A*01) | |
| I50(Integrase) | M | 0.93 | 0.91 | 152 | 280 | 0.010 | 0.25 | |||
| I50 | I | 0.91 | 0.93 | 253 | 179 | 0.027 | 0.30 | |||
| I84 | I | 0.93 | 0.90 | 363 | 97 | 0.001 | 0.10 | HA9 (B*54:01); IT10 (B*40:02) | ||
| I84 | M | 0.91 | 0.93 | 89 | 371 | 0.011 | 0.25 | |||
| Y100 | Y | 0.92 | 0.93 | 346 | 121 | 0.014 | 0.25 | 100Y (A*66); 100F (A*30:02) | ||
| Y100 | F | 0.93 | 0.92 | 118 | 349 | 0.027 | 0.30 | |||
| K111 | K | 0.92 | 0.97 | 469 | 11 | 0.022 | 0.28 | Flank HF8 (C*05) | ||
| V112 | I | 0.92 | 0.89 | 445 | 26 | 0.036 | 0.36 | Flank HF8 (C*05) | ||
| S119 | S | 0.92 | 0.93 | 403 | 75 | 0.015 | 0.25 | HF8 (C*05), Flank SW10 (B*57) | 119P (C*08:02); 834R (C*05:01) | |
| S119 | T | 0.95 | 0.92 | 33 | 445 | 0.016 | 0.25 | |||
| Q136 | K | 0.88 | 0.92 | 28 | 368 | 0.028 | 0.33 | IY9 (B*1503)g | 136K(B*1503) | |
| I201 | V | 0.83 | 0.92 | 5 | 404 | 0.040 | 0.36 | GI8 (B*4002) | ||
| K211 | R | 0.89 | 0.93 | 53 | 420 | 0.001 | 0.10 | IK9 (A*11) | ||
| K211 | K | 0.93 | 0.89 | 414 | 59 | 0.005 | 0.24 | |||
| K269 | K | 0.93 | 0.91 | 238 | 197 | 0.016 | 0.25 | RY9 (B*15:03), Flank VI9 (B*42) | ||
| K269 | R | 0.91 | 0.93 | 197 | 238 | 0.016 | 0.25 | 269R (B*42:01) | 269R, IM9-B*42:01(50) | |
A-list epitope as available from http://www.lanl.com.
HLA-associated polymorphism as described by Carlson et al. (39).
Confirmed and documented CD8+ T cell (CTL) escape as reported in published literature.
SL10 (B*8101), B-list epitope.
SM9 (B*8101, B*39), B-list epitope.
YL9 (B*57, B*81).
IY9 (B*39), B-list epitope.
Codons highlighted in boldface were identified in the combined analysis only, while the others were identified in either or both the chronic and combined analyses. In italics are instances where the consensus amino acids are associated with specific HLA alleles or were documented as the escaped form. Abbreviations: aa, amino acid. All associations for n (>5), P value (>0.05), and q value (<0.4) are listed.
DISCUSSION
Immune escape mutations in key HIV-1 proteins can reduce viral replication (3, 6, 7, 12). HIV-1 Gag function (as measured by Gag-protease-driven RC) influences HIV disease progression (29), and replicative costs of immune escape mutations in Gag have been demonstrated (3, 6–9). However, less is known about immune-mediated attenuation in the other HIV proteins (5).
Here, we investigated immune-mediated attenuation in RT-integrase in the world's most prevalent HIV-1 subtype (C) and demonstrate that RT-integrase-driven RC impacts HIV-1 disease progression. RT-integrase-driven RC in recent infection associated significantly with subsequent viral load set point and rate of CD4+ T cell decline: the lower the RT-integrase-driven RC in early infection, the lower the viral load set point and the slower the rate of CD4+ T cell decline. Furthermore, this effect remained detectable in chronic infection where RT-integrase RC correlated positively with cross-sectional viral loads and negatively with baseline CD4+ T cell counts. Previously, we reported a trend between Gag-protease-driven RC and viral load set point as well as rate of CD4+ T cell decline (27), and another study showed a significant relationship between Gag-driven RC in acute infection and drop in CD4+ T cell count to below 300 cells/mm3 (29). In the present analysis we show an additive effect of Gag-protease RC and RT-integrase RC on rate of CD4+ T cell decline and that RT-integrase RC is individually significantly associated with rate of CD4+ T cell decline. These observations support that RT-integrase is at least as suitable as Gag as a target of an attenuation-based vaccine. Consistent with this interpretation, more vulnerable peptides (those to which immune responses were associated with significantly lower viral loads in natural infection) were identified in Pol than Gag and deemed suitable for inclusion in a human immune data-informed vaccine (19, 20). Furthermore, following administration of a conserved-elements vaccine (largely comprised of Pol and Gag) to healthy volunteers, the immune responses elicited that inhibited viral replication in vitro were largely responses to conserved naturally subdominant Pol epitopes (17).
Our findings also provide evidence for immune-driven attenuation of RT-integrase function. First, we observed associations between certain HLA alleles and altered RT-integrase-driven RC. HLA-A*30:09 (in linkage disequilibrium with HLA-B*81), -B*81, and -B*07 associated with lower RC, suggesting that these alleles restrict CTL responses that drive attenuating mutations in RT-integrase. Indeed, HLA-B*81 (the most protective HLA allele in the context of subtype C in sub-Saharan Africa) was the only individual allele consistently linked with lower RC in both recent and chronic infection, although this was less pronounced in chronic infection, likely due to partial compensation of replicative fitness costs by this stage of infection. This observation parallels a previous report in HIV-1 subtype B, which showed evidence for HLA-B*57-mediated attenuation of RT-integrase function (where B*57 is the most protective HLA allele in the context of subtype B in Western populations [25]) in early but not chronic infection. This suggests that the replicative fitness costs selected by this allele were largely compensated for by the chronic stage of infection. Furthermore, it was notable that there was significantly more amino acid variation in the HLA-B*81-restricted epitope SL10 among HLA-B*81 expressing early in HIV-1 infection than could be expected by chance, suggesting these are fitness-reducing mutations. In addition, the HLA-B*81-associated E6K mutation in RT was significantly associated with lower RC in our codon-by-codon analysis. Moreover, the protective HLA-B alleles B*81, B*57:03, B*39:10, B*13:02, B*15:16, and B*14, when analyzed together as a group, also associated with lower RT-integrase-driven RC. Here, it is important to note that the association of protective alleles, particularly HLA-B*81, with lower viral RC was more pronounced for Gag-protease (9, 13, 27, 31) than with RT-integrase shown here; nevertheless, results from the present study support modest yet potentially biologically relevant effects of HLA-mediated immune pressures on Pol.
We also provide evidence for immune-driven mutations decreasing RT-integrase-driven RC, observations which corroborate reports in HIV-1 subtype B (24, 25). In early HIV-1 infection, we observed a positive correlation between RC and the number of HLA-associated polymorphisms in RT and a weak negative relationship between RC and the number of HLA-associated polymorphisms in integrase; similar trends were observed in chronic HIV-1 infection. Taken together with previous reports of a weak inverse correlation between the number of HLA-B-associated polymorphisms in integrase and RC in HIV-1 subtype B (24), our findings suggest that integrase is particularly susceptible to immune-driven attenuation. Unlike RT, integrase, besides its enzymatic activities, interacts with other proteins, like LEDGF/p75 (41), and this likely makes it more difficult for integrase to accommodate mutations without functional consequence. It has also been reported that integrase displays significantly decreased inter- and intrasubtype diversity and a lower Shannon's entropy than RT (42), which may also partly account for our findings. Furthermore, our observations of opposing effects on RC by HLA-associated polymorphisms in RT versus integrase are reminiscent of our previous observation that the majority of Gag p24 mutations lower RC, while accumulation of mutations in p17 correlates with increased RC (13). These observations raise the intriguing possibility that fitness-costly mutations in one protein could be offset (or even compensated by) mutations in another, a notion that merits investigation in future studies.
Using our combined genotype/RC data set, we also identified specific mutations, several of which are likely immune driven, that are associated with reduced RC. Specifically, the variants associated with the most pronounced decreases in RC were all rare (<1.5% frequency) variants: V241I (RT), I257V (RT), P272K (RT), E297K (RT), and I201V (integrase). Moreover, their location within or immediately flanking optimal epitopes (Table 2) support these as immune-driven polymorphisms. In particular, sequences harboring V241I (RT) and I257V (RT) exhibited the lowest RC; both of these flank the B*57-restricted epitope IW9. Similarly, integrase residue 201 also occurs in an epitope-rich region. Therefore, RT 241-257 and epitopes in the region surrounding integrase 201 could represent vaccine targets in Pol. RT codon 297 is also well conserved (although E297K did not remain associated with reduced RC in the combined data set); however, RT codon 272 is not. P272K is a rare variant, while common escape mutations at this codon did not alter replication capacity, suggesting that this residue is not an ideal vaccine target.
Our observations corroborate a study in HIV-1 subtype B demonstrating that uncommon HLA-associated polymorphisms in integrase were associated with reduced RC (24). Of note, variants at integrase codon S119 (S is the consensus in both subtypes) were significantly associated with altered RC in both subtype B and the present study; however, whereas the subtype B study identified 119R as being detrimental to RC (24), in the present analysis 119T and 119P were associated with increased RC (Table 2). Codon 119 is highly mutable, and there is a possibility that only 119R decreases RC. It is also interesting that in some instances, for example, A36E (RT) and A200E (RT), the subtype consensus represents the adapted (escaped) form for a particular HLA class I allele and is associated with modest reductions in RC, whereas variants at these codons are associated with a marginal increase in RC. Site-directed mutagenesis experiments are required to directly confirm the replicative fitness consequences of these mutations that are statistically associated with altered RC.
The present study utilized recombinant viruses incorporating participant-derived RT-integrase, and therefore potential interactions with other viral proteins encoded in the NL4-3 backbone that could affect RC cannot be ruled out. Similarly, the insertion of subtype C RT-integrase sequences into the subtype B (NL4-3) backbone, chosen because it is a well-studied laboratory strain of HIV that replicates well in cell culture, could conceivably influence viral RC. Indeed, we observed that longer integrase sequences (with respect to HXB2), which is a phenomenon largely observed in subtype C and non-B subtypes, were associated with lower in vitro RC, and it is possible that this is due to lower compatibility of these sequences with the subtype B NL4-3 backbone. Further studies, for example, to test integrase activity and HIV integration in cell culture or perform modeling to predict the effect of extended integrase on functional domains, will be required to assess the functional significance of the longer integrase sequences. Nevertheless, previous studies incorporating subtype C sequences into an NL4-3 backbone revealed no significant effects on viral RC attributable to backbone incompatibilities and further supported the RC data obtained in this system as biologically meaningful (13, 43). Prior studies have also demonstrated that replacing a subtype B viral test vector with a subtype C vector does not impact Pol-driven RC (44). Another limitation of the in vitro RC assay used in the present study is that not all variants are captured, resulting in a lower diversity of the recombinant virus sequences than that of their plasma counterparts. However, this approach preserves, at least to some extent, the naturally occurring quasispecies diversity within the original patient and may mimic more closely the conditions present in vivo than a cloning approach.
In summary, our observation that RT-integrase-driven RC associates significantly with markers of disease progression strongly supports RT-integrase-driven viral RC as a biologically and clinically relevant viral attribute. Furthermore, associations between specific HLA alleles and reduced RT-integrase RC, correlations between HLA-associated polymorphisms and RC, and our identification of specific immune-driven mutations that are associated with significantly reduced RC together indicate that immune-mediated mutations in Pol can reduce HIV replicative fitness in some cases. It is particularly notable that HLA-B*81, a protective HLA allele in subtype C infection, is associated with reduced RT-integrase RC. Polymorphisms at conserved codons were associated with the most pronounced attenuation of RC, and some of these mutations (RT 241I and 257V and integrase 201V) are within or flanking optimal CTL epitopes, identifying these regions as potential vaccine targets.
MATERIALS AND METHODS
Study participants.
The study population included 81 individuals with recent infection, recruited at a median of 49 (interquartile range [IQR], 34 to 69) days postinfection, from the Botswana Tshedimoso cohort (n = 30) (45), the HIV Pathogenesis Programme (HPP) acute infection cohort (n = 41) (46), and the Females Rising through Education, Support and Health (FRESH), cohort (n = 10) in Durban, South Africa (47, 48). For these individuals, plasma samples from the earliest available time point postinfection were studied. An additional 406 antiretroviral-naive individuals chronically infected with HIV-1 subtype C infection from the Sinikithemba cohort in Durban, South Africa (13, 33), were also studied. The time of infection of the Sinikithemba cohort participants was unknown, and plasma from the baseline time point (first presentation) was analyzed. Gag-protease-mediated RC data have been previously collected (27) for a subset of both the chronic (n = 388) and recently (n = 65) infected individuals described in this study. HLA class I profiles of the participants, generated via molecular methods, were available for analysis (33, 46, 47). Written informed consent was obtained from all participants, and ethical approval was granted by the biomedical ethics review committees of the relevant institutions.
Amplification and sequencing of RT-integrase.
RT-integrase products were generated from plasma RNA and sequenced using previously described methods (13). Briefly, HIV-1 RT-integrase was RT-PCR amplified from plasma-derived HIV RNA using subtype C sequence-specific primers (5′ GCCCCTAGGAAAAARGGCTGTTGG 3′, HXB2 nucleotides 2008 to 2031, and 5′ TCTCCTGTTTGCAAACCCCAATATGT 3′, HXB2 nucleotides 5267 to 5242). To enable the homologous recombination of the PCR product and the pNL4-3 reference strain backbone, a second round of nested PCR was performed using primers complementary to NL4-3 on either side of RT-integrase. The RT-integrase PCR products were sequenced using the BigDye ready reaction terminator kit v3.1 (Applied Biosystems, Foster City, CA) on an ABI 3130xl genetic analyzer (Applied Biosystems). Sequence chromatograms were edited, and nucleotide mixtures were identified using Sequencher v5.1 (Gene Codes, Ann Arbor, MI).
To confirm that the recombinant viruses matched the original plasma HIV RNA sequences, RNA from the culture supernatant was extracted from 10% of randomly selected viral stocks using the QIAamp viral RNA kit (Qiagen). RT-integrase was amplified and sequenced as previously described (13). All of the plasma and viral sequences were aligned to HXB2 using Gene Cutter and confirmed as subtype C using the recombinant identification program (RIP), and maximum-likelihood phylogenetic trees were generated using PhyML 3.0 (all available at http://www.hiv.lanl.gov) Phylogenetic trees were visualized using FigTree v.1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/).
Generation of RT-integrase recombinant viruses and RC measurement.
Recombinant viruses encoding participant-derived RT-integrase sequences were generated in an pNL4-3 (HIV-1 subtype B) backbone as previously described (25). Briefly, BstEII sites were engineered upstream of reverse transcriptase (HXB2 nucleotide position 2550) and downstream of integrase (HXB2 nucleotide position 5096) in the plasmid backbone, after which the intervening nucleotides were deleted and the plasmid religated. A CEM-derived GXR25 green fluorescent protein (GFP)-reporter T cell line (GXR cells [26]) was cotransfected with 10 μg of BstEII-linearized RT-integrase-deleted NL4-3 plasmid and 43 μl of the RT-integrase second-round nested PCR product via electroporation (exponential protocol at 250 V, 950 μF, infinite resistance) in 4-mm cuvettes using a Bio-Rad GenePulser electroporation system (Bio-Rad, New York, NY). Homologous recombination of insert and vector produces infectious recombinant HIV-1. Upon infection of new GXR cells in culture, Tat produced by these recombinant viruses drives GFP reporter expression in infected cells, allowing their monitoring by flow cytometry (FACSCalibur; BD Biosciences, CA). Once the level of GFP-positive cells reached approximately 25 to 30%, supernatants containing the recombinant viruses were harvested and aliquots stored at −80°C for subsequent experiments.
Viral titration and RC assays were performed as previously reported (13). Briefly, titers of viral stocks were determined by infecting GXR cells with a known volume of virus stock, and the percentage of GFP-expressing cells was quantified after 48 h. Assuming a linear relationship between virus concentration and cellular infection, the volume of virus stock needed to infect 0.3% of GXR cells (i.e., multiplicity of infection [MOI] of 0.003) in subsequent replication assays was then calculated. The target MOI was set at 0.003, as this allows the reproducible observation of exponential viral spread in culture over a 7-day assay period. To calculate RC, the mean slope of percent GFP-expressing cells from days 3 to 6 was calculated using the semilog method in Excel; this value was then normalized to the slope of growth of the wild-type NL4-3 control included in each assay (such that an RC of 1 indicates recombinant virus spread equivalent to that of NL4-3). Each recombinant virus was assayed in duplicate in independent experiments, and RC results are reported as the means from these two measurements.
Statistical analysis.
Relationships between RT-integrase-driven RC and clinical markers of HIV infection (plasma viral loads and CD4+ T cell counts) were assessed using Pearson's or Spearman's correlation for normally and nonnormally distributed data, respectively. For the recently infected individuals, the influence of RT-integrase-driven RC on the rate of CD4+ T cell count decline was assessed using generalized estimating equation (GEE) models with exchangeable correlation structure. In the model, cohort, baseline viral load, and baseline CD4+ T cell count were considered potential confounders and adjusted for if their inclusion resulted in a 10% or greater change in the estimated effect of RC. Similar analyses were conducted for Gag-protease-driven RC. To facilitate data interpretation, RC was subsequently dichotomized at the population median, and predicted values of CD4+ T cell count were calculated at the mean level of other covariates for each RC group at each time point. CD4+ T cell counts were square root transformed, and all GEE analyses were conducted in Stata v14. The viral load set point for these participants was determined as the mean viral load from 90 to 360 days postinfection.
The relationship between RT-integrase-mediated RC and the presence of HLA-associated polymorphisms in the bulk sequences was investigated using Spearman's correlation. Here, HLA-associated polymorphisms were defined as all adapted (i.e., inferred escape) associations originally defined at the stringent statistical threshold of a q value of ≤0.05 in a published analysis of 1,866 HIV-1 subtype C chronically infected individuals from southern Africa (which included the Sinikithemba cohort participants studied here) (39). For all HLA class I alleles expressed in a minimum of 5 individuals, the Student t test was used to compare RT-integrase-mediated RC values among individuals expressing, or not expressing, each HLA class I allele. Finally, Mann-Whitney U tests were used to identify specific Pol amino acids associated with increased or decreased RC. The significance cutoff for all analyses was a P value of <0.05; however, for the codon/function analyses, multiple comparisons were addressed using q values (the P value analogue of the false-discovery rate [49]).
Accession number(s).
RT-integrase sequences obtained in this study are available in the GenBank database under accession numbers MH486981 to MH487467.
ACKNOWLEDGMENTS
This work was funded by the Sub-Saharan African Network for TB/HIV Research Excellence (SANTHE), a DELTAS Africa Initiative [grant DEL-15-006]. The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)'s Alliance for Accelerating Excellence in Science in Africa (AESA) and supported by the New Partnership for Africa's Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust [grant 107752/Z/15/Z] and the UK government. T.N. received additional funding from the South African Department of Science and Technology through the National Research Foundation (South African Research Chairs Initiative), the Victor Daitz Foundation, and the Howard Hughes Medical Institute. This work was also partially supported by the Bill and Melinda Gates Foundation, the International AIDS Vaccine Initiative (IAVI) (UKZNRSA1001), the NIAID (R37AI067073), and Gilead Sciences (grant 00406). Research reported in this publication was supported by the South African Medical Research Council under a Self-Initiated Research Grant to J.M. D.B.A.O. received funding from the National Research Foundation (NRF-SA) and the College of Health sciences, University of KwaZulu-Natal. Z.L.B. is supported by a Scholar Award from the Michael Smith Foundation for Health Research. M.A.B. holds a Canada Research Chair in Viral Pathogenesis and Immunity. Z.L.B. and M.A.B. received additional funding from the Canadian Institutes of Health Research (CIHR; PJT-148621).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The views expressed in this publication are those of the author(s) and not necessarily those of AAS, NEPAD Agency, Wellcome Trust, or the UK government.
We thank Bruce Walker and Philip Goulder for their support and providing access to the samples. We are grateful to all study participants and support staff.
REFERENCES
- 1.McMichael AJ, Borrow P, Tomaras GD, Goonetilleke N, Haynes BF. 2010. The immune response during acute HIV-1 infection: clues for vaccine development. Nat Rev Immunol 10:11–23. doi: 10.1038/nri2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, Lifton MA, Racz P, Tenner-Racz K, Dalesandro M, Scallon BJ. 1999. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283:857–860. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- 3.Troyer RM, McNevin J, Liu Y, Zhang SC, Krizan RW, Abraha A, Tebit DM, Zhao H, Avila S, Lobritz MA. 2009. Variable fitness impact of HIV-1 escape mutations to cytotoxic T lymphocyte (CTL) response. PLoS Pathog 5:e1000365. doi: 10.1371/journal.ppat.1000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allen TM, Altfeld M. 2008. Crippling HIV one mutation at a time. J Exp Med 205:1003–1007. doi: 10.1084/jem.20080569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chopera DR, Wright JK, Brockman MA, Brumme ZL. 2011. Immune-mediated attenuation of HIV-1. Future Virol 6:917–928. doi: 10.2217/fvl.11.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brockman MA, Schneidewind A, Lahaie M, Schmidt A, Miura T, DeSouza I, Ryvkin F, Derdeyn CA, Allen S, Hunter E. 2007. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. J Virol 81:12608–12618. doi: 10.1128/JVI.01369-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinez-Picado J, Prado JG, Fry EE, Pfafferott K, Leslie A, Chetty S, Thobakgale C, Honeyborne I, Crawford H, Matthews P. 2006. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J Virol 80:3617–3623. doi: 10.1128/JVI.80.7.3617-3623.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schneidewind A, Brockman MA, Yang R, Adam RI, Li B, Le Gall S, Rinaldo CR, Craggs SL, Allgaier RL, Power KA. 2007. Escape from the dominant HLA-B27-restricted cytotoxic T-lymphocyte response in Gag is associated with a dramatic reduction in human immunodeficiency virus type 1 replication. J Virol 81:12382–12393. doi: 10.1128/JVI.01543-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wright JK, Naidoo VL, Brumme ZL, Prince JL, Claiborne DT, Goulder PJ, Brockman MA, Hunter E, Ndung'u T. 2012. Impact of HLA-B* 81-associated mutations in HIV-1 Gag on viral replication capacity. J Virol 86:3193–3199. doi: 10.1128/JVI.06682-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med 13:46–53. doi: 10.1038/nm1520. [DOI] [PubMed] [Google Scholar]
- 11.Rolland M, Heckerman D, Deng W, Rousseau CM, Coovadia H, Bishop K, Goulder P, Walker BD, Brander C, Mullins JI. 2008. Broad and Gag-biased HIV-1 epitope repertoires are associated with lower viral loads. PLoS One 3:e1424. doi: 10.1371/journal.pone.0001424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crawford H, Lumm W, Leslie A, Schaefer M, Boeras D, Prado JG, Tang J, Farmer P, Ndung'u T, Lakhi S. 2009. Evolution of HLA-B* 5703 HIV-1 escape mutations in HLA-B* 5703–positive individuals and their transmission recipients. J Exp Med 206:909–921. doi: 10.1084/jem.20081984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wright JK, Brumme ZL, Carlson JM, Heckerman D, Kadie CM, Brumme CJ, Wang B, Losina E, Miura T, Chonco F. 2010. Gag-protease-mediated replication capacity in HIV-1 subtype C chronic infection: associations with HLA type and clinical parameters. J Virol 84:10820–10831. doi: 10.1128/JVI.01084-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brumme ZL, John M, Carlson JM, Brumme CJ, Chan D, Brockman MA, Swenson LC, Tao I, Szeto S, Rosato P. 2009. HLA-associated immune escape pathways in HIV-1 subtype B Gag, Pol and Nef proteins. PLoS One 4:e6687. doi: 10.1371/journal.pone.0006687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campbell TB, Schneider K, Wrin T, Petropoulos CJ, Connick E. 2003. Relationship between in vitro human immunodeficiency virus type 1 replication rate and virus load in plasma. J Virol 77:12105–12112. doi: 10.1128/JVI.77.22.12105-12112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu MK, Hawkins N, Ritchie AJ, Ganusov VV, Whale V, Brackenridge S, Li H, Pavlicek JW, Cai F, Rose-Abrahams M. 2013. Vertical T cell immunodominance and epitope entropy determine HIV-1 escape. J Clin Investig 123:380–393. doi: 10.1172/JCI65330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahmed T, Borthwick NJ, Gilmour J, Hayes P, Dorrell L, Hanke T. 2016. Control of HIV-1 replication in vitro by vaccine-induced human CD8(+) T cells through conserved subdominant Pol epitopes. Vaccine 34:1215–1224. doi: 10.1016/j.vaccine.2015.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borthwick N, Ahmed T, Ondondo B, Hayes P, Rose A, Ebrahimsa U, Hayton E-J, Black A, Bridgeman A, Rosario M. 2014. Vaccine-elicited human T cells recognizing conserved protein regions inhibit HIV-1. Mol Ther 22:464–475. doi: 10.1038/mt.2013.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mothe B, Hu X, Llano A, Rosati M, Olvera A, Kulkarni V, Valentin A, Alicea C, Pilkington GR, Sardesai NY. 2015. A human immune data-informed vaccine concept elicits strong and broad T-cell specificities associated with HIV-1 control in mice and macaques. J Transl Med 13:60. doi: 10.1186/s12967-015-0392-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mothe B, Llano A, Ibarrondo J, Daniels M, Miranda C, Zamarreño J, Bach V, Zuniga R, Pérez-Álvarez S, Berger CT. 2011. Definition of the viral targets of protective HIV-1-specific T cell responses. J Transl Med 9:208. doi: 10.1186/1479-5876-9-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gandhi RT, Wurcel A, Rosenberg ES, Johnston MN, Hellmann N, Bates M, Hirsch MS, Walker BD. 2003. Progressive reversion of human immunodeficiency virus type 1 resistance mutations in vivo after transmission of a multiply drug-resistant virus. Clin Infect Dis 37:1693–1698. doi: 10.1086/379773. [DOI] [PubMed] [Google Scholar]
- 22.Hu Z, Kuritzkes DR. 2010. Effect of raltegravir resistance mutations in HIV-1 integrase on viral fitness. J Acquired Immune Defic Syndr 55:148. doi: 10.1097/QAI.0b013e3181e9a87a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weber J, Chakraborty B, Weberova J, Miller MD, Quinones-Mateu ME. 2005. Diminished replicative fitness of primary human immunodeficiency virus type 1 isolates harboring the K65R mutation. J Clin Microbiol 43:1395–1400. doi: 10.1128/JCM.43.3.1395-1400.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brockman MA, Chopera DR, Olvera A, Brumme CJ, Sela J, Markle TJ, Martin E, Carlson JM, Le AQ, McGovern R. 2012. Uncommon pathways of immune escape attenuate HIV-1 integrase replication capacity. J Virol 86:6913–6923. doi: 10.1128/JVI.07133-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brumme ZL, Li C, Miura T, Sela J, Rosato PC, Brumme CJ, Markle TJ, Martin E, Block BL, Trocha A. 2011. Reduced replication capacity of NL4-3 recombinant viruses encoding RT-Integrase sequences from HIV-1 elite controllers. J Acquired Immune Defic Syndr 56: doi: 10.1097/QAI.0b013e3181fe9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brockman MA, Tanzi GO, Walker BD, Allen TM. 2006. Use of a novel GFP reporter cell line to examine replication capacity of CXCR4-and CCR5-tropic HIV-1 by flow cytometry. J Virol Methods 131:134–142. doi: 10.1016/j.jviromet.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 27.Wright JK, Novitsky V, Brockman MA, Brumme ZL, Brumme CJ, Carlson JM, Heckerman D, Wang B, Losina E, Leshwedi M. 2011. Influence of Gag-protease-mediated replication capacity on disease progression in individuals recently infected with HIV-1 subtype C. J Virol 85:3996–4006. doi: 10.1128/JVI.02520-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rousseau CM, Daniels MG, Carlson JM, Kadie C, Crawford H, Prendergast A, Matthews P, Payne R, Rolland M, Raugi DN. 2008. HLA class I-driven evolution of human immunodeficiency virus type 1 subtype c proteome: immune escape and viral load. J Virol 82:6434–6446. doi: 10.1128/JVI.02455-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prince JL, Claiborne DT, Carlson JM, Schaefer M, Yu T, Lahki S, Prentice HA, Yue L, Vishwanathan SA, Kilembe W. 2012. Role of transmitted Gag CTL polymorphisms in defining replicative capacity and early HIV-1 pathogenesis. PLoS Pathog 8:e1003041. doi: 10.1371/journal.ppat.1003041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fraser C, Lythgoe K, Leventhal GE, Shirreff G, Hollingsworth TD, Alizon S, Bonhoeffer S. 2014. Virulence and pathogenesis of HIV-1 infection: an evolutionary perspective. Science 343:1243727. doi: 10.1126/science.1243727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brockman MA, Brumme ZL, Brumme CJ, Miura T, Sela J, Rosato PC, Kadie CM, Carlson JM, Markle TJ, Streeck H. 2010. Early selection in Gag by protective HLA alleles contributes to reduced HIV-1 replication capacity that may be largely compensated for in chronic infection. J Virol 84:11937–11949. doi: 10.1128/JVI.01086-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang KH, Goedhals D, Carlson JM, Brockman MA, Mishra S, Brumme ZL, Hickling S, Tang CS, Miura T, Seebregts C, Heckerman D, Ndung'u T, Walker B, Klenerman P, Steyn D, Goulder P, Phillips R, van Vuuren C, Frater J. 2011. Progression to AIDS in South Africa is associated with both reverting and compensatory viral mutations. PLoS One 6:e19018. doi: 10.1371/journal.pone.0019018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kiepiela P, Leslie AJ, Honeyborne I, Ramduth D, Thobakgale C, Chetty S, Rathnavalu P, Moore C, Pfafferott KJ, Hilton L. 2004. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature 432:769–775. doi: 10.1038/nature03113. [DOI] [PubMed] [Google Scholar]
- 34.Leslie A, Price DA, Mkhize P, Bishop K, Rathod A, Day C, Crawford H, Honeyborne I, Asher TE, Luzzi G, Edwards A, Rosseau CM, Mullins JI, Tudor-Williams G, Novelli V, Brander C, Douek DC, Kiepiela P, Walker BD, Goulder PJR. 2006. Differential selection pressure exerted on HIV by CTL targeting identical epitopes but restricted by distinct HLA alleles from the same HLA supertype. J Immunol 177:4699–4708. doi: 10.4049/jimmunol.177.7.4699. [DOI] [PubMed] [Google Scholar]
- 35.Tang J, Tang S, Lobashevsky E, Myracle AD, Fideli U, Aldrovandi G, Allen S, Musonda R, Kaslow RA. 2002. Favorable and unfavorable HLA class I alleles and haplotypes in Zambians predominantly infected with clade C human immunodeficiency virus type 1. J Virol 76:8276–8284. doi: 10.1128/JVI.76.16.8276-8284.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frahm N, Adams S, Kiepiela P, Linde CH, Hewitt HS, Lichterfeld M, Sango K, Brown NV, Pae E, Wurcel AG, Altfeld M, Feeney ME, Allen TM, Roach T, St John MA, Daar ES, Rosenberg E, Korber B, Marincola F, Walker BD, Goulder PJ, Brander C. 2005. HLA-B63 presents HLA-B57/B58-restricted cytotoxic T-lymphocyte epitopes and is associated with low human immunodeficiency virus load. J Virol 79:10218–10225. doi: 10.1128/JVI.79.16.10218-10225.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lazaryan A, Song W, Lobashevsky E, Tang J, Shrestha S, Zhang K, McNicholl JM, Gardner LI, Wilson CM, Klein RS. 2011. The influence of human leukocyte antigen class I alleles and their population frequencies on human immunodeficiency virus type 1 control among African Americans. Hum Immunol 72:312–318. doi: 10.1016/j.humimm.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, Walker BD, Ripke S, Brumme CJ, Pulit SL, Carrington M, Kadie CM, Carlson JM, Heckerman D, Graham RR, Plenge RM, Deeks SG, Gianniny L, Crawford G, Sullivan J, Gonzalez E, Davies L, Camargo A, Moore JM, Beattie N, Gupta S, Crenshaw A, Burtt NP, Guiducci C, Gupta N, Gao X, Qi Y, Yuki Y, Piechocka-Trocha A, Cutrell E, Rosenberg R, Moss KL, Lemay P, O'Leary J, Schaefer T, Verma P, Toth I, Block B, Baker B, Rothchild A, Lian J, Proudfoot J, Alvino DM, Vine S, Addo MM, Allen TM, et al. 2010. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 330:1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carlson JM, Schaefer M, Monaco DC, Batorsky R, Claiborne DT, Prince J, Deymier MJ, Ende ZS, Klatt NR, DeZiel CE. 2014. Selection bias at the heterosexual HIV-1 transmission bottleneck. Science 345:1254031. doi: 10.1126/science.1254031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frater AJ, Brown H, Oxenius A, Günthard H, Hirschel B, Robinson N, Leslie A, Payne R, Crawford H, Prendergast A. 2007. Effective T-cell responses select human immunodeficiency virus mutants and slow disease progression. J Virol 81:6742–6751. doi: 10.1128/JVI.00022-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ceccherini-Silberstein F, Malet I, D'Arrigo R, Antinori A, Marcelin A-G, Perno C-F. 2009. Characterization and structural analysis of HIV-1 integrase conservation. AIDS Rev 11:17–29. [PubMed] [Google Scholar]
- 42.Rhee S-Y, Liu TF, Kiuchi M, Zioni R, Gifford RJ, Holmes SP, Shafer RW. 2008. Natural variation of HIV-1 group M integrase: implications for a new class of antiretroviral inhibitors. Retrovirology 5:74. doi: 10.1186/1742-4690-5-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kiguoya MW, Mann JK, Chopera D, Gounder K, Lee GQ, Hunt PW, Martin JN, Ball TB, Kimani J, Brumme ZL, Brockman MA. 2017. Subtype-specific differences in Gag-protease-driven replication capacity are consistent with intersubtype differences in HIV-1. Dis Progr 91:e00253-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cho S, Stawiski E, Parkin N. 2007. Interpretation of drug-susceptibility and replication-capacity results from subtype CHIV-1 protease/RT is not influenced by the subtype of the resistance test vector. Antiviral Ther 12:S118–S118. [Google Scholar]
- 45.Novitsky V, Woldegabriel E, Wester C, McDonald E, Rossenkhan R, Ketunuti M, Makhema J, Seage GR III, Essex M. 2008. Identification of primary HIV-1C infection in Botswana. AIDS Care 20:806–811. doi: 10.1080/09540120701694055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Radebe M, Nair K, Chonco F, Bishop K, Wright JK, van der Stok M, Bassett IV, Mncube Z, Altfeld M, Walker BD. 2011. Limited immunogenicity of HIV CD8+ T-cell epitopes in acute clade C virus infection. J Infect Dis 204:768–776. doi: 10.1093/infdis/jir394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ndhlovu ZM, Kamya P, Mewalal N, Kløverpris HN, Nkosi T, Pretorius K, Laher F, Ogunshola F, Chopera D, Shekhar K. 2015. Magnitude and kinetics of CD8+ T cell activation during hyperacute HIV infection impact viral set point. Immunity 43:591–604. doi: 10.1016/j.immuni.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dong KL, Moodley A, Kwon DS, Ghebremichael MS, Dong M, Ismail N, Ndhlovu ZM, Mabuka JM, Muema DM, Pretorius K. 2018. Detection and treatment of Fiebig stage I HIV-1 infection in young at-risk women in South Africa: a prospective cohort study. Lancet HIV 5:e35–e44. doi: 10.1016/S2352-3018(17)30146-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Storey JD, Tibshirani R. 2003. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kløverpris HN, McGregor R, McLaren JE, Ladell K, Harndahl M, Stryhn A, Carlson JM, Koofhethile C, Gerritsen B, Keşmir C. 2015. CD8+ TCR bias and immunodominance in HIV-1 infection. J Immunol 194:5329–5345. doi: 10.4049/jimmunol.1400854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oxenius A, Price DA, Trkola A, Edwards C, Gostick E, Zhang H-T, Easterbrook PJ, Tun T, Johnson A, Waters A. 2004. Loss of viral control in early HIV-1 infection is temporally associated with sequential escape from CD8+ T cell responses and decrease in HIV-1-specific CD4+ and CD8+ T cell frequencies. J Infect Dis 190:713–721. doi: 10.1086/422760. [DOI] [PubMed] [Google Scholar]
- 52.Jamieson BD, Yang OO, Hultin L, Hausner MA, Hultin P, Matud J, Kunstman K, Killian S, Altman J, Kommander K, Korber B, Giorgi J, Wolinsky S. 2003. Epitope escape mutation and decay of human immunodeficiency virus type 1-specific CTL responses. J Immunol 171:5372–5379. doi: 10.4049/jimmunol.171.10.5372. [DOI] [PubMed] [Google Scholar]