

The accumulation of orally acquired prions within Peyer's patches in the small intestine is essential for the efficient spread of disease to the brain. Little is known of how the prions initially establish infection within Peyer's patches. Some gastrointestinal pathogens utilize molecules, such as the cellular prion protein PrPC, expressed on gut epithelial cells to enter Peyer's patches. Acute mucosal inflammation can enhance PrPC expression in the intestine, implying the potential to enhance oral prion disease susceptibility. We used transgenic mice to determine whether the uptake of prions into Peyer's patches was dependent upon PrPC expression in the gut epithelium. We show that orally acquired prions can establish infection in Peyer's patches independently of PrPC expression in gut epithelial cells. Our data suggest that the magnitude of PrPC expression in the epithelium lining the small intestine is unlikely to be an important factor which influences oral prion disease susceptibility.
KEYWORDS: Peyer's patches, PrP, gut epithelium, intestine, prions, transmissible spongiform encephalopathies
ABSTRACT
The early replication of certain prion strains within Peyer's patches in the small intestine is essential for the efficient spread of disease to the brain after oral exposure. Our data show that orally acquired prions utilize specialized gut epithelial cells known as M cells to enter Peyer's patches. M cells express the cellular isoform of the prion protein, PrPC, and this may be exploited by some pathogens as an uptake receptor to enter Peyer's patches. This suggested that PrPC might also mediate the uptake and transfer of prions across the gut epithelium into Peyer's patches in order to establish infection. Furthermore, the expression level of PrPC in the gut epithelium could influence the uptake of prions from the lumen of the small intestine. To test this hypothesis, transgenic mice were created in which deficiency in PrPC was specifically restricted to epithelial cells throughout the lining of the small intestine. Our data clearly show that efficient prion neuroinvasion after oral exposure occurred independently of PrPC expression in small intestinal epithelial cells. The specific absence of PrPC in the gut epithelium did not influence the early replication of prions in Peyer's patches or disease susceptibility. Acute mucosal inflammation can enhance PrPC expression in the intestine, implying the potential to enhance oral prion disease pathogenesis and susceptibility. However, our data suggest that the magnitude of PrPC expression in the epithelium lining the small intestine is unlikely to be an important factor which influences the risk of oral prion disease susceptibility.
IMPORTANCE The accumulation of orally acquired prions within Peyer's patches in the small intestine is essential for the efficient spread of disease to the brain. Little is known of how the prions initially establish infection within Peyer's patches. Some gastrointestinal pathogens utilize molecules, such as the cellular prion protein PrPC, expressed on gut epithelial cells to enter Peyer's patches. Acute mucosal inflammation can enhance PrPC expression in the intestine, implying the potential to enhance oral prion disease susceptibility. We used transgenic mice to determine whether the uptake of prions into Peyer's patches was dependent upon PrPC expression in the gut epithelium. We show that orally acquired prions can establish infection in Peyer's patches independently of PrPC expression in gut epithelial cells. Our data suggest that the magnitude of PrPC expression in the epithelium lining the small intestine is unlikely to be an important factor which influences oral prion disease susceptibility.
INTRODUCTION
Prions cause chronic neurodegenerative diseases that affect humans and some domesticated and free-ranging animal species for which there are no treatments. Bovine spongiform encephalopathy (BSE) prions also have zoonotic potential (1), exerting high societal and economic costs. The precise nature of the infectious prion is uncertain, but an abnormal, relatively proteinase-resistant isoform (PrPSc) of the host cellular prion protein (PrPC) copurifies with prion infectivity in diseased tissues (2), and host cells must express cellular PrPC to sustain prion infection (3).
Many natural prion diseases are acquired by oral consumption of contaminated food or pasture. The gut-associated lymphoid tissues (GALT) within the lining of the intestine, such as the tonsils, Peyer's patches, appendix, and colonic and cecal patches, together with the mesenteric lymph nodes (MLN), help to provide protection against intestinal pathogens. However, orally acquired prions exploit the GALT to achieve host infection (4–8). The early replication of prions within Peyer's patches in the small intestine is essential for their efficient spread from the gut to the brain (termed neuroinvasion), as oral prion disease susceptibility is blocked in their absence (5, 9–11).
Orally acquired prions utilize an elegant cellular relay in the GALT in order to establish host infection. After ingestion, the prions are first transported across the follicle-associated epithelium (FAE), which covers the lumenal surface of Peyer's patches by M cells (12–16). The prions are then acquired by mononuclear phagocytes within the GALT, which they appear to use as “Trojan horses” to shuttle them toward the follicular dendritic cells (FDC) in the B cell follicles (17–19). The subsequent replication of the prions on FDC is essential for efficient neuroinvasion from the intestine (4, 5, 17, 20). The prions then infect nearby enteric nerves before spreading along fibers of the sympathetic and parasympathetic nervous systems to the brain, where they ultimately cause neurodegeneration and death (17, 21).
M cells are specialized, highly phagocytic, intestinal epithelial cells that facilitate the uptake and transepithelial transfer of particulate antigens and microorganisms into the GALT from the gut lumen (22). The transcytosis of particulate antigens by M cells is an important initial step in the induction of efficient mucosal immune responses against certain pathogenic bacteria (23, 24) and the commensal bacterial flora (25). However, some orally acquired bacterial (26–28) and viral (29, 30) pathogens utilize M cells to achieve host infection. Prions also exploit M cells in order to enter Peyer's patches and establish host infection (13, 16). Furthermore, the density of M cells in the gut epithelium directly limits or enhances disease susceptibility. In the specific absence of M cells, the accumulation of prions in Peyer's patches and subsequent spread of the disease to the brain are blocked (13, 16). In contrast, increased M cell density at the time of oral exposure enhances prion disease susceptibility approximately 10-fold by increasing the uptake of prions from the gut lumen (16).
M cells are considered to express a variety of “immunosurveillance” receptors on their apical surfaces, which enable them to acquire certain pathogens and antigens. For example, glycoprotein 2 (GP2) can act as a receptor for FimH+ bacteria such as Escherichia coli and Salmonella enterica serovar Typhimurium (23). Uromodulin (also known as Tamm-Horsfall protein) may similarly mediate the uptake of certain strains of Lactobacillus acidophilus (31). Some pathogenic microorganisms appear to use receptors on M cells to aid host infection. The complement C5a receptor is expressed on the apical surface of M cells and aids the uptake of Yersinia enterocolitica to establish infection (32). Interactions between the type A1 botulinum neurotoxin complex and GP2 on the M cell surface have also been shown to mediate the intestinal translocation of the toxin in order to exert its toxic effects (33). M cells express the cellular isoform of the prion protein, PrPC, on their apical surfaces (26, 34). Data suggest that the pathogenic Gram-negative bacterium Brucella abortus utilizes PrPC on the M cell surface as an uptake receptor to enter Peyer's patches (26).
Whether the uptake and transcytosis of prions across the gut epithelium into Peyer's patches in order to establish infection predominantly occurs via constitutive sampling of the lumenal contents or via binding to specific receptors such as PrPC is not known. Treatments that impede the early accumulation of prions within the GALT can impede their spread to the brain and reduce disease susceptibility (4, 13, 16, 18). Thus, the identification of the molecular factors that facilitate the uptake of prions into the GALT will help the design of novel intervention targets and enhance our understanding of the factors that influence the risk of infection. Therefore, in the current study, transgenic mice were created in which Prnp expression (encoding PrPC) was specifically ablated in epithelial cells throughout the lining of the small intestine. These mice were then used to determine whether the absence of PrPC expression in the epithelium lining the small intestine influences oral prion disease susceptibility and the early replication of prions in the GALT.
RESULTS
Conditional ablation of Prnp throughout the small intestinal epithelium.
The expression of Cre recombinase under the control of the rat Cyp1a1 promoter element in Cyp1a1-Cre mice has been used in a series of studies to inducibly ablate the expression of LoxP site-flanked target genes in small intestinal progenitor cells and intestinal epithelial cells (IEC) following β-naphthoflavone (βNF) treatment (35–37). The FANTOM5 project of the FANTOM consortium (38) has collated a large collection of cap analysis of gene expression (CAGE) data from multiple mouse tissues and cells (http://fantom.gsc.riken.jp/zenbu). We used this publicly available data resource to compare the expression levels of Cyp1a1, Gp2, and Prnp in multiple data sets derived from mouse FAE, M cells, lymphocytes, leukocytes, and brain-derived cells. This analysis confirmed that Cyp1a1 and Prnp were expressed highly in the FAE and in GP2+ M cells (Fig. 1). However, Cyp1a1 expression was absent in B cells, T cells, and macrophages as well as in brain-derived microglia, astrocytes, and neurons (Fig. 1).
FIG 1.
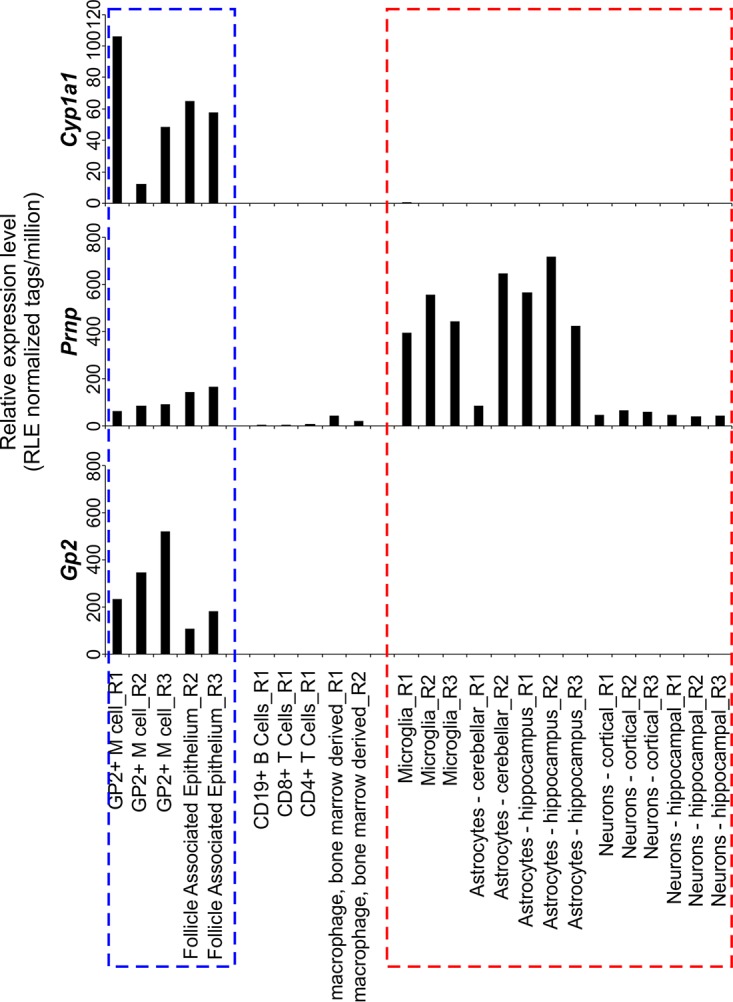
Cyp1a1 is expressed in the follicle-associated epithelium and in M cells in the small intestine. Shown is a comparison of Cyp1a1, Prnp, and Gp2 mRNA expression levels in individual cell populations in deep cap analysis of gene expression (CAGE) sequence data from the FANTOM5 project of the FANTOM consortium (38). Each bar shows the relative expression level of each gene per million reads in each sample [RLE normalized tags/million]. The blue-hatched box highlights the small intestine-derived glycoprotein 2-expressing (GP2+) M cell and the follicle-associated epithelium data sets. The red-hatched box highlights the brain-derived data sets.
Here, Cyp1a1-Cre mice were crossed with PrnpF/F mice, which carry a “floxed” Prnp gene (39), to enable the inducible ablation of Prnp specifically in IEC. Since the reliable detection of PrPC in the gut epithelium by immunohistochemistry (IHC) is technically challenging, these mice were additionally crossed with ROSA26F/F reporter mice (40) to enable the cellular specificity of the Cre-mediated gene ablation to be readily assessed by histological assessment of β-galactosidase (LacZ) expression. The resultant progeny Cyp1a1-Cre ROSA26F/F PrnpF/F mice were termed PrnpΔIEC mice here.
Female PrnpΔIEC mice were treated with βNF (or the vehicle alone as a control) for 5 days to specifically ablate Prnp expression in IEC, and tissues were analyzed 14 days later. Whole-mount histological analysis showed LacZ expression indicative of efficient Cre-mediated gene recombination throughout the small and large intestines of βNF-treated PrnpΔIEC mice (Fig. 2a). Analysis of tissue sections showed strong LacZ expression in IEC and crypts throughout the small intestine (Fig. 2c). The Cre-mediated gene recombination in the small intestinal crypts of βNF-treated PrnpΔIEC mice was highly efficient (99.5% ± 1.1%) (Fig. 2e). In contrast, the Cre-mediated gene recombination in colonic crypts and IEC in the large intestine was less efficient (64.1% ± 8.6%) (Fig. 2f) and presented as a mosaic pattern (Fig. 2c). No other cellular sites of Cre-mediated recombination were observed throughout the intestines of βNF-treated PrnpΔIEC mice. LacZ expression was absent within the submucosa (Fig. 2c) and also in the subepithelial dome and FDC-containing B cell follicle regions of the GALT (Fig. 2g). As anticipated, no LacZ expression was detected throughout the small and large intestines of vehicle-treated PrnpΔIEC control mice (Fig. 2b, d to f, and h). LacZ expression was also undetectable throughout the small and large intestines of untreated PrnpΔIEC control mice and βNF-treated PrnpF/F (Cre-deficient) control mice (Fig. 2e and f). These data clearly demonstrate that Cre-mediated gene recombination is restricted to IEC in the small intestines of βNF-treated PrnpΔIEC mice.
FIG 2.

Cre-mediated gene recombination is restricted to IEC in the small intestines of βNF-treated PrnpΔIEC mice. Female PrnpΔIEC mice were treated with β-naphthoflavone (βNF) for 5 days to specifically ablate Prnp expression in intestinal epithelial cells, and tissues were analyzed 14 days later. PrnpΔIEC mice treated with the vehicle alone (Veh.) were used as controls. (a and b) Whole-mount histological analysis of LacZ expression (blue) in the intestines of βNF-treated PrnpΔIEC mice (a) or vehicle-treated PrnpΔIEC control mice (b). S, small intestine; L, large intestine. (c and d) Histological analysis of LacZ expression (blue) in IEC and crypts in the intestines of βNF-treated PrnpΔIEC mice (c) or vehicle-treated PrnpΔIEC control mice (d). Sections were counterstained with nuclear fast red to detect cell nuclei (red). SM, submucosa. (e and f) Comparison of the percentages of LacZ-expressing crypts in the small (e) and large (f) intestines of βNF-treated PrnpF/F control mice. Untreated PrnpΔIEC mice, vehicle-treated PrnpΔIEC mice, and βNF-treated PrnpF/F mice were used as controls. Data represent mean percentages of LacZ-expressing crypts/mouse (n = 5 mice/group; 50 to 105 crypts/mouse). (g and h) Histological analysis of LacZ expression (blue) in Peyer's patches and colonic patches of βNF-treated PrnpΔIEC mice (g) or vehicle-treated PrnpΔIEC control mice (h). SED, subepithelial dome; Fo, follicle.
Effect of IEC-restricted Prnp ablation on prion accumulation in lymphoid tissues.
To determine the effects of IEC-specific PrPC deficiency on oral prion disease pathogenesis, groups of female PrnpΔIEC mice were treated with βNF for 5 days to specifically ablate Prnp expression in IEC. Untreated PrnpΔIEC mice, vehicle-treated PrnpΔIEC mice, and βNF-treated PrnpF/F (Cre-deficient) mice were used as controls. Fourteen days later, 10 mice/group were subsequently orally exposed to ME7 scrapie prions, and tissues were collected at 70 days postinfection. The presence of prion disease-specific, abnormal accumulations of PrP (referred to as PrPd), which occur only in the tissues of affected animals, was detected by IHC (4, 5, 11, 13, 16, 19, 41–43). However, since the IHC analysis cannot unequivocally discriminate between PrPSc and cellular PrPC, paraffin-embedded tissue immunoblot analysis of adjacent membrane-bound sections was also used to confirm that these PrPd aggregates contained prion disease-specific, relatively proteinase K (PK)-resistant PrPSc. As anticipated, abundant PrPSc accumulations were detected in association with FDC (CD21/35+ cells) in Peyer's patches of control PrnpΔIEC mice (Fig. 3a, arrows). Abundant FDC-associated PrPSc accumulations were also detected in Peyer's patches of βNF-treated PrnpΔIEC mice.
FIG 3.

Effect of intestinal epithelial cell-restricted Prnp ablation on prion accumulation in lymphoid tissues. Female PrnpΔIEC mice were treated with βNF for 5 days to specifically ablate Prnp expression in intestinal epithelial cells. Untreated PrnpΔIEC mice and PrnpΔIEC mice treated with the vehicle alone (Veh.) were used as controls. Fourteen days later, the mice were orally exposed to ME7 scrapie prions, and Peyer's patches, mesenteric lymph nodes (MLN), and spleens were collected at 70 days postinfection. (a) Immunohistochemical analysis reveals high levels of disease-specific PrP (PrPd) (middle row, red, arrows) detected in association with FDC (CD21/35+ cells) (top row, red) in Peyer's patches from mice from each group. Sections were counterstained with hematoxylin to detect cell nuclei (blue). Analysis of adjacent sections by paraffin-embedded tissue immunoblotting confirmed the presence of prion-specific PK-resistant PrPSc (blue/black). Data are representative of results for tissues from 6 mice/group. (b to d) Prion infectivity levels were assayed in Peyer's patches (b), MLN (c), and spleens (d) from mice from each group collected at 70 days postinfection. Prion infectivity titers (log10 i.c. ID50 per gram of tissue) were determined by injection of tissue homogenates into groups of C57BL/Dk indicator mice (n = 4 recipient mice/tissue). Each symbol represents data derived from an individual tissue. Red line, median prion infectivity titer for groups in which all samples contained >1 log10 i.c. ID50/g tissue. Data below the broken horizontal line indicate disease incidence in the recipient mice of <100%, which were considered to contain trace levels of prion infectivity.
Consistent with the IHC data (Fig. 3a), high levels of prion infectivity were detected in Peyer's patches of mice from each control group (median infectivity level, 6.0 to 6.6 log10 intracerebral [i.c.] 50% infectious dose [ID50] units/g; n = 2 to 4 mice/group) (Fig. 3b). IEC-restricted Prnp ablation did not influence the early accumulation of infectious prions within Peyer's patches, as high levels of prion infectivity were also detected in tissues from βNF-treated PrnpΔIEC mice (median infectivity level, 6.1 log10 i.c. ID50 units/g; n = 4 mice) (Fig. 3b).
Within weeks after oral exposure, high levels of ME7 scrapie prions first accumulate on FDC in Peyer's patches and subsequently are disseminated via the blood and lymph to most other lymphoid tissues, including the MLN and spleen (4, 5, 11, 13, 16, 18, 19, 44). The levels of prion infectivity detected in the MLN and spleens from mice from each treatment and control group were also similar (Fig. 3c and d, respectively).
These data clearly show that IEC-restricted Prnp ablation does not affect the early accumulation of orally acquired prions within Peyer's patches or their subsequent dissemination to the MLN or spleen.
IEC-restricted Prnp ablation does not influence oral prion disease susceptibility.
Female PrnpΔIEC mice were treated with βNF for 5 days to ablate Prnp expression in IEC, and 14 days later, they were subsequently orally exposed to ME7 scrapie prions. Untreated PrnpΔIEC mice, vehicle-treated PrnpΔIEC mice, and βNF-treated PrnpF/F (Cre-deficient) mice were used as controls. As anticipated, all of the orally exposed untreated PrnpΔIEC (control) mice succumbed to clinical prion disease (mean survival time of 307 ± 23 days; median of 300 days; n = 10/10) (Table 1). Furthermore, IEC-restricted Prnp ablation did not affect disease duration (survival times) or susceptibility, as all of the βNF-treated PrnpΔIEC mice also succumbed to clinical prion disease with similar survival times (mean of 306 ± 11 days; median of 306 days; n = 12/12; P = 0.673 by one-way analysis of variance [ANOVA] with Dunnett's posttest) (Table 1).
TABLE 1.
Prnp deficiency in the gut epithelium does not influence oral prion disease susceptibility
| Mouse modela | Mean survival time (days) ± SDb | Median survival time (days) | No. of animals with clinical disease/total no. of animals tested | No. of animals with histopathological signs of prion disease in brain (spongiform encephalopathy)/total no. of animals tested |
|---|---|---|---|---|
| PrnpΔIEC | 307 ± 23 | 300 | 10/10 | 10/10 |
| PrnpΔIEC + Veh | 303 ± 12 | 303 | 10/10 | 10/10 |
| PrnpΔIEC + βNF | 308 ± 11 | 306 | 12/12 | 12/12 |
| PrnpF/F + βNF | 313 ± 19 | 305 | 9/9 | 9/9 |
Where indicated, mice were given daily intraperitonal injections of β-napthoflavone (βNF) or corn oil (vehicle control [Veh]) for 5 days. Mice were orally exposed to ME7 scrapie prions 14 days after the last treatment.
Duration from the time of injection with prions to culling at the clinical endpoint. No statistical differences in survival times were observed between groups (P = 0.673 by one-way ANOVA with Dunnett's posttest).
All the brains from the clinically affected mice in each group displayed the characteristic spongiform pathology (vacuolation), PrPSc accumulation, astrogliosis, and microgliosis, which is associated with terminal infection with ME7 scrapie prions (Fig. 4A and B). The severity and distribution of the spongiform pathology were also similar in the brains of the clinically affected mice from each group (Fig. 4C).
FIG 4.

Intestinal epithelial cell-restricted Prnp ablation does not influence development of the histopathological signs of prion disease in the brains of clinically affected mice. Female PrnpΔIEC mice were treated with βNF for 5 days to specifically ablate Prnp expression in intestinal epithelial cells. Untreated PrnpΔIEC mice and PrnpΔIEC mice treated with the vehicle alone (Veh.) were used as controls. Fourteen days later, the mice were orally exposed to ME7 scrapie prions and culled when they succumbed to clinical prion disease. (A) High levels of spongiform pathology (hematoxylin and eosin [H&E] stain), heavy accumulations of disease-specific PrP (PrPd) (brown), reactive astrocytes expressing GFAP (brown), and active microglia expressing Iba-1 (brown) were detected in the brains of all orally exposed mice with clinical prion disease. Clin., clinical prion disease status; pos., clinically positive. Individual survival times are shown (dpi, days postinfection). Sections were counterstained with hematoxylin to detect cell nuclei (blue). (B) Immunoblot analysis of brain tissue homogenates confirms the presence of high levels of prion-specific, relatively proteinase K (PK)-resistant PrPSc within the brains of the clinically affected mice from each group. Samples were treated in the presence (+) or absence (−) of PK before electrophoresis. After PK treatment, a typical three-band pattern was observed between molecular mass values of 20 and 30 kDa, representing unglycosylated, monoglycosylated, and diglycosylated isomers of PrP (in order of increasing molecular mass). (C) The severity and distribution of the spongiform pathology (vacuolation) within each brain were scored on a scale of 1 to 5 in nine gray matter areas: dorsal medulla (G1), cerebellar cortex (G2), superior colliculus (G3), hypothalamus (G4), thalamus (G5), hippocampus (G6), septum (G7), retrosplenial and adjacent motor cortex (G8), and cingulate and adjacent motor cortex (G9). Each point represents the mean vacuolation score ± SD (n = 10 to 12 mice/group).
Together, these data clearly show that efficient prion neuroinvasion after oral exposure occurs independently of PrPC expression in IEC in the small intestine.
DISCUSSION
The initial transport of prions across the gut epithelium by M cells into small intestinal Peyer's patches is essential to establish efficient infection after oral exposure (13, 16). However, whether the uptake and translocation of prions across the gut epithelium involves a specific receptor is uncertain. Treatments that prevent the initial replication of prions within the GALT impede the spread of prions to the brain and reduce disease susceptibility (4, 13, 16, 18). Thus, the identification of the molecular factors that facilitate the uptake of prions into the GALT will help the design of novel intervention strategies and enhance our understanding of the factors that influence the risk of infection. Small intestinal M cells express cellular PrPC on their apical surfaces, and this may be used by certain gastrointestinal pathogens as an uptake receptor to infect Peyer's patches (26, 34). Independent IHC-based tracing studies have suggested that orally administered prion protein can be transported across the gut epithelium of PrPC-deficient mice (14, 17), but whether the expression of PrPC on IEC populations contributed to the establishment of host infection had not been assessed. Data in the current study clearly show that prion neuroinvasion after oral exposure occurs independently of PrPC expression in small intestinal IEC.
Orally acquired prions first replicate in the small intestinal GALT and subsequently spread to most other secondary lymphoid tissues, including the large intestinal GALT. Since oral prion disease susceptibility is substantially reduced in the specific absence of the small intestinal GALT (11), this suggests that the early replication of prions within Peyer's patches is essential to establish efficient host infection after oral exposure. The small intestinal GALT also appear to be the important early sites of prion replication in natural host species (45–47). Although we observed highly efficient Cre-mediated gene recombination in intestinal crypts and IEC throughout the small intestines of βNF-treated PrnpΔIEC mice, the efficiency in the colon was lower and presented a mosaic pattern (Fig. 2c) (35). The less efficient Prnp ablation in the large intestine was unlikely to have influenced oral prion disease pathogenesis in the current study, as the large intestinal GALT, such as the colonic patches, are not important early sites of prion replication and neuroinvasion (11).
Despite the potentially widespread exposure of the United Kingdom population to BSE-contaminated food in the 1980s, there have fortunately been many fewer clinical cases of variant Creutzfeldt-Jakob disease in humans than the original estimates suggested (48) (178 definite or probable cases, as of 4 May 2018 [http://www.cjd.ed.ac.uk/]). This implies that additional factors could potentially influence an individual's susceptibility to oral prion infection by enhancing or impeding the initial uptake of prions from the gut lumen. In support of this hypothesis, we have shown that stimuli that increase the density of M cells in the gut epithelium also increase oral prion disease susceptibility approximately 10-fold by enhancing the uptake of prions into Peyer's patches (16). The expression level of PrPC in host cells such as neurons and FDC directly influences survival times of prion-infected mice (43, 49–51). Acute mucosal inflammation following oral infection with S. Typhimurium and treatment with dextran sodium sulfate have each been shown to enhance PrPC expression in the large intestine, implying the potential to enhance oral prion disease pathogenesis and susceptibility (52, 53). Conversely, PrPC expression was reported to be downregulated in the small intestines of mice treated with the nonsteroidal anti-inflammatory drug indomethacin and coincided with a modest increase in survival time after oral exposure to ME7 scrapie prions (54). Although the cellular sites of PrPC expression were not determined in the above-mentioned studies, our data suggest that the magnitude of PrPC expression in IEC throughout the small intestine is unlikely to be an important factor which influences the risk of oral prion disease susceptibility.
In sheep with natural scrapie (55) or orally exposed to BSE prions (56), prion accumulation is first detected in the palatine tonsils in addition to Peyer's patches. Natural prion disease-susceptible host species such as sheep and cervids also have highly developed olfactory systems, which they use to detect food, select mates, and sense predators. A series of experimental studies in rodents and sheep showed that prion infections can be established via the nasal cavity (57–59). Thus, it cannot be excluded that soil-bound prions might also be inhaled and infect the host as the animal forages for food. Although M cells are present in the epithelia covering the nasal-associated lymphoid tissue (60), studies in hamsters indicate that this prion uptake across the nasal epithelium occurs independently of M cells (61). Whether prion uptake across the mucosal surfaces in the upper gastrointestinal and upper respiratory tracts of natural host species is also PrPC independent remains to be determined.
In conclusion, we show that oral prion disease neuroinvasion occurs independently of PrPC expression in IEC in the small intestine. Whether prions exploit other receptors on the apical surfaces of M cells to establish host infection is uncertain. The specific targeting of vaccine antigens to M cells has been shown to be an effective method to induce protective antigen-specific mucosal immunity (62). Mucosal immunization has also been shown to provide promising protection against oral prion infections in mice (63) and white-tailed deer (64). Thus, a thorough understanding of the mechanisms that prions exploit to establish infection within the GALT may help to identify important factors which influence disease susceptibility or identify novel targets for prophylactic intervention.
MATERIALS AND METHODS
Mice.
The following mouse strains were used in this study where indicated: Cyp1a1-Cre (35); the ROSA26F/F reporter strain (40); and PrnpF/F mice (strain Prnptm2Tuzi), which have LoxP sites flanking exon 3 of the Prnp gene (39). C57BL/Dk mice were also used where indicated. All mice were bred and maintained under specific-pathogen-free (SPF) conditions. All studies and regulatory licenses were approved by the institute's ethics committee and carried out under the authority of a United Kingdom Home Office project license. Prior to the use of mice in experiments, the genotype of each mouse was confirmed by PCR analysis of tail DNA (Table 2).
TABLE 2.
PCR primers used to confirm mouse genotypesa
| Allele | Description | Primer sequence | Product size (bp) |
|---|---|---|---|
| Cre | Fwd | CGAGTGATGAGGTTCGCAAGAACC | 786 |
| Rev | GCTAAGTGCCTTCTCTACACCTGC | ||
| LacZ | Fwd | TACCACAGCGGATGGTTCGG | 300 |
| Rev | GTGGTGGTTATGCCGATCGC | Recombined PrnpF, 344 | |
| Prnpflox | 1 | AATGGTTAAACTTTCGTTAAGGAT | PrnpF, 210 |
| 2 | GCCGACATCAGTCCACATAG | Prnp+, 167 | |
| 3 | GGTTGACGCCATGACTTTC | ||
| Prnp+ | Fwd | TCATCCCACGATCAGGAAGATGAG | 600 |
| Rev | ATGGCGAACCTTGGCTACTGGCTG |
Fwd, forward primer; Rev, reverse primer; Recombined PrnpF, Cre-mediated DNA recombined allele.
β-Naphthoflavone treatment.
Where indicated, mice were given five daily intraperitoneal injections of β-naphthoflavone (80 mg/kg of body weight; Sigma-Aldrich, Poole, UK) dissolved in corn oil (Sigma-Aldrich) and analyzed 14 days after the last injection or used in subsequent experiments. Where indicated, some mice received either corn oil alone (vehicle) or no treatment as controls.
Histological assessment of LacZ expression.
Tissues were first immersed in LacZ fixative (phosphate-buffered saline [PBS] [pH 7.4] containing 2% paraformaldehyde, 0.2% glutaraldehyde, 0.02% Nonidet P-40, 0.01% sodium deoxycholate, 5 mM EGTA, and 2 mM MgCl2) and washed in LacZ wash buffer (PBS [pH 7.4] containing 0.02% Nonidet P-40, 0.01% sodium deoxycholate, and 2 mM MgCl2). Tissues were subsequently incubated in 15% (wt/vol) sucrose in PBS overnight, followed by a further overnight incubation in 30% (wt/vol) sucrose in PBS, and embedded in Tissue-Tek OCT compound (Bayer PLC, Newbury, UK). Serial sections (thickness, 8 mm) were cut on a cryostat and stained overnight with LacZ staining solution (Glycosynth, Warrington, UK). The staining reaction was stopped by washing in LacZ wash buffer followed by distilled water. Sections were counterstained with nuclear fast red (Vector Laboratories, Peterborough, UK). Intestinal whole mounts were prepared luminal side up, as described previously (65), and fixed in ice-cold 2% formaldehyde–0.2% glutaraldehyde in PBS (pH 7.4) for 1 h before overnight incubation in LacZ staining solution.
Prion exposure and disease monitoring.
For oral exposure, mice were fed individual food pellets dosed with 50 μl of a 1.0% (wt/vol) dilution of scrapie brain homogenate (containing approximately 4.6 log10 i.c. ID50 units) prepared from mice terminally affected with ME7 scrapie prions according to our standard protocol (11, 16, 19). During the dosing period, mice were individually housed in bedding- and food-free cages, with water provided ad libitum. A single prion-dosed food pellet was then placed in the cage. The mice were returned to their original cages (with bedding and food ad libitum) as soon as the food pellet was observed to have been completely ingested. The use of bedding- and additional food-free cages ensured easy monitoring of consumption of the prion-contaminated food pellet. Following prion exposure, mice were coded, assessed weekly for signs of clinical disease, and culled at a standard clinical endpoint. The clinical endpoint of disease was determined by rating the severity of clinical signs of prion disease exhibited by the mice. Mice were clinically scored as “unaffected,” “possibly affected,” and “definitely affected” using standard criteria that typically are present in mice with terminal ME7 scrapie prion disease. Clinical signs following infection with the ME7 scrapie prions may include weight loss; starry coat; hunched, jumpy behavior (at early onset) progressing to limited movement; upright tail; wet genitals; decreased awareness; discharge from eyes/blinking eyes; and ataxia of hind legs. The clinical endpoint of disease was defined in one of the following ways: (i) the day on which a mouse received a second consecutive “definite” rating, (ii) the day on which a mouse received a third “definite” rating within four consecutive weeks, or (iii) the day on which a mouse was culled in extremis. Prion diagnosis was confirmed by histopathological assessment of the magnitude of the spongiform pathology (vacuolation) in nine distinct gray matter regions of the brain as described previously (66).
For bioassays of prion infectivity, individual tissues were prepared as 10% (wt/vol) homogenates, and 20 μl was injected i.c. into each of 4 recipient C57BL/Dk indicator mice. The prion infectivity titer in each sample was determined from the mean incubation period in the indicator mice, by reference to a dose/incubation period-response curve for ME7 scrapie-infected spleen tissue serially titrated in C57BL/Dk indicator mice (67).
Immunohistochemistry.
For the detection of disease-specific PrP (PrPd) in intestines and brains, tissues were fixed in periodate-lysine-paraformaldehyde fixative and embedded in paraffin wax. Sections (thickness, 6 μm) were deparaffinized and pretreated to enhance the detection of PrPd by hydrated autoclaving (15 min, 121°C, hydration) and subsequent immersion in formic acid (98%) for 5 min. Sections were then immunostained with 1B3 PrP-specific polyclonal antibody (pAb). For the detection of FDC in intestines, deparaffinized sections were first pretreated with target retrieval solution (Dako) and subsequently immunostained with anti-CD21/35 (clone 7G6; BD Biosciences). Paraffin-embedded tissue immunoblot analysis was used to confirm that the PrPd detected by immunohistochemistry was proteinase K-resistant PrPSc (68). Membranes were subsequently immunostained with 1B3 PrP-specific pAb.
For the detection of astrocytes, brain sections were immunostained with anti-glial fibrillary acidic protein (GFAP; Dako, Ely, UK), and to detect microglia sections, they were immunostained with anti-ionized calcium-binding adaptor molecule 1 (Iba-1; Wako Chemicals GmbH, Neuss, Germany).
Following the addition of primary antibodies, biotin-conjugated species-specific secondary antibodies (Stratech, Soham, UK) were applied, and immunolabeling was revealed using either alkaline phosphatase conjugated to the avidin-biotin complex (Vector Laboratories, Peterborough, UK), visualized using Vector red, or horseradish peroxidase (HRP) conjugated to the avidin-biotin complex (Vector Laboratories), visualized with 3,3'-diaminobenzidine (Sigma). Sections were counterstained with hematoxylin to distinguish cell nuclei.
Immunoblot detection of PrPSc.
Brain homogenates (10%, wt/vol) were prepared in NP-40 lysis buffer (1% NP-40, 0.5% sodium deoxycholate, 150 mM NaCl, 50 mM Tris HCl [pH 7.5]) and incubated at 37°C for 1 h with 20 μg/ml PK. Digestions were halted by the addition of 1 mM phenylmethylsulfonyl fluoride. Samples were then subjected to electrophoresis through 12% Tris-glycine polyacrylamide gels (NuPAGE; Life Technologies) and transferred to polyvinylidene difluoride (PVDF) membranes by semidry blotting. PrP was detected using anti-mouse PrP-specific monoclonal antibody (mAb) 7A12 (69), followed by horseradish peroxidase-conjugated goat anti-mouse antibody (Jackson ImmunoResearch), and visualized by chemiluminescence (BM chemiluminescent substrate kit; Roche, Burgess Hill, UK).
Statistical analyses.
Unless indicated otherwise, data are presented as means ± standard deviations (SD), and significant differences between groups were sought by Student's t test. P values of <0.05 were accepted as significant.
ACKNOWLEDGMENTS
We thank Nadia Tuzi, Dorothy Kisielewski, Rebecca Greenan, Simon Cumming, Val Thomson, Kris Hogan, and the Pathology Services Group (University of Edinburgh, UK) for excellent technical support and Abigail Diack (University of Edinburgh) for helpful discussions.
This work was supported by project funding from the Department of Health (PR-IP-0807-0070161) and Institute Strategic Programme grant funding from the Biotechnology and Biological Sciences Research Council (grant numbers BBS/E/D/05241339, BBS/E/D/20251968, and BBS/E/D/20002174).
We dedicate this paper to Alan Clarke (1963–2015) in recognition of his inspiration and support in this study.
REFERENCES
- 1.Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H, Bostock CJ. 1997. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature 389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 2.Legname G, Baskakov IV, Nguyen H-OB, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. 2004. Synthetic mammalian prions. Science 305:673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 3.Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 1994. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol 8:121–127. doi: 10.1007/BF02780662. [DOI] [PubMed] [Google Scholar]
- 4.Mabbott NA, Young J, McConnell I, Bruce ME. 2003. Follicular dendritic cell dedifferentiation by treatment with an inhibitor of the lymphotoxin pathway dramatically reduces scrapie susceptibility. J Virol 77:6845–6854. doi: 10.1128/JVI.77.12.6845-6854.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glaysher BR, Mabbott NA. 2007. Role of the GALT in scrapie agent neuroinvasion from the intestine. J Immunol 178:3757–3766. doi: 10.4049/jimmunol.178.6.3757. [DOI] [PubMed] [Google Scholar]
- 6.Andreoletti O, Berthon P, Marc D, Sarradin P, Grosclaude J, van Keulen L, Schelcher F, Elsen J-M, Lantier F. 2000. Early accumulation of PrPSc in gut-associated lymphoid and nervous tissues of susceptible sheep from a Romanov flock with natural scrapie. J Gen Virol 81:3115–3126. doi: 10.1099/0022-1317-81-12-3115. [DOI] [PubMed] [Google Scholar]
- 7.Sigurdson CJ, Williams ES, Miller MW, Spraker TR, O'Rourke KI, Hoover EA. 1999. Oral transmission and early lymphoid tropism of chronic wasting disease PrPres in mule deer fawns (Odocoileus hemionus). J Gen Virol 80:2757–2764. doi: 10.1099/0022-1317-80-10-2757. [DOI] [PubMed] [Google Scholar]
- 8.Hilton D, Fathers E, Edwards P, Ironside J, Zajicek J. 1998. Prion immunoreactivity in appendix before clinical onset of variant Creutzfeldt-Jakob disease. Lancet 352:703–704. doi: 10.1016/S0140-6736(98)24035-9. [DOI] [PubMed] [Google Scholar]
- 9.Prinz M, Huber G, Macpherson AJS, Heppner FL, Glatzel M, Eugster H-P, Wagner N, Aguzzi A. 2003. Oral prion infection requires normal numbers of Peyer's patches but not of enteric lymphocytes. Am J Pathol 162:1103–1111. doi: 10.1016/S0002-9440(10)63907-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horiuchi M, Furuoka H, Kitamura N, Shinagawa M. 2006. Alymphoplasia mice are resistant to prion infection via oral route. Jpn J Vet Res 53:149–157. [PubMed] [Google Scholar]
- 11.Donaldson DS, Else KJ, Mabbott NA. 2015. The gut-associated lymphoid tissues in the small intestine, not the large intestine, play a major role in oral prion disease pathogenesis. J Virol 15:9532–9547. doi: 10.1128/JVI.01544-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heppner FL, Christ AD, Klein MA, Prinz M, Fried M, Kraehenbuhl J-P, Aguzzi A. 2001. Transepithelial prion transport by M cells. Nat Med 7:976–977. doi: 10.1038/nm0901-976. [DOI] [PubMed] [Google Scholar]
- 13.Donaldson DS, Kobayashi A, Ohno H, Yagita H, Williams IR, Mabbott NA. 2012. M cell depletion blocks oral prion disease pathogenesis. Mucosal Immunol 5:216–225. doi: 10.1038/mi.2011.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takakura I, Miyazawa K, Kanaya T, Itani W, Watanabe K, Ohwada S, Watanabe H, Hondo T, Rose MT, Mori T, Sakaguchi S, Nishida N, Katamine S, Yamaguchi T, Aso H. 2011. Orally administered prion protein is incorporated by M cells and spreads to lymphoid tissues with macrophages in prion protein knockout mice. Am J Pathol 179:1301–1309. doi: 10.1016/j.ajpath.2011.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyazawa K, Kanaya T, Takakura I, Tanaka S, Hondo T, Watanabe H, Rose MT, Kitazawa H, Yamaguchi T, Katamine S, Nishida N, Aso H. 2010. Transcytosis of murine-adapted bovine spongiform encephalopathy agents in an in vitro bovine M cell model. J Virol 84:12285–12291. doi: 10.1128/JVI.00969-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donaldson DS, Sehgal A, Rios D, Williams IR, Mabbott NA. 2016. Increased abundance of M cells in the gut epithelium dramatically enhances oral prion disease susceptibility. PLoS Pathog 12:e1006075. doi: 10.1371/journal.ppat.1006075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kujala P, Raymond C, Romeijn M, Godsave SF, van Kasteren SI, Wille H, Prusiner SB, Mabbott NA, Peters PJ. 2011. Prion uptake in the gut: identification of the first uptake and replication sites. PLoS Pathog 7:e1002449. doi: 10.1371/journal.ppat.1002449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raymond CR, Aucouturier P, Mabbott NA. 2007. In vivo depletion of CD11c+ cells impairs scrapie agent neuroinvasion from the intestine. J Immunol 179:7758–7766. doi: 10.4049/jimmunol.179.11.7758. [DOI] [PubMed] [Google Scholar]
- 19.Bradford BM, Reizis B, Mabbott NA. 2017. Oral prion disease pathogenesis is impeded in the specific absence of CXCR5-expressing dendritic cells. J Virol 91:e00124-17. doi: 10.1128/JVI.00124-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCulloch L, Brown KL, Mabbott NA. 2013. Ablation of the cellular prion protein, PrPC, specifically on follicular dendritic cells has no effect on their maturation or function. Immunology 138:246–257. doi: 10.1111/imm.12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beekes M, McBride PA. 2007. The spread of prions through the body in naturally acquired transmissible spongiform encephalopathies. FEBS J 274:588–605. doi: 10.1111/j.1742-4658.2007.05631.x. [DOI] [PubMed] [Google Scholar]
- 22.Mabbott NA, Donaldson DS, Ohno H, Williams IR, Mahajan A. 2013. Microfold (M) cells: important immunosurveillance posts in the intestinal epithelium. Mucosal Immunol 6:666–677. doi: 10.1038/mi.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hase K, Kawano K, Nochi T, Pontes GS, Fukuda S, Ebisawa M, Kadokura K, Tobe T, Fujimura Y, Kawano S, Yabashi A, Waguri S, Nakato G, Kimura S, Murakami T, Iimura M, Hamura K, Fukuoka S-I, Lowe AW, Itoh K, Kiyono H, Ohno H. 2009. Uptake through glycoprotein 2 of FimH+ bacteria by M cells initiates mucosal immune responses. Nature 462:226–231. doi: 10.1038/nature08529. [DOI] [PubMed] [Google Scholar]
- 24.Kanaya T, Hase K, Takahashi D, Fukuda S, Hoshino K, Sasaki I, Hemmi H, Knoop KA, Kumar N, Sato M, Katsuno T, Yokosuka O, Toyooka K, Nakai K, Sakamoto A, Kitahara Y, Jinnohara T, McSorley SJ, Kaisho T, Williams IR, Ohno H. 2012. The Ets transcription factor Spi-B is essential for the differentiation of intestinal microfold cells. Nat Immunol 13:729–736. doi: 10.1038/ni.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rios D, Wood MB, Li J, Chassaing B, Gewirtz AT, Williams IR. 2016. Antigen sampling by intestinal M cells is the principal pathway initiating mucosal IgA production to commensal enteric bacteria. Mucosal Immunol 9:907–916. doi: 10.1038/mi.2015.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakato G, Hase K, Suzuki M, Kimura M, Ato M, Hanazato M, Tobiume M, Horiuchi M, Atarashi R, Nishida N, Watarai H, Imaoka K, Ohno H. 2012. Cutting edge: Brucella abortus exploits a cellular prion protein on intestinal M cells as an invasive receptor. J Immunol 189:1540–1544. doi: 10.4049/jimmunol.1103332. [DOI] [PubMed] [Google Scholar]
- 27.Tahoun A, Mahajan S, Paxton E, Malterer G, Donaldson DS, Wang D, Tan A, Gillespie TL, O'Shea M, Rose A, Shaw DJ, Gally DL, Lengeling A, Mabbott NA, Haas J, Mahajan A. 2012. Salmonella transforms follicle-associated epithelial cells into M cells to promote intestinal invasion. Cell Host Microbe 12:645–666. doi: 10.1016/j.chom.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 28.Westphal S, Lugering A, von Wedel J, von Eiff C, Maaser C, Spahn T, Heusipp G, Schmidt MA, Herbst H, Williams IR, Domschke W, Kucharzik T. 2008. Resistance of chemokine receptor 6-deficient mice to Yersinia enterocolitica infection: evidence on defective M-cell formation in vivo. Am J Pathol 172:671–680. doi: 10.2353/ajpath.2008.070393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kolawole AO, Gonzalez-Hernandez MB, Turula H, Yu C, Elftman MD, Wobus CE. 2015. Oral norovirus infection is blocked in mice lacking Peyer's patches and mature M cells. J Virol 90:1499–1506. doi: 10.1128/JVI.02872-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonzalez-Hernandez MB, Liu T, Payne HC, Stencel-Baerenwald JE, Ikizler M, Yagita H, Dermody TS, Williams IR, Wobus CE. 2014. Efficient norovirus and reovirus replication in the mouse intestine requires microfold (M) cells. J Virol 88:6934–6943. doi: 10.1128/JVI.00204-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yanagihara S, Kanaya T, Fukuda S, Nakato G, Hanazato M, Wu XR, Yamamoto N, Ohno H. 2017. Uromodulin-SlpA binding dictates Lactobacillus acidophilus uptake by intestinal M cells. Int Immunol 29:357–363. doi: 10.1093/intimm/dxx043. [DOI] [PubMed] [Google Scholar]
- 32.Kim S-H, Jang Y-S. 2017. Yersinia enterocolitica exploits signal crosstalk between complement C5a receptor and Toll-like receptor 1/2 to avoid the bacterial clearance in M cells. Immune Netw 17:228–236. doi: 10.4110/in.2017.17.4.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsumura T, Sugawara Y, Yutani M, Amatsu S, Yagita H, Kohda T, Fukuoka S-I, Nakamura Y, Fukuda S, Hase K, Ohno H, Fujinaga Y. 2015. Botulinum toxin A complex exploits intestinal M cells to enter the host and exert neurotoxicity. Nat Commun 6:6255. doi: 10.1038/ncomms7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakato G, Fukuda S, Hase K, Goitsuka R, Cooper MD, Ohno H. 2009. New approach for M-cell-specific molecules by screening comprehensive transcriptome analysis. DNA Res 16:227–235. doi: 10.1093/dnares/dsp013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ireland H, Kemp R, Houghton C, Howard L, Clarke AR, Sansom OJ, Winton DJ. 2004. Inducible Cre-mediated control of gene expression in the murine gastrointestinal tract: effect of loss of β-catenin. Gastroenterology 126:1236–1246. doi: 10.1053/j.gastro.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 36.Gonneaud A, Turgeon N, Boudreau F, Perrault N, Rivard N, Asselin C. 2016. Distinct roles for intestinal epithelial cell-specific Hdac1 and Hdac2 in the regulation of murine intestinal homeostasis. J Cell Physiol 231:436–448. doi: 10.1002/jcp.25090. [DOI] [PubMed] [Google Scholar]
- 37.Asano J, Sato T, Ichinose S, Kajita M, Onai N, Shimizu S, Ohteki T. 2017. Intrinsic autophagy is required for the maintenance of intestinal stem cells and for irradiation-induced intestinal regeneration. Cell Rep 20:1050–1060. doi: 10.1016/j.celrep.2017.07.019. [DOI] [PubMed] [Google Scholar]
- 38.FANTOM Consortium and the RIKEN PMI and CLST (DGT), Forrest AR, Kawaji H, Rehli M, Baillie JK, de Hoon MJ, Haberle V, Lassmann T, Kulakovskiy IV, Lizio M, Itoh M, Andersson R, Mungall CJ, Meehan TF, Schmeier S, Bertin N, Jørgensen M, Dimont E, Arner E, Schmidl C, Schaefer U, Medvedeva YA, Plessy C, Vitezic M, Severin J, Semple C, Ishizu Y, Young RS, Francescatto M, Alam I, Albanese D, Altschuler GM, Arakawa T, Archer JA, Arner P, Babina M, Rennie S, Balwierz PJ, Beckhouse AG, Pradhan-Bhatt S, Blake JA, Blumenthal A, Bodega B, Bonetti A, Briggs J, Brombacher F, Burroughs AM, Califano A, Cannistraci CV, Carbajo D, et al. 2014. A promoter-level mammalian expression atlas. Nature 507:462–470. doi: 10.1038/nature13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tuzi NL, Clarke AR, Bradford B, Aitchison L, Thomson V, Manson JC. 2004. Cre-loxP mediated control of PrP to study transmissible spongiform encephalopathy diseases. Genesis 40:1–6. doi: 10.1002/gene.20046. [DOI] [PubMed] [Google Scholar]
- 40.Mao X, Fujiwara Y, Orkin SH. 1999. Improved reporter strain for monitoring Cre recombinase-mediated DNA excisions in mice. Proc Natl Acad Sci U S A 96:5037–5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McBride P, Eikelenboom P, Kraal G, Fraser H, Bruce ME. 1992. PrP protein is associated with follicular dendritic cells of spleens and lymph nodes in uninfected and scrapie-infected mice. J Pathol 168:413–418. doi: 10.1002/path.1711680412. [DOI] [PubMed] [Google Scholar]
- 42.Mabbott NA, Mackay F, Minns F, Bruce ME. 2000. Temporary inactivation of follicular dendritic cells delays neuroinvasion of scrapie. Nat Med 6:719–720. doi: 10.1038/77401. [DOI] [PubMed] [Google Scholar]
- 43.Brown KL, Stewart K, Ritchie D, Mabbott NA, Williams A, Fraser H, Morrison WI, Bruce ME. 1999. Scrapie replication in lymphoid tissues depends on PrP-expressing follicular dendritic cells. Nat Med 5:1308–1312. doi: 10.1038/15264. [DOI] [PubMed] [Google Scholar]
- 44.Mok SW, Proia RL, Brinkmann V, Mabbott NA. 2012. B cell-specific S1PR1 deficiency blocks prion dissemination between secondary lymphoid organs. J Immunol 188:5032–5040. doi: 10.4049/jimmunol.1200349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Keulen LJ, Schreuder BE, Vromans ME, Langeveld JP, Smits MA. 2000. Pathogenesis of natural scrapie in sheep. Arch Virol Suppl 16:57–71. [DOI] [PubMed] [Google Scholar]
- 46.Gonzalez L, Martin S, Siso S, Konold T, Ortiz-Pelaez A, Phelan L, Goldmann W, Stewart P, Saunders G, Windl O, Jeffrey M, Hawkins SAC, Dawson M, Hope J. 2009. High prevalence of scrapie in a dairy goat herd: tissue distribution of disease-associated PrP and effect of PRNP genotype and age. Vet Res 40:65. doi: 10.1051/vetres/2009048. [DOI] [PubMed] [Google Scholar]
- 47.Thomsen BV, Schneider DA, O'Rourke KI, Gidlewski T, McLane J, Allen RW, McIsaac AA, Mitchell GB, Keane DP, Spraker TR, Balachandran A. 2012. Diagnostic accuracy of rectal mucosa biopsy testing for chronic wasting disease within white-tailed deer (Odocoileus virginianus) herds in North America: effects of age, sex, polymorphism at PRNP codon 96, and disease progression. J Vet Diagn Invest 24:878–887. doi: 10.1177/1040638712453582. [DOI] [PubMed] [Google Scholar]
- 48.Diack AB, Head MW, McCutcheon S, Boyle A, Knight R, Ironside JW, Manson JC, Will RG. 2014. Variant CJD. 18 years of research and surveillance. Prion 2014:286–295. doi: 10.4161/pri.29237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manson JC, Clarke AR, McBride PA, McConnell I, Hope J. 1994. PrP gene dosage determines the timing but not the final intensity or distribution of lesions in scrapie pathology. Neurodegeneration 3:331–340. [PubMed] [Google Scholar]
- 50.Klein MA, Frigg R, Raeber AJ, Flechsig E, Hegyi I, Zinkernagel RM, Weissmann C, Aguzzi A. 1998. PrP expression in B lymphocytes is not required for prion neuroinvasion. Nat Med 4:1429–1433. doi: 10.1038/4022. [DOI] [PubMed] [Google Scholar]
- 51.McCulloch L, Brown KL, Bradford BM, Hopkins J, Bailey M, Rajewsky K, Manson JC, Mabbott NA. 2011. Follicular dendritic cell-specific prion protein (PrPC) expression alone is sufficient to sustain prion infection in the spleen. PLoS Pathog 7:e1002402. doi: 10.1371/journal.ppat.1002402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martin GR, Keenan CM, Sharkey KA, Jirik FR. 2011. Endogenous prion protein attenuates experimentally induced colitis. Am J Pathol 179:2290–2301. doi: 10.1016/j.ajpath.2011.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sigurdson CJ, Heikenwalder M, Manco G, Barthel M, Schwarz P, Stecher B, Krautler NJ, Hardt W-D, Seifert B, MacPherson AJS, Corthesy I, Aguzzi A. 2009. Bacterial colitis increases susceptibility to oral prion pathogenesis. J Infect Dis 199:243–252. doi: 10.1086/595791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin GR, Sharkey KA, Jirik FR. 2015. Orally administered indomethacin acutely reduces cellular prion protein in the small intestine and modestkt increases survival of mice exposed to infectious prions. Scand J Gastroenterol 50:542–549. doi: 10.3109/00365521.2014.1003400. [DOI] [PubMed] [Google Scholar]
- 55.van Keulen LJM, Vromans MEW, van Zijderveld FG. 2002. Early and late pathogenesis of natural scrapie infection in sheep. APMIS 110:23–32. doi: 10.1034/j.1600-0463.2002.100104.x. [DOI] [PubMed] [Google Scholar]
- 56.Van Keulen LJM, Vromans MEW, Dolstra CH, Bossers A, van Zijderveld FG. 2008. Pathogenesis of bovine spongiform encephalopathy in sheep. Arch Virol 153:445–453. doi: 10.1007/s00705-007-0007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kincaid AE, Bartz JC. 2007. The nasal cavity is a route for prion infection in hamsters. J Virol 81:4482–4491. doi: 10.1128/JVI.02649-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hamir AN, Kunkle RA, Richt JA, Miller JM, Greenlee JJ. 2008. Experimental transmission of US scrapie agent by nasal, peritoneal, and conjunctival routes to genetically susceptible sheep. Vet Pathol 45:7–11. doi: 10.1354/vp.45-1-7. [DOI] [PubMed] [Google Scholar]
- 59.Denkers ND, Seelig DM, Telling GC, Hoover EA. 2010. Aerosol and nasal transmission of chronic wasting disease in cervidized mice. J Gen Virol 91:1651–1658. doi: 10.1099/vir.0.017335-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mutoh M, Kimura S, Takashi-Iwanaga H, Hisamoto M, Iwanaga T, Iida J. 2016. RANKL regulates differentiation of microfold cells in mouse nasopharynx-associated lymphoid tissue (NALT). Cell Tissue Res 364:175–184. doi: 10.1007/s00441-015-2309-2. [DOI] [PubMed] [Google Scholar]
- 61.Kincaid AE, Ayers JI, Bartz JC. 2016. Specificity, size, and frequency of spaces that characterize the mechanism of bulk transepithelial transport of prions in the nasal cavities of hamsters and mice. J Virol 90:8293–8301. doi: 10.1128/JVI.01103-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shima H, Watanabe T, Fukuda S, Fukuoka S-I, Ohara O, Ohno H. 2014. A novel vaccine targeting Peyer's patch M cells induces protective antigen-specific IgA responses. Int Immunol 26:619–625. doi: 10.1093/intimm/dxu061. [DOI] [PubMed] [Google Scholar]
- 63.Goñi F, Prelli F, Schreiber F, Scholtzova H, Chung E, Kascsak R, Brown DR, Sigurdsson EM, Chabalgoity JA, Wisniewski T. 2008. High titers of mucosal and systemic anti-PrP antibodies abrogate oral prion infection in mucosal-vaccinated mice. Neuroscience 153:679–686. doi: 10.1016/j.neuroscience.2008.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Goñi F, Mathiason CK, Yim L, Wong K, Hayes-Klug J, Nalls A, Peyser D, Estevez V, Denkers N, Xu J, Osborn DA, Miller KV, Warren RJ, Brown DR, Chabalgoity JA, Hoover EA, Wisniewski T. 2015. Mucosal immunization with an attenuated Salmonella vaccine partially protects white-tailed deer from chronic wasting disease. Vaccine 33:726–733. doi: 10.1016/j.vaccine.2014.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Winton DJ, Ponder BAJ. 1990. Stem cell organisation in mouse small intestine. Proc Biol Sci 241:13–18. doi: 10.1098/rspb.1990.0059. [DOI] [PubMed] [Google Scholar]
- 66.Fraser H, Dickinson AG. 1968. The sequential development of the brain lesions of scrapie in three strains of mice. J Comp Pathol 78:301–311. doi: 10.1016/0021-9975(68)90006-6. [DOI] [PubMed] [Google Scholar]
- 67.Brown KL, Wathne GJ, Sales J, Bruce ME, Mabbott NA. 2009. The effects of host age on follicular dendritic cell status dramatically impair scrapie agent neuroinvasion in aged mice. J Immunol 183:5199–5207. doi: 10.4049/jimmunol.0802695. [DOI] [PubMed] [Google Scholar]
- 68.Schulz-Schaeffer WJ, Tschoke S, Kranefuss N, Drose W, Hause-Reitner D, Giese A, Groschup MH, Kretzschmar HA. 2000. The paraffin-embedded tissue blot detects PrPsc early in the incubation time in prion diseases. Am J Pathol 156:51–56. doi: 10.1016/S0002-9440(10)64705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yin S, Pham N, Yu S, Li C, Wong P, Chang B, Kang S-C, Biasini E, Tien P, Harris DA, Sy M-S. 2007. Human prion proteins with pathogenic mutations share common conformational changes resulting in enhanced binding to glycosaminoglycans. Proc Natl Acad Sci U S A 104:7546–7551. doi: 10.1073/pnas.0610827104. [DOI] [PMC free article] [PubMed] [Google Scholar]