

Previous studies using the furin inhibitor dec-RVKR-CMK suggest that MERS-CoV utilizes a cellular protease, furin, to activate viral glycoproteins during cell entry. However, we found that dec-RVKR-CMK inhibits not only furin but also other proteases. Furthermore, we found no evidence that MERS-CoV uses furin. These findings suggest that previous studies in the virology field based on dec-RVKR-CMK should be reexamined carefully. Here we describe appropriate experiments that can be used to assess the effect of protease inhibitors on virus cell entry.
KEYWORDS: TMPRSS2, cathepsin L, coronavirus, dec-RVKR-CMK, furin
ABSTRACT
Middle East respiratory syndrome coronavirus (MERS-CoV) utilizes host cellular proteases to enter cells. A previous report shows that furin, which is distributed mainly in the Golgi apparatus and cycled to the cell surface and endosomes, proteolytically activates the MERS-CoV spike (S) protein following receptor binding to mediate fusion between the viral and cellular membranes. In this study, we reexamined furin usage by MERS-CoV using a real-time PCR-based virus cell entry assay after inhibition of cellular proteases. We found that the furin inhibitor dec-RVKR-CMK blocked entry of MERS-CoV harboring an S protein lacking furin cleavage sites; it even blocked entry into furin-deficient LoVo cells. In addition, dec-RVKR-CMK inhibited not only the enzymatic activity of furin but also those of cathepsin L, cathepsin B, trypsin, papain, and TMPRSS2. Furthermore, a virus cell entry assay and a cell-cell fusion assay provided no evidence that the S protein was activated by exogenous furin. Therefore, we conclude that furin does not play a role in entry of MERS-CoV into cells and that the inhibitory effect of dec-RVKR-CMK is specific for TMPRSS2 and cathepsin L rather than furin.
IMPORTANCE Previous studies using the furin inhibitor dec-RVKR-CMK suggest that MERS-CoV utilizes a cellular protease, furin, to activate viral glycoproteins during cell entry. However, we found that dec-RVKR-CMK inhibits not only furin but also other proteases. Furthermore, we found no evidence that MERS-CoV uses furin. These findings suggest that previous studies in the virology field based on dec-RVKR-CMK should be reexamined carefully. Here we describe appropriate experiments that can be used to assess the effect of protease inhibitors on virus cell entry.
INTRODUCTION
Many species of enveloped virus utilize host cellular proteases to infect cells (1). Two major mechanisms are responsible for proteolytic activation of viral spike (S) glycoproteins. In the case of human immunodeficiency virus and highly pathogenic avian influenza viruses, a cellular protease called furin cleaves the viral glycoprotein during biogenesis, thereby converting the precursor glycoprotein to its fusion-competent state (1–3). Alternatively, the S protein of viruses such as severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) is cleaved by cell surface or endosomal proteases such as transmembrane protease serine 2 (TMPRSS2), HAT, furin, trypsin, elastase, or cathepsin L. This cleavage triggers conformational changes during viral entry after the receptor-binding step (2–14). In the presence of extracellular or cell surface proteases such as elastase or TMPRSS2, MERS-CoV enters cells after binding to the cell surface receptor; however, in their absence, MERS-CoV utilizes cathepsin L in the late endosome (10). Cleavage releases the receptor-binding S1 subunit from the membrane fusion S2 subunit, which triggers conformational changes in the S2 subunit to induce membrane fusion (2).
The role of furin in activating the MERS-CoV S protein is controversial. Furin is a proprotein convertase responsible for maturation of a huge number of inactive proteins; it is localized principally in the trans-Golgi network, from which it is cycled to the cell surface and the endosomes, which are organelles used by MERS-CoV for cell entry (11). Of the furin inhibitors reported previously (15–17), dec-RVKR-CMK and a proprotein convertase inhibitor (PCI) are often used in virology experiments. Gierer et al. used a PCI to show that enzymatic processing during biogenesis of the MERS-CoV S protein by proprotein convertases such as furin is not required for infectivity; however, they stated that the host cell protease is required for S protein activation during viral uptake into target cells (14). In contrast, Park et al. used dec-RVKR-CMK to show that mutated MERS-CoV (in which the furin cleavage site was masked) tended to enter cells via the endosomal pathway, while the wild-type (wt) virus entered at the cell via cell surface proteases; this indicates that cleavage of the S protein during biogenesis determines the tropism of the virus for different cell types (8). Furthermore, Millet and Whittaker reported that the MERS-CoV S protein harbors furin cleavage sequences at the S1/S2 and S2′ sites; these sites could be targeted by host cellular furin during cell entry. Indeed, noncytotoxic concentrations (2.5 to 100 μM) of dec-RVKR-CMK prevented entry of pseudotyped and authentic MERS-CoV (13). Burkard et al. used dec-RVKR-CMK to show that murine coronavirus mouse hepatitis virus (MHV) also uses furin for viral cell entry (12). However, furin is thought to make only a minor contribution to viral cell entry because MERS-CoV entry into some cell lines (even furin-expressing cells) is almost completely blocked by simultaneous treatment with a TMPRSS2 inhibitor (camostat mesylate) and a cathepsin inhibitor (E64d) (10). In this study, we reexamined the role of furin during MERS-CoV infection to clarify when and how it is involved in virus cell entry. The findings raise questions about our current understanding of host protease usage underlying MERS-CoV infection.
RESULTS
Evaluation of an appropriate assay to monitor virus cell entry.
First, we examined cell entry by three types of virus: (i) a pseudotyped vesicular stomatitis virus (VSV) bearing a MERS-CoV S protein in which the VSV glycoproteing (VSV-G) gene was replaced by the green fluorescent protein (GFP) gene (VSV-ΔG/GFP-MERS-S), (ii) the same virus in which the GFP gene was replaced by the luciferase (Luc) gene (VSV-ΔG/Luc-MERS-S), and (iii) authentic MERS-CoV. These viruses were used to infect Vero/TMPRSS2 cells (18), which are highly susceptible to MERS-CoV (10). Entry of these viruses into cells was measured using appropriate assays (see Materials and methods). The range for VSV-ΔG/GFP-MERS-S was narrow (1 to 3 log10 GFP-cell counts) (Fig. 1A), whereas that for luciferase was broad (1 to 5 log10 Luc units) (Fig. 1B); however, the range for authentic MERS-CoV (assessed by real-time PCR) was broader still (2 to 7 log10 copies of viral mRNA) (Fig. 1C).
FIG 1.

Comparison of three assays to quantify virus cell entry. (A to C) Cell entry by pseudotyped or authentic MERS-CoV. Vero/TMPRSS2 cells in 96-well plates were infected with the serially diluted viruses indicated at the top. The relationship between inoculated virus titer (x axis) and data values (average of two experiments) for each assay (y axis) is shown. (A) GFP-positive cells were counted at 20 h postinfection. (B) Luciferase activity in cells was measured at 20 h postinfection. (C) The amount of viral mRNA in cells at 6 h postinfection was measured by real-time PCR. (D to F) Effect of a furin inhibitor on cell entry by pseudotyped or authentic MERS-CoV. Vero/TMPRSS2 cells were inoculated with viruses in the presence or absence of the furin inhibitor dec-RVKR-CMK. Virus entry was measured using appropriate assays. (D) Effect of dec-RVKR-CMK on cell entry by VSV-ΔG/GFP-MERS-S. Cells were inoculated with 103 infectious units of VSV-ΔG/GFP-MERS-S (MOI = 0.01). GFP-positive cells were counted at 20 h postinfection (n = 4). (E) Effect of dec-RVKR-CMK on cell entry by VSV-ΔG/Luc-MERS-S. Cells were inoculated with 104 infectious units of VSV-ΔG/Luc-MERS-S (MOI = 0.1). Luciferase activity in cells was measured at 20 h postinfection (n = 4). Data are presented on linear (left) and logarithmic (right) scales. (F) Effect of dec-RVKR-CMK on cell entry by authentic MERS-CoV. Cells were inoculated with 105 infectious units of authentic MERS-CoV (MOI = 1). The amount of viral mRNA in cells at 6 h postinfection was measured by real-time PCR (n = 4). A two-tailed Student t test was used to analyze statistical significance.
To evaluate the suitability of the assays for the furin inhibition experiments, we examined the effect of a furin inhibitor (dec-RVKR-CMK; 100 μM) on viral entry into cells. Infection with VSV-ΔG/GFP-MERS-S in the presence of dec-RVKR-CMK led to a fall in the number of GFP-positive cells by 60% (0.38 log) (Fig. 1D). Infection by VSV-ΔG/Luc-MERS-S in the presence of the inhibitor led to a fall of 40% (0.21 log) (Fig. 1E, left graph). In contrast, infection by authentic MERS-CoV led to a 97% (1.53 log) reduction in the viral mRNA copy number (Fig. 1F). The pseudotyped virus encoding GFP is suitable for measuring the percent infection within a narrow range. However, we surmised that VSV-ΔG/Luc-MERS-S would not provide reliable results because the 40% reduction observed in the assay is negligible on a logarithmic scale (Fig. 1E, right graph); the quantifiable range for this virus was 4 logs, and the luminometer used in the assay has a dynamic range of 9 logs. Therefore, we used authentic MERS-CoV for experiments designed to measure the inhibitory effects of dec-RVKR-CMK.
Entry of MERS-CoV into Calu-3 cells in the presence of a furin inhibitor.
To examine the effect of cellular furin on MERS-CoV infection, we infected Calu-3 cells (which are derived from human bronchial epithelial cells) with authentic MERS-CoV in the presence of inhibitors. After 6 h of incubation at 37°C, cellular RNA was isolated and real-time PCR was carried out to quantify the amount of viral mRNA. As shown in Fig. 2, a TMPRSS2 inhibitor (camostat mesylate; 10 μM) suppressed virus entry 100-fold; similar results were obtained after treatment with 20 μM dec-RVKR-CMK. E64d had little effect on virus entry, suggesting that MERS-CoV mainly uses the TMPRSS2/cell surface pathway rather than the cathepsin/endosome pathway to enter Calu-3 cells. Taken together, the results suggest that either furin is essential for S protein activation or dec-RVKR-CMK suppresses both furin and TMPRSS2.
FIG 2.
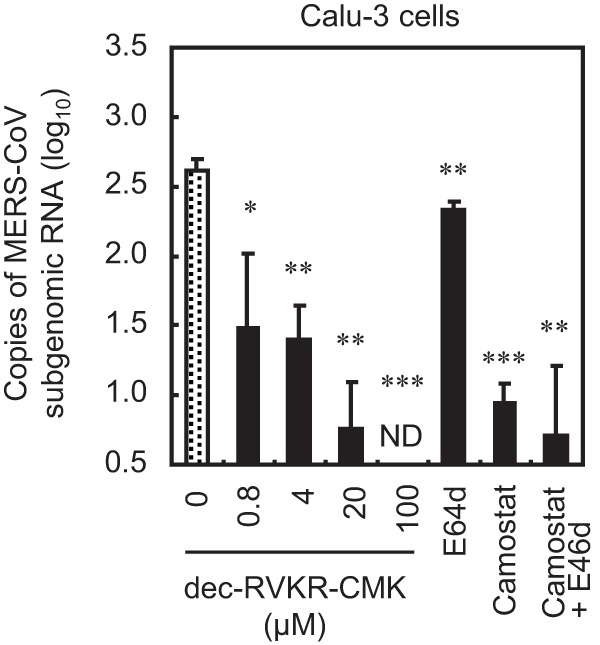
Effect of a furin inhibitor on MERS-CoV entry into Calu-3 cells. Calu-3 human bronchial epithelial cells were pretreated for 30 min with increasing concentrations (0 to 100 μM) of the furin inhibitor dec-RVKR-CMK. E64d (10 μM), camostat (10 μM), or a combination of both was used as a comparison control. The cells were then infected with 105 PFU of MERS-CoV (MOI = 1) in the presence of inhibitor. The amount of viral mRNA in Calu-3 cells at 6 h postinfection was measured by real-time PCR (n = 4). A two-tailed Student t test was used to analyze statistical significance.
Susceptibility of furin-deficient LoVo cells to infection by MERS-CoV.
LoVo cells (derived from human colon adenocarcinoma cells) lack furin activity because they harbor two distinct mutant alleles of the furin gene (1286T and 1639C) (19, 20). We purchased LoVo cells from the American Type Culture Collection (ATCC) and quantified expression of mRNA encoding proteins involved in cell entry by MERS-CoV. We confirmed that like Calu-3 and Huh-7 (derived from human liver carcinoma) cells, LoVo cells expressed DPP4, furin, cathepsin L, and TMPRSS2; however, unlike Calu-3 and Huh-7 cells, LoVo cells expressed HAT (Fig. 3A). Furin expression was highest in Huh-7 cells. We also confirmed deletion and substitution (1286T and 1639C) mutations within the furin mRNA sequence (Fig. 3B) (19, 20). Next, we inoculated MERS-CoV onto LoVo cells and measured virus propagation at 24 h. As reported previously (21), MERS-CoV infected and replicated in LoVo and Calu-3 cells (Fig. 3C). This indicates that furin is not essential for MERS-CoV infection.
FIG 3.

Comparison of transcripts in LoVo cells. (A) Expression of mRNA in Calu-3, LoVo, and Huh-7 cells. Total cellular RNA (0.1 μg) was evaluated for expression of GAPDH, DPP4, furin, cathepsin L, TMPRSS2, and HAT transcripts using real-time PCR (n = 4). ND, no transcripts were detected. (B) Electropherograms of furin cDNA. The mRNAs isolated from Calu-3 and LoVo cells were reverse transcribed and amplified using a thermal cycler and used for DNA sequencing. (C) Viral yield in LoVo cells. Confluent Calu-3 cells and LoVo cells were grown in 96-well plates and infected with MERS-CoV at an MOI of 0.001 for 24 h. Cell-free supernatants were harvested, and infectious viral titers were measured in a standard plaque assay using Vero/TMPRSS2 cells (n = 4). A two-tailed Student t test was used to analyze statistical significance.
Entry of MERS-CoV into furin-deficient LoVo cells.
Next, we examined the effect of protease inhibitors on MERS-CoV entry into LoVo cells. E64d suppressed entry of MERS-CoV, but camostat did not, suggesting that the cathepsin/endosomal pathway rather than the TMPRSS2/cell surface pathway is dominant in LoVo cells (Fig. 4A). Treatment with 100 μM dec-RVKR-CMK completely suppressed entry of MERS-CoV, similar to cotreatment with camostat and E64d (Fig. 4A). Furthermore, cell entry by SARS-CoV, in which the S protein lacks furin cleavage sites (13), was also suppressed by dec-RVKR-CMK (Fig. 4B).
FIG 4.
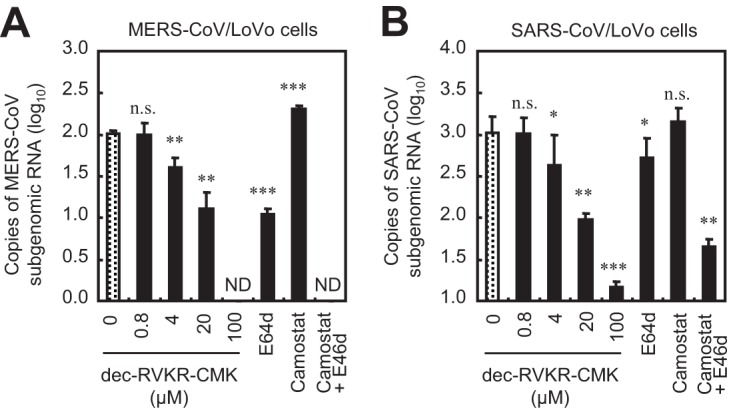
Effect of furin inhibitor on MERS-CoV and SARS-CoV entry into LoVo cells. (A) Effect of inhibitors on MERS-CoV entry. LoVo cells were pretreated for 30 min with the furin inhibitor dec-RVKR-CMK (concentration, 0 to 100 μM). E64d (10 μM), camostat (10 μM), or a combination of both was used as a comparison control. The cells were then infected with 105 PFU of MERS-CoV (MOI = 1) in the presence of inhibitor. The amount of viral mRNA in LoVo cells at 6 h postinfection was measured by real-time PCR (n = 4). (B) Effects of inhibitors on SARS-CoV entry. SARS-CoV was used instead of MERS-CoV; all experiments were carried out as described for panel A. A two-tailed Student t test was used to analyze statistical significance.
MERS-CoV mutants in which the S protein lacks furin cleavage sites at R748 (the S1/S2 site) and/or R884 (the S2′ site) were generated using a recently developed reverse genetics system (22). Western blot analysis of S proteins harboring mutations at R748 (R748S or R748S/R884S) detected no 80-kDa cleavage product on virions (Fig. 5A, lanes 2 and 4). To characterize the cleavability of these S proteins within cells, Vero/TMPRSS2, Huh-7 (expressing high levels of furin), and LoVo (lacking furin) cells were infected with viruses and cell lysates were examined by Western blotting. The S proteins of wt and R884 mutant viruses in Vero/TMPRSS2 and Huh-7 cells were cleaved, but those of R748 mutant viruses (R748S or R748S/R884S) were not (Fig. 5B). In contrast, none of the S proteins were cleaved in LoVo cells (Fig. 5B). This confirmed that LoVo cells lack furin activity and that the R748 mutation in S protein lies within the furin cleavage site. To assess the furin-dependent cell entry of these mutant viruses, they were inoculated onto Huh-7 or LoVo cells and the amount of viral mRNA at 6 h postinfection was measured by real-time PCR. No significant difference was observed between wt and mutant viruses in Huh-7 cells (Fig. 5C), indicating that high levels of furin expressed by these cells did not affect S protein activation during viral entry. Treatment with dec-RVKR-CMK suppressed cell entry by both wt MERS-CoV and mutant MERS-CoV in which the S protein lacks furin cleavage sites (Fig. 5D). These results indicate that entry of furin-deficient cells by virus lacking furin cleavage sites is blocked by the furin inhibitor, suggesting that the molecule targeted by dec-RVKR-CMK during MERS-CoV entry is not furin.
FIG 5.

Effect of mutations in the furin cleavage site of the MERS-CoV S protein. (A) Cleavage of the S protein on virions. Wild-type (wt) and mutant MERS-CoV lacking furin cleavage sites within the S protein (generated in Vero/TMPRSS2 cells) was subjected to Western blot analysis with an anti-S polyclonal antibody. (B) Cleavage of S protein in cells. The wt and mutant viruses shown in panel A were used to infect Vero/TMPRSS2, Huh-7, and LoVo cells, and cell lysates prepared at 20 h postinfection were subjected to Western blot analysis. (C) Entry of mutant viruses into LoVo and Huh-7 cells. The cells were infected with 104 PFU of MERS-CoV (MOI = 0.1). The amount of viral mRNA in cells at 6 h postinfection was measured by real-time PCR (n = 4). (D) Effect of mutations on virus entry. LoVo cells were pretreated for 30 min with the furin inhibitor dec-RVKR-CMK (concentration of 100 μM). Then cells were infected with 104 PFU of MERS-CoV (MOI = 0.1) in the presence of inhibitor. The amount of viral mRNA in cells at 6 h postinfection was measured by real-time PCR (n = 4). Data are expressed as the fold change in viral mRNA levels relative to that in the absence of dec-RVKR-CMK. A two-tailed Student t test was used to analyze statistical significance.
Exogenous furin does not activate the S protein.
Next, we examined direct activation of the S protein by exogenous furin. First, we confirmed that commercial furin had the advertised level of activity by testing it using a furin substrate, BOC-RVRR-AMC (Fig. 6A). Treatment with exogenous trypsin, but not exogenous furin, increased virus entry into LoVo cells (Fig. 6B) and cell-cell fusion by MERS-CoV-infected LoVo cells (Fig. 6C). Therefore, we concluded that furin does not activate the MERS-CoV S protein. In addition, Western blot analysis detected very small amounts of cleaved S protein (80 kDa) in non-protease-treated viruses propagated in Vero cells (Fig. 6D, lane 1); similar results were obtained for viruses propagated in Vero/TMPRSS2 cells (Fig. 5A). No 80-kDa S protein was detected when cells were treated with exogenous furin (2,000 units/ml), although the 150-kDa and 50-kDa products were detected (Fig. 6D, lane 5). Of note, the S protein-harboring virions used for the experiments in Fig. 6D were propagated in Vero cells cultured in medium lacking trypsin (a nonenzymatic cell dissociation solution [C5914; Sigma] was used for cell passage). The intensity of the 80-kDa band increased only when cells were exposed to exogenous trypsin (0.1 μg/ml) (Fig. 6D, lanes 3 and 7). Therefore, we conclude that the S protein is cleaved by furin during biogenesis, not on virions after they exit the cells.
FIG 6.
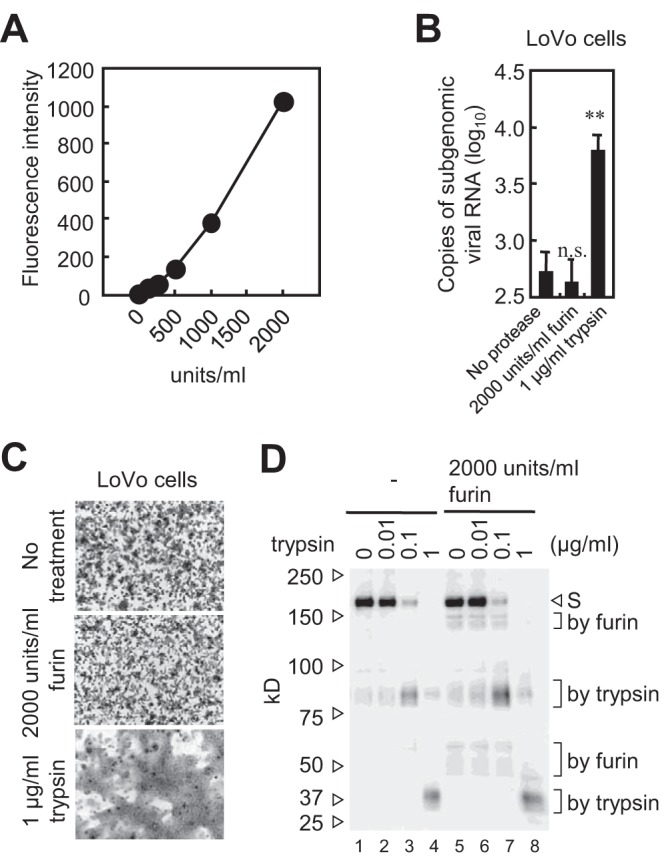
S protein activation by exogenous furin. (A) Activity of commercial furin. The enzymatic activity of recombinant human furin used in the experiment was confirmed using the fluorescent substrate BOC-RVRR-AMC. (B) Exogenous furin does not trigger cell entry by virus. LoVo cells in 96-well plates were inoculated with MERS-CoV (MOI of 0.1) for 30 min and then treated with furin (2,000 units/ml) or trypsin (10 μg/ml) at 37°C for 10 min. Cells were then cultured at 37°C for 6 h. The amount of viral mRNA was measured by real-time PCR (n = 4). A two-tailed Student t test was used to analyze statistical significance. (C) Exogenous furin does not induce syncytium formation. LoVo cells infected with MERS-CoV (MOI of 0.01) were cultured for 20 h and then treated with furin (2,000 units/ml) or trypsin (10 μg/ml) for 5 h. (D) Cleavage of S protein by exogenous furin. MERS-CoV propagated in Vero cells was treated with the indicated concentrations of trypsin or furin at 37°C for 60 min. Western blot analysis was performed using an anti-S polyclonal antibody.
The inhibitor dec-RVKR-CMK targets the early stage of MERS-CoV infection.
Our next task was to identify the true target of dec-RVKR-CMK during MERS-CoV infection. First, we examined the effect of dec-RVKR-CMK on endocytosis. pHrodo dextran, which is nonfluorescent at neutral pH but exhibits fluorescence as the pH becomes acidic, was used as a tracker of endocytic internalization and lysosomal sequestration within live cells. In Calu-3 cells, bafilomycin A1, which blocks acidification of endosomes, clearly inhibited development of pHrodo red, but dec-RVKR-CMK, camostat, and E64d did not (data not shown); this suggests that dec-RVKR-CMK has no effect on endocytosis.
Next, to clarify the time point at which inhibitors block MERS-CoV infection, LoVo cells were treated with 100 μM dec-RVKR-CMK or a mixture of 10 μM E64d and 10 μM camostat (E64d/camostat) during infection with MERS-CoV. Lopinavir, which targets MERS-CoV 3CL protease and blocks viral RNA replication, was used for comparison. The inhibitors were added at the desired time points, and cellular RNA was isolated 6 h after infection. Samples collected at 6 h postinfection were used as a control for no inhibitor treatment. The amount of viral mRNA was quantified by real-time PCR. Dec-RVKR-CMK and E64d/camostat showed inhibitory effects within 30 min after infection (Fig. 7A). In comparison, addition of lopinavir at 2 h postinfection still inhibited viral RNA replication (Fig. 7A). In Calu-3 cells, dec-RVKR-CMK and E64d/camostat showed similar effects: both of these inhibitors act at the very early stage of infection (within 30 min) (Fig. 7B). Taken together, the data indicate that dec-RVKR-CMK targets an early step during MERS-CoV entry, potentially S protein activation by TMPRSS2 and cathepsin L.
FIG 7.

Timing of inhibitor addition to block entry of MERS-CoV into cells. (A) Dec-RVKR-CMK (100 μM), camostat (10 μM) plus E64d (10 μM), or lopinavir (30 μM) was added to LoVo cells (for 30 min on ice) at the indicated times before and after MERS-CoV inoculation. The amount of viral mRNA in cells at 6 h postinfection was measured by real-time PCR (n = 1). (B) Calu-3 cells were used instead of LoVo cells; all experiments were carried out as described for panel A (excluding lopinavir treatment) (n = 1).
Inhibitory effect of dec-RVKR-CMK on commercial proteases and TMPRSS2.
Next, we examined the inhibitory effect of dec-RVKR-CMK on various proteases. We purchased proteases from commercial sources and tested them using a fluorescent protease assay kit which measures degradation products of fluorescein-labeled casein. Unfortunately, the kit was unable to detect furin activity because casein does not contain the furin cleavage site. First, 10-fold serially diluted protease was incubated with the substrate to identify the appropriate concentration representing the linear phase of the reaction at 30 min; this was used for the experiments described below. Next, appropriate concentrations of various proteases required to degrade the substrate were mixed with dec-RVKR-CMK and inhibition kinetics were measured. Figure 8A shows that a high concentration (100 μM) of dec-RVKR-CMK completely suppressed the activities of cathepsin L, cathepsin B, trypsin, and papain, partially suppressed those of proteinase K and dispase, and slightly suppressed those of elastase and chymotrypsin. Neither E64d nor bafilomycin A1 (used as a control) suppressed the activity of trypsin, chymotrypsin, or elastase (Fig. 8B).
FIG 8.

Effect of furin inhibitor on commercial proteases and cell surface TMPRSS2. (A) Inhibitory effect of dec-RVKR-CMK on commercial proteases. Degradation products of fluorescein-labeled casein generated by treatment with commercial proteases, cathepsin L (20 μg/ml), cathepsin B (20 μg/ml), trypsin (50 μg/ml), papain (0.3 units/ml), proteinase K (0.3 units/ml), dispase (3 units/ml), elastase (200 μg/ml), and chymotrypsin (10 μg/ml), at 37°C for 30 min in the presence of dec-RVKR-CMK (serially diluted 10-fold) were quantified by fluorometry (n = 3). Data are expressed as average percentages relative to that in the absence of dec-RVKR-CMK. N, no inhibitor treatment. (B) E64d and bafilomycin A1 do not inhibit commercial proteases. E64d or bafilomycin A1 was used instead of dec-RVKR-CMK; all experiments were carried out as described for panel A (proteases tested were trypsin, chymotrypsin, and elastase). (C) Inhibitory effect of dec-RVKR-CMK against TMPRSS2, as assessed in the FFWO assay. A high titer of MERS-CoV (MOI = 10) was adsorbed onto Vero/TMPRSS2 or Vero cells (on ice for 1 h). Cells were then incubated at 37°C in the presence of the inhibitor. After 5 h, cells were fixed and stained with crystal violet. DMSO, dimethyl sulfoxide.
To examine the inhibitory effect of dec-RVKR-CMK on TMPRSS2, we performed a fusion-from-without (FFWO) assay as described previously (10). This assay detects viral S protein-mediated cell-cell fusion activated by TMPRSS2 on the cell surface; the assay excludes the effects of inhibitors on virus replication, meaning that it detects TMPRSS2 activity directly. Briefly, a high titer (multiplicity of infection [MOI] = 10) of MERS-CoV was adsorbed onto Vero/TMPRSS2 cells on ice for 1 h. The cells were then shifted to 37°C in the presence or absence of inhibitors. Cell-cell fusion was first observed at 3 h after warming. The dec-RVKR-CMK inhibitor (100 μM) suppressed cell-cell fusion completely, as did camostat (10 μM; used as the control) (Fig. 8C). These results strongly suggest that dec-RVKR-CMK targets cell surface TMPRSS2, which plays a role in cell entry by MERS-CoV.
DISCUSSION
Cellular furin plays a role in virus infection and numerous other biological phenomena, most of which were identified by experimental observations using the furin inhibitor dec-RVKR-CMK (11, 13, 23–25). However, the results presented herein indicate that dec-RVKR-CMK inhibited not only furin but also cathepsin L and TMPRSS2 (Fig. 8A and C). In addition, we observed that dec-RVKR-CMK blocked entry of viruses harboring an S protein lacking furin cleavage sites; it even blocked entry into furin-deficient LoVo cells (Fig. 5D). This suggests that the results of previous studies using dec-RVKR-CMK may have to be reexamined.
In addition, virus entry or cell-cell fusion assays provided no evidence that the S protein was activated by exogenous furin (Fig. 6B and C). A previous study claims to show direct evidence of S protein activation by furin because induction of cell-cell fusion in S protein-expressing Huh-7 cells was observed after removal of dec-RVKR-CMK from the culture medium and addition of exogenous furin (13). However, exogenous furin may not be required to induce cell-cell fusion of Huh-7 cells because these cells induce cell-cell fusion after MERS-CoV infection in the absence of proteases (26).
Here we discuss the use of host proteases by MERS-CoV for cell entry. Studies show clearly that TMPRSS2 activates the MERS-CoV S protein (4, 10, 27). Expression of TMPRSS2 at the cell surface induces both virus entry into cells and cell-cell fusion of S protein-expressing cells (4, 10, 27). Furthermore, these phenomena are suppressed by the serine protease inhibitors camostat mesylate and nafamostat mesylate (4, 10, 28). In addition, other extracellular proteases, such as trypsin and elastase, activate the MERS-CoV S protein in a manner similar to that of TMPRSS2 (10).
However, cathepsin L usage by MERS-CoV remains controversial because there is no clear evidence that the S protein is activated by exposure to exogenous cathepsin L. A previous study shows that cell-cell fusion induced by exogenous cathepsin L in SARS-CoV S protein-expressing cells is insufficient (29). Also, our previous study shows that cathepsin L requires chlorpromazine, a membrane-permeable cationic drug that lowers the energy requirement for membrane fusion, to induce a small increase in cell entry by murine coronavirus (30). Therefore, it is unclear whether cathepsin L plays a role in activating the coronavirus S protein. However, it is at least certain that the enzymatic activity of cathepsin is necessary for proteolytic activation of the MERS-CoV cell entry because expression of TMPRSS2 overcomes blockade of viral cell entry by cathepsin inhibitors (10).
Taken together, the results presented herein do not support a role for cellular furin during direct activation of MERS-CoV S protein for viral cell entry. These findings are compatible with those of a previous study by Gierer et al. showing that MERS-CoV does not require furin for infectivity (14). Park et al. report that the only role played by furin during MERS-CoV infection is to determine the cell tropism of the virus (8). What is certain is that the MERS-CoV S protein precleaved by furin at the S1/S2 site still requires cell surface/extracellular and endosomal proteases, such as TMPRSS2, elastase, and cathepsin L, for cleavage at the S2′ site.
MATERIALS AND METHODS
Cells and viruses.
LoVo and Calu-3 cell lines were cultured as recommended by the ATCC. Vero cells obtained from the ATCC and Vero cells expressing TMPRSS2 (Vero/TMPRSS2) (18) were maintained in Dulbecco's modified Eagle medium (DMEM; Sigma-Aldrich, USA) supplemented with 5% heat-inactivated fetal bovine serum (Gibco-BRL, USA). MERS-CoV strain EMC and SARS-CoV strain Frankfurt 1 were propagated in Vero cells.
Generation of a pseudotyped virus.
The VSV-pseudotyped virus expressing GFP or luciferase and harboring the MERS-CoV S protein was prepared as previously described (31). Briefly, at 24 h after transfection with pKS-MERS-St16, 293T cells were infected with VSVΔG-G/GFP or VSVΔG-G/Luc at an MOI of 0.1. After absorption for 1 h, the inoculum was replaced with culture medium. After a further incubation for 24 h, the culture supernatants were collected and stored at −80°C. The titer of VSV-pseudotyped viruses was determined in Vero cells. For the VSV-pseudotyped virus expressing GFP, images were captured under a BZ8000 microscope (Keyence, Japan) and GFP-positive cells were counted using image measurement and analysis software (VH-H1A5 version 2.6; Keyence). For the VSV-pseudotyped virus expressing luciferase, cells were lysed and assayed for luciferase activity using a luciferase assay kit (Promega, USA) and a Glomax 20/20 luminometer (Promega).
Generation of recombinant MERS-CoV from BAC plasmids.
A bacterial artificial chromosome (BAC) clone carrying the full-length infectious genome of the MERS-CoV EMC2012 strain (termed pBAC-MERS-wt) was used to generate recombinant MERS-CoV (22). Amino acid substitutions at the P1 site within the fusion cleavage sites in the S protein were generated by modification of the pBAC-MERS-wt (the template) using a Red/ET recombination system counterselection BAC modification kit (Gene Bridges, Heidelberg, Germany). This yielded pBAC-MERS-S/R748S, pBAC-MERS-S/R884S, and pBAC-MERS-S/R748S/R884S. Huh-7 cells were grown to approximately 60% confluence in a 6-well plate (VIOLAMO, Japan) and then transfected with 4 μg of the desired BAC DNA using the X-tremeGENE 9 DNA transfection reagent (Roche). Transfected cells were cultured at 37°C for the appropriate times, and the culture supernatants and cell pellets were collected. The culture supernatants were inoculated onto a 10-cm dish (VIOLAMO) containing Huh-7 cells and cultured overnight. Infected cells were incubated at 37°C for 3 or 4 days until a cytopathic effect (CPE) was observed. Culture supernatants were collected, centrifuged at 2,500 × g for 5 min at 4°C, and stored at −80°C.
Proteases and protease inhibitors.
The following proteases were used: cathepsin L (16-12-030112; Athens, USA), cathepsin B (16-12-030102; Athens), trypsin (T8802; Sigma, USA), papain (53J6521; Worthington, USA), proteinase K (166-21051; Wako, Japan), dispase (1 276 921; Roche, Switzerland), elastase (Sigma; E-0258), chymotrypsin (Sigma; C-3142), and furin (P8077; New England BioLabs [NEB], UK). The following inhibitors were used: dec-RVKR-CMK (3501; Tocris Bioscience, UK), E64d (330005; Calbiochem, USA), camostat (3193; Tocris Bioscience), bafilomycin A1 (B1793; Sigma), and lopinavir (SML1222; Sigma).
Cell entry assay for authentic MERS-CoV.
Confluent cells in 96-well plates were pretreated for 30 min with inhibitors. Cells were then inoculated with MERS-CoV and incubated (with the inhibitors) for 1 h on ice, followed by culture at 37°C for 6 h. Cellular RNA was isolated by addition of Isogen reagent (315-02504; Nippon Gene, Japan). A real-time PCR assay was performed to quantify the amount of newly synthesized subgenomic MERS-CoV RNA using the primers and probes described previously (10). PCR analysis was performed in a LightCycler-Nano instrument (Roche Diagnostics, Switzerland).
Quantification of transcripts in LoVo cells.
Total RNA was isolated from LoVo cells using the Isogen reagent. Real-time PCR was performed to quantify expression of mRNAs encoding glyceraldehyde-3-phosphate dehydrogenase (GAPDH), DPP4, furin, cathepsin L, TMPRSS2, and HAT using the primers and probes described previously (10). Comparative expression of mRNA was calculated from a calibration line obtained by stepwise dilution (10-fold) of RNA samples.
Quantification of furin activity.
Recombinant human furin (P8077; New England BioLabs, UK) was mixed with 100 μM furin substrate (boc-RVRR-AMC; I-1645; Bachem, Switzerland) in phosphate-buffered saline containing 1 mM CaCl2 (32). After 30 min at 37°C, fluorescence analysis was performed using a Power Scan HT instrument (DS Pharma, Japan) fitted with fluorescein excitation/emission filters (360/480 nm).
Western blot analysis.
To detect cleaved S protein in virions, viral supernatant was mixed with recombinant human furin (P8077; New England BioLabs) or trypsin (T-8802; Sigma) in furin reaction buffer (20 mM HEPES [pH 7.5], 0.1% Triton X-100, 0.2 mM CaCl2, and 0.2 mM β-mercaptoethanol) and incubated at 37°C for 1 h. A one-quarter volume of sample buffer (30% glycerol, 250 mM Tris [pH 6.8], 2.5% SDS, a small amount of bromophenol blue, 100 mM dithiothreitol [DTT], and 1 mM Pefabloc SC) was added to the reaction mixture and boiled for 5 min. Samples were loaded onto SDS-PAGE (3 to 10% gradient) gels (e-PAGEL; ATTO, Japan), transferred to a polyvinylidene difluoride (PVDF) membrane (Immobilon-P; Millipore, USA), and soaked in ImmunoBlock (CTKN001; DS Pharma Biomedical, Japan). Western blot analysis was carried out using an anti-S2 antibody and anti-rabbit IgG (sc-2054; Santa Cruz Biotech, USA). Immunoreactive bands were visualized with an enhanced chemiluminescence kit (ECL; RPN2232; GE Healthcare, USA) and a LAS-3000 instrument (Fuji, Japan).
To detect the S protein in MERS-CoV-infected cells, cells were dissolved in sample buffer and subjected to Western blot analysis as described above. After detection by an anti-S2 antibody, the membrane was soaked in stripping buffer (46428; Thermo Fisher, USA) for 5 min at room temperature to remove the antibodies and then rinsed 10 times with rinse buffer (20845; Millipore). The membrane was then blocked and reprobed with an anti-GAPDH antibody (IMG-5143A; IMGENEX, USA), followed by anti-rabbit IgG.
Fluorescent protease assay for commercial proteases.
The activities of the proteases listed above were quantified using a fluorescent protease assay kit (23266; Pierce, USA), which measures the degradation products of fluorescein-labeled casein. To measure the activity of neutral pH-dependent proteases (trypsin, papain, proteinase K, dispase, elastase, and chymotrypsin), experiments were carried out according to the manufacturer's protocol. To measure the activities of low-pH-dependent cathepsin L and cathepsin B, the substrate (which has neutral pH-dependent fluorescence) was first dissolved in low-pH buffer (50 mM sodium acetate [pH 5].0, 1 mM EDTA, and 5 mM DTT). After a 30-min incubation with cathepsin at 37°C, 1/20 volume of 1 M Tris-HCl buffer (pH 8.0) was added to render the fluorescein detectable at neutral pH. Fluorescence analysis was performed using a Power Scan HT instrument (DS Pharma, Japan) fitted with fluorescein excitation/emission filters (485/528 nm).
FFWO assay.
The FFWO assay was performed as previously described (10). Briefly, Vero/TMPRSS2 cells (105) in 96-well plates were inoculated with a high titer (106 PFU) of authentic MERS-CoV. Cells were then incubated at 37°C in the presence of inhibitors. After incubation for 5 h, cells were fixed with 4% formaldehyde and stained with crystal violet.
Statistical analysis.
Statistical significance was assessed using a two-tailed Student t test. A P value of <0.05 was considered statistically significant. In figures, significance is indicated as follows: n.s., not significant; *, significant (P ≤ 0.05); **, highly significant (P ≤ 0.01); and ***, very highly significant (P ≤ 0.001). Error bars indicate standard deviations (SD).
ACKNOWLEDGMENTS
We thank Ron A. M. Fouchier (Erasmus Medical Center, Rotterdam, The Netherlands) for providing the MERS-CoV EMC strain and John Ziebuhr (University of Würzburg, Würzburg, Germany) for providing the SARS-CoV Frankfurt 1 strain. We also thank Aiko Fukuma and Hideki Tani (NIID, Japan) for valuable suggestions.
This study was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (grant no. 17K08868).
REFERENCES
- 1.White JM, Whittaker GR. 2016. Fusion of enveloped viruses in endosomes. Traffic 17:593–614. doi: 10.1111/tra.12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heald-Sargent T, Gallagher T. 2012. Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses 4:557–580. doi: 10.3390/v4040557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Millet JK, Whittaker GR. 2015. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res 202:120–134. doi: 10.1016/j.virusres.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gierer S, Bertram S, Kaup F, Wrensch F, Heurich A, Krämer-Kühl A, Welsch K, Winkler M, Meyer B, Drosten C, Dittmer U, von Hahn T, Simmons G, Hofmann H, Pöhlmann S. 2013. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. J Virol 87:5502–5511. doi: 10.1128/JVI.00128-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simmons G, Reeves JD, Rennekamp AJ, Amberg SM, Piefer AJ, Bates P. 2004. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc Natl Acad Sci U S A 101:4240–4245. doi: 10.1073/pnas.0306446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belouzard S, Chu VC, Whittaker GR. 2009. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci U S A 106:5871–5876. doi: 10.1073/pnas.0809524106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Watanabe R, Matsuyama S, Shirato K, Maejima M, Fukushi S, Morikawa S, Taguchi F. 2008. Entry from the cell surface of severe acute respiratory syndrome coronavirus with cleaved S protein as revealed by pseudotype virus bearing cleaved S protein. J Virol 82:11985–11991. doi: 10.1128/JVI.01412-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park J-E, Li K, Barlan A, Fehr AR, Perlman S, McCray PB, Gallagher T. 2016. Proteolytic processing of Middle East respiratory syndrome coronavirus spikes expands virus tropism. Proc Natl Acad Sci U S A 113:12262–12267. doi: 10.1073/pnas.1608147113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reinke LM, Spiegel M, Plegge T, Hartleib A, Nehlmeier I, Gierer S, Hoffmann M, Hofmann-Winkler H, Winkler M, Pöhlmann S. 2017. Different residues in the SARS-CoV spike protein determine cleavage and activation by the host cell protease TMPRSS2. PLoS One 12(6):e0179177. doi: 10.1371/journal.pone.0179177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shirato K, Kawase M, Matsuyama S. 2013. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J Virol 87:12552–12561. doi: 10.1128/JVI.01890-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas G. 2002. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol 3:753–766. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burkard C, Verheije MH, Wicht O, van Kasteren SI, van Kuppeveld FJ, Haagmans BL, Pelkmans L, Rottier PJM, Bosch BJ, de Haan CAM. 2014. Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog 10:e1004502. doi: 10.1371/journal.ppat.1004502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Millet JK, Whittaker GR. 2014. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc Natl Acad Sci U S A 111:15214–15219. doi: 10.1073/pnas.1407087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gierer S, Muller MA, Heurich A, Ritz D, Springstein BL, Karsten CB, Schendzielorz A, Gnirss K, Drosten C, Pohlmann S. 2015. Inhibition of proprotein convertases abrogates processing of the Middle Eastern respiratory syndrome coronavirus spike protein in infected cells but does not reduce viral infectivity. J Infect Dis 211:889–897. doi: 10.1093/infdis/jiu407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coppola JM, Bhojani MS, Ross BD, Rehemtulla A. 2008. A small-molecule furin inhibitor inhibits cancer cell motility and invasiveness. Neoplasia 10:363–370. doi: 10.1593/neo.08166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Becker GL, Lu Y, Hardes K, Strehlow B, Levesque C, Lindberg I, Sandvig K, Bakowsky U, Day R, Garten W, Steinmetzer T. 2012. Highly potent inhibitors of proprotein convertase furin as potential drugs for treatment of infectious diseases. J Biol Chem 287:21992–22003. doi: 10.1074/jbc.M111.332643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basak A. 2005. Inhibitors of proprotein convertases. J Mol Med 83:844–855. doi: 10.1007/s00109-005-0710-0. [DOI] [PubMed] [Google Scholar]
- 18.Shirogane Y, Takeda M, Iwasaki M, Ishiguro N, Takeuchi H, Nakatsu Y, Tahara M, Kikuta H, Yanagi Y. 2008. Efficient multiplication of human metapneumovirus in Vero cells expressing the transmembrane serine protease TMPRSS2. J Virol 82:8942–8946. doi: 10.1128/JVI.00676-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takahashi S, Kasai K, Hatsuzawa K, Kitamura N, Misumi Y, Ikehara Y, Murakami K, Nakayama K. 1993. A mutation of furin causes the lack of precursor-processing activity in human colon carcinoma LoVo cells. Biochem Biophys Res Commun 195:1019–1026. doi: 10.1006/bbrc.1993.2146. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi S, Nakagawa T, Kasai K, Banno T, Duguay SJ, Ven Van De WJM, Murakami K, Nakayama K. 1995. A second mutant allele of furin in the processing-incompetent cell line, LoVo. J Biol Chem 270:26565–26569. doi: 10.1074/jbc.270.44.26565. [DOI] [PubMed] [Google Scholar]
- 21.Tao X, Hill TE, Morimoto C, Peters CJ, Ksiazek TG, Tseng C-TK. 2013. Bilateral entry and release of Middle East respiratory syndrome coronavirus induces profound apoptosis of human bronchial epithelial cells. J Virol 87:9953–9958. doi: 10.1128/JVI.01562-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Terada Y, Kawachi K, Matsuura Y, Kamitani W. 2017. MERS coronavirus nsp1 participates in an efficient propagation through a specific interaction with viral RNA. Virology 511:95–105. doi: 10.1016/j.virol.2017.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Haan CAM, Stadler K, Godeke G, Bosch BJ, Rottier PJM. 2004. Cleavage inhibition of the murine coronavirus spike protein by a furin-like enzyme affects cell-cell but not virus-cell fusion. J Virol 78:6048–6054. doi: 10.1128/JVI.78.11.6048-6054.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Couture F, D'Anjou F, Day R. 2011. On the cutting edge of proprotein convertase pharmacology: from molecular concepts to clinical applications. Biomol Concepts 2:421–438. doi: 10.1515/BMC.2011.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klein-Szanto AJ, Bassi DE. 2017. Proprotein convertase inhibition: paralyzing the cell's master switches. Biochem Pharmacol 140:8–15. doi: 10.1016/j.bcp.2017.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Wilde AH, Raj VS, Oudshoorn D, Bestebroer TM, van Nieuwkoop S, Limpens RWAL, Posthuma CC, van der Meer Y, Bárcena M, Haagmans BL, Snijder EJ, van den Hoogen BG. 2013. MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-α treatment. J Gen Virol 94:1749–1760. doi: 10.1099/vir.0.052910-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barlan A, Zhao J, Sarkar MK, Li K, McCray PB, Perlman S, Gallagher T. 2014. Receptor variation and susceptibility to Middle East respiratory syndrome coronavirus infection. J Virol 88:4953–4961. doi: 10.1128/JVI.00161-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamamoto M, Matsuyama S, Li X, Takeda M, Kawaguchi Y, Inoue J, Matsuda Z. 2016. Identification of nafamostat as a potent inhibitor of Middle East respiratory syndrome coronavirus S protein-mediated membrane fusion using the split-protein-based cell-cell fusion assay. Antimicrob Agents Chemother 60:6532–6539. doi: 10.1128/AAC.01043-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bosch BJ, Bartelink W, Rottier PJM. 2008. Cathepsin L functionally cleaves the severe acute respiratory syndrome coronavirus class I fusion protein upstream of rather than adjacent to the fusion peptide. J Virol 82:8887–8890. doi: 10.1128/JVI.00415-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsuyama S, Taguchi F. 2009. Two-step conformational changes in a coronavirus envelope glycoprotein mediated by receptor binding and proteolysis. J Virol 83:11133–11141. doi: 10.1128/JVI.00959-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fukuma A, Tani H, Taniguchi S, Shimojima M, Saijo M, Fukushi S. 2015. Inability of rat DPP4 to allow MERS-CoV infection revealed by using a VSV pseudotype bearing truncated MERS-CoV spike protein. Arch Virol 160:2293–2300. doi: 10.1007/s00705-015-2506-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Komiyama T, Coppola JM, Larsen MJ, van Dort ME, Ross BD, Day R, Rehemtulla A, Fuller RS. 2009. Inhibition of furin/proprotein convertase-catalyzed surface and intracellular processing by small molecules. J Biol Chem 284:15729–15738. doi: 10.1074/jbc.M901540200. [DOI] [PMC free article] [PubMed] [Google Scholar]