

Fragments of the TAR DNA-binding protein 43 (TDP43) are major components of intracellular aggregates associated with amyotrophic lateral sclerosis and frontotemporal dementia. A variety of C-terminal fragments (CTFs) exist, with distinct N termini; however, little is known regarding their differences in metabolism and aggregation dynamics.
KEYWORDS: TAR DNA-binding protein, aggregation, amyotrophic lateral sclerosis, proteasome, autophagy
ABSTRACT
Fragments of the TAR DNA-binding protein 43 (TDP43) are major components of intracellular aggregates associated with amyotrophic lateral sclerosis and frontotemporal dementia. A variety of C-terminal fragments (CTFs) exist, with distinct N termini; however, little is known regarding their differences in metabolism and aggregation dynamics. Previously, we found that specific CTFs accumulate in the absence of the Arg/N-end rule pathway of the ubiquitin proteasome system (UPS) and that their degradation requires arginyl-tRNA protein transferase 1 (ATE1). Here, we examined two specific CTFs of TDP43 (TDP43219 and TDP43247), which are ∼85% identical and differ at their N termini by 28 amino acids. We found that TDP43247 is degraded primarily by the Arg/N-end rule pathway, whereas degradation of TDP43219 continues in the absence of ATE1. These fragments also differ in their aggregation propensities and form morphologically distinct aggregates. This work reveals that the N termini of otherwise similar CTFs have profound effects on fragment behavior and may influence clinical outcomes in neurodegeneration associated with aggregation.
INTRODUCTION
The TAR DNA-binding protein 43 (TDP43) is a DNA- and RNA-binding protein originally identified through its ability to bind to the HIV type 1 TAR DNA sequence motifs and later shown to play a role in mRNA splicing, transport, and translational regulation, including the formation of stress granules (1–13). Although found primarily in the nucleus due to a nuclear localization signal (NLS) near its N terminus, TDP43 shuttles between the nucleus and the cytoplasm (14). TDP43 also contains a glycine-rich C terminus that includes a prion-like domain important for interaction with a number of RNA-binding proteins (Fig. 1) (14–17). TDP43 binds to UG-rich regions in the 3′ untranslated regions (UTRs) of mRNAs through tandem RNA recognition motifs (RRMs) that adopt a typical RRM architecture consisting of a β-sheet made up of antiparallel β-strands, stacked on two α-helix motifs (15). Although both RRMs are required for high-affinity RNA binding, each one is capable of binding nucleic acid individually (15). RRM2 is unique in that it contains an additional β-strand (β4) that forms a number of intramolecular contacts when bound to RNA and intermolecular contacts in the absence of RNA (15). The β4-strand also forms part of a cryptic nuclear export signal that becomes exposed in C-terminal fragments (CTFs) following proteolytic cleavage resulting from pathological conditions, including oxidative stress (18, 19), endoplasmic reticulum (ER) stress (20, 21), thermal stress (22), loss of RNA binding (23), or acetylation (24). Numerous studies have shown that CTFs of TDP43 form toxic, insoluble, cytoplasmic aggregates in patients with amyotrophic lateral sclerosis (ALS) and frontotemporal lobar dementia (FTLD) (16, 23, 25–30). The link between TDP43 and neurodegeneration is further strengthened by a number of disease-related mutations found within the C-terminal domain of TDP43 (31) and the occurrence of TDP43-related proteinopathies for other neurodegenerative disorders as well, including Alzheimer's disease (32, 33). Despite its strong association with neurodegenerative disease, it is unclear if cytotoxicity results from a loss of normal TDP43 function (e.g., mRNA processing) due to cleavage or from a toxic gain of function resulting from the accumulation and aggregation of CTFs. CTFs can apparently also act in a dominant negative fashion by nucleating cytoplasmic inclusions that include full-length TDP43 (23, 30, 34–38).
FIG 1.

Features of TDP43 and two of its C-terminal fragments associated with neurodegeneration. (Top) Primary and secondary structures of human TDP43 RNA recognition motif 2 with locations of endoproteolytic cleavage occurring between Met218 and Asp219 and between Glu246 and Asp247 under conditions associated with amyotrophic lateral sclerosis (ALS) and frontal temporal lobar dementia (FTLD) (37). (Middle) Human TDP43 consists of 414 amino acids and contains a tandem set of RNA recognition motifs (RRM1 and RRM2) as well as a nuclear localization signal (NLS) and a glycine-rich C-terminal domain that includes a prion-like region. (Bottom) TDP43219 and TDP43247 generated by the cleavages shown at the top. Also shown is a Kyte-Doolittle plot showing the relative hydrophobicity throughout the fragments. The window size is 9.
Although soluble TDP43 is thought to be degraded primarily by the ubiquitin (Ub) proteasome system (UPS), autophagy is thought to be required for the removal of insoluble aggregates (39–41). Intracellular protein homeostasis is established by both the UPS and autophagy, and defects in either contribute to a number of mammalian diseases (42–47). Previous work has shown an age-related decline in the ability of cells to maintain proper protein homeostasis, and a number of neurodegenerative disorders have been associated with decreased autophagy (48, 49) or UPS activity (50–54). The regulated degradation of protein fragments is achieved through a specific UPS pathway called the Arg/N-end rule pathway (55–57). Fragments bearing N-terminal basic amino acids (Arg, Lys, or His) or bulky hydrophobic amino acids (Phe, Leu, Trp, Tyr, or Ile) can be directly recognized by the Ub ligases of the N-end rule pathway (Ubr1, Ubr2, Ubr4, and Ubr5), which facilitate their polyubiquitylation and subsequent degradation by the proteasome. N-terminal Val, Gly, Pro, Ser, Thr, and Met (if not followed by a bulky hydrophobic residue [58]) are not recognized by the Arg/N-end rule pathway and are therefore nondestabilizing (55, 56). N-terminal Asn, Gln, Asp, Glu, and Cys are also destabilizing but require enzymatic modifications, including deamidation of Asn and Gln by Ntan1 and Ntaq1, respectively (59, 60), and N-terminal arginylation of Asp, Glu, or oxidized Cys by the Ate1-encoded arginyl-transferase (arginyl-tRNA protein transferase 1 [ATE1]) (61–63). ATE1-dependent posttranslational arginylation of proteins is emerging as an important player in maintaining nervous system function and in preventing neurodegeneration (64–68).
Previous studies using protein chemical analysis determined that the predominant TDP43-derived CTFs in cytoplasmic inclusions isolated from the brains of patients suffering from ALS and FTLD initiate with Arg208, Asp219, or Asp247 (37, 69, 70). Previously, we showed that these CTFs were short-lived substrates of the Arg/N-end rule pathway and that those bearing N-terminal Asp (TDP43219 and TDP43247) require ATE1 for their degradation by the N-end rule pathway (65). Here, we compared TDP43219 and TDP43247, which are ∼85% identical and differ by a hydrophobic N terminus of 28 amino acids (Fig. 1), and found that they have different fates in the absence of ATE1. While TDP43247 is degraded primarily by the Arg/N-end rule pathway in a manner that requires ATE1, an additional UPS pathway(s) is capable of degrading TDP43219 in the absence of ATE1. Consequently, in contrast to TDP43247, which forms aggregates in the majority of ATE1-lacking cells, TDP43219 has a low aggregation propensity in the absence of ATE1 and, under conditions that promote its accumulation, assembles into morphologically distinct aggregates. This work provides evidence that relatively small differences in the N termini of otherwise similar aggregation-prone fragments can have profound effects on fragment metabolization, aggregation propensity, and morphology and may influence clinical outcomes in neurodegeneration.
RESULTS
TDP43219 and TDP43247 are differentially degraded in the absence of ATE1.
To confirm the role of ATE1 in the degradation of CTFs of TDP43, we compared the steady-state levels of TDP43219 and TDP43247 in the presence and absence of ATE1 in yeast (Saccharomyces cerevisiae) as well as in a murine neuroblastoma cell line (N2a). In yeast, the inducible PMet promoter was used to induce the expression of TDP43219 and TDP43247 in wild-type and ate1Δ yeast strains when grown in medium lacking methionine. CTFs were expressed as Ub fusions (Fig. 2A) so that cotranslational cleavage by cellular deubiquitylases would produce TDP43219 or TDP43247 bearing its natural N-terminal Asp amino acid (71). Neither CTF was detected in wild-type extracts, indicating that they are degraded to negligible levels at steady state. In extracts from ate1Δ yeast, however, TDP43219 and (especially) TDP43247 were stabilized (Fig. 2B). We saw a similar pattern when TDP43219 and TDP43247 were expressed in wild-type N2a cells and N2a cells that had undergone CRISPR/Cas9-mediated knockout of the endogenous ATE1 gene (ATE1-KO cells) (Fig. 2C). At steady state, TDP43219 and TDP43247 (both bearing N-terminal Asp) were not detected in extracts from wild-type N2a cells but were readily detected in extracts from ATE1-KO cells (Fig. 2E). Consistent with numerous studies showing that aggregation-prone CTFs of TDP43 form insoluble proteinaceous inclusions in the cytoplasm, the bulk of TDP43219 and TDP43247 detected in ATE1-KO cells was found in the detergent-insoluble fractions (Fig. 2E, bottom). We also detected a slight increase in the amount of endogenous, full-length mouse TDP43 in the detergent-soluble fraction (conditions which do not lyse the nucleus) in ATE1-KO cells (Fig. 2E, lanes 4 and 6), which is consistent with previous reports that TDP43-derived CTFs are capable of seeding cytoplasmic aggregates containing mislocalized, full-length TDP43 (72, 73).
FIG 2.

Differential degradation of TDP43219 and TDP43247 in the absence of ATE1 and the Arg/N-end rule pathway. (A) TDP43219 and TDP43247 were expressed in yeast by using the ubiquitin fusion technique (71). Cotranslational cleavage by cellular deubiquitylases (DUBs) yields UbR48 (which contains a K48R mutation to prevent its participation in polyubiquitin chains that direct proteins to the proteasome for degradation) and a test fragment (TDP43219 or TDP43247) bearing a specified N-terminal amino acid (red X) and a C-terminal FLAG epitope tag. The PMet promoter was used to induce expression when grown in methionine-lacking medium. (B) SDS-PAGE and immunoblotting (IB) using an anti-TDP43 antibody to detect steady-state levels of TDP43219 and TDP43247 expressed in wild-type (wt) and ATE1-lacking (ate1Δ) yeast. Expression was induced by growth in medium lacking methionine (Met). (C) ATE1 is detected by immunoblotting at ∼55 kDa in lysates of wild-type N2a cells but not in N2a cells that have undergone CRISPR/Cas9-mediated ATE1 ablation. The bottom panel shows actin used as a loading control. (D) The ubiquitin reference technique (URT), derived from the Ub fusion technique (71). Cotranslational cleavage by DUBs of a URT-based fusion produces, at an initially equimolar ratio, a stable internal reference protein such as FLAGDHFR-UbR48, a FLAG-tagged derivative of the mouse dihydrofolate reductase, and a C-terminally FLAG-tagged test fragment (TDP43219 or TDP43247) bearing a specified N-terminal amino acid (red X). Moderate expression in mammalian cells was achieved by using the PSV40 promoter. (E) Steady-state levels of TDP43219 and TDP43247 expressed in wild-type N2a cells and those that had undergone CRISPR-mediated knockout of ATE1 (ATE1-KO). Full-length (FL), endogenous mouse TDP43 as well as exogenously added TDP43219 and TDP43247 were detected in detergent-soluble (Sol) and urea-soluble (Ins) fractions by SDS-PAGE and immunoblotting using an anti-TDP43 antibody. (F) Partitioning of detergent-soluble and -insoluble TDP43219 and TDP43247 bearing N-terminal Asp or N-terminal Val (not arginylated by ATE1), which were expressed in HEK293T cells. Cells were treated with 5 μM MG132 for 24 h, as indicated, prior to lysis. Fragments were immunoprecipitated (IP) from the soluble (S) and insoluble (In) fractions using an anti-FLAG antibody and detected by immunoblotting using an anti-TDP43 antibody. Asterisks represent the antibody light chain. (G) Graphical representation of the densitometric analysis of bands in panel F to show relative expression levels and the percentages of the total found in the soluble or insoluble fraction. (H) Pulse-chase analysis of TDP43219 and TDP43247 bearing either N-terminal Asp or Val in HEK293T cells. Fragments were labeled with [35S]Met-Cys, followed by a chase, preparation of extracts, immunoprecipitation with anti-FLAG, SDS-PAGE, and autoradiography. (I) Quantification of N-terminal Val-bearing fragments in panel H.
In order to investigate the effects of the Arg/N-end rule pathway and the UPS on CTF partitioning between the detergent-soluble and -insoluble fractions, TDP43219 and TDP43247 were expressed in HEK293T cells (which have robust N-end rule activity) bearing either N-terminal Asp or N-terminal Val (which is not arginylated by ATE1) in the presence or absence of the proteasome inhibitor MG132 for 24 h. Soluble and insoluble fractions were prepared, and CTFs were detected by anti-FLAG immunoprecipitation followed by SDS-PAGE and immunoblotting using an anti-TDP43 antibody (Fig. 2F). Similar to their metabolism in yeast and in N2a cells, TDP43219 and TDP43247 bearing their natural N-terminal Asp were not detected in soluble fractions of HEK293T cells but were detected in the insoluble fractions (Fig. 2F and G), indicating that CTFs that are not normally degraded by the proteasome ultimately form insoluble aggregates. The presence of MG132 resulted not only in a significant increase in the total amount of CTFs detected (∼3-fold and ∼1.6-fold for Asp-TDP43219 and Asp-TDP43247, respectively) but also in a significant shift in partitioning to the soluble fraction (∼22% and ∼26% of the total for Asp-TDP43219 and Asp-TDP43247, respectively) (Fig. 2F and G). N-terminal Val, which is not recognized by ATE1, had different effects on the overall levels and the partitioning of TDP43219 compared to TDP43247. In the absence of MG132, the overall levels of Val-TDP43247 increased 1.6-fold relative to those of Asp-TDP43247, and ∼20% of the total Val-TDP43247 was detected in the soluble fraction. MG132 did not significantly change the overall levels of Val-TDP43247 or its relative partitioning, suggesting that TDP43247 is degraded primarily by the Arg/N-end rule pathway. In contrast, Val-TDP43219 is not detected in the soluble fraction unless MG132 is present, indicating that soluble TDP43219 is degraded by the UPS even in the absence of its recognition by ATE1 and degradation by the Arg/N-end rule pathway.
In order to determine the fate of newly formed TDP43 CTFs, we utilized the ubiquitin reference technique (URT) to carry out in vivo pulse-chase analysis in HEK293T cells (71). URT-based fusion proteins generate, at initially equimolar concentrations, a stable reference protein (FLAGDHFR-UbR48) and CTFs bearing specified N-terminal amino acids (Fig. 2D). Using this method, the fates of [35S]Met-Cys-labeled CTFs of TDP43 were monitored by measuring CTF levels relative to the level of FLAGDHFR-UbR48 at various time points after the label was washed out and translation was blocked by cycloheximide. Asp-TDP43219 and Asp-TDP43247 were detected at low levels even at the zero time point, consistent with our previous study showing that they are ATE1-dependent substrates of the Arg/N-end rule pathway (Fig. 2H) (65). The otherwise identical fragments bearing N-terminal Val, however, were degraded at different rates. The half-life of Val-TDP43247 extended beyond 60 min, whereas Val-TDP43219 was significantly less stable and degraded with a half-life of ∼10 min (Fig. 2H and I). These data indicate that soluble TDP43247 largely depends on the Arg/N-end rule pathway for its degradation, whereas an additional pathway(s) participates in the degradation of TDP43219.
TDP43219 and TDP43247 differ in their propensities to form aggregates.
Previous studies have shown that CTFs of TDP43 have a higher propensity to aggregate in vivo than full-length TDP43 (65, 74). In order to examine the relative aggregation propensities of TDP43219 and TDP43247 in cells, we used a modified version of the URT where the cDNA encoding dihydrofolate reductase (DHFR) was replaced with cDNA encoding the red-fluorescent mCherry protein (Fig. 3A). Cotranslational cleavage of this fusion construct yields stable mCherry-UbR48, which “marks” transfected cells, and a C-terminally FLAG epitope-tagged fragment whose fate can be monitored by using indirect immunofluorescence with an anti-FLAG primary antibody and a fluorescein-conjugated secondary antibody (Fig. 3C and D).
FIG 3.

In vivo aggregation of TDP43219 and TDP43247. (A) Modified version of the URT whereby DHFR is replaced by mCherry in order to identify cells expressing aggregation-prone fragments. (B) HEK293T cells were transiently transfected with TDP43219 and TDP43247 and then treated with either 10 μM MG132 for 6 h or 50 μM chloroquine for 24 h prior to fixation. At 48 h posttransfection, cells were fixed, and aggregates were detected by using anti-FLAG primary antibody and an Alexa Fluor 488-conjugated secondary antibody. The percentages of mCherry-positive cells containing aggregates were quantified. Error bars indicate standard errors of the means (SEM). Statistical analyses were done by employing the unpaired t test. Asterisks represent bars that are significantly different from results with no treatment (No Tx) (P < 0.05). (C) Representative images of either wild-type (top) or ATE1-KO (bottom) mouse N2a cells transfected with a plasmid expressing mCherry-UbR48-TDP43219 as seen in panel A. mCherry-UbR48 was detected by red fluorescence, and TDP43219 was detected by indirect immunofluorescence, using anti-FLAG antibody and an Alexa Fluor 488-conjugated secondary antibody. Bars, 10 μm. (D) Similar to panel C except that cells were expressing TDP43247. (E) Schematic of TDP43219 and TDP43247 indicating relative positions of lysine residues. (F) Percentages of mCherry-positive wild-type and ATE1-KO N2a cells containing aggregates of TDP43219 or TDP43247. Error bars indicate SEM. Statistical analyses employed the unpaired t test (P = 0.0021). (G) Percentages of mCherry-positive wild-type and ATE1-KO N2a cells containing aggregates of wild-type TDP43219 or the indicated K→R mutations. (H) Same as panel G except with wild-type TDP43247 or the indicated K→R mutations. Error bars indicate SEM. Statistical analyses were done by using the unpaired t test. Asterisks represent bars that are significantly different from the wild-type fragment in the same cell type (P < 0.05).
Although our results using MG132 suggested that the UPS is involved in the ATE1-independent degradation of TDP43219 (Fig. 2F and G), we compared aggregate formation in HEK293T cells treated with either MG132 or chloroquine, a neutralizer of lysosomal pH which blocks degradation through autophagy. We found a far-greater increase in the aggregation propensities of both TDP43219 and TDP43247 as a result of MG132 treatment than with chloroquine treatment (Fig. 3B), suggesting that aggregation propensity is determined primarily by the UPS-mediated degradation of soluble CTFs.
In order to compare their relative aggregation propensities in the presence and absence of ATE1, TDP43219 and TDP43247 were expressed in both wild-type N2a cells and ATE1-KO N2a cells. Consistent with our previous finding that the Arg/N-end rule pathway can degrade CTFs of TDP43 (65), very few cells containing aggregates were detected in mCherry-positive, wild-type N2a cells. On the other hand, cytoplasmic, perinuclear aggregates of TDP43 CTFs could be detected in ATE1-KO cells (Fig. 3C and D). Interestingly, while aggregates of TDP43247 could be detected in ∼60% of mCherry-positive ATE1-KO cells, aggregates of TDP43219 could be detected in only ∼10% of mCherry-positive ATE1-KO cells (Fig. 3F). This is yet further evidence indicating that an additional pathway participates in the degradation of TDP43219 and, in doing so, prevents its aggregation even in the absence of ATE1.
TDP43219 and TDP43247 are identical throughout 85% of their sequences. However, TDP43219 possesses a hydrophobic, N-terminal, 28-amino-acid extension that includes a lysine residue at position 224 (relative to full-length human TDP43), which is not shared with TDP43247 (Fig. 3E). To determine if K224 played a role in aggregation propensity, we converted it to arginine (K224R), a conservative but nonubiquitylated residue, and examined its aggregation propensity in wild-type and ATE1-KO N2a cells. Surprisingly, the aggregation propensity of TDP43219-K224R in ATE1-KO N2a cells was not significantly increased, suggesting that K224 does not play a significant role in the UPS-mediated degradation of TDP43219 (Fig. 3G). The finding that TDP43219-K224R is not significantly more aggregated in wild-type cells also suggests that K224 is not a preferred site of ubiquitylation by the Arg/N-end rule pathway (Fig. 3G).
The amino acid sequence common to both TDP43219 and TDP43247 contains three additional lysine residues: K251 and K263 are located near the N terminus, and K408 is located at the extreme C terminus (Fig. 3E). In attempts to determine which lysine residues play an important role in the UPS-mediated degradation of CTFs and thereby prevent aggregate formation, they were each individually and collectively converted to arginine in both TDP43219 and TDP43247. Because TDP43247 is degraded primarily by the Arg/N-end rule pathway, no single mutation of any lysine residue or their collective mutation (TDP43247-tripleR) resulted in a significant increase in aggregation relative to nonmutated TDP43247 in ATE1-KO cells (Fig. 3H, red bars). In wild-type N2a cells, the aggregation propensity of TDP43247 should increase if a lysine normally ubiquitylated by the Arg/N-end rule pathway is mutated to arginine (Fig. 3H, black bars). Interestingly, mutation of K251, which is 6 residues from the N terminus of arginylated TDP43247, did not affect aggregation propensity, implying that K251 is sterically inaccessible to the ubiquitin ligases of the N-end rule of the Arg/N-end rule pathway. Although K263R had the greatest effect on the aggregation propensity of TDP43247, the effect was modest, suggesting that the Arg/N-end rule pathway is capable of ubiquitylating multiple lysine residues. This was substantiated by a dramatic increase in the aggregation of lysineless TDP43247-tripleR in wild-type N2a cells (Fig. 3H).
Although TDP43219 is capable of being degraded by the Arg/N-end rule pathway, our data indicate that an additional pathway, which is not capable of degrading TDP43247, participates in the degradation of TDP43219. To determine the relative contributions of the Arg/N-end rule pathway and the non-Arg/N-end rule pathway in the metabolism of TDP43219, we examined the aggregation propensity of TDP43219 harboring specific lysine mutations. In wild-type N2a cells, the K263R and K251R mutants had the greatest (albeit weak) effect on the aggregation propensity of TDP43219 (Fig. 3G, black bars). On the other hand, K408R had the greatest effect on the aggregation propensity of TDP43219 in ATE1-KO N2a cells (Fig. 3G, red bars). These results are consistent with the notion that the degradation of soluble TDP43219 is carried out predominantly by the Arg/N-end rule pathway, with a preference for ubiquitylation at K263 and/or K251, but in the absence of ATE1, an alternative UPS pathway, with a preference for K408, can facilitate its turnover. The finding that TDP43247, which also contains K408, is stable in the absence of ATE1 suggests that the 28-amino-acid N-terminal extension in TDP43219 harbors a degradation signal required for recognition by the alternative UPS pathway.
Posttranslational modifications of TDP43219 and TDP43247.
Aggregation-prone CTFs of TDP43 have been shown to be highly phosphorylated and ubiquitylated (29, 75, 76). We found that aggregates of TDP43219 completely colocalized with ubiquitin (Fig. 4A), whereas some TDP43247 aggregates either were not associated with ubiquitin or were associated to a lesser extent (Fig. 4A, arrows). In order to examine the relative associations of TDP43219 and TDP43247 with ubiquitin, Val-TDP43219 and Val-TDP43247 were expressed by using the URT (Fig. 2D), except that the highly expressing PCMV promoter was used and the UPS-mediated degradation was blocked by MG132 to promote accumulation and aggregation of CTFs. CTFs from the insoluble fractions were then isolated by immunoprecipitation using an anti-FLAG antibody, and ubiquitylation was evaluated by immunoblotting using an anti-Ub antibody. Ubiquitylation associated with insoluble CTFs were seen as a high-molecular-weight smear at >250 kDa (Fig. 4B). Interestingly, when samples were normalized for the amount of CTF, insoluble TDP43219 was ∼12-fold more heavily ubiquitylated than insoluble TDP43247 (Fig. 4B, compare lane 3 to lanes 4 through 7). The finding that Val-TDP43247 is less ubiquitylated than Val-TDP43219 is consistent with the existence of an additional UPS pathway capable of ubiquitylating TDP43219 but not TDP43247.
FIG 4.

Posttranslational modifications of TDP43219, TDP43247, and their aggregates. (A) Immunocytochemistry reveals the colocalization of ubiquitin and Val-TDP43219 or Val-TDP43247 in aggregates formed in HEK293T cells treated with 5 μM MG132 for 24 h prior to fixation. Ubiquitin was detected by using an antiubiquitin antibody, and CTFs were detected by using an anti-FLAG antibody. Bar, 10 μm. (B) Relative polyubiquitylation of detergent-insoluble (urea-solubilized) Val-TDP43219 and Val-TDP43247 expressed in cells treated with 5 μM MG132 for 24 h. Polyubiquitylation was detected by immunoprecipitation of CTFs using an anti-FLAG antibody, followed by SDS-PAGE and immunoblotting using an antiubiquitin antibody. Note the increase in the polyubiquitylation of TDP43219 versus similar levels of TDP43247 (compare lanes 3 and 5). LC, antibody light chain. (C, top and middle) Monomeric, oligomeric, and high-molecular-weight (HMW) species of TDP43219 and TDP43247 were detected in detergent-insoluble fractions of HEK293T cells expressing CTFs by immunoprecipitation using an anti-FLAG antibody, followed by SDS-PAGE and immunoblotting using an anti-TDP43 antibody. (Bottom) Phosphorylated monomers of TDP43219 and TDP43247 were detected only in insoluble fractions. Phosphorylation of oligomers and high-molecular-weight species was not detected (not shown).
The bulk of soluble TDP43219 and TDP43247 was detected as monomers, whereas a significant portion of TDP43219 and TDP43247 isolated from insoluble fractions was recovered as oligomeric and high-molecular-weight species (∼250 kDa) that were resistant to urea as well as SDS-PAGE (Fig. 4C). Interestingly, we also found that soluble TDP43219 was processed (cleaved) into a fragment that migrated more similarly to TDP43247 in SDS-PAGE gels (Fig. 4C, lanes 3 and 4). Since our anti-TDP43 antibody recognizes epitopes in the C terminus of human TDP43, this species is a result of N-terminal cleavage of TDP43219. Remarkably, this processing was inhibited by MG132. In order to determine the phosphorylation status of TDP43219 and TDP43247, we carried out immunoblotting on both soluble and insoluble TDP43219 and TDP43247 using an anti-Ser409/410 phosphorylation-specific TDP43 antibody. Phosphorylated TDP43 was not detected in the soluble fractions (Fig. 4C, lanes 1 to 6); however, monomeric CTFs were phosphorylated in the insoluble fractions (Fig. 4C, lanes 7 to 12), consistent with phosphorylation playing a role in aggregation dynamics of insoluble TDP43 species (76).
TDP43219 and TDP43247 form morphologically distinct cytoplasmic aggregates.
Previous studies reported spherical (both large and small) and filamentous (skein-like) TDP43-containing inclusions in upper and lower motor neurons as well as in specific brain cell populations associated with ALS and FTLD (75, 77). Although TDP43219 aggregates were scarce relative to TDP43247 aggregates (Fig. 3F), we noticed that TDP43219 formed aggregates that were small and spherical (dot-like), whereas TDP43247 formed larger spherical and often filamentous aggregates (Fig. 5A to D). In order to determine if spherical aggregates mature into filamentous aggregates over time, we transfected HEK293T cells with Val-TDP43219 or Val-TDP43247 and quantified cells containing spherical, filamentous, or a combination of aggregates at 24 h and 48 h posttransfection. We were unable to detect filamentous aggregates of Val-TDP43219 at either time point (Fig. 5E) but found that the number of cells containing spherical TDP43247 aggregates decreased (∼30% versus ∼12.5% at 24 and 48 h, respectively) with a concomitant increase in filamentous TDP43247 aggregates over time (∼61.2% and ∼82.6% at 24 and 48 h, respectively) (Fig. 5F). In order to rule out the possibility that the nonnatural N-terminal Val residue influences aggregate morphology, we compared aggregate morphologies of TDP43219 and TDP43247 bearing either N-terminal Val or their natural N-terminal Asp at 24 h posttransfection (Fig. 5G and H). One possibility is that filamentous TDP43 aggregates form only after a critical concentration of soluble CTFs is reached. Since TDP43247 is degraded primarily by the Arg/N-end rule pathway, whereas TDP43219 is degraded by the Arg/N-end rule pathway and an additional UPS pathway (Fig. 2 and 3), steady-state levels of TDP43219, even in the absence of its degradation by the Arg/N-end rule pathway, may be insufficient to form filamentous aggregates. Therefore, we used MG132 to prevent all UPS-mediated degradation and promote the accumulation of CTFs (Fig. 2F and G). Even in the presence of MG132 and with high-level expression using the PCMV promoter, only small spherical aggregates were detected for both Asp-TDP43219 and Val-TDP43219 (Fig. 5G). On the other hand, both Asp-TDP43247 and Val-TDP43247 were capable of forming filamentous aggregates (Fig. 5H). Collectively, these results indicate that the N-terminal 28 amino acids of TDP43219 limit its aggregation morphology to smaller, dot-like, spherical aggregates, whereas TDP43247 is capable of forming spherical aggregates (which tended to be larger) as well as filamentous aggregates.
FIG 5.
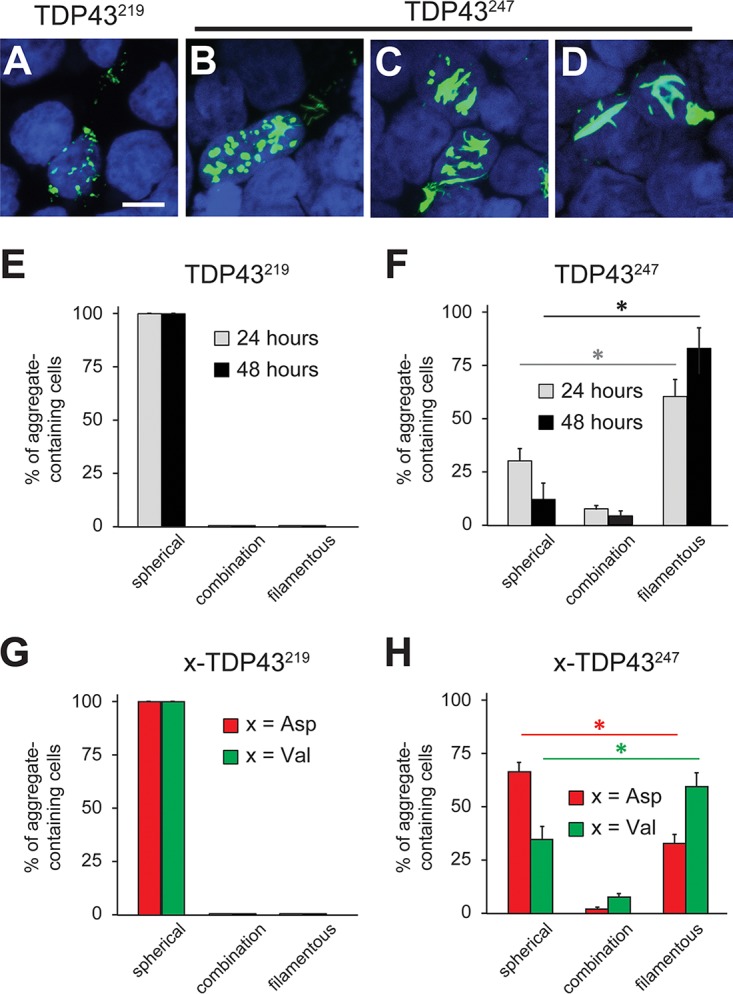
Aggregate morphology of TDP43219 and TDP43247. (A to D) Representative images of cytoplasmic aggregates formed in HEK293T cells by TDP43219 (A) and TDP43247 (B to D). Bar, 10 μm. (E) Quantification of spherical versus filamentous aggregates formed by Val-TDP43219 at 24 and 48 h posttransfection. (F) Same as panel E except that Val-TDP43247 was used. Error bars indicate SEM. Statistical analyses were done by employing the unpaired t test (P < 0.05) to compare spherical versus filamentous aggregates at 24 and 48 h. (G) Comparison of aggregate morphology with TDP43219 bearing N-terminal Asp versus N-terminal Val at 24 h posttransfection. Cells were treated with 10 μM MG132 for 6 h prior to fixation. (H) Same as panel G except that TDP43247 was used. Error bars indicate SEM. Statistical analyses were done by employing the unpaired t test (P < 0.05) to compare spherical versus filamentous aggregates formed by TDP43247 bearing N-terminal Asp and N-terminal Val.
DISCUSSION
In this study, we provide evidence that differences in the N termini of otherwise similar aggregation-prone C-terminal fragments of TDP43 can have a profound influence on fragment metabolism, on the propensity to form insoluble aggregates, and on aggregate morphology. We found that TDP43247 is degraded primarily by the Arg/N-end rule pathway in a manner that requires ATE1 but that an additional pathway(s) is capable of degrading TDP43219 even in the absence of ATE1. This is supported by multiple lines of evidence. First, steady-state levels of TDP43219 and TDP43247 in yeast and N2a cells lacking ATE1 show a greater accumulation of TDP43247 than of TDP43219 (Fig. 2B and E). Second, soluble Val-TDP43247, which is not recognized by ATE1 and therefore is not degraded by the Arg/N-end rule pathway, is stable, whereas soluble Val-TDP43219 is unstable unless the proteasome is inhibited (Fig. 2F and G). Third, experiments involving the metabolic labeling of CTFs reveal that in the absence of degradation by the Arg/N-end rule pathway, the half-life of TDP43219 is significantly shorter than that of TDP43247 (Fig. 2H and I). Fourth, aggregates of TDP43247 are detected in the majority of ATE1-lacking N2a cells, whereas aggregates of TDP43219 are detected in only ∼10% of ATE1-lacking N2a cells (Fig. 3F). Finally, insoluble TDP43219 is highly ubiquitylated, whereas insoluble TDP43247 is relatively free of ubiquitin (Fig. 4A and B). A number of proteins have been reported to play a role in TDP43 clearance (65, 78–80). Recently, the β4-strand of the RRM2 domain of human TDP43 was shown to harbor a degradation signal recognized by the VHL/ElonginB/C/Cul2/Rbx1 complex (80). Studies to identify the ATE1-independent UPS pathway contributing to the degradation of TDP43219 are under way.
We also found that despite being ∼85% similar, TDP43219 and TDP43247 form morphologically distinct intracellular aggregates (Fig. 5). Recently, Guenther et al. reported that the 247DLIIKGISVHI257 segment from RRM2 of TDP43 is capable of forming a range of amyloid polymorphs (81). We found that TDP43219 formed only small spherical aggregates, whereas TDP43247, which contains the 247DLIIKGISVHI257 segment directly at its N terminus, formed both spherical aggregates and filamentous aggregates over time (Fig. 5). The finding that TDP43219 does not form filamentous aggregates indicates that the additional N-terminal 28 amino acids (not included in TDP43247) function to prevent filamentous morphology. Previous studies analyzing the distribution of TDP43-immunoreactive neuronal inclusions in patients with ALS identified both spherical and skein-like inclusions in lower motor neurons of the spinal cord as well as in the motor cortex and the substantia nigra; however, TDP43-containing aggregates in the neostriatum and dentate gyrus were neither spherical nor skein-like (75, 77). In light of our findings, we suggest that distinct cleavages of TDP43 may occur in different neuronal populations under pathological conditions, resulting in fragment populations that differ in metabolism, aggregation propensity, and aggregate morphology, as determined (at least in part) by their N termini. Interestingly, Mori et al. detected spherical inclusions in motor neurons of ALS or FTLD patients with a disease duration of less than 100 months but only filamentous aggregates in motor neurons of patients with a disease duration of more than 100 months (77). Our data indicate that aggregates of at least some CTFs of TDP43 (e.g., TDP43247) undergo a time-dependent maturation process to form filamentous aggregates (Fig. 5F).
Multiple cleavage sites have been described for TDP43; therefore, a heterologous mixture of fragments can be generated under pathological conditions. Previous work has shown that TDP43 can be sequentially processed (21). As such, a given CTF can be a derivative of either full-length TDP43 or a larger “precursor” fragment. Of note, we detected a soluble CTF species resulting from N-terminal cleavage of TDP43219 migrating similarly to TDP43247 in SDS-PAGE gels (Fig. 4C). Although the protease responsible for this species is unknown, this cleavage is blocked by the proteasome inhibitor MG132 (Fig. 4C). It remains to be determined if this processing is catalyzed by the proteasome itself or by a separate cellular protease inhibited by MG132.
Collectively, our results suggest that cells may use N-terminal processing as a means to convert aggregation-prone fragments into species with altered properties. Such processing may alter the manner in which a fragment is degraded. If so, then the vulnerability of a given cell to the effects of a specific aggregation-prone fragment would be established by the complement of cellular proteases and the activity of particular degradation pathways within that cell. This may explain why some neuronal populations are more vulnerable than others to protein aggregates (e.g., Lewy bodies in neurons of the substantia nigra).
In sum, we found that the N termini of otherwise similar aggregation-prone C-terminal fragments of TDP43 have profound effects on its (i) metabolism, (ii) aggregation propensity, and (iii) aggregate morphology. This suggests that the N termini of aggregation-prone fragments may influence clinical outcomes in neurodegeneration. Ultimately, characterization of such differences may provide prognostic indicators useful in outcome predictions and may assist in the design of therapeutics for neurodegenerative diseases.
MATERIALS AND METHODS
Miscellaneous reagents.
Cycloheximide was obtained from Sigma-Aldrich. MG132 was obtained from Cayman Chemical. Chloroquine was obtained from Invitrogen/Thermo Scientific. Anti-C-terminal TDP43 (catalog number 12892-1-AP) and anti-phospho-TDP43 (catalog number 66318-1-Ig) were obtained from Proteintech. Anti-FLAG M2 (catalog number F1804) and anti-FLAG M2 magnetic beads (catalog number M8823) were obtained from Sigma-Aldrich. Anti-ATE1 (catalog number sc-398805), antiactin (catalog number sc-47778), and anti-ubiquitin P4D1 (catalog number sc-8017) were obtained from Santa Cruz Biotechnology. Detection was carried out by using the following goat secondary antibodies from Thermo Scientific: anti-mouse IgG(H+L)–Alexa Fluor 488 (catalog number A-11001), anti-rabbit IgG(H+L)–Alexa Fluor 546 (catalog number A11010), anti-mouse IgG–DyLight 680 (catalog number PI35519), anti-rabbit IgG–DyLight 680 (catalog number SA5-10176), anti-mouse IgG–DyLight 800 (catalog number PI35569), and anti-rabbit IgG–DyLight 680 (catalog number SA5-10036).
Synthesis of plasmids.
To construct the plasmids pCB398 and pCB399 used in in vivo aggregation assays (Fig. 3), DNA encoding mCherry was amplified from pKP551 (see Table S1 in the supplemental material) by using primers CB383F and CB384R. The resulting PCR product was used to replace the Flag-DHFR moiety in pCB334 and pCB336, respectively, after digestion with HindIII/AgeI. The Flag-DHFR moiety in pCB340 was replaced with the mCherry cDNA to produce pCB401. For the generation of pCB541, pCB542, pCB543, and pCB544, UbK48R-X-TDP43219 or UbK48R-X-TDP43247 (where X is Asp or Val) was amplified by PCR and inserted into pRS416 (ATCC 87521) after digestion with HindIII/XhoI. For the generation of plasmids encoding K→R mutants of TDP43219 and TDP43247 (pYK002 to pYK016) as well as control vectors expressing mCherry-UbK48R only (pYK001) or FLAGDHFR-UbK48R only (pYK027), the Q5 site-directed mutagenesis kit (New England BioLabs) was used according to the manufacturer's protocol. Briefly, plasmids were made by amplifying specific template plasmids with specific primer sets. Primer sets were designed to be nonoverlapping, and one primer contained specified mutations in the middle of the oligonucleotide sequence (Table S2). pYK001 was made by amplifying pCB400 with primers YK006R and YK007F. pYK004 was made by amplifying pCB398 with primers YK009R and YK010F. pYK005 was made by amplifying pCB398 with primers YK013F and YK014R. pYK006 was made by amplifying pCB398 with primers YK015F and YK016R. pYK007 was made by amplifying pCB398 with primers YK017F and YK018R. pYK009 was made by amplifying pYK007 with primers YK013F and YK014R. pYK010 was made by amplifying pCB400 with primers YK017F and YK018R. pYK011 was made by amplifying pYK005 with primers YK025F and YK027R. pYK012 was made by amplifying pYK006 with primers YK026F and YK027R. pYK015 was made by amplifying pYK009 with primers YK015F and YK016R. pYK016 was made by amplifying pYK015 with primers YK025F and YK027R. pYK027 was made by amplifying pCB328 with primers YK027R and YK032F. PCR was carried out for 25 cycles, and 1 μl of the resulting reaction mix was then used in a kinase-ligase-DpnI (KLD) reaction for 5 min at room temperature to obtain ligation and template removal. NEB5α competent cells (New England BioLabs) were transformed with 5 μl of the KLD reaction mixture and selected on LB plates supplemented with ampicillin (100 μg/ml).
Expression of TDP43219 and TDP43247 in S. cerevisiae.
S. cerevisiae JD52 (MATa trp1-Δ63 ura3-52 his3-Δ200 leu2-3112 lys2-801) and CHY21 (ate1Δ::KanMX6 in JD52) were described previously (65). S. cerevisiae media included YPD medium (1% yeast extract, 2% peptone, and 2% glucose; only the most relevant medium components are cited), SD medium (0.17% yeast nitrogen base, 0.5% ammonium sulfate, 2% glucose), and synthetic complete (SC) medium (0.17% yeast nitrogen base, 0.5% ammonium sulfate, and 2% glucose plus a dropout mixture of compounds required by auxotrophic strains). S. cerevisiae was transformed with the low-copy-number pCB541, pCB542, pCB543, or pCB544 plasmid by using standard techniques (82). Expression of TDP43219 and TDP43247 was induced by growth in synthetic defined medium (lacking Ura and Met) for 24 h at 30°C. Equivalent numbers of yeast cells were resuspended in protein-loading buffer (PLB) (80 mM Tris [pH 6.8], 2% SDS, 10% glycerol, 0.0006% bromophenol blue, and 0.1 M dithiothreitol [DTT]) and lysed by sonication. The resulting samples were centrifuged at 13,000 rpm at room temperature, and proteins from the supernatant were boiled at 95°C and separated by SDS–4 to 15% PAGE followed by transfer to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad) in Towbin buffer (25 mM Tris, 192 mM glycine, and 20% methanol). Membranes were blocked in phosphate-buffered saline (PBS) containing 5% milk and 0.1% Tween 20 at room temperature for 1 h. Primary anti-TDP43 C-terminal antibody and goat anti-rabbit secondary antibody were used to detect TDP43219 and TDP43247. Blots were developed by using the Licor Odyssey CLx system.
Cell culture and transfection.
Human embryonic kidney HEK293T cells and neuroblastoma (N2a) cells were maintained at 37°C with 5% CO2 in Dulbecco modified Eagle medium (DMEM) (Corning Cellgro) containing 15% fetal bovine serum (Gemini Bio-products) supplemented with 20 mM glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin (Fischer Bioreagents). Cells were transfected by using the BioT transfection reagent (Bioland Scientific) according to the manufacturer's protocol.
Generation of ATE1-lacking N2a cells.
Mouse neuroblastoma cells (N2a) were generated by using the CRISPR/Cas9 system (83). Briefly, pCB431, a plasmid encoding human codon-optimized Cas9 derived from Streptococcus pyogenes and a chimeric guide RNA targeting exon 2 of the mouse ATE1 gene, was constructed by the ligation of a double-strand oligomer (made by the denaturation and renaturation of CB431 and CB432) into BbsI-digested pX330 (pX330-U6-Chimeric_BB-CBh-hSpCas9 was a gift from Feng Zhang) (Addgene plasmid 42230). Cells were transfected with pCB431, and individual clones were selected and screened for the loss of the ATE1 protein by immunoblotting (Fig. 2C). ATE1-KO cells were also found to lack arginylation activity (data not shown).
Pulse-chase assay.
HEK293T cells (∼75% confluent) were transfected with 0.8 μg plasmid per 35-mm well using BioT according to the manufacturer's protocol. At 24 h posttransfection, cells were pulse labeled for 15 min with 0.1 mCi/ml 35S-labeled l-methionine (MP Biomedicals) in DMEM lacking Met and Cys. Labeling was quenched by the addition of 0.1 mg/ml cycloheximide (VWR) in complete DMEM containing Met and Cys. Cells contained in an entire 35-mm well were lysed at the indicated time points of a chase by rapid scraping in 150 μl of TSD buffer (50 mM Tris-HCl [pH 7.4], 1% SDS, 5 mM DTT) and snap-freezing in liquid nitrogen. Samples were then heated at 95°C for 10 min and diluted with 10 volumes of TNN buffer (0.5% NP-40, 0.25 M NaCl, 5 mM EDTA, 50 mM Tris-HCl [pH 7.4]) containing the complete protease inhibitor mixture (Roche). Total 35S (in the 10% CCl3COOH-insoluble fraction) in samples was then measured, and volumes were adjusted to equalize 35S among different samples. Normalized samples were then immunoprecipitated by the addition of 5 μl of anti-FLAG-M2 magnetic beads (Sigma) and incubation with rocking at 4°C for 3 h. Immunoprecipitated proteins were then washed 3 times in TNN buffer and once in phosphate-buffered saline, followed by resuspension in 20 μl of SDS sample buffer, heating at 95°C for 10 min, 4 to 15% SDS-PAGE, and autoradiography.
Immunocytochemistry.
Cells were seeded in poly-d-lysine-coated chamber slides at a density of 2 × 103 cells/well and grown until they reached 70 to 80% confluence for transfection. Twenty-four hours after transfection, cells were fixed for 10 min in 4% formaldehyde and then permeabilized in 0.5% Triton X-100 in PBS at room temperature. Slides were blocked in 10% goat serum (ThermoFisher Scientific) at 37°C for 1 h, followed by incubation with anti-FLAG, anti-TDP43, or anti-ubiquitin P4D1 antibody, as indicated in the figures, at a 1:1,000 dilution in 1× PBS containing 5% goat serum and 0.1% Tween 20 for 2 h at 37°C. Slides were washed three times with 1× PBS containing 0.1% Tween 20 at room temperature and then incubated with secondary antibodies (goat anti-mouse antibody–Alexa Fluor 488 and goat anti-rabbit antibody–Alexa Fluor 546) at a 1:500 dilution for 1 h. Finally, the slides were washed three times with PBS containing 0.1% Tween 20 and once with PBS. The slides were then mounted with 4′,6-diamidino-2-phenylindole (DAPI)-containing Vectashield H-1200 mounting medium (Vector Laboratories). Aggregates were quantified by using a Nikon A1T-A1 confocal system with a Nikon Eclipse Ti inverted microscope and Nikon Instrument Software (NIS) elements AR-3.2 imaging software (Nikon Instruments, Melville, NY).
In vivo aggregation.
Wild-type and ATE1-lacking N2a cells at ∼70% confluence were transfected in chamber slides with plasmids encoding wild-type TDP43219 (pCB398), TDP43219 K→R mutants (pYK004, pYK005, pYK006, pYK007, and pYK015), wild-type TDP43247 (pCB400), or TDP43247 K→R mutants (pYK010, pYK011, pYK012, and pYK016), as indicated in the figures. At 24 h posttransfection, cells were fixed in 4% formaldehyde for 10 min at room temperature and permeabilized in 0.5% Triton X-100 in PBS for 10 min at room temperature. Indirect immunocytochemistry was performed (as described above) by using an anti-FLAG primary antibody and goat anti-mouse Alexa Fluor 488-conjugated secondary antibody to detect fragments. Cells were examined for fluorescence with mCherry, DAPI, and Alexa Fluor 488-conjugated secondary antibody by using a Nikon A1 confocal microscope.
Qualitative analysis of TDP43219 and TDP43247 aggregates.
In vivo aggregation was achieved as described above, except that HEK293T cells were transfected with plasmids expressing TDP43219 (pCB398 and pCB399) and TDP43247 (pCB400 and pCB401) bearing either N-terminal Asp or Val. pCB328 and pCB332 were used to analyze aggregate morphology resulting from the high-level expression of Val-bearing CTFs from the PCMV promoter. Cells were also treated with 10 μM MG132 for 6 h to inhibit proteasomal degradation. Aggregate-containing cells were scored for spherical or filamentous morphology or a combination of both. Spherical aggregates with a diameter of <1 μm were considered small, and those with a diameter of >1 μm were considered large.
Ubiquitylation of TDP43219 and TDP43247 fragments.
HEK293T cells were transfected with plasmids expressing TDP43219 (pCB328) and TDP43247 (pCB332) bearing N-terminal Val from the PCMV promoter and treated with 5 μM MG132 for 24 h the following day. At 48 h posttransfection, cells were fixed, permeabilized, and blocked as described above. Ubiquitylation and TDP43 were detected by using mouse antiubiquitin and rabbit anti-TDP43 primary antibodies, followed by goat anti-mouse Alexa Fluor 488-conjugated and goat anti-rabbit Alexa Fluor 546-conjugated antibodies. Images of ubiquitin-positive TDP43 aggregates were taken with a Nikon A1 confocal microscope.
Lysate preparation, immunoprecipitation, and immunoblotting.
Cells were harvested and lysed in tissue lysis buffer (TLB) (50 mM HEPES, 10% glycerol, 0.05% NP-40, 150 mM NaCl, 1 mM DTT, and 1 mM phenylmethylsulfonyl fluoride [PMSF] containing the complete protease inhibitor mixture) by freezing-thawing. The lysate was then centrifuged at 13,000 rpm for 20 min at 4°C, and the supernatant was collected as the soluble fraction. The pellet was washed twice in TLB, resuspended by sonication in 8 M urea buffer (8 M urea, 50 mM Tris, 1 mM DTT, and 1 mM PMSF), and centrifuged at 13,000 rpm at room temperature. The final supernatant was collected as the “insoluble” fraction. Protein concentrations for both soluble and insoluble fractions were determined by using the Bio-Rad protein assay reagent according to the manufacturer's protocol. For immunoprecipitation and immunoblotting, sample protein concentrations were normalized. For immunoprecipitation, protein G magnetic beads (Bio-Rad) were incubated with 0.5 μg mouse anti-FLAG-M2 antibody per sample for 30 min at 4°C. This antibody-bead mixture was added to normalized protein lysates and rotated for 2 h at 4°C. Beads were then separated from the lysate by using a magnetic rack, rinsed three times with TLB, and resuspended in 2× SDS-PAGE PLB. Prior to SDS-PAGE, samples were boiled at 95°C for 5 min to denature and elute bound proteins. For immunoblotting, normalized protein samples were separated on 4-to-12% gradient NuPage Bis-Tris premade gels (Invitrogen) and transferred onto a methanol-activated PVDF membrane (Bio-Rad) in Towbin buffer (25 mM Tris, 192 mM glycine, and 20% methanol). Membranes were blocked in 5% milk in PBS containing 0.1% Tween 20 at room temperature for 1 h. Membranes were then incubated with primary antibodies (1:1,000 dilution in 5% milk in PBS containing 0.1% Tween 20) overnight at 4°C or for 3 h at room temperature, washed three times in PBS containing 0.1% Tween 20 for 5 min, and then incubated with secondary antibodies (1:7,000 dilution), as indicated, for 1 h at room temperature. Thereafter, blots were washed three times with PBS containing 0.1% Tween 20 and twice in PBS and developed by using the Licor Odyssey CLx system.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under award number R15NS095317 (to C.S.B.) and by the TWU Research Enhancement Program (to C.S.B.).
We thank Sarah Beisner, Joel Lukose, and Jacy Haynes for their technical assistance.
All authors have full access to the data in this study and take responsibility for the integrity of the data and accuracy of analysis. Y.A.T.K. and C.S.B. were responsible for the study design and concept. Y.A.T.K., S.A., A.L., N.L., and C.S.B. performed data acquisition. Y.A.T.K., S.A., A.L., N.L., and C.S.B. performed data analysis and interpretation. Y.A.T.K., S.A., and C.S.B. drafted the manuscript.
We declare no conflicts of interests.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00243-18.
REFERENCES
- 1.Aulas A, Vande Velde C. 2015. Alterations in stress granule dynamics driven by TDP-43 and FUS: a link to pathological inclusions in ALS? Front Cell Neurosci 9:423. doi: 10.3389/fncel.2015.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ling SC, Polymenidou M, Cleveland DW. 2013. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan Q, Yalamanchili HK, Park J, De Maio A, Lu HC, Wan YW, White JJ, Bondar VV, Sayegh LS, Liu X, Gao Y, Sillitoe RV, Orr HT, Liu Z, Zoghbi HY. 2016. Extensive cryptic splicing upon loss of RBM17 and TDP43 in neurodegeneration models. Hum Mol Genet 25:5083–5093. doi: 10.1093/hmg/ddw337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. 1995. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 69:3584–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li YR, King OD, Shorter J, Gitler AD. 2013. Stress granules as crucibles of ALS pathogenesis. J Cell Biol 201:361–372. doi: 10.1083/jcb.201302044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu LS, Cheng WC, Hou SC, Yan YT, Jiang ST, Shen CK. 2010. TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis 48:56–62. doi: 10.1002/dvg.20584. [DOI] [PubMed] [Google Scholar]
- 7.Ayala YM, Misteli T, Baralle FE. 2008. TDP-43 regulates retinoblastoma protein phosphorylation through the repression of cyclin-dependent kinase 6 expression. Proc Natl Acad Sci U S A 105:3785–3789. doi: 10.1073/pnas.0800546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bose JK, Wang IF, Hung L, Tarn WY, Shen CK. 2008. TDP-43 overexpression enhances exon 7 inclusion during the survival of motor neuron pre-mRNA splicing. J Biol Chem 283:28852–28859. doi: 10.1074/jbc.M805376200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iguchi Y, Katsuno M, Niwa J, Yamada S, Sone J, Waza M, Adachi H, Tanaka F, Nagata K, Arimura N, Watanabe T, Kaibuchi K, Sobue G. 2009. TDP-43 depletion induces neuronal cell damage through dysregulation of Rho family GTPases. J Biol Chem 284:22059–22066. doi: 10.1074/jbc.M109.012195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ling JP, Pletnikova O, Troncoso JC, Wong PC. 2015. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349:650–655. doi: 10.1126/science.aab0983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amlie-Wolf A, Ryvkin P, Tong R, Dragomir I, Suh E, Xu Y, Van Deerlin VM, Gregory BD, Kwong LK, Trojanowski JQ, Lee VM, Wang LS, Lee EB. 2015. Transcriptomic changes due to cytoplasmic TDP-43 expression reveal dysregulation of histone transcripts and nuclear chromatin. PLoS One 10:e0141836. doi: 10.1371/journal.pone.0141836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fiesel FC, Schurr C, Weber SS, Kahle PJ. 2011. TDP-43 knockdown impairs neurite outgrowth dependent on its target histone deacetylase 6. Mol Neurodegener 6:64. doi: 10.1186/1750-1326-6-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nehls J, Koppensteiner H, Brack-Werner R, Floss T, Schindler M. 2014. HIV-1 replication in human immune cells is independent of TAR DNA binding protein 43 (TDP-43) expression. PLoS One 9:e105478. doi: 10.1371/journal.pone.0105478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ayala YM, Zago P, D'Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE. 2008. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci 121:3778–3785. doi: 10.1242/jcs.038950. [DOI] [PubMed] [Google Scholar]
- 15.Lukavsky PJ, Daujotyte D, Tollervey JR, Ule J, Stuani C, Buratti E, Baralle FE, Damberger FF, Allain FH. 2013. Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat Struct Mol Biol 20:1443–1449. doi: 10.1038/nsmb.2698. [DOI] [PubMed] [Google Scholar]
- 16.D'Ambrogio A, Buratti E, Stuani C, Guarnaccia C, Romano M, Ayala YM, Baralle FE. 2009. Functional mapping of the interaction between TDP-43 and hnRNP A2 in vivo. Nucleic Acids Res 37:4116–4126. doi: 10.1093/nar/gkp342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buratti E, Baralle FE. 2012. TDP-43: gumming up neurons through protein-protein and protein-RNA interactions. Trends Biochem Sci 37:237–247. doi: 10.1016/j.tibs.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 18.Rabdano SO, Izmailov SA, Luzik DA, Groves A, Podkorytov IS, Skrynnikov NR. 2017. Onset of disorder and protein aggregation due to oxidation-induced intermolecular disulfide bonds: case study of RRM2 domain from TDP-43. Sci Rep 7:11161. doi: 10.1038/s41598-017-10574-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang CK, Chiang MH, Toh EK, Chang CF, Huang TH. 2013. Molecular mechanism of oxidation-induced TDP-43 RRM1 aggregation and loss of function. FEBS Lett 587:575–582. doi: 10.1016/j.febslet.2013.01.038. [DOI] [PubMed] [Google Scholar]
- 20.Tadic V, Prell T, Lautenschlaeger J, Grosskreutz J. 2014. The ER mitochondria calcium cycle and ER stress response as therapeutic targets in amyotrophic lateral sclerosis. Front Cell Neurosci 8:147. doi: 10.3389/fncel.2014.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Q, Yokoshi M, Okada H, Kawahara Y. 2015. The cleavage pattern of TDP-43 determines its rate of clearance and cytotoxicity. Nat Commun 6:6183. doi: 10.1038/ncomms7183. [DOI] [PubMed] [Google Scholar]
- 22.Xu G, Stevens SM Jr, Kobeissy F, Brown H, McClung S, Gold MS, Borchelt DR. 2012. Identification of proteins sensitive to thermal stress in human neuroblastoma and glioma cell lines. PLoS One 7:e49021. doi: 10.1371/journal.pone.0049021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitamura A, Nakayama Y, Shibasaki A, Taki A, Yuno S, Takeda K, Yahara M, Tanabe N, Kinjo M. 2016. Interaction of RNA with a C-terminal fragment of the amyotrophic lateral sclerosis-associated TDP43 reduces cytotoxicity. Sci Rep 6:19230. doi: 10.1038/srep19230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cohen TJ, Hwang AW, Restrepo CR, Yuan CX, Trojanowski JQ, Lee VM. 2015. An acetylation switch controls TDP-43 function and aggregation propensity. Nat Commun 6:5845. doi: 10.1038/ncomms6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. 2006. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 26.Budini M, Buratti E, Stuani C, Guarnaccia C, Romano V, De Conti L, Baralle FE. 2012. Cellular model of TAR DNA-binding protein 43 (TDP-43) aggregation based on its C-terminal Gln/Asn-rich region. J Biol Chem 287:7512–7525. doi: 10.1074/jbc.M111.288720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Romano M, Feiguin F, Buratti E. 2012. Drosophila answers to TDP-43 proteinopathies. J Amino Acids 2012:356081. doi: 10.1155/2012/356081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kametani F, Obi T, Shishido T, Akatsu H, Murayama S, Saito Y, Yoshida M, Hasegawa M. 2016. Mass spectrometric analysis of accumulated TDP-43 in amyotrophic lateral sclerosis brains. Sci Rep 6:23281. doi: 10.1038/srep23281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nonaka T, Arai T, Buratti E, Baralle FE, Akiyama H, Hasegawa M. 2009. Phosphorylated and ubiquitinated TDP-43 pathological inclusions in ALS and FTLD-U are recapitulated in SH-SY5Y cells. FEBS Lett 583:394–400. doi: 10.1016/j.febslet.2008.12.031. [DOI] [PubMed] [Google Scholar]
- 30.Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, Lin WL, Tong J, Castanedes-Casey M, Ash P, Gass J, Rangachari V, Buratti E, Baralle F, Golde TE, Dickson DW, Petrucelli L. 2009. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A 106:7607–7612. doi: 10.1073/pnas.0900688106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pesiridis GS, Lee VM, Trojanowski JQ. 2009. Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum Mol Genet 18:R156–R162. doi: 10.1093/hmg/ddp303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mackenzie IR, Rademakers R, Neumann M. 2010. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9:995–1007. doi: 10.1016/S1474-4422(10)70195-2. [DOI] [PubMed] [Google Scholar]
- 33.Geser F, Lee VM, Trojanowski JQ. 2010. Amyotrophic lateral sclerosis and frontotemporal lobar degeneration: a spectrum of TDP-43 proteinopathies. Neuropathology 30:103–112. doi: 10.1111/j.1440-1789.2009.01091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caccamo A, Majumder S, Oddo S. 2012. Cognitive decline typical of frontotemporal lobar degeneration in transgenic mice expressing the 25-kDa C-terminal fragment of TDP-43. Am J Pathol 180:293–302. doi: 10.1016/j.ajpath.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M, Clark CM, Elman LB, Miller BL, Grossman M, McCluskey LF, Trojanowski JQ, Lee VM. 2008. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol 173:182–194. doi: 10.2353/ajpath.2008.080003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dormann D, Capell A, Carlson AM, Shankaran SS, Rodde R, Neumann M, Kremmer E, Matsuwaki T, Yamanouchi K, Nishihara M, Haass C. 2009. Proteolytic processing of TAR DNA binding protein-43 by caspases produces C-terminal fragments with disease defining properties independent of progranulin. J Neurochem 110:1082–1094. doi: 10.1111/j.1471-4159.2009.06211.x. [DOI] [PubMed] [Google Scholar]
- 37.Nonaka T, Kametani F, Arai T, Akiyama H, Hasegawa M. 2009. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum Mol Genet 18:3353–3364. doi: 10.1093/hmg/ddp275. [DOI] [PubMed] [Google Scholar]
- 38.Yang C, Tan W, Whittle C, Qiu L, Cao L, Akbarian S, Xu Z. 2010. The C-terminal TDP-43 fragments have a high aggregation propensity and harm neurons by a dominant-negative mechanism. PLoS One 5:e15878. doi: 10.1371/journal.pone.0015878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scotter EL, Vance C, Nishimura AL, Lee YB, Chen HJ, Urwin H, Sardone V, Mitchell JC, Rogelj B, Rubinsztein DC, Shaw CE. 2014. Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J Cell Sci 127:1263–1278. doi: 10.1242/jcs.140087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shintani T, Klionsky DJ. 2004. Autophagy in health and disease: a double-edged sword. Science 306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klionsky DJ, Emr SD. 2000. Autophagy as a regulated pathway of cellular degradation. Science 290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park C, Cuervo AM. 2013. Selective autophagy: talking with the UPS. Cell Biochem Biophys 67:3–13. doi: 10.1007/s12013-013-9623-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. 2014. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205:143–153. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamano K, Youle RJ. 2013. PINK1 is degraded through the N-end rule pathway. Autophagy 9:1758–1769. doi: 10.4161/auto.24633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haller M, Hock AK, Giampazolias E, Oberst A, Green DR, Debnath J, Ryan KM, Vousden KH, Tait SW. 2014. Ubiquitination and proteasomal degradation of ATG12 regulates its proapoptotic activity. Autophagy 10:2269–2278. doi: 10.4161/15548627.2014.981914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kageyama S, Sou YS, Uemura T, Kametaka S, Saito T, Ishimura R, Kouno T, Bedford L, Mayer RJ, Lee MS, Yamamoto M, Waguri S, Tanaka K, Komatsu M. 2014. Proteasome dysfunction activates autophagy and the Keap1-Nrf2 pathway. J Biol Chem 289:24944–24955. doi: 10.1074/jbc.M114.580357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Korolchuk VI, Menzies FM, Rubinsztein DC. 2010. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 584:1393–1398. doi: 10.1016/j.febslet.2009.12.047. [DOI] [PubMed] [Google Scholar]
- 48.Harris H, Rubinsztein DC. 2012. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol 8:108–117. doi: 10.1038/nrneurol.2011.200. [DOI] [PubMed] [Google Scholar]
- 49.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. 2006. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 50.Douglas PM, Dillin A. 2010. Protein homeostasis and aging in neurodegeneration. J Cell Biol 190:719–729. doi: 10.1083/jcb.201005144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kourtis N, Tavernarakis N. 2011. Cellular stress response pathways and ageing: intricate molecular relationships. EMBO J 30:2520–2531. doi: 10.1038/emboj.2011.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoppe T. 2010. Life and destruction: ubiquitin-mediated proteolysis in aging and longevity. F1000 Biol Rep 2:79. doi: 10.3410/B2-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rubinsztein DC. 2006. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443:780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 54.Tashiro Y, Urushitani M, Inoue H, Koike M, Uchiyama Y, Komatsu M, Tanaka K, Yamazaki M, Abe M, Misawa H, Sakimura K, Ito H, Takahashi R. 2012. Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J Biol Chem 287:42984–42994. doi: 10.1074/jbc.M112.417600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Varshavsky A. 2011. The N-end rule pathway and regulation by proteolysis. Protein Sci 20:1298–1345. doi: 10.1002/pro.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tasaki T, Sriram SM, Park KS, Kwon YT. 2012. The N-end rule pathway. Annu Rev Biochem 81:261–289. doi: 10.1146/annurev-biochem-051710-093308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gibbs DJ, Bacardit J, Bachmair A, Holdsworth MJ. 2014. The eukaryotic N-end rule pathway: conserved mechanisms and diverse functions. Trends Cell Biol 24:603–611. doi: 10.1016/j.tcb.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 58.Kim HK, Kim RR, Oh JH, Cho H, Varshavsky A, Hwang CS. 2014. The N-terminal methionine of cellular proteins as a degradation signal. Cell 156:158–169. doi: 10.1016/j.cell.2013.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grigoryev S, Stewart AE, Kwon YT, Arfin SM, Bradshaw RA, Jenkins NA, Copeland NG, Varshavsky A. 1996. A mouse amidase specific for N-terminal asparagine. The gene, the enzyme, and their function in the N-end rule pathway. J Biol Chem 271:28521–28532. doi: 10.1074/jbc.271.45.28521. [DOI] [PubMed] [Google Scholar]
- 60.Wang H, Piatkov KI, Brower CS, Varshavsky A. 2009. Glutamine-specific N-terminal amidase, a component of the N-end rule pathway. Mol Cell 34:686–695. doi: 10.1016/j.molcel.2009.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kwon YT, Kashina AS, Davydov IV, Hu R-G, An JY, Seo JW, Du F, Varshavsky A. 2002. An essential role of N-terminal arginylation in cardiovascular development. Science 297:96–99. doi: 10.1126/science.1069531. [DOI] [PubMed] [Google Scholar]
- 62.Hu RG, Sheng J, Qi X, Xu Z, Takahashi TT, Varshavsky A. 2005. The N-end rule pathway as a nitric oxide sensor controlling the levels of multiple regulators. Nature 437:981–986. doi: 10.1038/nature04027. [DOI] [PubMed] [Google Scholar]
- 63.Hu RG, Brower CS, Wang H, Davydov IV, Sheng J, Zhou J, Kwon YT, Varshavsky A. 2006. Arginyltransferase, its specificity, putative substrates, bidirectional promoter, and splicing-derived isoforms. J Biol Chem 281:32559–32573. doi: 10.1074/jbc.M604355200. [DOI] [PubMed] [Google Scholar]
- 64.Bongiovanni G, Fidelio GD, Barra HS, Hallak ME. 1995. The post-translational incorporation of arginine into a beta-amyloid peptide increases the probability of alpha-helix formation. Neuroreport 7:326–328. doi: 10.1097/00001756-199512290-00078. [DOI] [PubMed] [Google Scholar]
- 65.Brower CS, Piatkov KI, Varshavsky A. 2013. Neurodegeneration-associated protein fragments as short-lived substrates of the N-end rule pathway. Mol Cell 50:161–171. doi: 10.1016/j.molcel.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brower CS, Varshavsky A. 2009. Ablation of arginylation in the mouse N-end rule pathway: loss of fat, higher metabolic rate, damaged spermatogenesis, and neurological perturbations. PLoS One 4:e7757. doi: 10.1371/journal.pone.0007757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Galiano MR, Goitea VE, Hallak ME. 2016. Post-translational protein arginylation in the normal nervous system and in neurodegeneration. J Neurochem 138:506–517. doi: 10.1111/jnc.13708. [DOI] [PubMed] [Google Scholar]
- 68.Wang J, Han X, Leu NA, Sterling S, Kurosaka S, Fina M, Lee VM, Dong DW, Yates JR III, Kashina A. 2017. Protein arginylation targets alpha synuclein, facilitates normal brain health, and prevents neurodegeneration. Sci Rep 7:11323. doi: 10.1038/s41598-017-11713-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee EB, Lee VM, Trojanowski JQ. 2012. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci 13:38–50. doi: 10.1038/nrn3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Igaz LM, Kwong LK, Chen-Plotkin A, Winton MJ, Unger TL, Xu Y, Neumann M, Trojanowski JQ, Lee VM. 2009. Expression of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 proteinopathies. J Biol Chem 284:8516–8524. doi: 10.1074/jbc.M809462200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Varshavsky A. 2005. Ubiquitin fusion technique and related methods. Methods Enzymol 399:777–799. doi: 10.1016/S0076-6879(05)99051-4. [DOI] [PubMed] [Google Scholar]
- 72.Igaz LM, Kwong LK, Lee EB, Chen-Plotkin A, Swanson E, Unger T, Malunda J, Xu Y, Winton MJ, Trojanowski JQ, Lee VM. 2011. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J Clin Invest 121:726–738. doi: 10.1172/JCI44867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ayala YM, De Conti L, Avendano-Vazquez SE, Dhir A, Romano M, D'Ambrogio A, Tollervey J, Ule J, Baralle M, Buratti E, Baralle FE. 2011. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J 30:277–288. doi: 10.1038/emboj.2010.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wegorzewska I, Baloh RH. 2011. TDP-43-based animal models of neurodegeneration: new insights into ALS pathology and pathophysiology. Neurodegener Dis 8:262–274. doi: 10.1159/000321547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Braak H, Ludolph A, Thal DR, Del Tredici K. 2010. Amyotrophic lateral sclerosis: dash-like accumulation of phosphorylated TDP-43 in somatodendritic and axonal compartments of somatomotor neurons of the lower brainstem and spinal cord. Acta Neuropathol 120:67–74. doi: 10.1007/s00401-010-0683-0. [DOI] [PubMed] [Google Scholar]
- 76.Li HY, Yeh PA, Chiu HC, Tang CY, Tu BP. 2011. Hyperphosphorylation as a defense mechanism to reduce TDP-43 aggregation. PLoS One 6:e23075. doi: 10.1371/journal.pone.0023075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mori F, Tanji K, Zhang HX, Nishihira Y, Tan CF, Takahashi H, Wakabayashi K. 2008. Maturation process of TDP-43-positive neuronal cytoplasmic inclusions in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol 116:193–203. doi: 10.1007/s00401-008-0396-9. [DOI] [PubMed] [Google Scholar]
- 78.Hebron ML, Lonskaya I, Sharpe K, Weerasinghe PP, Algarzae NK, Shekoyan AR, Moussa CE. 2013. Parkin ubiquitinates Tar-DNA binding protein-43 (TDP-43) and promotes its cytosolic accumulation via interaction with histone deacetylase 6 (HDAC6). J Biol Chem 288:4103–4115. doi: 10.1074/jbc.M112.419945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Uechi H, Kuranaga E, Iriki T, Takano K, Hirayama S, Miura M, Hamazaki J, Murata S. 2018. Ubiquitin-binding protein CG5445 suppresses aggregation and cytotoxicity of amyotrophic lateral sclerosis-linked TDP-43 in Drosophila. Mol Cell Biol 38:e00195-17. doi: 10.1128/MCB.00195-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Uchida T, Tamaki Y, Ayaki T, Shodai A, Kaji S, Morimura T, Banno Y, Nishitsuji K, Sakashita N, Maki T, Yamashita H, Ito H, Takahashi R, Urushitani M. 2016. CUL2-mediated clearance of misfolded TDP-43 is paradoxically affected by VHL in oligodendrocytes in ALS. Sci Rep 6:19118. doi: 10.1038/srep19118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Guenther EL, Ge P, Trinh H, Sawaya MR, Cascio D, Boyer DR, Gonen T, Zhou ZH, Eisenberg DS. 12 March 2018. Atomic-level evidence for packing and positional amyloid polymorphism by segment from TDP-43 RRM2. Nat Struct Mol Biol doi: 10.1038/s41594-018-0045-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ausubel FM, Brent R, Kingston RE, Moore DD, Smith JA, Seidman JG, Struhl K (ed). 2010. Current protocols in molecular biology. Wiley-Interscience, New York, NY. [Google Scholar]
- 83.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.