

Abstract
Here, we report how the stability of polyion complex (PIC) particles containing Pseudomonas aeruginosa’s elastase (LasB) degradable peptides and antimicrobial poly(ethylene imine) is significantly improved by careful design of the peptide component. Three LasB‐degradable peptides are reported herein, all of them carrying the LasB‐degradable sequence −GLA− and for which the number of anionic amino acids and cysteine units per peptide were systematically varied. Our results suggest that while net charge and potential to cross‐link via disulfide bond formation do not have a predictable effect on the ability of LasB to degrade these peptides, a significant effect of these two parameters on particle preparation and stability is observed. A range of techniques has been used to characterize these new materials and demonstrates that increasing the charge and cross‐linking potential of the peptides results in PIC particles with better stability in physiological conditions and upon storage. These results highlight the importance of molecular design for the preparation of PIC particles and should underpin the future development of these materials for responsive drug delivery.
Keywords: Nanoparticles, Enzymes, Polyelectrolytes, Self-assembly, Drug delivery
Introduction
Polyion complex (PIC) (nano)particles, also known as polyelectrolyte complexes (PECs)1 or interpolyelectrolyte complexes (IPECs),2 are soft colloids obtained from the self‐assembly of oppositely charged polyelectrolytes in solution.3 These nanomaterials are attractive vehicles for the delivery of charged drugs such as antineoplastics,4, 5, 6, 7 antimicrobials8, 9, 10, 11, 12, 13, 14 and nucleic acids,15 which can be complexed with oppositely charged polymers to form PIC (nano)particles that reduce the toxicity and/or control the activity of these drugs. Given the key role of enzymes in many diseases and their remarkable specificity,16 PIC (nano)particles prepared from polyelectrolytes that degrade in the presence of these enzymes have a great potential for biomedical applications. This approach is particularly interesting to tackle infections, since many pathogens secrete enzymes (e. g. proteases) to overcome the host defenses.17 By incorporating enzyme‐responsive components into the PIC (nano)particle, the release of the antimicrobial can be localized to the surroundings of the pathogenic organism, leading to improved therapeutic profiles.
With these principles in mind, we have recently reported the preparation of enzyme‐responsive PIC nanoparticles for the targeted delivery of antimicrobial branched poly(ethylene imine) (B‐PEI), which was released upon exposure to Pseudomonas aeruginosa’s elastase LasB.18 To this end, anionic peptide P1SH (Figure 1) was designed, that contained the amino acid sequence glycine‐leucine‐alanine (−GLA−), which is hydrolyzed by LasB. This sequence was inserted in between two glutamic acids (E), responsible for the electrostatic interaction with B‐PEI, and two cysteines (C), to cross‐link via disulfide formation once the nanoparticle is formed. The reported PIC nanoparticles showed excellent potential for the targeted delivery of B‐PEI to P. aeruginosa, being specifically degraded by the bacterial elastase and displaying a LasB‐specific activity against P. aeruginosa. However, the overall antimicrobial activity of the particles was low, with only 20% of the activity of free B‐PEI recovered in the presence of P. aeruginosa. Due to the low multivalency of peptide P1SH, only a small range of formulations resulted in the formation of stable PIC nanoparticles, thus compromising optimization of the delivery system.
Figure 1.
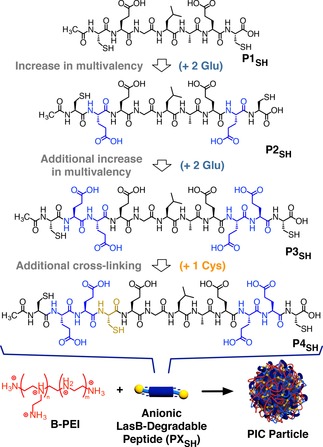
Structures of the LasB‐degradable peptides evaluated in this work and their self‐assembly with branched poly(ethylene imine) (B‐PEI) to form antimicrobial PIC particles.
In this article, we present our efforts to improve the stability of these LasB‐responsive PIC particles by optimizing the structure of the peptide component. Three new LasB‐responsive peptides were prepared with increasing number of anionic residues and cross‐linking groups (Figure 1, P2SH‐P4SH). The hydrolysis of peptides P2SH‐P4SH by LasB was evaluated and compared to that caused by a model human elastase. While the extent and specificity of hydrolysis was influenced by their amino acid sequence, no clear correlation between multivalency and susceptibility to LasB was observed. When mixed with the antimicrobial B‐PEI, all these peptides were able to give PIC particles across a wide range of formulations. Representative PIC particles were characterized using a range of light scattering techniques, suggesting that no significant structural differences could be observed between nanoparticles. Finally, the stability of these nanoparticles under simulated physiological conditions or under storage was also evaluated. Overall, particles prepared with the new peptides P2SH‐P4SH showed better stability than those obtained from P1SH, with increasing stability in simulated physiological conditions as the multivalency of the peptides was increased.
Results and Discussion
Peptide Design and Synthesis
Based on the structure of LasB‐responsive peptide P1SH (Ac‐C‐E‐GLA‐E‐C‐OH), previously reported by our group,18 two modifications were proposed to enhance the saline stability of the resulting PIC nanoparticles: i) Additional glutamic acids were introduced to increase the number of anionic residues (Figure 1, P2SH and P3SH), potentially leading to a stronger affinity for antimicrobial B‐PEI; and ii) the incorporation of a third cysteine (Figure 1, P4SH), which is expected to increase the cross‐linking density via disulfide formation once the PIC nanoparticles are formed. Both charge density and cross‐linking density have been reported as factors contributing to the stability of this type of nanoparticles.3 Overall, the peptides prepared included either two additional glutamic acids (P2SH), four additional glutamic acids (P3SH), or four additional glutamic acids and an extra cysteine (P4SH) compared to the parent peptide P1SH. All peptides were synthesized by solid‐phase chemistry in good to excellent yields and high purity without any chromatographic purification (see SI: Figures S1–S3† for experimental details and characterization).
Enzymatic Degradation of Peptides
Next, the degradation of peptides P1SH–P4SH by P. aeruginosa’s elastase (LasB) was evaluated. This experiment involved quantification of the number of primary amines formed as a result of peptide hydrolysis, using the fluorescent reporter fluorescamine.19 P. aeruginosa’s LasB hydrolyses peptides containing the −GLA− sequence between the glycine and leucine residues.20 In these experiments, the fluorescent intensity of all fluorescamine adducts (Figure S4) was compared to that observed in the presence of H2N‐LAE‐OH (P5). P5’s sequence should be formed following LasB‐mediated hydrolysis of the −GLAE− sequence, present in all the peptides reported. Thus, 100% hydrolysis was assigned to the fluorescent intensity of the fluorescamine adduct of P5 and all other intensities reported as a percentage of this one (Figure 2). The degradation of peptides P1SH–P4SH in the presence of Human Leukocyte Elastase (HLE), a protease released by white blood cells during P. aeruginosa infections,21 was also evaluated. This comparison allowed us to assess if these peptides were selectively hydrolyzed by the bacterial elastase over a relevant human enzyme.
Figure 2.
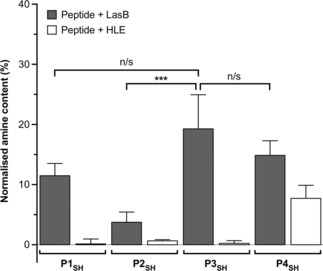
Relative amine content in samples of LasB responsive anionic peptides evaluated in this work. Relative amine content was calculated from fluorescamine conjugates formed following incubation with enzymes for 4 hours and normalized to the fluorescence observed with a model degradation peptide (P5, H2N‐LA‐E‐OH) (Figure S4). n=3, mean values± standard deviation. One‐way ANOVA, followed by Tukey's test (CI=95%,) was used to test for significance. Statistical significance was determined ‘n/s’=not significant, *** p<0.001.
All peptides were hydrolyzed by LasB, in agreement with our previous work that demonstrated that the addition of extra amino acids around the −GLA− tripeptide did not seriously compromise the activity of LasB.18 We anticipated that increasing the number of glutamic acids could have a detrimental effect on the activity of LasB, due to the potential sequestration of Ca2+ by carboxylic acids. Ca2+ is a LasB cofactor and polycarboxylated compounds, in our case ethylenediaminetetraacetic acid (EDTA), are commonly used to quench the activity of the enzyme. However, no obvious trend was observed for our peptides. P1SH, P3SH and P4SH displayed the highest susceptibility to LasB hydrolysis, with no statistical difference between them (Figure 2). Interestingly, P2SH was the least active of the peptides tested, despite having an intermediate number of glutamic acid residues. The potential of these peptides to adopt different conformations in solution that could explain this difference in activity was evaluated using circular dichroism (CD). However, CD suggested that all peptides had a random/extended conformation in solution without any significant difference across the collection (Figure S5A).22 Finally, introducing an additional cysteine did not have a big effect on the activity of LasB, with the number of amines obtained from the hydrolysis of P4SH only 5% smaller than those obtained from P3SH. However, this extra cysteine had a significant effect on the specificity of the peptide with P4SH being the only peptide significantly degraded by HLE (ca. 50% of the hydrolysis observed with LasB, Figure 2). This loss of specificity is in agreement with the ability of HLE to cleave at cysteine residues,23,24 while LasB's activity is moderately inhibited by thiols and cysteine residues.25, 26, 27
Self‐assembly and Characterization of PIC Particles
Having established that all peptides were hydrolyzed by LasB, we then explored their suitability for the preparation of B‐PEI containing PIC nanoparticles. These nanoparticles were prepared by mixing peptides P1SH–P4SH with B‐PEI in aqueous medium at physiological pH as previously reported for P1SH.18 The colloidal stability of PIC particles is highly dependent on their charge ratio (i. e. the relative number of positive and negative charges mixed in the formulation). Therefore, several formulations were explored for each peptide, where the relative number of cationic amines in B‐PEI versus anionic carboxylic acids in peptides P1SH–P4SH (N : COOH ratio) was systematically varied (Figure 3, Table S1). From their ζ‐potentials, formulations of these LasB‐responsive peptides could be split into 3 groups: a) Formulations resulting in the formation of cationic nanoparticles: While this was the case for all of the formulations made from P1SH (Figure 3, top), only those formulations with the smallest amounts of P2SH–P4SH peptides resulted in particles with a positively charged corona (Figure 3, 0.4 and 0.3 N : COOH ratios). b) Formulations that yielded negatively charged nanoparticles: We had previously reported that P1SH was unable to form negatively charged particles (Figure 3, top),18 possibly because of its small size and multivalency. We were therefore very pleased to see that increasing the multivalency for P2SH–P4SH, resulted in particles with a negatively charged corona for a broad range of formulations (Figure 3). This was particularly the case for P3SH and P4SH, the two peptides with the highest multivalency of the ones reported, which yielded negatively charged particles for formulations with N : COOH ratios ≥0.6. c) A region where no PIC nanoparticles formed in agreement with the mechanism of nucleation for this type of supramolecular aggregates.2,3,28 When mixed, polyelectrolytes will interact to form a neutral core made of a stoichiometric mixture of oppositely charged polyelectrolytes, surrounded by a corona of whichever polyion is present in excess. Neutral complexes that lack a charged stabilizing corona will not be colloidally stable and flocculation of particles will occur. While this phenomenon was observed at 1 : 1 N : COOH ratios or above for P1SH,18 unstable particles were formed at a 1 : 0.5 N : COOH ratio for all the new peptides. Interestingly, P2SH showed the smallest range of formulation that yielded colloidally stable particles (Figure 3), suggesting again that this peptide may adopt a different conformation in solution. The deviation from the theoretical neutral point at N : COOH=1 : 1 towards B‐PEI‐rich mixtures had been previously described for other B‐PEI‐containing PIC nanoparticles,29 and it is likely a result of the incomplete protonation of all amines in B‐PEI due to Coulombic interactions between neighboring ammonium groups.30 Similarly, since peptides with higher multivalencies should have stronger affinities for B‐PEI, the exchange of the peptides with higher multivalency (i. e. P3SH and P4SH) between PIC nanoparticles and the solution should be slower than for the smaller peptides, thus trapping colloidally stable intermediates even at N : COOH ratios that should favor the formation of neutral PIC nanoparticles. Additionally, P3SH and P4SH showed no significant difference in the size or charge of any of the resulting PIC nanoparticles (Figure 3), suggesting that multivalency is the predominant factor that governs the self‐assembly of this collection of peptides. Regardless of the peptide used, no PIC nanoparticles could be detected at a 1 : 0.2 N : COOH ratio, probably as a result of the incomplete complexation of polyelectrolytes at this low concentration of peptide. The polydispersity indices (PDIs) of all PIC nanoparticles ranged between 0.01–0.06 with the exception of the formulations prepared at a 1 : 0.3 N : COOH ratio, which displayed significantly higher PDI values of up to 0.17 (Table S1). It was clear from dynamic light scattering (DLS) studies that formulations with lower peptide contents led to broader size distributions, although this phenomenon was less pronounced as the net charge of the peptide increased (Figure S6).
Figure 3.
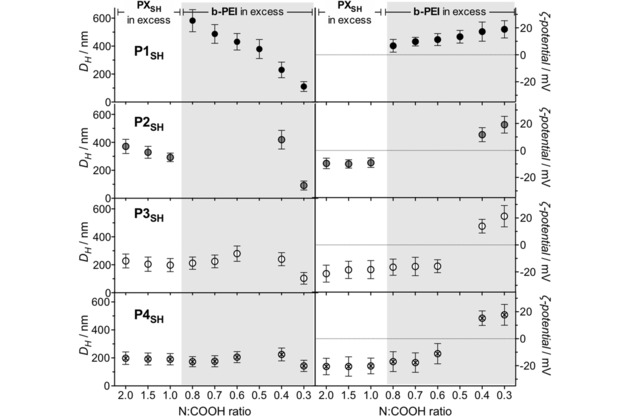
Hydrodynamic diameter (DH, left) and ζ‐potential (right) of PIC nanoparticles prepared from peptides P1SH–P4SH at different N : COOH ratios. Each value represents the mean size and charge of the only population fitted by the software for each sample ± its standard deviation. Empty spaces indicate formulations that did not form PIC particles. Further details can be found in Table S1. Results obtained directly after the assembly of the nanoparticles without prior filtration. Data for P1SH reproduced from I. Insua, E. Liamas, Z. Zhang, A. F. Peacock, A. M. Krachler, F. Fernandez‐Trillo, Polym Chem 2016, 7, 2684–2690 – Published by The Royal Society of Chemistry.
Size‐wise, P2SH–P4SH formulations showed a smaller variability in size than those reported for P1SH (Figure 3). Particles with larger hydrodynamic diameters (DH) were obtained for P2SH (ca. 300–400 nm) than for P3SH and P4SH (ca. 100–300 nm), regardless of the N : COOH ratio tested (Figure 3). Nanoparticles of smaller size for these two peptides could be the result of their higher multivalency (7 COOH groups for P3SH and P4SH versus 5 for P2SH and 3 for P1SH), which would result in more strongly and tightly bound polyelectrolyte networks within these nanoparticles. For all peptides, formulations prepared at a 1 : 0.3 N : COOH ratio gave the smallest PIC nanoparticles, with an average DH of ca. 110 nm and ζ‐potential of +19 mV – in agreement with the values previously found for P1SH at this N : COOH ratio.18
Structural Characterization of PIC Particles
To further understand the impact that the multivalency and cross‐linking units of peptides P1SH–P4SH have on the structure of the PIC nanoparticles formed, these materials were characterized by Static Light Scattering (SLS). Multi‐angle SLS analysis of nanomaterials allows the calculation of their radius of gyration (Rg) by deconvolution of the Zimm equation (Eq. 1);31,32 where K is a constant containing the optical parameters, c is the concentration of the sample, R θ the Rayleigh ratio of the particles at a given scattering angle (θ), q is the scattering wave vector and MwPIC is the average molecular weight of the nanoparticles. Thus, the ratio between gyration and hydrodynamic radii (i. e. Rg/RH), calculated by SLS and DLS respectively, provides valuable information about the internal structure and shape of nanomaterials.31
| (1) |
PIC nanoparticles prepared from all four peptides at a 1 : 0.3 N : COOH ratio were selected for SLS analysis, being the most consistent formulation throughout the collection of peptides studied. The scattering intensity from a suspension of PIC nanoparticles was measured at different angles ranging from 20 to 100°, and their DLS profiles were simultaneously recorded. Unfortunately, non‐linear Zimm plots were obtained for all PIC particle samples with deviations at low angles, which made impossible the accurate measurement of MwPIC and Rg (Figure S7). Since larger particles have higher scattering contributions at low angles,32 we considered that this non‐linearity could be corrected by filtering the samples through membranes with a pore size of 0.45 μm, and thus remove any scatterers larger than the expected nanoparticles – which were all smaller than the filter's cut‐off size according to our previous DLS characterization (Figure 3). All filtered samples of PIC nanoparticles displayed linear Zimm plots that allowed the calculation of their Rg, which was then compared with the RH obtained by DLS under the same conditions (Figure 4). The average RH measured from filtered samples was 57.5 nm, which is in agreement with the DH previously obtained from unfiltered nanoparticles (ca. 110 nm), suggesting that the filtration did not affect the main composition of PIC nanoparticles in the samples.
Figure 4.
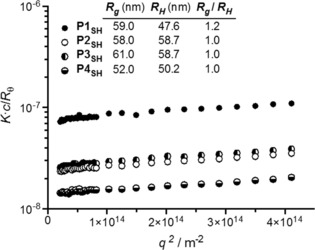
Partial Zimm plots obtained by SLS of filtered PIC particles prepared from peptides P1SH–P4SH at a 1 : 0.3 N : COOH ratio. The inset indicates the gyration (Rg) and hydrodynamic (RH) radii simultaneously calculated by SLS and DLS, respectively.
Nanoparticles prepared from peptides P2SH–P4SH consistently showed an Rg/RH value of 1.0, whereas those containing the peptide P1SH presented a slight deviation towards Rg/RH > 1, which is characteristic of anisotropic nanomaterials.31 This difference can be explained from the tendency of the latter nanoparticles to aggregate, as observed when their DH was monitored over time (Figure S8): Whereas PIC nanoparticles prepared from peptides P2SH–P4SH displayed the same size over time, the DH of the nanoparticles made from P1SH increased by 50% after 10 days, which was the time that passed between the preparation of these PIC nanoparticles and their SLS analysis. Nevertheless, the Rg/RH values found for this collection of nanoparticles are in agreement with those reported in the literature for other PIC nanoparticles (1.0–1.6).33,34 These Rg/RH ratios, which are higher than the value expected for a solid sphere (0.775),31 have been rationalized by the high polydispersity of PIC nanoparticles and their tendency to aggregate in some cases.33,35 Both of these factors act as a bias towards higher Rg/RH values, making the elucidation of the internal structure of these nanomaterials extremely difficult.
In summary, the structural characterization of these nanoparticles by SLS and DLS indicates that all formulations have very similar Rg and RH regardless of the peptide used, and therefore no distinct structural features should be expected in any of these complexes.
Stability under Physiological Conditions of PIC Particles
The electrostatic forces that keep PIC nanoparticles together can be shielded by small electrolytes, leading to swelling and ultimately breakdown of these nanoparticles.36 Hence, the integrity of PIC nanoparticles is often compromised by the concentrations of salts present in biological fluids. This lack of stability was the case for our previously reported nanoparticles, for which only one of the formulations was stable under simulated physiological conditions (Figure 5 and Figure S9, P1SH, 0.3 N : COOH ratio). The tolerance to physiological conditions of the new nanoparticles was evaluated by incubation in the presence of 154 mM NaCl at 37 °C, and the change of DH was monitored over four hours (Figure 5).
Figure 5.
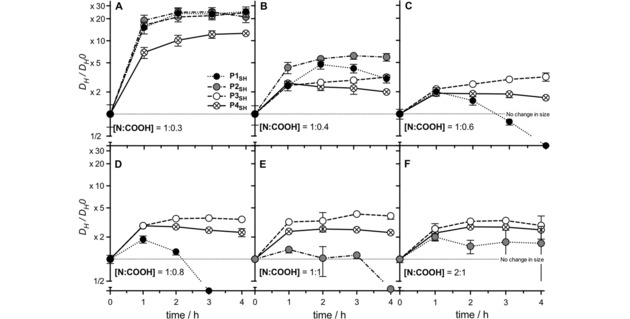
Relative change in size (DH/DH0) for PIC particles prepared from peptides P1SH–P4SH at representative N : COOH ratios under simulated physiological conditions (37 °C, 154 mM NaCl, pH 7.4) (A 1 : 0.3 [N : COOH] ratio, B 1 : 0.4 [N : COOH] ratio, C 1 : 0.6 [N : COOH] ratio, D 1 : 0.8 [N : COOH] ratio, E 1 : 1 [N : COOH] ratio and F 2 : 1 [N : COOH] ratio). Particle size (DH) was normalized to that of the PIC particles in the absence of NaCl (0 hours, No change in size). n=3, mean values±standard deviation. Results obtained directly after the assembly of the nanoparticles without prior filtration. Data for P1SH reproduced from I. Insua, E. Liamas, Z. Zhang, A. F. Peacock, A. M. Krachler, F. Fernandez‐Trillo, Polym Chem 2016, 7, 2684–2690 – Published by The Royal Society of Chemistry.
All PIC nanoparticles swelled when exposed to NaCl and this swelling was inversely proportional to the peptides’ multivalency and the degree of cross‐linking in the particle. As predicted, increasing the multivalency in the peptide resulted in tighter nanoparticles, that swelled less in the presence of NaCl (Figure 5). For all peptides, nanoparticles made at a 1 : 0.3 N : COOH ratio swelled the most, with particles prepared from P2SH and P3SH becoming over 20 times bigger after 4 hours of incubation (Figure 5A), a similar increase in size to that reported for P1SH. Interestingly, those prepared from P4SH, which carries an extra cysteine and should give higher cross‐linking densities at the same N : COOH ratio, were the least affected by this saline medium, regardless of the formulation employed (Figure 5, ⊗).
Overall, all the particles prepared with the new peptides P2SH–P4SH showed better physiological stabilities than those previously reported for trivalent P1SH (Figure 5, •),18 reinforcing the correlation between peptide multivalency and PIC nanoparticle stability (Figure 5 and Figure S9). Particles prepared from the least multivalent peptide P2SH showed more polydisperse aggregates after 2 and 3 hours and, for those prepared at a 1 : 1 N : COOH ratio, no particles could be detected after 4 hours of incubation under these conditions (Figure 5E, •). These results highlight the critical effect that multivalency and cross‐linking degree have on the salt tolerance of PIC particles, as evidenced by their physiological stability: P1SH<P2SH<P3SH<P4SH.
Stability Upon Freeze‐drying
Finally, we evaluated if the increase in stability observed under physiological conditions for the nanoparticles formed with the new peptides P2SH–P4SH could be correlated to an increased stability upon storage. Nanoparticles prepared at a 1 : 0.3 N : COOH ratio were selected for this evaluation, being a consistent formulation across all peptides evaluated. Also, nanoparticles prepared at this N : COOH ratio have the least amount of peptide and while stable, swelled more upon incubation in physiological conditions. This swelling may indicate that the cohesive forces in the core of these nanoparticles are weak enough to accommodate the presence of competing counterions, or a higher tendency to aggregate, making it an ideal formulation to test stability. To this end, freshly prepared nanoparticles at this N : COOH ratio were allowed to stand in a dark, cool and dry place, and their size monitored over time (Figure S8). As described before, no change in size was observed for any the nanoparticles prepared with the new peptides P2SH–P4SH, even after 10 days of storage. This was not the case for the nanoparticles prepared with P1SH which quickly increased in size.
Nanoparticles prepared from P3SH at a 0.3 N : COOH ratio, which showed excellent stability in solution, were then selected as a representative example to evaluate the potential of these formulations to be stored as a powder. Therefore, freshly prepared nanoparticles from P3SH at a 0.3 N : COOH ratio were freeze‐dried overnight to yield a white powder. Approximately 1.32 mg of powder were recovered for all 3 samples prepared (standard deviation 0.042), in close agreement with the 1.47 mg expected taking into account the amount of peptide and B‐PEI used, and how much solid content was in the buffer used to prepare the nanoparticle suspension. This powder was then reconstituted in deionized water to give the original volume (1 mL). Samples were then gently resuspended on a roller for 30 mins and characterized via DLS and ζ‐potential. A small increase in size was observed following resuspension (Figure 6A, •) in agreement with the small shift in the autocorrelation curve (Figure 6D), while no changes in the ζ‐potential of the particles were observed following resuspension on the rollers (Figure 6B, •). A bigger effect was observed on the number of counts, which decreased from 402.8±2.3 to 54.2±1.2 (Figure 6C, •) suggesting that less particles were available in suspension following reconstitution.
Figure 6.
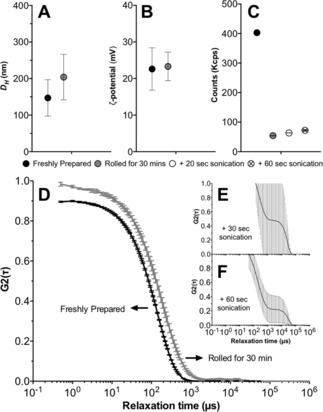
Effect of freeze‐drying and resuspension of the size (A), ζ‐potential (B) and number of counts (C) of nanoparticles prepared with P3SH at a 0.3 N : COOH ratio. Autocorrelation function curves for freshly prepared and nanoparticles following resuspension on a roller for 30 mins (D) and additional sonication for 30 sec (E) or 60 secs (F).
These samples were then sonicated for 30 or 60 seconds to try to increase the number of nanoparticles in suspension. While a small increase in the number of counts was observed (Figure 6C, ○ and ⊗ for 30 and 60 secs respectively), sonication had a detrimental effect on the nanoparticles and very noisy autocorrelation function curves were obtained (Figure 6E and F), which prevented accurate characterization of their size and ζ‐potential (Figure 6A and B).
Conclusions
Three new enzyme‐responsive anionic peptides (P2SH–P4SH) have been synthesized and their use in the preparation of PIC nanoparticles containing the antimicrobial polymer B‐PEI reported. These peptides were designed to incorporate the LasB‐degradable sequence −GLA−, and increasing amounts of glutamic acids for a stronger interaction with B‐PEI (P2SH and P3SH), and an additional cysteine (P4SH) to generate PIC nanoparticles with higher cross‐linking density. The enzymatic degradation of these peptides was assessed against bacterial (LasB) and human (HLE) elastases and, while all peptides were degraded by the bacterial elastase, no direct correlation with the multivalency of the peptides could be identified. All new peptides formed PIC nanoparticles when incubated with B‐PEI, and a broad range of formulations could be accessed by varying the ratio of polyelectrolytes (i. e. N : COOH ratio). Our results show that these new peptides allow the preparation of negatively charged nanoparticles, not previously accessible using the less multivalent peptide P1SH, previously reported by our group.18 More importantly, the stability of the new nanoparticles under simulated physiological conditions increased with increasing multivalency and cross‐linking degree of the peptides (i. e. P2SH<P3SH<P4SH) and displayed higher saline stability than those prepared using P1SH. The structural analysis of these peptides and their resulting PIC nanoparticles by CD and SLS, respectively, suggests that there are no differences in conformation or architecture across the whole collection of materials tested that could explain these differences in susceptibility against LasB and stability. Finally, the stability of representative formulations upon storage was evaluated. Our results demonstrate the importance of carefully optimizing peptide sequence and multivalency in the design of peptide‐based PIC particles and we believe these new peptides and formulations will underpin the future development of “smart” delivery systems for antimicrobials. Our efforts to identify LasB‐responsive nanoparticles with optimized release and antimicrobial activity, as well as the application of these peptides for the preparation of other delivery systems, will be reported in due course.
Experimental Section
Materials
Enzymes (Pseudomonas aeruginosa Elastase (LasB): EC 3.4.24.26 and Human Leucocyte Elastase (HLE): EC 3.4.21.37) were purchased from Merck Millipore. Branched poly(ethylene imine) 25 kDa average molecular weight (B‐PEI) and 4‐(2‐hydroxyethyl)piperazine‐1‐ethanesulfonic acid (HEPES) were bought from Sigma‐Aldrich®. Fluorescamine and dimethylsulphoxide (DMSO) were purchased from Acros OrganicsTM. Ethylenediaminetetraacetic acid (EDTA) was purchased from Alfa Aesar®. Nylon 0.45 μm syringe filters were purchased from Camlab.
Instrumentation
Dynamic Light Scattering (DLS) and ζ‐potential measurements were carried out with a Zetasizer Nano ZSP (Malvern Instruments Ltd.) stabilized at 37 °C. DLS was read at 173° (backscattering) for 60 seconds in triplicate and ζ‐potentials were averaged from 30 measurements at 140 V. DLS correlograms were processed with Malvern's General Purpose non‐negative least squares algorithm, and their DH values correspond to the mean size and standard deviation of the only population found in their size‐intensity plots. A FLUOstar Omega (BMGLabtech Gmbh) microplate reader was used to incubate and measure fluorescamine reactions. Static Light Scattering (SLS) and simultaneous DLS data was collected using an CGS‐3 Compact Goniometer System (ALV Gmbh) stabilized at 20 °C and operating at a wavelength of 633 nm against a toluene standard.
Preparation of PIC Nanoparticles
For nanoparticles prepared at a 1 : 0.3 N : COOH ratio (defined as the ratio between amines in B‐PEI and carboxylic acids in the peptides), stock solutions of B‐PEI (2.5 mM in amines) and peptide (P1SH–P4SH) (0.75 mM in carboxylate groups) in 5 mM HEPES buffer at pH 7.4 were prepared. Then, both solutions were filtered and mixed in equal volumes drop‐wise under stirring. The reaction mixture was stirred at room temperature for 24 hours open to air to allow thiol oxidation. PIC nanoparticles prepared at different N : COOH ratios were obtained by changing the concentration of peptide stock solution and mixing with the 2.5 mM B‐PEI stock following the same protocol (Table S1). After 24 hours, samples were analyzed directly by DLS and ζ‐potential without prior filtration.
Enzymatic Degradation of Peptides – Fluorescamine Assay
Stock solutions of peptide (1 mM) or succinyl casein (0.5 mg/mL) were prepared in 25 mM Na2B4O7 buffer at pH 8.0 with 10 mM CaCl2 and 10% v/v DMSO. 125 μL of these substrate solutions were added to a 96‐well black‐walled microplate and mixed with 125 μL of the same buffer without DMSO, containing 15 μg of enzyme (LasB or HLE). Solutions of enzymes and substrates alone were prepared as controls. Every sample was prepared in triplicate. The microplate was incubated at 37 °C for 4 hours under orbital shaking. After 4 hours, 50 μL of 0.1 M EDTA in water at pH 8.0 were added to each well to quench all enzymatic activity. Then, each sample was mixed in a 1 : 1 volume ratio with a 1 mM solution of fluorescamine19 in methanol. The microplate was incubated at 37 °C under orbital shaking for 30 minutes. After this time, fluorescence was measured exciting at 355 nm and reading the emission at 460±10 nm.
Stability of PIC Nanoparticles in Simulated Physiological Conditions
182 μL of a 1 M solution of NaCl in water was added to a sample of PIC nanoparticles (1 mL), prepared as described above. This mixture was then incubated at 37 °C to obtain physiological osmotic pressure and temperature. Every hour, the sample was analyzed by DLS as described above.
Freeze‐drying and Resuspension of PIC Nanoparticles
A sample (1 mL) of freshly made PIC nanoparticles from P3SH and B‐PEI at a 1 : 0.3 N : COOH ratio was frozen inside a tared 2 mL‐scintillation vial, to be then left overnight under vacuum at −80 °C. A white solid was thus obtained, which was weighed by difference inside the tared vial. Then, 1 mL of deionized water was added to the vial and the mixture was gently and simultaneously rocked and rolled for 30 min. After this time, the sample was characterized by DLS and ζ‐potential as indicated above. Replicates of this freeze‐dried and reconstituted sample were further processed by immersion in an ultrasonic bath at room temperature for 30 and 60 sec, and characterized likewise. For the measurement of light scattering intensity by DLS (i. e. counts, Figure 6C), the attenuator value of the instrument was kept constant across all measurements.
Author Contributions
All authors contributed to the experimental set‐up and discussed the results. II and MP synthesized and characterized the peptides. II and FFT designed the peptides, and the nanoparticle preparation and characterization. II, FFT and AMK designed the enzymatic assays. LDB, RK, APB and RKOR designed and performed structural characterization. II carried out all other experiments. FFT and AMK secured funding. II and FFT analyzed the data and wrote the paper, with all other authors contributing to the final version of the manuscript.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Wellcome Trust (177ISSFPP), the Engineering and Physical Sciences Research Council (EPSRC) (EP/N508755/1), the Birmingham Science City and the European Regional Development Fund, and the University of Birmingham (FFT and II, John Evans Fellowship; AMK, Birmingham Fellowship). The authors thank Dr Marie‐Christine Jones and Dr Hanene Ali‐Boucetta (School of Pharmacy, UoB) for the access to the DLS, and Nicolas Perez‐Soto for useful discussions.
I. Insua, M. Petit, L. D. Blackman, R. Keogh, A. Pitto-Barry, R. K. O'Reilly, A. F. A. Peacock, A. M. Krachler, F. Fernandez-Trillo, ChemNanoMat 2018, 4, 807.
References
- 1. Kulkarni A. D., Vanjari Y. H., Sancheti K. H., Patel H. M., Belgamwar V. S., Surana S. J., Pardeshi C. V., Artif. Cells, Nanomed., Biotechnol. 2016, 44, 1615–1625. [DOI] [PubMed] [Google Scholar]
- 2. Pergushov D. V., Müller A. H. E., Schacher F. H., Chem. Soc. Rev. 2012, 41, 6888–6901. [DOI] [PubMed] [Google Scholar]
- 3. Insua I., Wilkinson A., Fernández-Trillo F., Eur. Polym. J. 2016, 81, 198–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. How S. C., Chen Y. F., Hsieh P. L., Wang S. S. S., Jan J.-S., Colloids Surf. B 2017, 153, 244–252. [DOI] [PubMed] [Google Scholar]
- 5. Zhang L., Wang J., Ni C., Zhang Y., Shi G., Mater. Sci. Eng. C 2016, 58, 724–729. [DOI] [PubMed] [Google Scholar]
- 6. Yang L., Gao S., Asghar S., Liu G., Song J., Wang X., Ping Q., Zhang C., Xiao Y., Int. J. Biol. Macromol. 2015, 72, 1391–1401. [DOI] [PubMed] [Google Scholar]
- 7. Hsieh Y.-H., Hsiao Y.-T., Jan J.-S., Soft Matter 2014, 10, 9568–9576. [DOI] [PubMed] [Google Scholar]
- 8. Insua I., Majok S., Peacock A. F. A., Krachler A. M., Fernández-Trillo F., Eur. Polym. J. 2017, 87, 478–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Les K. A., Mohamed-Ahmed A. H. A., Balan S., Choi J.-W., Martin D., Yardley V., Powell K., Godwin A., Brocchini S. J., Brocchini S., Polym. Chem. 2014, 5, 1037–1048. [Google Scholar]
- 10. Niece K. L., Vaughan A. D., Devore D. I., J. Biomed. Mater. Res. A 2013, 101, 2548–2558. [DOI] [PubMed] [Google Scholar]
- 11. Insua I., Zizmare L., Peacock A. F. A., Krachler A. M., Fernández-Trillo F., Sci. Rep. 2017, 7, 9396–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carmona-Ribeiro A., Sampaio J., Santos H., Carrasco L., Drug Delivery Letters 2017, 7, 39–47. [Google Scholar]
- 13. Craparo E. F., Porsio B., Schillaci D., Cusimano M. G., Spigolon D., Giammona G., Cavallaro G., Nanomedicine 2017, 12, 25–42. [DOI] [PubMed] [Google Scholar]
- 14. Vehlow D., Schmidt R., Gebert A., Siebert M., Lips K. S., Müller M., Nanomaterials 2016, 6, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lächelt U., Wagner E., Chem. Rev. 2015, 115, 11043–11078. [DOI] [PubMed] [Google Scholar]
- 16. Ulijn R. V., J. Mater. Chem. 2006, 16, 2217–2225. [Google Scholar]
- 17. Kaman W. E., Hays J. P., Endtz H. P., Bikker F. J., Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1081–1087. [DOI] [PubMed] [Google Scholar]
- 18. Insua I., Liamas E., Zhang Z., Peacock A. F. A., Krachler A. M., Fernández-Trillo F., Polym. Chem. 2016, 7, 2684–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Udenfriend S., Stein S., Böhlen P., Dairman W., Leimgruber W., Weigele M., Science 1972, 178, 871–872. [DOI] [PubMed] [Google Scholar]
- 20. Morihara K., Tsuzuki H., Arch. Biochem. Biophys. 1971, 146, 291–296. [DOI] [PubMed] [Google Scholar]
- 21. Suter S., Schaad U. B., Roux L., Nydegger U. E., Waldvogel F. A., J. Infect. Dis. 1984, 149, 523–531. [DOI] [PubMed] [Google Scholar]
- 22. Greenfield N. J., Nat. Protoc. 2006, 1, 2876–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Korkmaz B., Attucci S., Moreau T., Godat E., Juliano L., Gauthier F., Am. J. Respir. Cell Mol. Biol. 2004, 30, 801–807. [DOI] [PubMed] [Google Scholar]
- 24. Korkmaz B., Attucci S., Hazouard E., Ferrandière M., Jourdan M. L., Brillard-Bourdet M., Juliano L., Gauthier F., J. Biol. Chem. 2002, 277, 39074–39081. [DOI] [PubMed] [Google Scholar]
- 25. Morihara K., Tsuzuki H., Oka T., Inoue H., Ebata M., J. Biol. Chem. 1965, 240, 3295–3304. [PubMed] [Google Scholar]
- 26. Wretlind B., Wadström T., J. Gen. Microbiol. 1977, 103, 319–327. [DOI] [PubMed] [Google Scholar]
- 27. Kessler E., Ohman D. E., in Handbook of Proteolytic Enzymes, Elsevier, 2013, pp. 582–592. [Google Scholar]
- 28. Müller M., Polyelectrolyte Complexes in the Dispersed and Solid State II, Springer, Berlin, 2013. [Google Scholar]
- 29. Sato H., Nakajima A., Polym. J. 1975, 7, 241–247. [Google Scholar]
- 30. Borkovec M., Koper G., Macromolecules 1997, 30, 2151–2158. [Google Scholar]
- 31. Patterson J. P., Robin M. P., Chassenieux C., Colombani O., O′Reilly R. K., Chem. Soc. Rev. 2014, 43, 2412–2425. [DOI] [PubMed] [Google Scholar]
- 32. Zimm B. H., J. Chem. Phys. 1948, 16, 1099–1116. [Google Scholar]
- 33. Ueno K., Ueno H., Sato T., Polym. J. 2012, 44, 59–64. [Google Scholar]
- 34. Fischer D., Dautzenberg H., Kunath K., Kissel T., Int. J. Pharm. 2004, 280, 253–269. [DOI] [PubMed] [Google Scholar]
- 35. Dautzenberg H., Kriz J., Langmuir 2003, 19, 5204–5211. [Google Scholar]
- 36. Dautzenberg H., Rother G., Macromol. Chem. Phys. 2004, 205, 114–121. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary