

Despite the increasing wealth of sequencing data, the health contributions of many bacteria found in the human gut microbiota have yet to be elucidated. This study applies a novel experimental approach to predict the ability of gut microbes to carry out a specific metabolic activity, i.e., gallate metabolism. The study showed that, while gallate-decarboxylating bacteria represented 47% of the bacterial genera detected previously in the same human fecal samples, no gallate decarboxylase homologs were identified from representatives of Bacteroidetes. The presence of functional gallate decarboxylases was demonstrated in representative Proteobacteria and Firmicutes strains from the human microbiota, an observation that could be of considerable relevance to the in vivo production of pyrogallol, a physiologically important bioactive compound.
KEYWORDS: HTS, antioxidant, human intestinal tract, microbiota, phenolic compounds, pyrogallol
ABSTRACT
The human gut microbiota contains a broad variety of bacteria that possess functional genes, with resultant metabolites that affect human physiology and therefore health. Dietary gallates are phenolic components that are present in many foods and beverages and are regarded as having health-promoting attributes. However, the potential for metabolism of these phenolic compounds by the human microbiota remains largely unknown. The emergence of high-throughput sequencing (HTS) technologies allows this issue to be addressed. In this study, HTS was used to assess the incidence of gallate-decarboxylating bacteria within the gut microbiota of healthy individuals for whom bacterial diversity was previously determined to be high. This process was facilitated by the design and application of degenerate PCR primers to amplify a region encoding the catalytic C subunit of gallate decarboxylase (LpdC) from total metagenomic DNA extracted from human fecal samples. HTS resulted in the generation of a total of 3,261,967 sequence reads and revealed that the primary gallate-decarboxylating microbial phyla in the intestinal microbiota were Firmicutes (74.6%), Proteobacteria (17.6%), and Actinobacteria (7.8%). These reads corresponded to 53 genera, i.e., 47% of the bacterial genera detected previously in these samples. Among these genera, Anaerostipes and Klebsiella accounted for the majority of reads (40%). The usefulness of the HTS-lpdC method was demonstrated by the production of pyrogallol from gallic acid, as expected for functional gallate decarboxylases, among representative strains belonging to species identified in the human gut microbiota by this method.
IMPORTANCE Despite the increasing wealth of sequencing data, the health contributions of many bacteria found in the human gut microbiota have yet to be elucidated. This study applies a novel experimental approach to predict the ability of gut microbes to carry out a specific metabolic activity, i.e., gallate metabolism. The study showed that, while gallate-decarboxylating bacteria represented 47% of the bacterial genera detected previously in the same human fecal samples, no gallate decarboxylase homologs were identified from representatives of Bacteroidetes. The presence of functional gallate decarboxylases was demonstrated in representative Proteobacteria and Firmicutes strains from the human microbiota, an observation that could be of considerable relevance to the in vivo production of pyrogallol, a physiologically important bioactive compound.
INTRODUCTION
Phenolic compounds are strong antioxidants present in plant-derived foods and beverages (1). Phenolic acids account for almost one-third of dietary phenols (2). Gallic acid is a phenolic acid that is widely distributed in edible plants and occurs in several legumes, fruits, vegetables, nuts, and beverages of plant origin (3, 4). Studies have found different properties associated with gallic acid, namely, anticarcinogenic, antimutagenic, and antioxidant properties. For these reasons, gallic acid and its esters have been used extensively as food additives (5). Although gallic acid is widely distributed in foods, it is not regarded as a preferred substrate for bacterial growth. In fact, only bacteria of the genus Pseudomonas have been reported to utilize free gallic acid as a sole carbon and energy source under aerobic conditions (6). In addition, there are microorganisms, such Lactobacillus plantarum, that nonoxidatively decarboxylate gallic acid but do not possess appropriate mechanisms to further degrade the pyrogallol produced by this dead-end pathway (7). The biochemical pathway followed by L. plantarum implies that gallate esters are hydrolyzed to gallic acid, and the gallic acid formed is subsequently decarboxylated to pyrogallol (8). This metabolic transformation implies the successive actions of esterase (tannase) and gallate decarboxylase enzymes. Generally, nonoxidative aromatic acid decarboxylases, such gallate decarboxylase, are encoded by a 3-gene operon that encodes the 3 subunits of decarboxylases (B, C, and D subunits) (9). L. plantarum is the only bacterium described in which the lpdC gene and the lpdB and lpdD genes are separated in the chromosome (7). Although LpdC is the only protein responsible for the catalytic activity in vitro, LpdB is also essential for decarboxylase activity in vivo (7). The L. plantarum gallate decarboxylase was genetically identified and characterized (7). Genes similar to L. plantarum gallate decarboxylase genes were found in other food-related lactic acid bacteria, such as Enterococcus faecium, Lactobacillus brevis, and Oenococcus oeni, among others. Moreover, the presence of decarboxylase genes was associated with gallate decarboxylation and pyrogallol production (7).
L. plantarum is one of the few species of lactic acid bacteria that has successfully adapted to food habitats and also is part of the human colonic microbiota. In the context of symbiosis with the human host, it may be that, as is the case for L. plantarum, other bacteria from the gut microbiota might have evolved to possess the biochemical pathways responsible for the bioactivation/degradation of dietary polyphenols. Therefore, the bioaccessibility and health effects of food containing gallic acid may depend on the activity of a subset of gut microbes that resist its antibacterial activity. In this scenario, novel approaches for analyzing the metabolism of dietary phenols by gut microbes must be developed. Particular benefits can now be gained from high-throughput sequencing (HTS) analysis. HTS has significantly enhanced the knowledge of the complex gut microbial ecosystem. While most such studies are based on 16S rRNA gene amplification, it is also possible to use HTS to sequence select non-16S-rRNA-based genes (10, 11). To date, however, there have not been studies of this nature to detect by HTS the presence of genes involved in the bacterial metabolism of dietary phenolic compounds in the human gut microbiota.
In this study, the presence of genes involved in gallate metabolism was screened for in healthy human gut microbiota samples. More specifically, optimized PCR primers for amplification of the C catalytic subunit of the gallate decarboxylase gene (lpdC) were designed to detect the presence of decarboxylases involved in gallate metabolism in the human gut (HTS-lpdC method). HTS analysis revealed the dominant and subdominant genera and species with potential gallate decarboxylase activities. The presence of functional gallate decarboxylase activity in the identified species demonstrated the usefulness of the proposed HTS method. In addition to achieving this specific goal, this work highlights the value of employing HTS to survey the potential metabolism of a dietary component in a complex microbial population.
RESULTS AND DISCUSSION
Distribution of gallate-utilization-associated proteins across the Lactobacillus genus.
Gallotannins are gallate esters that are present in healthy foods and beverages, such as pomegranate and teas (1). Food gallotannins are hydrolyzed to gallic acid by an esterase enzyme (tannase), and the gallic acid formed is decarboxylated to pyrogallol by the action of gallate decarboxylase (8, 12); these enzymes have recently been described for L. plantarum WCFS1 (7, 13, 14). Among lactic acid bacteria, L. plantarum is the prototypical species possessing tannase activity. However, some other lactic acid bacterial species, harboring similar lpdC genes, are also able to decarboxylate gallic acid (7).
Lactobacilli are lactic acid bacteria that are widely found in plant-derived food fermentations (15), in which gallic acid and gallotannins are present. To examine the abundance and conservation of gallate decarboxylase proteins in the genus Lactobacillus, a comparative analysis was used to identify enzymes homologous to the catalytic C subunit, LpdC. Amino acid sequences homologous to the functional LpdC from L. plantarum WCFS1 (7) were identified in type strains of 28 species in the genus Lactobacillus. Additionally, a comparative analysis to identify enzymes homologous to tannase was performed (Fig. 1). The analysis clearly revealed the extended presence and conservation of LpdC among lactobacilli, with >70% identity, in contrast to the scarcity of the tannase enzyme. These results are in agreement with previous studies in which tannase (12, 16) or gallate decarboxylase (7) activity among lactic acid bacteria was analyzed. Given that the latter study also revealed that, across the lactic acid bacteria assayed, gallate decarboxylase activity corresponded to the presence of the lpdC gene (7), we selected this gene to study the distribution and abundance of dietary gallate metabolism in the human gut microbiota.
FIG 1.

Heat map representing the distribution across the Lactobacillus genus of proteins homologous to the gallate decarboxylase C subunit (LpdC) and tannase from L. plantarum WCFS1. Gene products from the representative type strain genomes of all Lactobacillus species available online with significant homology of 70% iterative similarity over 50% of the protein length are represented in the matrix. This matrix employs a color code that represents the degree of sequence identity, from black (absent) to purple. Species are ordered by the degree of homology, and the isolation source for each strain is indicated.
Presence of gallate decarboxylase genes in the human gut microbiota revealed by HTS.
Determining the functional attributes of the microbiome is essential for understanding its role in host metabolism and disease (17). Shotgun metagenomic approaches provide direct assessment of the functional attributes of the microbiome (18, 19) but continue to be expensive. A less expensive and more targeted option to assess the distribution of the lpdC gene among human gut microbiome samples was to employ specific optimized PCR primers.
To examine the distribution of the lpdC gene among members of the human gut community, DNA previously isolated from fecal samples from elite athletes (n = 15) was used (20). These athlete samples were selected on the basis of their high levels of bacterial alpha diversity (20). The selected primers detected the presence of 237-bp amplicons, which were subjected to HTS. In total, 3,261,967 sequence reads were obtained (after quality and length filtering), representing an average of 217,464 sequences per sample. Phylogenetic assignment of HTS data allowed the successful allocation of 367 sequences at the species level (with an average of 127 species per sample) using Bowtie 2 (21), with a maximum of 1 mismatch in every read against a nucleotide database of 40,552 sequences for the 3-octaprenyl-4-hydroxybenzoate carboxy-lyase (UbiD) gene downloaded from the European Nucleotide Archive (ENA) website. These reads were assigned at the phylum (Fig. 2A), genus (Fig. 2B), and species (Fig. 2C) levels. At the phylum level, reads corresponded to members of the Firmicutes (74.6%), Proteobacteria (17.6%), and Actinobacteria (7.8%) phyla (Fig. 2A). These proportions across phyla were maintained among all of the samples analyzed (see Fig. S1 in the supplemental material). Notably, none of the reads was assigned to members of the Bacteroidetes phylum.
FIG 2.

Relative abundance of the gallate decarboxylase C subunit protein at the phylum (A), genus (B), or species (C) level in human gut microbiome samples from athletes, determined using HTS technologies.
The reads corresponded to 53 genera, i.e., 47% of the 113 genera detected previously in these samples (20). On average, Anaerostipes accounted for the majority of reads (19.7%), followed by Klebsiella (18.5%), Lachnospiraceae (12.2%), Dorea (11.7%), Clostridium (10.9%), Blautia (9.4%), Actinomyces (8.2%), and Lactobacillus (1.4%) (Fig. 2B). Indirectly, the HTS-lpdC method allowed the successful identification of 367 species in all of the samples (Fig. 2C). As shown in Fig. 2C, the greatest percentages of reads were assigned to Anaerostipes hadrus (20%) and Dorea longicatena (12%).
Strains from some species identified in this study, such as Pantoea agglomerans (22), Enterococcus faecalis (23), Klebsiella pneumoniae (23), and L. plantarum (8), were reported previously to decarboxylate gallic acid into pyrogallol. Apart from these human gut bacteria, the metabolism of gallate in Streptococcus gallolyticus has been also described (24, 25). This species is a common gallate-metabolizing inhabitant of the gut of herbivores, where plant gallates are abundant (26). This species has been described as an opportunistic pathogen in the healthy human gut and has been associated with colorectal cancer (27, 28). To our knowledge, no other human gut bacterial species with gallate decarboxylase activity have been described so far.
From the taxonomic composition shown in Fig. 2C and 3, gallate decarboxylase genes were identified in gut microbiota samples that were homologous to genes present in the genomes of Hungatella hathewayi DSM 13479, Erysipelotrichaceae bacterium 3_1_53, Enterobacter cloacae ATCC 13047, Actinomyces glycerinitolerans, Anaerostipes hadrus DSM 3319, Enterococcus raffinosus ATCC 49464, Pediococcus ethanolidurans DSM 22301, Klebsiella michiganensis KCTC 1686, Intestinibacter bartlettii DSM 16795, Blautia sp. strain KLE 1732, Dorea longicatena CAG 42, Clostridium butyricum 5521, Acetobacter tropicalis NBRC 101654, Turicibacter sanguinis PC909, and Azotobacter chroococcum NCIMB 8003, among other human gut bacteria, thereby demonstrating that this activity may be distributed among the major bacterial phylogenetic divisions of the human gut microbiota. The absence of specific databases, compared to the well-annotated 16S rRNA databases, could affect the identification by Bowtie 2 analysis. Moreover, the existence of less conserved genes that would not have been amplified by the degenerate primers could also contribute to underestimation of the distribution of these genes.
FIG 3.

Genetic organization of the chromosomal region of some bacterial genera identified by HTS as containing gallate decarboxylase-encoding genes. The genetic organization of the chromosomal region containing the corresponding L. plantarum WCFS1 genes (GenBank accession no. NC_004567) is also represented. Arrows indicate genes. Genes with identical putative functions are represented by identical colors. Genes coding for gallate esterase (tannase) (dark blue), gallate decarboxylase B (blue) and C (yellow) subunits, and the transcriptional regulator (green) are indicated.
Usefulness of the HTS-lpdC method for detection of bacterial gallate metabolism.
To confirm that detection of a lpdC gene with the proposed HTS-lpdC method is a functional marker for bacterial gallate metabolism, whether its presence in a genome was predictive of the ability to convert gallic acid into pyrogallol was tested. Six DSM collection strains belonging to Firmicutes (Streptococcus gallolyticus DSM 16831, Intestinibacter bartlettii DSM 16795, Anaerostipes hadrus DSM 3319, and Hungatella hathewayi DSM 13479) and Proteobacteria (Kosakonia radicincitans DSM 16656 and Shimwellia blattae DSM 4481) phyla were grown in the presence of gallic acid (3 mM) to detect functional gallate decarboxylase activity, as predicted by the HTS-lpdC method. After 7 days of incubation, samples were collected, and the gallic acid and pyrogallol present in the supernatants were extracted and analyzed by high-pressure liquid chromatography (HPLC) (Fig. 4). The identification of these compounds was carried out by comparing the retention times and spectral data of each peak with those of commercial standards. The results indicated that all of the assayed strains were able to decarboxylate to pyrogallol the gallic acid present in the medium. As shown in Fig. 4, the anaerobic strains (I. bartlettii, A. hadrus, and H. hathewayi) decarboxylated only partially the gallic acid present in the medium, probably due to their slight growth under the anaerobic conditions used.
FIG 4.

HPLC chromatograms showing the gallate decarboxylase activity of bacterial species identified in the microbiota by the HTS-lpdC method. Chromatograms of supernatants from bacteria from the Firmicutes phylum, i.e., S. gallolyticus DSM 16831 (A), I. bartlettii DSM 16795 (B), A. hadrus DSM 3319 (C), and H. hathewayi DSM 13479 (D), and from the Proteobacteria phylum, i.e., K. radicincitans DSM 16656 (E) and S. blattae DSM 4481 (F), grown for 7 days in the presence of 3 mM gallic acid, are shown. Control media containing gallic acid are also shown. The chromatograms were recorded at 280 nm. The gallic acid (GA) and pyrogallol (P) detected in the chromatograms are indicated.
In this work, the ability of I. bartlettii, A. hadrus, H. hathewayi, K. radicincitans, and S. blattae strains to decarboxylate gallate has been described. All of these species are inhabitants of the human gastrointestinal tract. Moreover, the results obtained confirmed that the HTS-lpdC method predicts gallate metabolism across a broad range of gut organisms belonging to different bacterial phyla.
Genetic organization of gallate decarboxylases in the gut microbiota.
Once genes apparently encoding LpdC proteins were found in different members of the human gut microbiota, a comparative analysis of those chromosomal regions was performed (Fig. 3). The gallate decarboxylase C subunit from L. plantarum WCFS1 (LpdC) is erroneously annotated as 3-octaprenyl-4-hydroxybenzoate carboxy-lyase (UbiD) (7). In gallate decarboxylases, ubiD-like genes are located within operons that encode partner proteins that are required for decarboxylation (7). These bacterial multisubunit decarboxylases are encoded by 3 clustered genes, encoding the B, C, and D subunits (9). To date, only in the genome of L. plantarum has it been noted that the genes encoding gallate decarboxylase are not clustered. In this instance, the gene encoding the C subunit, lpdC or lp_2945, is located close to the tannase-encoding gene (tanLp1 or lp_2956). However, the genes encoding the B (lpdB or lp_0271) and D (lpdD or lp_0272) subunits are located more than 1 Mb apart in the L. plantarum genome (7) (Fig. 3). Genomic sequence analysis of the species from the human gut microbiota harboring genes homologous to the lpdC gene revealed that, as in L. plantarum, the B subunit determinant is not clustered with the C subunit equivalent in T. sanguinis and A. chroococcum (Fig. 3). Furthermore, in the species in which the gene encoding the B subunit is clustered together with the C subunit determinant, this study revealed the existence of different gene organizations among these decarboxylases, i.e., a minority BC gene arrangement (such as in H. hathewayi, an Erysipelotrichaceae species, and Enterobacter cloacae) or a widely represented CB arrangement (in Enterococcus raffinosus, P. ethanolidurans, Lactobacillus rhamnosus, K. michiganensis, C. butyricum, and S. gallolyticus, among others) (Fig. 3). In this regard, the HTS-lpdC method allowed the identification of lpdC genes allocated in all different gene arrangements. As shown in Fig. 3, some of the human gut microbiota species containing gallate decarboxylase genes also contain a tannase-encoding gene in the same region of the genome (as observed in C. butyricum, A. tropicalis, and S. gallolyticus). Similar to findings for Lactobacillus species, gallate decarboxylase-encoding genes were more common than the corresponding esterase (tannase) activity in the athlete gut microbiota samples. The presence of gallate decarboxylase but not tannase activity in some bacteria could suggest a cooperative process among commensal bacteria in dietary tannin metabolism. In addition, in the human gut microbiota, it could be imagined that other bacterial species could possess appropriate mechanisms to further degrade the pyrogallol produced by these gallate decarboxylases. Therefore, all of these human gut bacteria could play important roles when tannins or gallates are present in food, having the ability to degrade and to detoxify these dietary constituents into simpler compounds.
Conservation of LpdC in the human gut microbiota.
The sequences of LpdC proteins recovered from the species from the human gut identified in this study were aligned (Fig. S2). This alignment allowed the identification of conserved amino acid domains within the C subunit. The domains selected for the design of degenerate oligonucleotides for HTS, i.e., MAGIPTEA and VDEDVDIF, were highly conserved. A phylogenetic analysis of LpdC subunits retrieved from each representative species found in the human gut microbiota was performed (Fig. 5). LpdC proteins from Lachnospiraceae bacterium (GenBank accession no. EFV16561.1) and Anaerostipes hadrus (GenBank accession no. WP_009204323.1) presented the highest level of identity (99.8%). LpdC proteins from Blautia sp. (GenBank accession no. WP_021650680.1), Firmicutes bacterium (GenBank accession no. CDD72954.1), A. hadrus, Lachnospiraceae bacterium, Dorea longicatena (GenBank accession no. CDE20644.1), Intestinibacter bartlettii (GenBank accession no. WP_007285641.1), and Clostridium butyricum (GenBank accession no. WP_058371993.1) exhibited 87% identity. Among genetically related bacteria, such as among members of the lactic acid bacteria, the identity levels ranged from 73 to 90% (7). Surprisingly, a high degree of conservation was found among LpdC proteins from phylogenetically unrelated genera such as Komagataeibacter intermedius (GenBank accession no. WP_039733191.1) and Acetobacter tropicalis (GenBank accession no. WP_006559787.1) (96% identity) (Fig. 5). These results could indicate that the LpdC domains selected for primer design are highly conserved and allowed the successful identification of a diverse range of these enzymes in the human gut.
FIG 5.
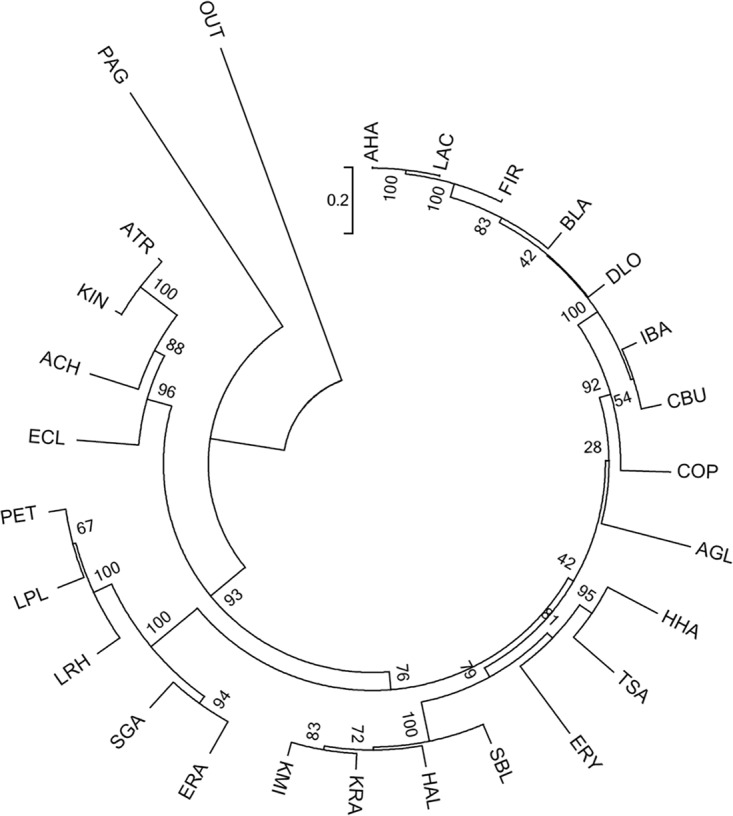
Phylogenetic analysis of gallate decarboxylase C subunit sequences. A cladogram of 26 gallate decarboxylase C subunit proteins from the human gut identified in this study, based on the amino acid sequences, is shown. The tree was built using the neighbor-joining method, visualizing the branch length information. The C subunits are from Acetobacter tropicalis (ATR) (GenBank accession no. WP_006559787.1), Actinomyces glycerinitolerans (AGL) (GenBank accession no. WP_073329259.1), Anaerostipes hadrus (AHA) (GenBank accession no. WP_009204323.1), Azotobacter chroococcum (ACH) (GenBank accession no. WP_052264016.1), Blautia sp. strain KLE 1732 (BLA) (GenBank accession no. WP_021650680.1), Clostridium butyricum (CBU) (GenBank accession no. WP_058371993.1), Coprobacillus (COP) (GenBank accession no. WP_008787659.1), Dorea longicatena CAG:42 (DLO) (GenBank accession no. CDE20644.1), Enterobacter cloacae subsp. cloacae ATCC 13047 (ECL) (GenBank accession no. YP_003612445.1), Enterococcus (ERA) (GenBank accession no. WP_010743655.1), Erysipelotrichaceae bacterium 3_1_53 (ERY) (GenBank accession no. EFP59895.1), Firmicutes bacterium CAG:270 (FIR) (GenBank accession no. CDD72954), Hafnia alvei ATCC 51873 (HAL) (GenBank accession no. WP_004092226.1), Hungatella hathewayi (HHA) (GenBank accession no. WP_006771947.1), Intestinibacter bartlettii (IBA) (GenBank accession no. WP_007285641.1), Klebsiella michiganensis KCTC 1686 (KMI) (GenBank accession no. AEX02211.1), Komagataeibacter intermedius (KIN) (GenBank accession no. WP_039733191.1), Kosakonia (KRA) (GenBank accession no. WP_071921386.1), Lachnospiraceae bacterium 5_163FAA (LAC) (GenBank accession no. EFV16561), Lactobacillus rhamnosus (LRH) (GenBank accession no. WP_005712295.1), Lactobacillus plantarum ATCC 14917T (LPL) (D7VDD5), Pantoea agglomerans strain FDAARGOS_160 (PAG) (GenBank accession no. AMG60167.1), Pediococcus ethanolidurans (PET) (GenBank accession no. WP_057806460.1), Shimwellia blattae (SBL) (GenBank accession no. WP_002441866.1), Streptococcus (SGA) (GenBank accession no. WP_003065832.1), and Turicibacter sanguinis (TSA) (GenBank accession no. WP_040763984.1). The amino acid sequence of Lp_2956 (tannase) from L. plantarum WCFS1 was used as the outgroup (OUT).
Changes in the human gut microbiome are associated with altered human metabolism and health. HTS has revolutionized human gut microbiome research, but most current applications concentrate on studying the microbial diversity of communities and have at best provided associations between specific gut bacteria and human health. However, little is known about the inner metabolic mechanisms in the gut ecosystem (29). This study represents a novel approach for analyzing gut microbes and their metabolism of dietary phenolic compounds. The results clearly show the successful use of the HTS-lpdC method to identify functional gallate decarboxylases in complex microbial communities and the subsequent use of this gene in a phylogenetic approach to investigate the gut microbiome according to activities involved in the metabolism of a relevant dietary component. Metagenomics can identify potential metabolic functions specially, not only when the function is encoded by a single gene or operon but also when the phenolic conversions may require a consortium of microbes, and can indicate the immediate catalytic potential of a microbial community (30). In the context of phenolic compound/diet-microbiota interactions, the identification of novel microorganisms involved in gallate metabolism could relate to how microbial communities adjust their metabolic activities to survive in the gut environment with dietary variations. A human gut microbial gene catalogue has been published (31), indicating that individuals share about 38% of their metagenomes, called the “minimal gut metagenome” (31). Among the encoded bacterial functions that are important for life in the gut is the functional category related to secondary metabolites, and it can be assumed that this category includes some genes involved in phenolic metabolism. In this regard, the results shown in this work indicate that gallate decarboxylase activity, which is key for metabolism of the gallates present in plant-derived foods, is present in members of the main genera in the human gut, and this feature could be an advantage for surviving in this environment. This methodology represents a great approximation, however, and further metatranscriptomic approaches are necessary to provide information about the expression of these genes in the human gut microbiota (30).
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains used in this study were purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ), and they were grown with the media and conditions recommended by the DSMZ. The assayed bacteria belonging to the Firmicutes phylum were Streptococcus gallolyticus DSM 16831, which was grown in DSM medium 215 (brain heart infusion [BHI] medium) at 37°C, Intestinibacter bartlettii DSM 16795 and Anaerostipes hadrus DSM 3319, which were grown anaerobically in DSM medium 110 (chopped meat medium with carbohydrates) at 37°C, and Hungatella hathewayi DSM 13479, which was grown anaerobically in DSM medium 104b (PY+X medium) at 30°C. Bacteria from the Proteobacteria phylum were Kosakonia radicincitans DSM 16656 and Shimwellia blattae DSM 4481, which were grown in DSM medium 1 (nutritive broth) at 30°C and 37°C, respectively.
To test gallate decarboxylase activity, the medium was supplemented with filter-sterilized gallic acid at a final concentration of 3 mM. The inoculated media were incubated for 7 days in the dark. Bacterial cultures incubated without gallic acid were used as controls. The gallic acid and pyrogallol present in the supernatant were extracted with a standard protocol involving two extraction steps with one-third of the reaction mixture volume of ethyl acetate. The extracted phenolic compounds were analyzed by HPLC with diode array detection, as described previously (7).
DNA isolation from fecal samples.
The samples used in this study were described previously (20). Briefly, fecal samples were collected from healthy male elite professional rugby players (n = 15), since it had been demonstrated previously that athletes had greater diversity of gut microorganisms (20). DNA was extracted from fresh stool samples, which were stored on ice until processing. DNA was purified from fresh stool samples using the QIAamp DNA stool minikit (Qiagen), according to the manufacturer's instructions but with the addition of a bead beating step (30 s × 3), and was stored at −20°C. The microbiota composition of the samples was established by amplicon sequencing of the 16S rRNA gene V4 region, as described previously (20).
PCR detection of lpdC gene fragments using selected primer sets.
A comparison of amino acid sequences of the C subunit of gallate decarboxylases from lactic acid bacteria was used to find conserved domains (7). Currently, in databases, the gallate decarboxylase C subunit from L. plantarum WCFS1 (GenBank accession no. YP_004890530) is erroneously annotated as 3-octaprenyl-4-hydroxybenzoate carboxy-lyase (UbiD), based on its similarity to this protein from Escherichia coli K-12 (GenBank accession no. NP_418285). Therefore, the design of degenerate primers to detect LpdC determinants needed to ensure the nonamplification of genes encoding UbiD proteins. The protein selection for primer design took into account the findings that L. plantarum LpdC is only 22% identical to E. coli UbiD and that UbiD genes do not form clusters within which a LpdB subunit is also encoded (9). Moreover, proteins showing high levels of identity to LpdC (although annotated as UbiD) from Anaerostipes hadrus DSM 3319 (L1PXM5), Clostridium bolteae ATCC BBA-613 (A8S1T4), Coriobacterium glomerans ATCC 49209 (F2N9Z4), and Desulfosporosinus orientis ATCC 19365 (G7WBX3), which are located contiguous to a decarboxylase B subunit, were also included in an alignment to find conserved regions to facilitate the design of degenerate LpdC PCR primers for HTS (data not shown). Taking into account the restriction on the amplicon size for HTS analysis, two conserved domains were chosen. Degenerate lpdC-Fw primer (5′-ATGGCNGGNATHCCNACNGARGC), coding for MAGIPTEA, and degenerate lpdC-Rev primer (5′-RAANADRTCNACRTCYTCRTCNAC), coding for VDEDVDIF, amplified a 237-bp fragment suitable for HTS analysis. The resultant lpdC-Fw and lpdC-Rev oligonucleotides amplify a 237-bp internal fragment of the gene encoding the C subunit and do not amplify the E. coli ubiD gene encoding a 3-octaprenyl-4-hydroxybenzoate carboxy-lyase.
PCRs were carried out in duplicate by using the KAPA2G Robust HotStart ReadyMix PCR kit (Kapa Biosystems) and contained 25 μl of mix, 5 μl of each primer (5 μM), 10 μl of DNA template (standardized to 100 ng DNA/reaction), and nuclease-free water to a final volume of 50 μl. PCR amplifications were carried out using a G-Storm thermal cycler (Gene Technologies). Amplification consisted of an initial denaturation step at 95°C for 3 min, 40 cycles of denaturation at 95°C for 15 s, annealing at 48°C for 15 s, and extension at 72°C for 30 s, and a final elongation step at 72°C for 7 min. PCR amplicons were pooled and cleaned using the AMPure XP magnetic bead-based purification system (Beckman Coulter).
High-throughput sequencing.
The Ion Plus fragment library kit (Life Technologies) was used to prepare lpdC amplicon libraries. Libraries were barcoded, prior to sequencing, using Ion Xpress barcode adaptors (Life Technologies). Amplicon libraries were assessed for size distribution and concentrations using a Bioanalyzer 2100 (Agilent Technologies). Following library quantification and equimolar pooling, the Ion OneTouch 2 system was used to prepare template-positive ion sphere particles (ISPs) containing the clonally amplified DNA libraries by using the Ion PGM Template OT2 400 kit, which allows <400-bp reads. Enrichment of the template-positive ISPs was performed using the Ion OneTouch ES system. Sequencing was performed with the Ion Torrent PGM system (Life Technologies) by using an Ion 318v2 chip and the Ion PGM Sequencing 400 kit (Life Technologies), according to the manufacturer's guidelines, at the Teagasc next-generation sequencing facility.
Bioinformatic analysis.
The reads from the Ion PGM sequencing were filtered on the basis of quality (removal of low-quality nucleotides at the 3′ end and removal of windows of 20 nucleotides with low average quality) and length (removal of sequences with less than 200 nucleotides) with PRINSEQ (32). The sequences were cleaned of dereplicates and unique sequences and chimeras were eliminated by using the closed-reference USEARCH v8.0 algorithm (33). The resulting sequences were taxonomically assigned against the database of lpdC gene found in the ENA with Bowtie 2 (21). The Bowtie 2 tool was used for the mapping of the filtered clean reads to the lpdC gene database, with a maximum of 1 mismatch in every read. The relative abundance of lpdC among different taxa was estimated by dividing the number of reads that uniquely mapped to that group by the total number of reads from the sample.
Amino acid sequences were aligned with MUSCLE (34). The presence or absence of LpdC and tannase proteins was analyzed using the Lactobacillus genomes available from the National Center for Biotechnology Information (NCBI) website. A BLASTP approach was used to represent the presence/absence matrix in a heatmap, employing a color code that represents the degree of sequence similarity (35). This BLASTP approach used 70% iterative similarity across the Lactobacillus species over 50% of the protein length, with an E value of 0.0001 as a significance cutoff value. The gene cluster characterization was performed with the genomic sequences available at the NCBI website, using BLAST. The phylogenetic tree was generated with MEGA v6.06 (36). Multiple alignments were made using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo), after retrieval of sequences from the GenBank and Swiss-Prot databases.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to M. V. Santamaría and J. M. Barcenilla for their assistance.
This work was supported by MINECO grant AGL2014-52911-R (AEI/FEDER, UE). L.S. is a recipient of a FPI Fellowship from the MINECO, and M.E.-T. is a recipient of an Irish Research Council postdoctoral grant (grant GOIPD/2017/1302).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01558-18.
REFERENCES
- 1.Shahidi F, Naczk M. 2003. Phenolics in food and nutraceuticals. CRC Press, London, United Kingdom. [Google Scholar]
- 2.Haminuik CWI, Maciel GM, Plata-Oviedo MSV, Peralta RM. 2012. Phenolic compounds in fruits: an overview. Int J Food Sci Technol 47:2023–2044. doi: 10.1111/j.1365-2621.2012.03067.x. [DOI] [Google Scholar]
- 3.Ghaani M, Nasirizadeh N, Ardakani SAY, Mehrjardi FZ, Scampicchio M, Farris S. 2016. Development of an electrochemical nanosensor for the determination of gallic acid in food. Anal Methods 8:1103–1110. doi: 10.1039/C5AY02747K. [DOI] [Google Scholar]
- 4.Newsome AG, Li Y, van Breemen RB. 2016. Improved quantification of free and ester-bound gallic acid in foods and beverages by UPLC-MS/MS. J Agric Food Chem 64:1326–1334. doi: 10.1021/acs.jafc.5b04966. [DOI] [PubMed] [Google Scholar]
- 5.Ow YY, Stupand I. 2003. Gallic acid and gallic acid derivatives: effects on drug metabolizing enzymes. Curr Drug Metab 3:241–248. doi: 10.2174/1389200033489479. [DOI] [PubMed] [Google Scholar]
- 6.Nogales J, Canales A, Jiménez-Barbero J, Serra B, Pingarrón JM, García JL, Díaz E. 2011. Unravelling the gallic acid degradation pathway in bacteria: the gal cluster from Pseudomonas putida. Mol Microbiol 79:359–374. doi: 10.1111/j.1365-2958.2010.07448.x. [DOI] [PubMed] [Google Scholar]
- 7.Jiménez N, Curiel JA, Reverón I, de las Rivas B, Muñoz R. 2013. Uncovering the Lactobacillus plantarum WCFS1 gallate decarboxylase involved in tannin degradation. Appl Environ Microbiol 79:4253–4263. doi: 10.1128/AEM.00840-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodríguez H, de las Rivas B, Gómez-Cordovés MC, Muñoz R. 2008. Degradation of tannic acid by cell-free extracts of Lactobacillus plantarum. Food Chem 107:664–670. doi: 10.1016/j.foodchem.2007.08.063. [DOI] [PubMed] [Google Scholar]
- 9.Lupa B, Lyon D, Gibbs MD, Reeves RA, Wiegel J. 2005. Distribution of genes encoding the microbial non-oxidative reversible hydroxyarylic acid decarboxylases/phenol carboxylases. Genomics 86:342–351. doi: 10.1016/j.ygeno.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 10.O'Sullivan DJ, Fallico V, O'Sullivan O, McSweeney PLH, Sheehan JJ, Cotter PD, Giblin L. 2015. High-throughput DNA sequencing to survey bacterial histidine and tyrosine decarboxylase in raw milk cheeses. BMC Microbiol 15:266. doi: 10.1186/s12866-015-0596-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frantzen CA, Kleppen HP, Holo H. 2018. Lactococcus lactis diversity in undefined mixed dairy starter cultures as revealed by comparative genome analyses and targeted amplicon sequencing of epsD. Appl Environ Microbiol 84:e02199-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Osawa R, Kuroiso K, Goto S, Shimizu A. 2000. Isolation of tannin-degrading lactobacilli from humans and fermented foods. Appl Environ Microbiol 66:3093–3097. doi: 10.1128/AEM.66.7.3093-3097.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Curiel JA, Rodríguez H, Acebrón I, Mancheño JM, de las Rivas Muñoz R. 2009. Production and physicochemical properties of recombinant Lactobacillus plantarum tannase. J Agric Food Chem 57:6224–6230. doi: 10.1021/jf901045s. [DOI] [PubMed] [Google Scholar]
- 14.Jiménez N, Esteban-Torres M, Mancheño JM, de las Rivas B, Muñoz R. 2014. Tannin degradation by a novel tannase enzyme present in some Lactobacillus plantarum strains. Appl Environ Microbiol 80:2991–2997. doi: 10.1128/AEM.00324-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun Z, Harris HMB, McCann A, Guo C, Argimón S, Zhang W, Yang X, Jeffery IB, Cooney JC, Kagawa TF, Liu W, Song Y, Salvetti E, Wrobel A, Rasinkangas P, Parkhill J, Rea MC, O'Sullivan O, Ritari J, Douillard FP, Ross RP, Yang R, Briner AE, Felis GE, de Vos WM, Barrangou R, Klaenhammer TR, Caufield PW, Cui Y, Zhang H, O'Toole PW. 2015. Expanding the biotechnology potential of lactobacilli through comparative genomics of 213 strains and associated genera. Nat Commun 6:8322. doi: 10.1038/ncomms9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaquero I, Marcobal A, Muñoz R. 2004. Tannase activity by lactic acid bacteria isolated from grape must and wine. Int J Food Microbiol 96:199–204. doi: 10.1016/j.ijfoodmicro.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 17.Joice R, Yasuda K, Shafquat A, Morgan XC, Huttenhower C. 2014. Determining microbial products and identifying molecular targets in the human microbiome. Cell Metab 20:731–741. doi: 10.1016/j.cmet.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riesenfeld CS, Schloss PD, Handelsman J. 2004. Metagenomics: genomic analysis of microbial communities. Annu Rev Genet 38:525–552. doi: 10.1146/annurev.genet.38.072902.091216. [DOI] [PubMed] [Google Scholar]
- 19.Knight R, Jansson J, Field D, Fierer N, Desai N, Fuhrman JA, Hugenholtz P, van der Lelie D, Meyer F, Stevens R, Bailey MJ, Gordon JI, Kowalchuk GA, Gilbert JA. 2012. Unlocking the potential of metagenomics through replicated experimental design. Nat Biotechnol 30:513–520. doi: 10.1038/nbt.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clarke SF, Murphy EF, O'Sullivan O, Lucey AJ, Humphreys M, Hogan A, Hayes P, O'Reilly M, Jeffery IB, Wood-Martin R, Kerins DM, Quigley E, Ross RP, O'Toole PW, Molloy MG, Falvey E, Shanahan F, Cotter PD. 2014. Exercise and associated dietary extremes impact on gut microbial diversity. Gut 63:1913–1920. doi: 10.1136/gutjnl-2013-306541. [DOI] [PubMed] [Google Scholar]
- 21.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeida M, Wieser M, Yoshida T, Sugio T, Nagasawa T. 1998. Purification and characterization of gallic acid decarboxylase from Pantoea agglomerans T71. Appl Environ Microbiol 64:4743–4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakajima H, Otani C, Niimura T. 1992. Decarboxylation of gallate by cell-free extracts of Streptococcus faecalis and Klebsiella pneumoniae isolated from rat feces. J Food Hyg Soc Jpn 33:371–377. doi: 10.3358/shokueishi.33.371. [DOI] [Google Scholar]
- 24.Chamkha M, Patel BKC, Traore A, García J-L, Labta M. 2002. Isolation from a shea cake digester of a tannin-degrading Streptococcus gallolyticus strain that decarboxylates protocatechuic acid hydroxycinnamic acids, and emendation of the species. Int J Syst Evol Microbiol 52:939–944. doi: 10.1099/00207713-52-3-939. [DOI] [PubMed] [Google Scholar]
- 25.Jiménez N, Reverón I, Esteban-Torres M, López de Felipe F, de las Rivas B, Muñoz R. 2014. Genetic and biochemical approaches towards unravelling the degradation of gallotannins by Streptococcus gallolyticus. Microb Cell Fact 13:154. doi: 10.1186/s12934-014-0154-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osawa R, Fujisawa T, Sly LI. 1995. Streptococcus gallolyticus sp. nov.: gallate degrading organisms formerly assigned to Streptococcus bovis. Syst Appl Microbiol 18:74–78. doi: 10.1016/S0723-2020(11)80451-0. [DOI] [Google Scholar]
- 27.Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. 2012. A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol 10:575–582. doi: 10.1038/nrmicro2819. [DOI] [PubMed] [Google Scholar]
- 28.López de Felipe F, de las Rivas B, Muñoz R. 2014. Bioactive compounds produced by gut microbial tannase: implications for colorectal cancer development. Front Microbiol 5:684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ji B, Nielsen J. 2015. From next-generation sequencing to systematic modeling of the gut microbiome. Front Genet 6:219. doi: 10.3389/fgene.2015.00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kemperman RA, Bolca S, Roger LC, Vaughan EE. 2010. Novel approaches for analysing gut microbes and dietary polyphenols: challenges and opportunities. Microbiology 156:3224–3231. doi: 10.1099/mic.0.042127-0. [DOI] [PubMed] [Google Scholar]
- 31.Qin J, Ruiqiang L, Raes J, Arumugan M, Solvsten Burgdorf K, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Doré J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, MetaHIT Consortium, Bork P, Ehrlich SD, Wang J. 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmieder R, Edwards R. 2011. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 34.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Altschul SF, Madden TL, Schäffer AA, Zhang J, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.