

Abstract
Formaldehyde (HCHO) is an important air pollutant from both an atmospheric chemistry and human health standpoint. This study uses an instrumented photochemical Air Quality Model, CMAQ-DDM, to identify the sensitivity of HCHO concentrations across the United States (US) to major source types and hydrocarbon speciation. In July, biogenic sources of hydrocarbons contribute the most (92% of total hydrocarbon sensitivity), split between isoprene and other alkenes. Among anthropogenic sources, mobile sources of hydrocarbons and nitrogen oxides (NOx) dominate. In January, HCHO is more sensitive to anthropogenic hydrocarbons than biogenic sources, especially mobile sources and residential wood combustion (36% of national hydrocarbon sensitivity). While ozone (O3) is three times more sensitive to NOx than hydrocarbons across most areas of the US, HCHO is six times more sensitive to hydrocarbons than NOx, largely due to sensitivity to biogenic precursors and the importance of low-NOx chemistry. In winter, both HCHO and O3 show negative sensitivity to NOx (increases with removal of NOx), although O3 increases are larger. Relative sensitivities do not change substantially across different regions of the country.
1. INTRODUCTION
Formaldehyde (HCHO) is a ubiquitous trace chemical in the ambient atmosphere. It plays an important role in atmospheric photochemistry because it reacts quickly and its photolysis provides new hydroxyl (OH) and hydroperoxy radicals (HO2) which drive ozone (O3) production.1 HCHO can also influence secondary organic aerosol (SOA) by providing radicals that increase gas phase oxidation of hydrocarbons and increasing surface-active organic material.2
HCHO also is important per se because it can adversely impact human health; it is a Hazardous Air Pollutant (HAP) under the 1990 US Clean Air Act. HCHO is reactive in upper airways with health impacts from acute and chronic inhalation exposure including irritation to the eyes, nose and throat.3 HCHO is a probable human carcinogen, based on limited evidence in humans and sufficient evidence in animals.4 In national studies of concentration and risk from 187 HAPs,5,6 HCHO contributes over half the total cancer risk and 9% of noncancer risk; it is identified as a national risk driver in 99% of US census tracts.7 Up to 12,600 people annually were estimated to develop cancer based on satellite-derived estimates of ambient HCHO exposures.8
HCHO can be emitted directly from many sources, with fuel combustion - including mobile sources and industrial fuel – being predominant anthropogenic sources. Direct emissions of HCHO from biogenic sources is the largest source of primary HCHO in the U.S.9
HCHO also is produced in the atmosphere from emissions of almost every volatile organic hydrocarbon (VOC; in this study defined to include ethane and acetone). Previous studies of HCHO attempting to unravel the role of secondary production versus direct emissions vary widely in their estimates. A recent study in Southeast Texas using the Community Multiscale Air Quality (CMAQ) model with a source-oriented mechanism estimated 30–50% of total HCHO resulting from atmospheric production, with the rest either primary (10%) or upwind (20–50%), and 30–50% of the total from biogenic sources.10 In contrast, measurements and emission estimates over the same area, using different techniques, determined 95% of HCHO from secondary production, mostly point sources (92%).11 In Canada, predicted secondary production of HCHO varied widely, ranging from 71–96% of total HCHO in summer and 10–67% in winter.12 In the Pearl River Delta, China, secondary sources contributed about 53% to total HCHO.13 Conclusions drawn from spatially- and temporally-limited studies are difficult to generalize to larger areas, such as the US.
Less work has been done to quantify specific anthropogenic source types or VOC precursors contributing to HCHO. Application of the Comprehensive Air Quality Model with Extensions (CAMX) with the path-integral method predicted an anthropogenic increment of HCHO of 25 and 40% in rural and urban locations, respectively, with about 40% of the anthropogenic increment from mobile sources.14 An early version of the CMAQ model was used to attribute HCHO to individual VOCs across a large domain, identifying alkenes, particularly isoprene, as important precursors.15 While alkenes were also identified as important contributors in China, they were mostly anthropogenic.13
A widely-used tool to study sensitivity in Air Quality Models (AQMs) is the Decoupled Direct Method (DDM)16 which uses a parallel set of local, semi-normalized sensitivities to perturbations in model parameters (usually emission changes) within the underlying atmospheric model.
The goal of this study is to use a photochemical model, CMAQ, instrumented with DDM, to quantify the relative role of emission sectors and individual classes of VOCs on ambient HCHO sensitivities across the continental US, addressing the following questions:
What types of anthropogenic emission sources have the greatest influence on HCHO concentrations?
Which anthropogenic VOCs have the largest influence on HCHO?
Does the importance of anthropogenic versus biogenic VOCs change across time and space?
Is HCHO more sensitive to emissions of VOC or NOx (nitrogen oxide (NO) and nitrogen dioxide (NO2))?
We contrast the sensitivities of HCHO, acetaldehyde and O3, to provide insights on whether similar emission control strategies could be used to reduce all three pollutants. We use an improved AQM, updated inventories and sensitivity tools, and a larger spatial domain, covering different chemical and meteorological conditions than previous studies.
This study builds on the 2011 National Air Toxics Assessment (NATA)5,17 to provide additional understanding and analysis of those findings. In the longer term, this information will aid the development of effective multipollutant control strategies for O3 or PM2.5 that might also reduce other harmful pollutants.
2. METHODS
2.1. Atmospheric formaldehyde formation in AQMs
AQMs must represent the chemistry of over 1000 emitted VOCs and their multi-generational products, resulting in tens of millions of VOCs.18 To condense this chemistry sufficiently for use in AQMs, multiple assumptions are made in the development of chemical mechanisms. Our simulations used the CB05 chemical mechanism consisting of CB05 with chlorine chemistry19, toluene updates20, and additional HAPs21. Secondary production of HAPs are tracked separately to quantify the contribution of chemistry and emissions. While some chemical species are represented explicitly in CB05, others are broken into constituent chemical groups, or model species, which have similar chemical reactivity. A definition of the model species for which sensitivities are reported in this study and the structures represented is given in Table 1.
Table 1.
Listing of sources and hydrocarbons tracked. Sources include all hydrocarbon or NOx emissions in each category; hydrocarbons tracked includes emissions combined across source categories.
| Source categories trackeda | Hydrocarbons trackedb |
|---|---|
| Mobile sources (onroad, nonroad, marine, rail) |
Alkanes: lumped singly-bonded carbons (model species PAR) and explicit ethane (ETHA) |
| Oil and gas (point and non-point) | Alcohols: methanol (model species MEOH) and ethanol (ETOH) |
| EGU and other point sources | Formaldehyde (model species FORM) |
| Fires (wild, prescribed and agricultural) |
Aldehydes with 2 or more carbons: Acetaldehyde (model species ALD2) and lumped aldehydes with more than 2 carbons (ALDX) |
| Residential wood combustion |
Alkenes: ethene (model species ETHE), lumped terminal carbon- carbon double bonds (OLE) and lumped internal carbon-carbon double bonds (IOLE) |
| Total Biogenic hydrocarbons, total biogenic NOx |
Isoprene (model species ISOP) |
| Boundary conditions | Monoterpenes: lumped terpenes (model species TERP) |
|
Aromatics: toluene and other monoalkyl aromatics (model species TOL) and xylene and polyalkyl aromatics (XYL) | |
| Methane (from constant background) |
Each source category was tracked separately for NOx (sum of NO, NO2; here we also add HONO when it is assumed to be a portion of NOx emissions) and hydrocarbons (sum of model species PAR, ETHA, MEOH, ETOH, FORM, ALD2, ALDX, ETHE, OLE, IOLE, ISOP, TERP, TOL and XYL)
Sum of each model specie emissions were tracked separately for sum of all anthropogenic sources and sum of all biogenic sources.
CB05 characterizes multiple routes for secondary photochemical formation of HCHO; a detailed description of these routes is presented in the Supplemental Information (SI); here we briefly summarize how HCHO formation is represented in the chemical mechanism. Because the chemistry in CB05 is highly condensed, with multiple reactions often collapsed into one pseudo-reaction, not all of the chemistry forming HCHO can be represented in the mechanism.
Alkenes react with O3 through addition of O3 to the double bond, forming an energy rich primary ozonide, which rapidly fragments, some forming HCHO.22 . In CB05, this series of steps is modeled as one reaction, with alkene ozonolysis promptly forming HCHO through reaction with ethene (yield=1.0), lumped terminal alkenes (0.74), lumped internal alkenes (0.25), isoprene (0.6), lumped terpenes (0.24), unsaturated isoprene products, including methacrolein and methylvinylketone (0.15) and lumped aromatic fragments (0.7).
Rapid unimolecular decomposition of methyl radicals containing an alcohol, hydroperoxide, or nitrate group can produce HCHO. These reactions are represented in CB05 by the reaction of OH with methanol (yield=1.0) and ethanol (0.10), with chlorine radical reacting with ethanol (1.0), and photolysis of methylhydroperoxide (1.0) and unsaturated isoprene products (0.9). The reaction of nitrate radical (NO3) with alkenes can yield HCHO from nitrooxy methyl radicals, as well as nitrooxy alkoxy radicals.22 In CB05, these pathways are represented by NO3 reaction with ethene (yield=2.0), terminal alkenes (1.0), unsaturated isoprene products (0.282) and cresols (0.24).
Alkoxy radicals are ubiquitous in atmospheric chemistry and can form HCHO through reaction with oxygen or decomposition. Alkoxy radicals are not represented explicitly in CB05 – their chemistry is collapsed and the expected end products are assigned as products to the reaction. For example, the reaction of OH with ethene produces a prompt yield of HCHO, incorporating the expected later reactions of the hydroxyperoxy radical, alkoxy radical and decomposition. CB05 also includes prompt HCHO formation from reactions of NO with methylperoxy radical (yield=1.0), aromatic peroxy radicals (0.336) and a generalized peroxy radical from unsaturated dicarbonyls (0.344).
Methylperoxy radical reactions with hydroperoxy radical (HO2) and self/cross-reactions can also form HCHO. In CB05, the methylperoxy radical self reaction produces HCHO by combining and collapsing the reaction channels producing closed-shell products and radicals,23 weighted by reaction rate to immediately produce HCHO (yield =1.37). Other reactions forming HCHO include methyl peroxy radical reaction with ethyl peroxy radicals (1.0), and larger peroxy radicals (0.1). Reactions of peroxy radicals with HO2 do not form HCHO in CB05. In areas with lower NOx concentrations, peroxy radical reactions with other peroxy radicals and HO2 can be important – recently estimated to be 15–58% of total peroxy radical fate.24 Almost every VOC can produce formaldehyde, some in greater than unit yield, so our ability to characterize the atmospheric chemistry of formaldehyde represents our understanding of overall atmospheric chemistry.
2.2. CMAQ model configuration
We applied CMAQ version 5.02, configured for the 2011 NATA modelling platform.17 This application uses a 12×12 km2 grid resolution covering the continental US and parts of Canada and Mexico, and 25 vertical layers, with finer layers near the surface, gradually increasing in size until the domain top, approximately 16 km. Boundary conditions were extracted from the GEOS-Chem model. Meteorological inputs were obtained from the MM5 meteorological model version 3.6.1. Simulations were performed for the months of January and July, 2011, after 10 days of model initialization.
Anthropogenic emissions of hydrocarbons and NOx were derived from the 2011 NEIv2, providing a comprehensive estimate of emissions from all US sources, offshore sources and marine vessels in Federal Waters. HAP emissions in the NEI are from state/local/tribal submissions, including reporting at the “facility total” level (stack and fugitive), augmented with data based on emissions-factor ratios. Emissions were allocated spatially and temporally to CMAQ grids. While total emissions were used to model the photochemical environment, individual source category emissions were retained to calculate sensitivity coefficients for different source types.
Biogenic emissions of individual VOCs were quantified with the BEIS3.60 model,25 which has been evaluated against field measurements and performs well in CMAQ, slightly underestimating isoprene (median bias=−6.0%). An alternative model, MEGAN, overestimated isoprene in CMAQ (median bias =203%).25
The CMAQ model has been widely used by research and regulatory groups, evaluated numerous times against observations, and found to perform well for O3, NOy species, and particulate components,26,27 and reasonably well for HAPs.17,21
2.3. Sensitivity calculations and Decoupled Direct Method
The purpose of sensitivity studies is to quantify the change in pollutant concentration or deposition due to a change in a model parameter of interest, in this case, emissions. Sensitivity studies commonly utilize the “brute force” method, where a separate simulation with a perturbed parameter is subtracted from a base simulation. While straightforward to implement and interpret, the brute force approach does not directly address the sensitivity of ambient concentratiosn to emission changes because the integration of concentrations can be dominated by nonlinear chemistry and reducing a large VOC source changes the chemical environment. At smaller emissions perturbations, brute force can be prone to numerical noise leading to nonphysical results, and separate simulations for each perturbation are typically computationally more expensive than using DDM.28
In this study, sensitivity coefficients were calculated using DDM-3D16,29 implemented in CMAQ.30 DDM-3D has been used previously in AQMs to predict the response of O3 and PM2.5 to changes in emissions of source types, source regions and individual VOCs.31 In this application, DDM-3D is used to study the sensitivity of individual pollutants to sources and hydrocarbon precursors.
DDM-3D outputs sensitivity coefficients, Sj(x,t), in space (x) and time (t), such as: Sj(x,t)=δCi(x,t)/δεj, where Ci(x,t) is the concentration field, and εj is the user-defined model input parameter of interest. The sensitivity coefficients can be used to predict concentrations as a function of any model perturbation, εj, using the Reduced Form Model derived from the Taylor Series Expansion:
| 1 |
Where Cj(x̄,t) is the concentration of pollutant resulting from perturbation j, and C0(x̄,t) is the base (unperturbed) concentration; Δεj is the fractional perturbation of parameter j; Sj(1)(x̄,t) and Sj,j(2)(x̄,,t) are the first and second-order sensitivity coefficients, and h.o.t. represents higher order terms. The Taylor Series can be used in this manner, because DDM-3D automatically semi-normalizes the sensitivity coefficients by the full magnitude of the input parameter. Furthermore, this enables direct comparison of different sensitivity coefficients, Sj(x,t) for all j, between one another in order to analyze their relative importance on the model output of interest (e.g., HCHO or O3 concentration).
DDM-3D was used to calculate the response of selected model species to perturbations in NOx and VOC emissions from different anthropogenic emission categories and natural emissions, and to emissions of eight VOC model species groupings and methane (Table 1). Emissions of VOC and NOx from each source sector were calculated separately.
3. RESULTS
3.1. Formaldehyde sensitivity and concentrations
Figure 1 shows hourly HCHO sensitivities to total NOx and total hydrocarbons, at the surface layer in the model, averaged over the months of July and January, 2011. Average HCHO concentrations are shown along with the fraction of total HCHO due to primary (from emissions) sources in SI Figure S1. Primary emissions include HCHO from both anthropogenic and biogenic sources; HCHO from secondary sources (from VOC chemical reactions) comprises the remainder. The relative amount of HCHO from direct emissions of HCHO varies across the domain, between 10–40%, with slightly higher values in areas with high anthropogenic activity. In January, the fraction of primary HCHO increases in urban areas compared to July.
Figure 1.
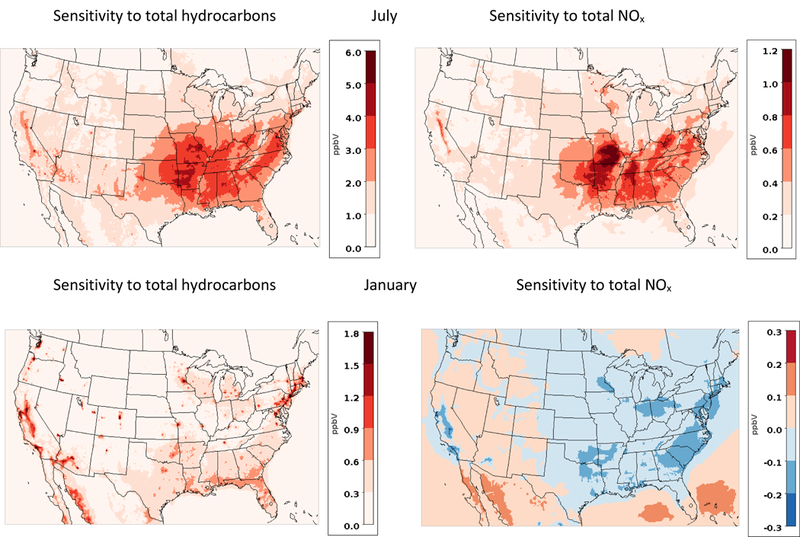
Monthly averaged sensitivity of HCHO concentrations, to total hydrocarbons (left) and total NOx emissions (right). Sensitivities are shown for July (top) and January (bottom).
HCHO concentrations predicted by CMAQ for 2011 in the NATA simulations were previously found to reasonably represent measurements at 110 sites in the Air Quality Subsystem (AQS). Overall, HCHO was underestimated, with an annual mean bias of −0.9 μg/m3 (−34.2%).17 For the months studied here, CMAQ underpredicts HCHO observations in both January (mean bias of −0.59 (−29%)) and July (mean bias of −1.44 (−34%)); with daily-averaged model predictions plotted against observations in SI Figure S2. HCHO is typically underpredicted by AQMs, which may be partially due to measurement uncertainties, including limited spatial and temporal coverage, instrument interferences and protocol differences. There are also incompatibilities when comparing grid-averaged model predictions (approximately 2880 m3 surface volumes) to point measurements. Difficulties in estimating emissions from high-emitting sources of HCHO is also a problem. Ozone is typically predicted much better by AQMs: for these simulations, the 8-hr maximum O3 is slightly overpredicted both in January (mean bias of 0.05 ppb (10%)) and in July (mean bias of 3.5 ppb (7.1%)) (Figure S3).
3.2. Sensitivity to Source Sector
Figure 2 shows the sensitivity of HCHO to both VOC and NOx emissions by source type, summed across the continental US. In July, HCHO is 6 times more sensitive to VOC than NOx emissions. Formaldehyde can be formed in both high-NOx and low-NOx conditions, although alkoxy radicals, which can form HCHO, have higher yields through reactions with NO (section 2.1). Yet, the concentrations of HCHO are much less sensitive to emissions of NOx than VOCs.
Figure 2.
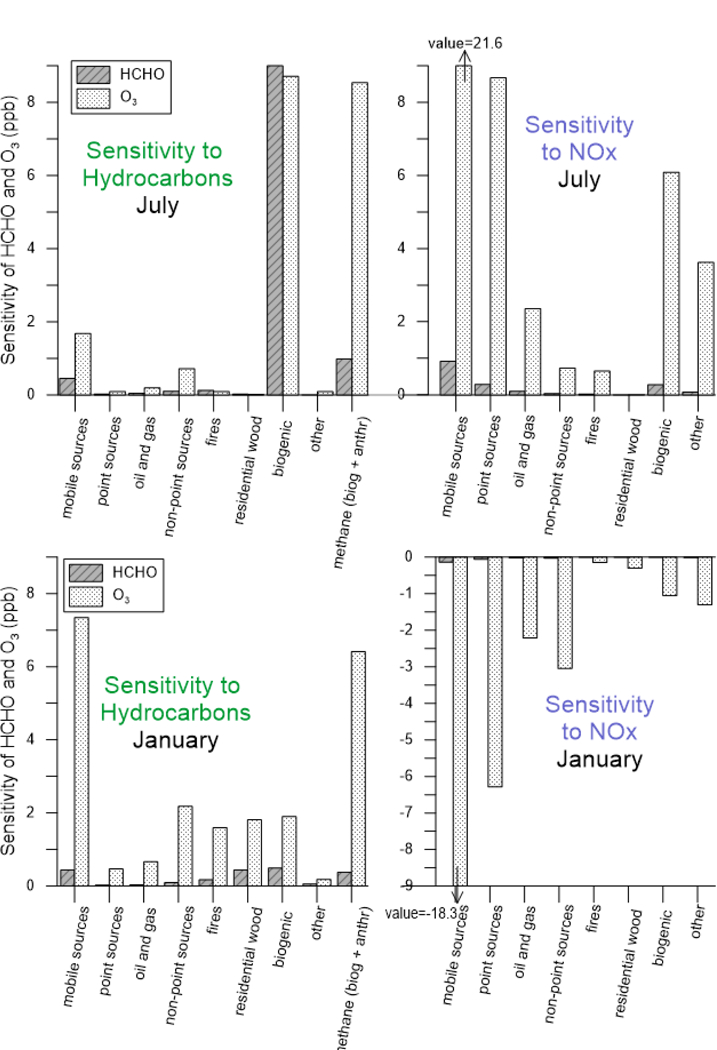
Sensitivity of HCHO and O3 to source types, separated by hydrocarbons and NOx emissions for the months of July (top) and January (bottom). Sensitivities are calculated on a national basis.
In July, HCHO sensitivity is driven by biogenic VOCs; 20–60% of which is from isoprene (SI Figure S4). The reaction of methane with OH accounts for 10% of the total hydrocarbon sensitivity. Because it is relatively unreactive, little attention has been paid to developing accurate methane inventories.
Among anthropogenic sources of VOC sensitivity in July, mobile sources contribute the most on a national basis (59%), followed by fires (16%).
The sensitivity of O3 is compared to HCHO on a national basis in Figure 3 and SI Table S1. In contrast to HCHO, O3 is 3.5 times more sensitive to NOx than VOC, and mobile sources dominate the NOx sensitivities, with point sources (primarily utilities) also substantial. Similar to HCHO, biogenic VOCs contribute 76% of total hydrocarbon sensitivity of O3, but because O3 is more sensitive to NOx than VOC in most of the US, the overall sensitivity is dominated by anthropogenic NOx. A recent study using source apportionment quantifies the overall sensitivity of O3 to biogenic sources at 13–18%, depending on region.32 The current study is not directly comparable, but by weighting the biogenic sensitivity by the sum of VOC and NOx sensitivities (Table S1), one can estimate ~17% biogenic emission contributions to O3.
Figure 3.
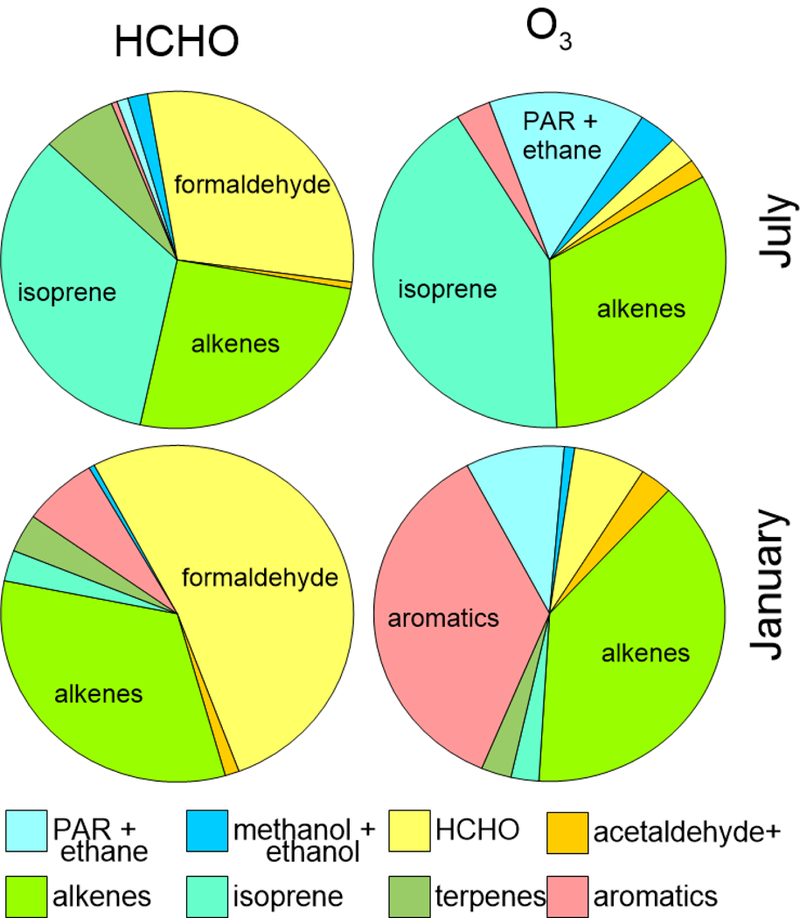
Relative distribution of sensitivityto individual VOCs, summed across all source sectors, for HCHO (left) and O3 (right) in July (top) and January (bottom). Results are summarized on a national basis and reflect relative sensitivities for each hydrocarbon to HCHO and O3.
Formation of O3 requires only a reaction that creates NO2, which occurs primarily through oxidation of NO by peroxy radicals, (every hydroperoxy and organic peroxy radical can participate). In contrast, formaldehyde production requires specific reaction pathways, previously described in Section 2.1. The ubiquity of HCHO as a product under both low- and high-NO chemistry, along with the direct emissions of HCHO, helps explain why HCHO is less sensitive to NOx than is O3. In addition, the lifetime of O3 is impacted by reaction with NO, while HCHO is not.
Methane also contributes to O3, having the second largest non-NOx sensitivity after biogenic VOCs.
In January, biogenic emissions are lower, so HCHO sensitivity to biogenic emissions is reduced, and residential wood combustion becomes relatively more important. Mobile sources remain large, contributing 30% to total sensitivity. The sensitivity of HCHO to NOx is smaller in absolute magnitude and negative (i.e. reducing NOx increases HCHO). The relative sensitivity of O3 to anthropogenic VOCs increases in winter, with mobile source VOCs increasing by a factor of 4, and other anthropogenic sources by 2–3. In January, O3 sensitivity to NOx is negative; mobile source sensitivities have similar magnitude as in July but opposite direction.
3.3. Sensitivity of formaldehyde and ozone to VOCs
Figure 3 shows contributions of modeled VOCs to both HCHO and O3 on a national basis. While both are sensitive to isoprene and other alkenes, HCHO is less sensitive to alkanes (model species PAR- representing singly bonded carbon atoms - and ethane). In January, atmospheric chemistry is slower, and HCHO is more sensitive to emissions. Biogenic emissions of isoprene are lower in winter, thus a smaller portion of the sensitivity, and anthropogenic aromatic emissions play a larger role.
Acetaldehyde is also a HAP and a national cancer risk driver.17 While acetaldehyde and formaldehyde have similar functionality, they have different precursor pathways. CMAQ generally overpredicts acetaldehyde, with an annual mean bias of 0.4 (27.1%) and mean error of 0.7 μg/m3 (41.4%).17 Figure 4 compares the relative sensitivity of acetaldehyde, HCHO and O3 to anthropogenic VOCs over the continental US. Each compound has different sensitivity to the same group of VOCs. Acetaldehyde concentrations are less influenced by direct emissions than HCHO, and more from alkanes (PAR+ethane). All three pollutants would respond differently to a cut in precursor VOCs. In January, acetaldehyde sensitivity is shifted towards primary emissions, with anthropogenic alkenes still important.
Figure 4.
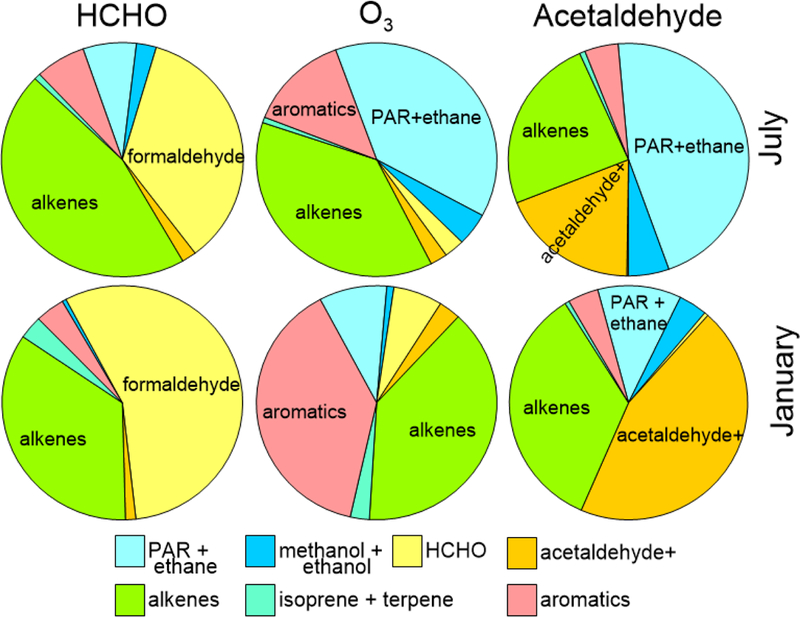
Comparison of Hcho, O3 and acetaldehyde senisitivites to modeled anthropogenic hydrocarbons on a national basis, July (top) and January (bottom)
O3 is formed in different yields from different VOCs, a concept used in reactivity scales that quantify the amount of O3 formed from a given quantity of individual VOC33,34 A similar reactivity scale for HCHO has not been developed. The total sensitivity of HCHO, O3 and acetaldehyde to different VOCs result from both “per-molecule” sensitivity (how much HCHO each VOC molecule produces) and total emissions of each VOC. When sensitivities are examined on a per-molecule basis, normalized by total emissions (SI Figure S5) some VOCs with high overall sensitivity, including model species PAR and ethane, are low on a per-molecule scale. In contrast, isoprene and terpenes have low anthropogenic emissions, resulting in low overall anthropogenic sensitivity (Figure 4), but high per-molecule sensitivity (Figure S3).
3.4. regional and anthropogenic/biogenic contributions
One goal of this study is to examine whether conclusions about contributors to HCHO sensitivity are valid across the whole US. To examine whether regional differences are substantial, we grouped states into five regions (SI Figure S6), based on the patterns of HCHO concentrations and sensitivities (Figures 1 and S1).
Despite large spatial variations in HCHO, there is little difference across the US in relative sensitivities to individual anthropogenic VOCs, with slightly larger differences for biogenic sources (Figure S7). Alkene species in CB05 (ETHE, OLE and IOLE) represent multiple biogenic VOCs, including ethene, propene, hexenol, methylbutenol, and other reactive VOCs. The Southeast is enriched in isoprene relative to other alkenes, whereas vegetation in California has high emissions of methylbutenol, resulting in larger alkene sensitivity in California and the West.
4. DISCUSSION
4.1. Potential role of uncertainty in the chemical mechanism representing VOCs
The chemistry condensations described in Section 2.1 are necessary to run an AQM efficiently with fine resolution or over large domains, but simplifying many details in the chemical reactions can introduce uncertainty into model predictions. These condensations are developed and evaluated primarily for accuracy in predicting O3,35 not as much for predictions of individual VOCs, and some pathways may be consolidated or eliminated without examining the impact on HCHO. Assumptions made on prompt yields of HCHO may reflect higher-NOx environments. Indeed, different techniques for condensing atmospheric chemistry can result in HCHO predictions that vary by 25–40%,36,37 due to differences in OH, HO2 or peroxy radicals,38 and VOC grouping. For example, 2-methyl-2-butene, represented by species OLE2 in the SAPRC07 chemical mechanism,39 yields 0.29 moles of HCHO after reaction with NO. In CB05, represented by IOLE, it yields acetaldehyde and larger aldehydes. We note that aside from the uncertainties related to condensation, there are still aspects of the detailed chemistry where HCHO yields are uncertain.
As discussed previously, CMAQ underestimates HCHO, as do other AQMs.12,15,40 Uncertainties in emissions may be a factor - an analysis from the 2005 National Emissions Inventory (NEI) found that HCHO emissions calculated from VOC speciation profiles can be different from reported emissions.41 One area of effort in recent NEI updates is identifying and improving sources where HCHO emissions may be in error. Quantifying emission uncertainties is difficult, however these uncertainties likely have less impact on the relative sensitivities, as done in this study, than on absolute concentrations.
Is the relative insensitivity of HCHO to NOx in CMAQ supported by previous research or an artifact of mechanism uncertainties? A recent analysis showed a link between NOx concentrations and HCHO formation from isoprene.42 In contrast, Valin et al.24 correlated HCHO to production of hydroxyl radical, or total VOC reactivity, or both, depending on OHOH concentrations, and found only 10–20% reductions in HCHO from a 90% anthropogenic NOx reduction. The influence of NOx on HCHO was attributed to feedback on production of OH, not from dominance of alkoxy radicals produced via NO to NO2 oxidation, as radical-radical reactions were sufficient to produce HCHO.24 Similarly, a GEOS-chem brute-force NOx zero-out simulation resulted in only 10–30% reductions in annual HCHO across the U.S.8 An examination of 10-year trends in HCHO column data, found that NOx decreases only explained about 20% of HCHO concentration changes; the trends were correlated to changes in activities which emit VOCs.43 Lastly, we note that O3 concentrations show weekday/weekend differences from generally lower NOx emissions on weekends. Pun et al.44 used routine observational measurements from 1995–2004 to search for a similar weekly trend in HCHO, but found no statistical differences between weekday and weekend HCHO concentrations at several cities across the US.44 This provides further evidence to support our conclusion of relatively low sensitivity of HCHO to NOx.
4.2. Atmospheric implications
The modeling study described here predicts that isoprene contributes about 1/3 of the total biogenic sensitivity of HCHO and O3 concentrations, but other alkenyl species, and direct HCHO emissions are equally important. Satellite column measurements of HCHO are used to evaluate and improve isoprene inventories,45,46 so it is important to accurately describe the role that isoprene plays in formation of HCHO, versus other biogenic VOCs. In January, neither HCHO nor O3 are sensitive to biogenic emissions, and anthropogenic precursors are important.
Our results showing that HCHO has much smaller sensitivity to NOx than VOCs indicates the importance of low NOx chemistry on HAP formation. When condensed chemical mechanisms such as CB05 are used to develop emission control strategies, these sensitivities must be confirmed so that impacts of NOx changes are not under-estimated. Dynamic evaluations would be useful to test out these sensitivities in “real life” atmospheres.
Multipollutant control strategies aim to develop emission reductions targeted at one important compound but with co-benefits for reducing other harmful pollutants. While both O3 and HCHO are formed under similar conditions (high actinic flux, availability of sufficient VOC and NOx), they show strikingly different sensitivities, although directionally similar. VOC-focused strategies to reduce O3 would be the most optimal for reducing HCHO, although NOx controls will have some positive impact. The sources with most similar relative contribution to anthropogenic sensitivity are mobile NOx in July and mobile VOC in both months.
Mechanism condensation can cause uncertainties in HCHO predictions. New techniques for chemical mechanism creation and condensation should consider a more multi-chemical perspective.47 Because HCHO is both a component of atmospheric chemistry and a HAP, a better understanding of both emissions and chemistry, including how to adequately represent HCHO formation in a condensed format, is essential.
Supplementary Material
ACKNOWLEDGEMENTS
The authors gratefully acknowledge Lukas Valin and Kirk Baker for their many helpful comments on this manuscript. Although this work was reviewed by EPA and approved for publication, it may not necessarily reflect official agency policy.
REFERENCES
- 1.Calvert JG; Orlando JJ; Stockwell WR; Wallington TJ The Mechanisms of Reactions Influencing Atmospheric Ozone: Oxford University Press: New York, NY, 2015. [Google Scholar]
- 2.Li Z; Schwier AN; Sareen N; McNeill VF Reactive processing of formaldehyde and acetaldehyde in aqueous aerosol mimics: surface tension depression and secondary organic products. Atmos. Chem. Phys 2011, 11, 11617–11629. [Google Scholar]
- 3.U.S. National Library of Medicine. Hazardous Substances Data Bank (HSDB): Formaldehyde; 2017; https://toxnet.nlm.nih.gov/newtoxnet/hsdb.htm.
- 4.Formaldehyde; https://cfpub.epa.gov/ncea/iris/iris_documents/documents/subst/0419_summary.pdf.
- 5.NATA overview; https://www.epa.gov/national-air-toxics-assessment/nata-overview.
- 6.Strum M; Scheffe R National review of ambient air toxics observations. Journal of the Air & Waste Management Association 2016, 66, 120–133. [DOI] [PubMed] [Google Scholar]
- 7.Scheffe RD; Strum M; Phillips SB; Thurman J; Eyth A; Fudge S; Morris M; Palma T; Cook R Hybrid Modeling Approach to Estimate Exposures of Hazardous Air Pollutants (HAPs) for the National Air Toxics Assessment (NATA). Environ. Sci. Technol 2016. 50, 12356–12364. [DOI] [PubMed] [Google Scholar]
- 8.Zhu L; Jacob DJ; Keutsch FN; Mickley LJ; Scheffe R; Strum M; Abad GG; Chance K; Yang K; Rappengluck B; Millet DB; Baasandorj M; Jaegle L; Shah V Formaldehyde (HCHO) as a Hazardous Air Pollutant: Mapping surface air concentrations from satellite and inferring cancer risks in the United States. Environ. Sci. Technol 2017, 51, 5650–5657. [DOI] [PubMed] [Google Scholar]
- 9.2011. National Emissions Inventory (NEI) Data; https://www.epa.gov/air-emissions-inventories/2011-national-emissions-inventory-nei-data
- 10.Zhang H; Li J; Ying Q; Guven BB; Olaguer EP Source apportionment of formaldehyde during TexAQS 2006 using a source-oriented chemical transport model. J. Geophys. Res.: Atmos 2013, 118, 1525–1535. [Google Scholar]
- 11.Parrish DD; Ryerson TB; Mellqvist J; Johansson J; Fried A; Richter D; Walega JG; Washenfelder RA; de Gouw JA; Peischl J; Aikin KC; McKeen SA; Frost GJ; Fehsenfeld FC; Herndon SC Primary and secondary sources of formaldehyde in urban atmospheres: Houston Texas region. Atmos. Chem. Phys 2012, 12, 3273–3288. [Google Scholar]
- 12.Stroud CA, Zaganescu C, Chen J, McLinden CA, Zhang J, Wang D Toxic volatile organic air pollutants across Canada: multi-year concentration trends, regional air quality modelling and source apportionment. J. Atmos. Chem 2016, 73, 137–164. [Google Scholar]
- 13.Ling ZH, Zhao J, Fan SJ, Wang XM Sources of formaldehyde and their contributions to photochemical O3 formation at an urban site in the Pearl River Delta, southern China. Chemosphere 2017, 168, 1293–1301. [DOI] [PubMed] [Google Scholar]
- 14.Dunker AM; Koo B.; Yarwood G. Source Apportionment of the Anthropogenic Increment to Ozone, Formaldehyde, and Nitrogen Dioxide by the Path-Integral Method. Environ. Sci. Technol 2015, 49, 6751−67. [DOI] [PubMed] [Google Scholar]
- 15.Luecken DJ; Hutzell WT; Strum ML; Pouliot GA Regional sources of atmospheric formaldehyde and acetaldehyde, and implications for atmospheric modeling. Atmos. Env 2012, 47, 477–490. [Google Scholar]
- 16.Hakami A; Odman MT; Russell AG High-Order, Direct Sensitivity Analysis of Multidimensional Air Quality Models. Env. Sci.Tech 2003, 37, 2442–2452. [DOI] [PubMed] [Google Scholar]
- 17.Technical Support Document, EPA’s 2011. National-scale Air Toxics Assessment; U.S. Environmental Protection Agency , Office of Air Quality, Planning, and Standards, Research Triangle Park, NC, 2015; https://www.epa.gov/sites/production/files/2015-12/documents/2011-nata-tsd.pdf [Google Scholar]
- 18.Aumont B; Szopa S; Madronich S Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach. Atmos. Chem. Phys 2005, 5, 2497–2517. [Google Scholar]
- 19.Yarwood G, Rao S, Yocke M, Whitten GZ Updates to the Carbon Bond Mechanism: CB05. Final Report to the US EPA, RT-0400675 Yocke and Company, Novato, CA, 2005. [Google Scholar]
- 20.Whitten GZ; Heo G; Kimura Y; McDonald-Buller E; Allen DT; Carter WPL; Yarwood G A new condensed toluene mechanism for Carbon Bond: CB05-TU. Atmos. Env 2010, 44, 5346–5355. [Google Scholar]
- 21.Luecken DJ; Hutzell WT; Gipson GL Development and analysis of air quality modeling simulations for hazardous air pollutants. Atmos. Env 2006, 40, 5087–5096. [Google Scholar]
- 22.Calvert JG; Atkinson R; Kerr JA; Madronich S; Moortgat GK; Wallington TJ; Yarwood G The mechanisms of atmospheric oxidation of the alkenes; Oxford University Press: New York/Oxford, 2000. [Google Scholar]
- 23.Orlando JJ; Tyndall GS Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance. Chem. Soc. Rev 2012, 41, 6294–6317. [DOI] [PubMed] [Google Scholar]
- 24.Valin LC; Fiore AM; Chance K; González Abad G The role of OH production in interpreting the variability of CH2O columns in the southeast U.S. J. Geophys. Res.: Atmos 2016, 121, 478–493. [Google Scholar]
- 25.Bash JO; Baker KR; Beaver MR Evaluation of improved land use and canopy representation in BEIS v3. 61 with biogenic VOC measurements in California. Geosci. Model Dev 2016, 9, 2191. [Google Scholar]
- 26.Appel KW; Napelenok SL; Foley KM; Pye HOT; Hogrefe C; Luecken DJ; Bash JO; Roselle SJ; Pleim JE; Foroutan H; Hutzell WT; Pouliot GA; Sarwar G; Fahey KM; Gantt B; Gilliam RC; Kang D; Mathur R; Schwede DB; Spero TL; Wong DC; Young JO Overview and evaluation of the Community Multiscale Air Quality (CMAQ) model version 5.1. Geosci. Model Dev 2016, 10, 1703–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nolte CG; Appel KW; Kelly JT; Bhave PV; Fahey KM; Collett JL Jr; Zhang L; Young JO Evaluation of the Community Multiscale Air Quality (CMAQ) model v5.0 against size-resolved measurements of inorganic particle composition across sites in North America. Geosci. Model Dev 2015, 8, 2877–2892. [Google Scholar]
- 28.Napelenok SL; Cohan DS; Hu Y; Russell AG Decoupled direct 3D sensitivity analysis for particulate matter (DDM-3D/PM). Atmos. Env 2006, 40, 6112–6121. [Google Scholar]
- 29.Yang YJ; Wilkinson JG; Russell AG Fast, direct sensitivity analysis of multidimensional photochemical models. Environ. Sci. Tech 1997, 31, 2859–2868. [Google Scholar]
- 30.Napelenok SL; Cohan DS; Odman MT; Tonse S Extension and evaluation of sensitivity analysis capabilities in a photochemical model. Environ. Mod. Software 2008, 23, 994–999. [Google Scholar]
- 31.Cohan DS; Hakami A; Hu Y; Russell AG Nonlinear response of ozone to emissions: source apportionment and sensitivity analysis. Environ. Sci. Tech 2005, 39, 6739–6748. [DOI] [PubMed] [Google Scholar]
- 32.Zhang R; Cohan A; Pour Biazar A; Cohan DS Source apportionment of biogenic contributions to ozone formation over the United States. Atmos. Env 2017, 164, 8–19. [Google Scholar]
- 33.Carter WPL Development of ozone reactivity scales for volatile organic compounds. J. Air Waste Man. Assoc 1994, 44, 881–899. [Google Scholar]
- 34.Derwent RG; Jenkin ME; Pilling MJ; Carter WPL; Kaduwela A, 2010. Reactivity Scales as Comparative Tools for Chemical Mechanisms. J. Air Waste Man. Assoc 2010, 60, 914–924. [DOI] [PubMed] [Google Scholar]
- 35.Carter WPL Development of a condensed SAPRC-07 chemical mechanism. Atmos. Env 2010, 44, 5336–5345. [Google Scholar]
- 36.Knote C; Tuccella P; Curci G; Emmons L; Orlando JJ; Madronich S; Baró R; Jiménez-Guerrero P; Luecken D; Hogrefe C; Forkel R; Werhahn J; Hirtl M; Pérez JL; San José R; Giordano L; Brunner D; Yahya K; Zhang Y Influence of the choice of gas-phase mechanism on predictions of key gaseous pollutants during the AQMEII phase-2 intercomparison. Atmos. Env 2015, 115, 553–568. [Google Scholar]
- 37.Luecken DJ; Phillips S; Sarwar G; Jang C Effects of using the CB05 vs. SAPRC99 vs. CB4 chemical mechanism on model predictions: Ozone and gas-phase photochemical precursor concentrations. Atmos. Env 2008, 42, 5805–5820. [Google Scholar]
- 38.Derwent R Intercomparison of chemical mechanisms for air quality policy formulation and assessment under North American conditions. J. Air Waste Man. Assoc 2017, 67, 789–796. [DOI] [PubMed] [Google Scholar]
- 39.Carter WPL Development of the SAPRC-07 chemical mechanism. Atmos. Env 2010, 44, 5324–5335. [Google Scholar]
- 40.Marvin MR; Wolfe GM; Salawitch RJ; Canty TP; Roberts SJ; Travis KR; Aikin KC; de Gouw JA; Graus M; Hanisco TF; Holloway JS; Hübler G; Kaiser J; Keutsch FN; Peischl J; Pollack IB; Roberts JM; Ryerson TB; Veres PR; Warneke C Impact of evolving isoprene mechanisms on simulated formaldehyde: An inter-comparison supported by in situ observations from SENEX. Atmos. Env 2017, 164, 325–336. [Google Scholar]
- 41.Simon H; Beck L; Bhave P; Divita F; Hsu Y; Luecken D; Mobley D; Pouliot G; Reff A; Sarwar G; Strum M The development and uses of EPA’s SPECIATE database Atmos. Poll. Res 2010, 1, 196–206 [Google Scholar]
- 42.Wolfe GM; Kaiser J; Hanisco TF; Keutsch FN; de Gouw JA; Gilman JB; Graus M; Hatch CD; Holloway J; Horowitz LW; Lee BH; Lerner BM; Lopez-Hilifiker F; Mao J; Marvin MR; Peischl J; Pollack IB; Roberts JM; Ryerson TB; Thornton JA; Veres PR; Warneke C Formaldehyde production from isoprene oxidation across NOx regimes. Atmos. Chem. Phys 2016, 16, 2597–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu L; Mickley LJ; Jacob DJ; Marais EA; Sheng J; Hu L; Abad GG; Chance K 2017. Long-term (2005–2014) trends in formaldehyde (HCHO) columns across North America as seen by the OMI satellite instrument: Evidence of changing emissions of volatile organic compounds. Geophys. Res. Lett 2017, 44, 7079–7086. [Google Scholar]
- 44.Pun BK; Lohman K; Seigneur C Weekday/weekend differences in concentrations of air toxics in New York, NY; Philadelphia, PA; and Houston, TX: Task 2 Report, Project A-49, prepared for Coordinating Research Council, Atmospheric and Environmental Research, Inc., San Ramon, CA, 2005; https://www.crcao.org/reports/recentstudies2006/A-49%20Task%202%20Final%20Report.pdf [Google Scholar]
- 45.Palmer PI; Abbot DS; Fu T-M; Jacob DJ; Chance K; Kurosu TP; Guenther A; Wiedinmyer C; Stanton JC; Pilling MJ; Pressley SN; Lamb B; Sumner AL Quantifying the seasonal and interannual variability of North American isoprene emissions using satellite observations of the formaldehyde column. J. Geophys. Res 2006, 111, D12315. [Google Scholar]
- 46.Bauwens M; Stavrakou T; Müller JF; De Smedt I; Van Roozendael M; van der Werf GR; Wiedinmyer C; Kaiser JW; Sindelarova K; Guenther A Nine years of global hydrocarbon emissions based on source inversion of OMI formaldehyde observations. Atmos. Chem. Phys 2016, 16, 10133–10158. [Google Scholar]
- 47.Kaduwela A; Luecken D; Carter W; Derwent R New directions: Atmospheric chemical mechanisms for the future. Atmos. Env 2015, 122, 609–610. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.