

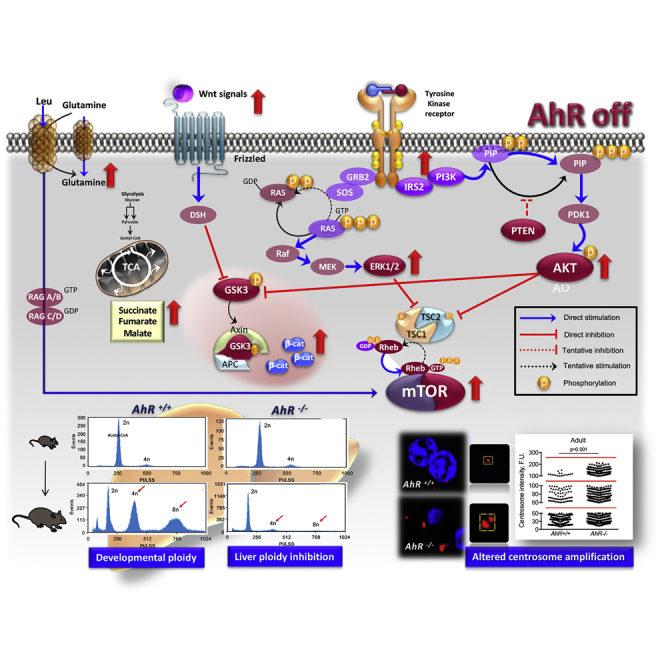
Summary
Aryl hydrocarbon receptor (AhR) deficiency alters tissue homeostasis. However, how AhR regulates organ maturation and differentiation remains mostly unknown. Liver differentiation entails a polyploidization process fundamental for cell growth, metabolism, and stress responses. Here, we report that AhR regulates polyploidization during the preweaning-to-adult mouse liver maturation. Preweaning AhR-null (AhR−/−) livers had smaller hepatocytes, hypercellularity, altered cell cycle regulation, and enhanced proliferation. Those phenotypes persisted in adult AhR−/− mice and correlated with compromised polyploidy, predominance of diploid hepatocytes, and enlarged centrosomes. Phosphatidylinositol-3-phosphate kinase (PI3K), extracellular signal-regulated kinase (ERK), and Wnt/β-catenin signaling remained upregulated from preweaning to adult AhR-null liver, likely increasing mammalian target of rapamycin (mTOR) activation. Metabolomics revealed the deregulation of mitochondrial oxidative phosphorylation intermediates succinate and fumarate in AhR−/− liver. Consistently, PI3K, ERK, and Wnt/β-catenin inhibition partially rescued polyploidy in AhR−/− mice. Thus, AhR may integrate survival, proliferation, and metabolism for liver polyploidization. Since tumor cells tend to be polyploid, AhR modulation could have therapeutic value in the liver.
Subject Areas: Developmental Biology, Cancer Systems Biology, Metabolomics
Graphical Abstract
Highlights
-
•
AhR is required for liver polyploidization during preweaning-to-adult transition
-
•
INS-R/PI3K/AKT, ERK, Wnt/β-Cat and mTOR are downregulated during liver polyploidization
-
•
Reduced polyploidy relates with enhanced mitochondrial metabolism in AhR-null liver
-
•
Understanding how AhR modulates polyploidy may provide strategies against cancer
Developmental Biology; Cancer Systems Biology; Metabolomics
Introduction
Most eukaryotic cells have a diploid cell cycle in which chromosomes are replicated only once during each cell division to generate two identical 2n daughter cells. Nevertheless, certain organisms can undergo successive rounds of genome duplication in the absence of cytokinesis to acquire a polyploid status that can involve the whole organism or just specific tissues and organs (Edgar et al., 2014, Fox and Duronio, 2013, Schoenfelder and Fox, 2015). Among mammals, including humans, polyploidy is particularly relevant in the liver hepatocytes, megakaryocytes, and placental giant trophoblast cells, although it also takes place in the heart and muscle (Gentric et al., 2012, Hannibal et al., 2014). In the liver, the percentage of polyploid hepatocytes ranges from 80% to 90% in rats, to 30% in humans, and to close to 50% in mice (Celton-Morizur et al., 2010, Duncan et al., 2010). Polyploidy does not seem to be a uniform phenomenon, and usually aneuploid cells with altered number of chromosomes co-exist with cells having ≥2n DNA content (Schoenfelder and Fox, 2015). Several mechanisms have been proposed to explain physiological polyploidy, including failure to complete cytokinesis after mitosis of diploid hepatocytes (Guidotti et al., 2003), cell fusion of placental cells, or endomitosis in megakaryocytes (Ullah et al., 2009, Zielke et al., 2013). Recent reports have suggested that polyploidization may not be an irreversible process since mouse hepatocytes with one-half chromosomal content can be obtained from polyploid liver cells by artificial cell fusion (Duncan et al., 2009, Wang et al., 2003). Such phenomena would then generate mixed cell populations with dissimilar number of chromosomes by a process named ploidy conveyor (Duncan et al., 2010).
Physiological polyploidy takes place in the mouse liver right after the transition from preweaning (approximately 3 weeks after birth) to adulthood (Marques et al., 2008, Pandit et al., 2012, Pandit et al., 2013). During this period, immature hepatoblasts start to differentiate into mature hepatocytes concomitantly with a reduction in their proliferative ability (Germain et al., 1988, Shiojiri et al., 1991). From a functional perspective, polyploidy induces a terminal differentiated phenotype that increases cell size, amplifies gene expression, helps tissue organization, and modifies hepatic metabolism (Schoenfelder and Fox, 2015, Zielke et al., 2013). In fact, transcriptomic studies have revealed that larger polyploid hepatocytes switch their metabolism from mitochondrial oxidative phosphorylation to glycolysis to adapt to metabolic stresses, although some controversy exists on the relative contribution of ploidy versus cell size in their metabolic reprogramming (Miettinen et al., 2014). The liver is exposed to the deleterious effects of endo- and xenobiotics and, as such, has the remarkable property to regenerate upon injury (Taub, 2004). Polyploid hepatocytes can therefore trigger a response against toxic compounds by entering mitosis and generating proliferative hepatocytes that will reconstitute the damaged parenchyma (Duncan et al., 2012). Importantly, several human cancers seem to contain polyploid cells that can re-enter cell cycle acquiring Warburg-like glycolytic metabolism and contributing to tumor growth (Davoli and de Lange, 2011, Ganem et al., 2007, Zack et al., 2013).
The aryl hydrocarbon/dioxin receptor (AhR) has many different physiological and homeostatic functions some of which are now beginning to emerge. Mouse models have shown that complete AhR depletion alters the development and function of several organs, including the liver, heart, skin, and immune system (Esser and Rannug, 2015, Mulero-Navarro and Fernandez-Salguero, 2016, Pohjanvirta, 2012, Puga et al., 2009, Schmidt and Bradfield, 1996). Despite early studies showing that adult AhR-null mice (AhR−/−) have smaller livers (Fernandez-Salguero et al., 1995, Fernandez-Salguero et al., 1997, Schmidt et al., 1996) with embryo-derived intrahepatic portosystemic shunt (Lahvis et al., 2000, Schmidt et al., 1996), there are no studies exploring how AhR affects liver maturation and differentiation during the critical postnatal to adulthood developmental window.
In this work, we have investigated whether AhR is needed for the diploid-to-polyploid conversion that takes place during the transition from an immature to an adult liver. AhR deficiency severely compromised the generation of polyploid hepatocytes and maintained a more proliferative liver under physiological conditions. Persistent upregulation of signaling pathways controlling survival, proliferation, and metabolism in adult AhR-null liver likely compromised polyploidization and favored a diploid and undifferentiated phenotype. Such attributes could greatly influence the regenerative competence of the AhR−/− liver in the short term as well as its increased ability to develop hepatocarcinomas upon carcinogen exposure in the long term (Moreno-Marín et al., 2017). Thus, AhR is a relevant component of a complex signaling network controlling physiological liver polyploidy and differentiation. Selective AhR modulators may be useful to regulate ploidy-related liver responses such as those required for regeneration after toxic damage or surgical intervention or for inhibition of tumor progression.
Results
AhR Deficiency Increases Liver Cellularity and Proliferation and Impairs Adult Polyploidy
Early reports showed that AhR-null mice (AhR−/−) have developmental hepatic alterations, including a reduced organ size (Fernandez-Salguero et al., 1995, Schmidt et al., 1996). In this work, we have used mice around the time of weaning (25 days of age, hereafter preweaning) and adult mice (9–10 weeks of age) having or lacking AhR expression. Histological examination of liver sections from preweaning and adult AhR−/− mice suggested an increase in cellularity with respect to age-matched AhR+/+ mice (Figure 1A), despite the smaller liver size of AhR-null preweaning mice (Figure 1B). Adult AhR−/− mice also have a significant reduction in liver size and weight as previously reported (Fernandez-Salguero et al., 1995, Schmidt et al., 1996). This de visu observation was confirmed by cell counting and, indeed, AhR−/− livers had significantly higher numbers of hepatocytes than AhR+/+ livers at preweaning and adult age (Figure 1C). Cellularity in adults was reduced with respect to preweaning livers regardless of mice genotype, possibly because of a normal developmental process that decreased cell proliferation and increased cell growth (Figure 1C). Accordingly, the average nuclear area of 4',6-diamidino-2-phenylindole (DAPI)-stained AhR−/− hepatocytes was significantly smaller in both preweaning and adult livers (Figure 1D). In addition, the nuclei of preweaning hepatocytes were smaller than those of adult mice, irrespective of AhR expression (Figure 1D). Liver maturation involves several important cytological changes, including the appearance of binucleated and mononucleated polyploid hepatocytes (Gerlyng et al., 1993, Schoenfelder and Fox, 2015, Zielke et al., 2013). Interestingly, confocal fluorescence microscopy of DAPI-stained liver sections revealed that whereas binucleated hepatocytes accounted for 25%–30% of liver cells in adult AhR+/+ mice, they represented only about 10% of liver cells in aged-matched AhR−/− mice (Figure 1E). Binucleated hepatocytes were less abundant in preweaning mice and, in particular, in those lacking AhR expression (Figure 1E).
Figure 1.

AhR-Null Liver has Hypercellularity, Increased Nuclear Size, and Reduced Number of Binucleated Hepatocytes
(A) Livers were collected from preweaning and adult AhR+/+ and AhR−/− mice, fixed, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E).
(B) Relative liver weight was obtained in preweaning mice relative to the total body weight of each animal.
(C) Liver cellularity was quantified in tissue sections obtained from preweaning and adult AhR mice of each genotype.
(D) Nuclear area of hepatocytes was determined in AhR+/+ and AhR−/− liver sections from preweaning and adult mice after staining with DAPI. ImageJ software was used.
(E) Binucleated cells in preweaning and adult AhR+/+ and AhR−/− livers were quantified by confocal fluorescence microscopy using DAPI-stained sections. Six mice for each developmental time and genotype and three technical replicates were analyzed.
Data are shown as mean ± SD. Scale bar, 50 μm; scale bar in inset, 50 μm. Nuclear area is represented as the integrated density (IntDen) measured by the ImageJ software in micrographs taken at the same magnification and resolution. SD, standard deviation.
Altogether, these results suggested that lack of AhR could compromise physiological control of hepatocyte proliferation and the preweaning-to-adult transition in mouse liver. We then decided to analyze if the AhR-null phenotype could involve altered polyploidization since an increase in ploidy reduces proliferation and induces differentiation in the liver (Davoli and de Lange, 2011, Gentric and Desdouets, 2014, Ullah et al., 2009). Flow cytometry analysis of the DNA content of primary hepatocytes isolated from preweaning mice revealed that most cells were diploid (2c) in both AhR+/+ and AhR−/− livers and that the amount of tetraploid (4c) and octaploid (8c) cells was minimal (Figures 2A, 2C, and S1). By contrast, adult AhR+/+ livers became significantly enriched in 4c and 8c hepatocytes, whereas such enrichment was not found in AhR−/− hepatocytes, which remained mostly diploid (Figures 2B and 2D). A marked asymmetry in polyploidy was therefore observed between adult AhR+/+ and AhR−/− livers (Figure 2E) that did not appear to be due to a significant level of endogenous apoptotic cell death (Figure 2F). Therefore, AhR controls normal liver architecture and cellularity and the nuclear content and ploidy of hepatocytes. Since polyploidization is related to liver differentiation, we next examined changes in albumin levels in preweaning and adult mice. Indeed, albumin messenger RNA (mRNA) (Figure 2G) and protein (Figure 2H) levels were significantly downregulated in AhR−/− livers at either developmental stage, suggesting that AhR expression is needed for physiological liver maturation.
Figure 2.

AhR Deficiency Impairs Liver Polyploidy in the Absence of Increased Apoptosis
(A–D) Primary hepatocytes were isolated from preweaning (A and C) and adult (B and D) AhR+/+ and AhR−/− mice by collagenase liver perfusion. Cells were fixed, stained with propidium iodide, and their DNA content analyzed by flow cytometry in a MACSQuant VYB flow cytometer. Peaks correspond to diploid (2c), tetraploid (4c), and octaploid (8c) hepatocytes (red arrows).
(E) Cell subpopulations with different ploidy status were quantified and their percentages represented. Percentage of cells at the S phase (2c-to-4c and 4c-to-8c) transitions are also indicated.
(F) Apoptotic cells in adult AhR−/− livers were quantified and normalized by those of AhR+/+ mice.
(G and H) Albumin mRNA (G) and protein (H) expression was determined in liver tissue from preweaning and adult AhR+/+ and AhR−/− mice. mRNA gene expression was normalized by Gapdh and represented as 2−ΔΔCt.
Six AhR+/+ and seven AhR−/− mice were analyzed for each developmental time and four technical replicates were performed. Data are shown as mean ± SD. n.s., Not statistically significant; SD, standard deviation. Antibodies and oligonucleotides used are indicated in Tables S1 and S2, respectively. See also Figure S1.
AhR is known to regulate cell cycle progression by interacting with retinoblastoma protein (RB) and blocking E2F-dependent transcription of target genes such as Cyclin E (Gao et al., 2016, Mitchell et al., 2006, Pohjanvirta, 2012, Puga et al., 2002). AhR activation by TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin) also increases tumor suppressor p27Kip1, which, in turn, inactivates Cyclin E (Kolluri et al., 1999, Siu et al., 2012). We then decided to analyze cell proliferation in mice liver in vivo. Ki67 staining of preweaning livers revealed high rates of cell proliferation in both genotypes, the number of proliferating cells being much higher in the absence of AhR than in control mice (Figure 3A). For adults, multiphoton confocal microscopy was used on tissue sections stained for the proliferating cell nuclear antigen (PCNA) because of the low rates of cell proliferation usually present in the matured liver. Although cell proliferation was markedly reduced with aging, AhR−/− livers still had more proliferative cells than AhR+/+ livers (Figure 3A). Such increased proliferative potential of AhR−/− hepatocytes was associated with a higher number of cells passing through the G0/G1 (2c) and G2/M (4c) phases of the cell cycle, as determined by flow cytometry examination of freshly isolated primary hepatocytes (Figure 3B). Surprisingly, however, cell cycle regulators Cyclin B1 (G2/M) and Cyclin E (G1/S) were downregulated in liver from preweaning AhR−/− mice when compared with AhR+/+ mice (Figures 3C and 3D). An opposite pattern of expression was observed in adult mice because both Cyclin B1 and Cyclin E were upregulated in AhR-null livers (Figures 3C and 3D). In addition, the expression of cyclin-dependent kinase inhibitory protein p27Kip1 was markedly reduced in preweaning and adult AhR−/− livers (Figure 3E). No significant differences in Cyclin D1 (G1/S) were found between AhR+/+ and AhR−/− livers at any developmental stage (not shown). These results suggest that high Cyclin B1 and Cyclin E expression, in parallel to reduced p27Kip1 levels, could maintain proliferation and inhibit polyploidy in AhR−/− liver, in agreement with the role of Cip/Kip proteins in promoting differentiation and polyploidy in mammalian cells (Ullah et al., 2009). We next determined if the preweaning-to-adult transition involved changes in AhR expression. Immunoblotting analysis revealed that AhR protein levels were significantly lower in adult livers than in AhR+/+ preweaning livers; AhR protein remained undetectable at any developmental stage in AhR−/− mice (Figure 3F).
Figure 3.

AhR-Null Livers Have Increased Proliferation and Changes in Cell Cycle Regulators Cyclin E and p27Kip1
(A) In vivo proliferation was determined in Ki67-stained tissue sections from preweaning AhR+/+ and AhR−/− mouse liver. Adult livers from AhR+/+ and AhR−/− mice were analyzed by confocal multiphoton microscopy using tissue sections stained for the proliferating cell nuclear antigen (PCNA). An Olympus FV1000 confocal microscope (Olympus) equipped with a multiphoton unit was used. Arrowheads mark proliferation positive hepatocytes.
(B) Primary hepatocytes were isolated from adult AhR wild-type and AhR-null mice by collagenase perfusion and freshly analyzed for cell cycle distribution by flow cytometry after staining with propidium iodide. Fraction of cells in G0/G1 (2c), S (2 < c > 4), and G2/M (4c) are represented. A Cytomics FC500 equipment was used.
(C–E) Total protein was obtained from preweaning and adult AhR+/+ and AhR−/− livers and analyzed by immunoblotting for Cyclin B1 (C), Cyclin E (D), and p27Kip1 (D) using specific antibodies.
(F) AhR protein expression was also analyzed in preweaning (PW) and adult (AD) liver extracts from AhR+/+ and AhR−/− by immunoblotting. β-Actin was used to confirm protein integrity and equal loading.
Two representative mice for each experimental condition are shown. Seven mice for each developmental time and genotype and three technical replicates were analyzed. Data are shown as mean ± SD. n.s., Not statistically significant; SD, standard deviation. Scale bar, 50 μm. Antibodies used are indicated in Table S1.
Persistent INS-R/PI3K-Dependent Signaling and Sustained ERK1/2 Activation in Non-polyploid AhR-Null Livers
To investigate the possible signaling networks that could mediate the AhR-null liver phenotype, we first focused on the insulin receptor (INS-R) and its downstream PI3K (phosphatidylinositol-3-phosphate kinase) pathway since it is a critical regulator of cell viability, proliferation, and eventually, ploidy (Celton-Morizur et al., 2010, Yu and Cui, 2016). INS-R protein levels did not significantly differ between AhR+/+ and AhR−/− livers in preweaning or adult mice (Figure 4A). Although liver maturation seemed to involve a reduction in INS-R expression, it was AhR independent (Figure 4A). Notably, activation of the major INS-R intermediate protein in the liver phospho-IRS-2 (insulin receptor substrate-2) was significantly higher in AhR−/− than in AhR+/+ mice at both preweaning and adult age (Figure 4B), suggesting that despite similar receptor levels INS-R signaling might be increased in the absence of AhR. We next decided to address whether INS-R/IRS-2 overactivation was functionally relevant in modulating PI3K signaling in AhR−/−liver. The expression levels of the PI3K regulatory subunit, which interacts with phospho-IRS-2, p85α, were significantly increased in preweaning and adult AhR−/− livers when compared with AhR+/+ livers (Figure 4C).
Figure 4.

Insulin Receptor Signaling Is Altered in AhR-null Livers and the INS-R/PI3K Pathway May Contribute to the Increased Proliferative Potential of AhR−/− Livers
(A) Protein expression of the insulin receptor (INS-R) was analyzed by immunoblotting in liver extracts from preweaning (PW) and adult (AD) AhR+/+ and AhR−/− mice.
(B) Activation of the INS-R signaling intermediate IRS-2 was quantified by an enzyme-linked immunosorbent assay (ELISA) kit that detects the phosphorylated form of the protein. A positive control was performed using mice treated with insulin.
(C) Protein levels of IRS-2 downstream intermediate p85-PI3K were determined by immunoblotting using the experimental conditions indicated above.
(D and E) Immunoblotting using specific antibodies was used to analyze activation levels of molecular intermediates of the INS-R/PI3K pathway: AKT (total AKT and p-AKTSer473) (D) and GSK3β (total GSK3β and p-GSK3βSer9) (E).
(F) Protein levels of the PI3K-negative regulator PTEN were also analyzed by immunoblotting. β-Actin was used to confirm protein integrity and equal loading.
Two representative mice for each experimental condition are shown. Ten mice for each developmental time and genotype and three to four technical replicates were analyzed. Data are shown as mean ± SD. n.s., Not statistically significant; SD, standard deviation. Antibodies used are indicated in Table S1.
Serine-threonine protein kinase-B/AKT (hereinafter AKT) is the required PI3K signaling intermediate in most cell types (Yu and Cui, 2016). Total protein levels of AKT were not affected by AhR expression at any of the developmental times analyzed. However, active phospho-AKT (p-AKTSer473) was significantly upregulated by AhR deficiency in both preweaning and adult livers, and total levels in adults exceed those in preweaning mice (Figure 4D). Consistently, p-AKT target protein glycogen synthase kinase-3β (GSK3β) was more efficiently phosphorylated (p-GSK3βSer9) in preweaning and adult AhR−/− livers than in their AhR+/+ counterparts with a pattern closely resembling that of p-AKT (Figure 4E). PI3K activity and AKT phosphorylation are negatively regulated by the phosphatase and tensin homolog (PTEN) (Bunney and Katan, 2010). Accordingly, PTEN was significantly downregulated in preweaning and adult AhR−/− livers with a pattern inverse to that found for p-AKT (Figure 4F). Altogether, these results suggest that the increased proliferative potential of AhR−/− livers may be associated with a sustained overactivation of the INS-R/PI3K pathway. PI3K also signals to the Ras pathway, ultimately regulating the activation of mitogen-activated protein kinases (MAPKs) involved in cell proliferation, including the extracellular signal-regulated kinases 1/2 (ERK1/2) (Busca et al., 2016, Sturgill, 2008). We observed that the total levels of ERK1 (ERK1/p44) did not significantly change between AhR+/+ and AhR−/− livers in preweaning or adult mice. However, normalized phosphorylated ERK1/p44 levels (pERK/ERK ratio) increased in the absence of AhR under both developmental conditions (Figures 5A and 5B). The ERK2/p42 isoform did not show significant differences in normalized phosphorylation levels between AhR wild-type and AhR-null preweaning mice, although it was upregulated in adult AhR−/− livers (Figures 5A and 5C). Thus, sustained PI3K-dependent ERK1/2 signaling may also contribute to altered preweaning-to-adult transition in AhR-deficient liver.
Figure 5.

Liver Maturation Involves Changes in ERK1/2, p53, and p21Cip1
(A–E) Preweaning (PW) and adult (AD) AhR+/+ and AhR−/− livers were processed to obtain total protein extracts that were analyzed by immunoblotting for total and phosphorylated ERK1/p44 and ERK2/p42 (ERK1/2, p-ERK1/2Thr202/Tyr204) (A–C), p53 (D), and p21Cip1 (E). β-Actin was used to confirm protein integrity and equal loading. Two representative mice for each experimental condition are shown. Ten mice for each developmental time and genotype and three technical replicates were analyzed. Data are shown as mean ± SD. n.s., Not statistically significant; SD, standard deviation. Antibodies used are indicated in Table S1.
PI3K also signals to inhibit the p53 tumor suppressor to block apoptosis in proliferating cells (Sabbatini and McCormick, 1999, Yamaguchi et al., 2001). Notably, recent studies have also shown that p53 has relevant functions in preventing polyploidy in mature cells (Aylon and Oren, 2011, Kurinna et al., 2013). We then sought to analyze whether liver maturation involved changes in p53 expression in an AhR-dependent manner. Immunoblotting experiments showed that p53 levels were markedly reduced in preweaning AhR−/− livers with respect to AhR+/+ livers (Figure 5D). One of the most relevant targets of active p53 is the p21Cip1 protein (p21Cip1), also involved in repressing cell proliferation (Jung et al., 2010, Karimian et al., 2016). Accordingly, its expression closely followed that of p53 in preweaning AhR−/− and AhR+/+ livers, being significantly downregulated in AhR-null mice (Figure 5E). Surprisingly, however, p53 expression was higher in adult AhR−/− livers than in age-matched AhR+/+ livers (Figure 5D), despite the persistent p21Cip1 repression present in AhR-lacking hepatocytes (Figure 5E). Thus, low p53 levels may allow higher proliferation rates during preweaning in AhR−/− mice, whereas its increasing expression in adults could block polyploidy in the absence of a significant inhibitory effect on proliferation.
Lack of Polyploidy in Adult AhR−/− Liver Involves Altered Wnt/β-Cat Signaling
The PI3K pathway is also linked to Wnt/β-Cat signaling since the AKT downstream target GSK3β is a component of the Wnt/β-Cat degradation complex (Nusse and Clevers, 2017). Immunofluorescence analysis by confocal microscopy of liver sections from preweaning mice revealed increased levels and a more abundant nuclear localization of β-Cat in the centrilobular areas of the hepatic parenchyma in AhR−/− than in AhR+/+ mice (Figure 6A), in agreement with the zonation of β-Cat in the liver (Benhamouche et al., 2006, Burke et al., 2009). Immunoblotting of nuclear extracts also showed increased levels of β-Cat in AhR-null preweaning mice (Figure 6A). The difference in β-Cat expression between AhR+/+ and AhR−/− livers was milder in adult mice, and its overall levels were reduced with respect to preweaning mice of either phenotype (Figure 6A). Nevertheless, β-Cat was active in adult AhR−/− liver since the mRNA expression of its target genes Axin, Cyclin D1, c-Myc, and Lef1 was increased with respect to AhR+/+ adult liver (Figure 6B). Interestingly, co-immunoprecipitation experiments showed that, in fact, AhR may be a component of a protein complex that also includes β-Cat in both preweaning and adult livers (Figure 6C); no significant co-immunoprecipitation between AhR and β-Cat was detected in preweaning or adult AhR−/− livers (Figure 6C). In addition, β-Cat seemed to also interact in a common protein complex with activated phospho-AKT in preweaning and adult livers and with increased efficiency in AhR−/− mice (Figure 6D). Thus, in vivo, upregulated AKT and β-Cat signaling may cooperate to maintain hepatocyte proliferation and to reduce their ploidy in the absence of AhR expression.
Figure 6.

β-Cat Expression and Signaling Are Upregulated in AhR-Deficient Liver and AhR Modulates Wnt/β-Cat-Dependent Transcription in Mouse Primary Hepatocytes
(A) Liver tissue was obtained from preweaning and adult AhR+/+ and AhR−/− mice, fixed, embedded in paraffin, sectioned, and analyzed by immunofluorescence for β-Cat expression using a specific primary antibody and an Alexa 633-labeled secondary antibody. Sections were visualized using an Olympus FV1000 confocal microscope (Olympus). DAPI was used to label cell nuclei. Nuclear extracts (A, left panel) and total liver protein (A, right panel) were also analyzed for β-Cat protein levels by immunoblotting using a specific antibody.
(B) Total RNA was purified from adult AhR+/+ and AhR−/− liver, reverse transcribed, and mRNA gene expression for the β-Cat target genes Axin2, Cyclin D1, and c-Myc and Lef1 were quantified by real-time quantitative polymerase chain reaction (qPCR) using the oligonucleotides indicated in Table S1.
(C and D) Total liver protein from preweaning (PW) and adult (AD) AhR+/+ and AhR−/− mice was immunoprecipitated for AhR (C) or pAKT (D) and the presence of β-Cat in the complexes detected by immunoblotting using a specific antibody. Adult primary hepatocytes were isolated from AhR+/+ and AhR−/− mice by collagenase perfusion and cultured in complete medium containing HGF and EGF.
(E) Adult AhR+/+ primary hepatocytes were treated with medium from control L1 cells (control), medium from L1-Wnt3a-producing cells (1:4 dilution), or 10 μM FICZ. AhR−/− primary hepatocytes were treated with medium from control L1 cells (control) or medium from L1-Wnt3a-producing cells (1:4 dilution). Total RNA was purified and analyzed for Axin2 or Cyp1a1 mRNA expression by reverse-transcriptase (RT)-qPCR.
(F) Adult primary hepatocytes from AhR+/+ and AhR−/− liver were transfected with the TOP/FOP luciferase system to determine β-Cat-dependent transcription under basal cell conditions or after treatment with L1-Wnt3a conditioned medium (1:4 dilution).
(G) The TOP/FOP luciferase assay was also performed in preweaning primary hepatocytes obtained from mice of either genotype. mRNA gene expression was normalized by Gapdh and represented as 2−ΔΔCt. β-Actin and histone H3 (H3) were used to confirm protein integrity and equal loading. Eight mice for each developmental time and genotype and at least three technical replicates were analyzed.
Data are shown as mean ± SD. n.s., Not statistically significant; SD, standard deviation. Scale bar, 50 μm. Antibodies and oligonucleotides used are indicated in Tables S1 and S2, respectively.
To further investigate if AhR modulates Wnt/β-Cat signaling in adult liver, primary hepatocytes were isolated from AhR+/+ and AhR−/− mice by collagenase perfusion and cultured in cell medium containing hepatocyte growth factor (HGF) and epidermal growth factor (EGF). Experiments were done in the presence of conditioned medium enriched in the Wnt/β-Cat ligand Wnt3a or after treatment with the AhR non-toxic agonist 6-formylindolo[3,2-b]carbazole (FICZ). mRNA expression of the β-Cat target gene Axin2 was moderately induced by Wnt3a but not by FICZ in AhR wild-type hepatocytes; by contrast, Wnt3a treatment markedly increased Axin2 mRNA in AhR-null primary hepatocytes (Figure 6E). AhR target gene Cyp1a1 was largely induced by FICZ and significantly by Wnt3a in AhR+/+ primary hepatocytes; no significant Cyp1a1 expression was detected in AhR−/− primary hepatocytes (Figure 6E). Thus, the transcriptional activity of AhR can be induced by Wnt/β-Cat signaling in AhR-expressing primary hepatocytes, but not vice versa, and interestingly, AhR-lacking primary hepatocytes were highly responsive to activation of the Wnt/β-Cat pathway. To give additional support to these data, primary hepatocytes from AhR+/+ and AhR−/− livers were transfected with a TOP-Flash reporter construct that allows quantification of β-Cat-dependent transcription. TOP-Flash luciferase activity was higher in adult AhR−/− primary hepatocytes under basal culture conditions (Figure 6F), and more notably after treatment with Wnt3a-enriched medium (Figure 6F). Transfection of the TOP-Flash reporter construct in preweaning primary hepatocytes also revealed an increase in β-Cat transcriptional activity in AhR−/− cells (Figure 6G).
Conserved mTOR Activation during the Preweaning-to-Adult Transition in the Liver
Different signaling pathways controlling cell proliferation, metabolism, and differentiation converge to the mammalian target of rapamycin (mTOR). Particularly relevant are those mediated by PI3K, ERK, and Wnt/β-Cat, which activate the mTORC1 complex through the guanosine triphosphate (GTP)-binding protein RHEB (Laplante and Sabatini, 2009, Laplante and Sabatini, 2012, Saxton and Sabatini, 2017). We therefore decided to determine whether the sustained activation of those pathways in the preweaning-to-adult transition in AhR−/− liver resulted in increased mTORC1 activation. Protein analysis showed that mTOR expression remained at a higher level in both preweaning and adult AhR−/− livers when compared with their counterpart AhR+/+ livers (Figure 7A). One major target of the mTORC1 complex is the ribosomal S6 kinase-1 (S6K1), which is activated by phosphorylation (Laplante and Sabatini, 2009, Laplante and Sabatini, 2012, Saxton and Sabatini, 2017). In addition to its functions in protein synthesis, S6K1 has been recently implicated in the control of polyploidy (Ma et al., 2009). The levels of phosphorylated S6K1 were increased in preweaning and adult AhR−/− livers when compared with AhR+/+ liver (Figure 7B), in agreement with the observed pattern of mTOR expression. Hence, the persistent activation of INS-R/PI3K/ERK and Wnt/β-Cat signaling pathways that takes place during liver maturation in AhR−/− mice might assemble at the mTORC1 complex, maintaining proliferation and inhibiting differentiation-related polyploidy.
Figure 7.

mTOR Expression, Ribosomal S6K1 Activation, and Metabolomic Changes during the Preweaning-to-Adult Transition in AhR−/− Liver
(A) Total protein obtained from preweaning (PW) and adult (AD) AhR+/+ and AhR−/− liver were analyzed for the expression of mTOR by immunoblotting using a specific antibody.
(B–D) (B) Protein extracts from the same mice were used to determine by immunoblotting the level of activation of mTORC1 target protein phospho-S6K1 (p-S6K1Tyr389). Serum samples were obtained from preweaning (PW) and adult (AD) AhR+/+ and AhR−/− mice, processed, and their metabolites analyzed by chromatographic separation and mass spectrometry. The levels of mTORC1-activating amino acids L-Leu (C) and L-Gln (D) were measured and quantified in both AhR genotypes and developmental stages.
(E) Metabolomics were also used to identify and quantify the accumulation of intermediates of the mitochondrial oxidative metabolism succinate, fumarate, and malate in the samples indicated above.
(F) Levels of azelaic acid were determined by chromatography and mass spectrometry in serum samples from preweaning AhR+/+ and AhR−/− mice.
(G) Protein levels of the hepatic carboxylesterase-3 (CES3) were determined in preweaning AhR+/+ and AhR−/− mice by immunoblotting using a specific antibody. β-Actin was used to confirm protein integrity and equal loading.
Two representative mice for each experimental condition are shown. Eight mice (A–D) or six mice (E–F) for each developmental time and genotype and three technical replicates were analyzed. Data are shown as mean ± SD. Antibodies used are indicated in Table S1. SD, standard deviation.
AhR Deficiency Alters Metabolic Parameters Associated with Polyploidy and mTOR Activity
The PI3K-mTOR pathway has relevant functions in metabolism and energy control (Saxton and Sabatini, 2017, Yu and Cui, 2016). Recent work suggests that mTOR inhibition reduces glycolytic metabolism in polyploid cells (Liu et al., 2013) and that amino acids such as L-Leu may regulate mTORC1 activity in certain forms of anemia (Boultwood et al., 2013, Payne et al., 2012). We then performed metabolome analysis for amino acid content in serum from preweaning and adult AhR+/+ and AhR−/− mice. Interestingly, L-Leu was significantly enriched in AhR−/− preweaning serum when compared with AhR+/+ mice, although no significant changes were observed in adult mice (Figure 7C). L-Gln, which cooperates in transporting L-Leu into the cell, had also significantly higher levels in preweaning AhR−/− mice serum and a tendency of accumulation in adult AhR-null mice (Figure 7D). It is thus possible that L-Leu/L-Gln may contribute to mTORC1 activation in preweaning liver. We next did metabolomics for intermediates of the mitochondrial oxidative phosphorylation to determine whether reduced polyploidy in AhR−/− liver favors oxidative versus glycolytic metabolism. The results showed significant increases in serum levels of aerobic metabolism intermediates succinate, fumarate, and malate during the preweaning-to-adult transition in AhR−/− mice when compared with AhR+/+ mice (Figure 7E), in agreement with the prominent roles of succinate (and to a lesser extent fumarate) in providing cellular energy and blocking senescence in replicative cells (Chen et al., 2015), and with the preferred glycolytic metabolism found in high-ploidy cells (Liu et al., 2013). Moreover, azelaic acid monoesters, which are negatively regulated by the hepatic carboxylesterase-3 (CES3) in an AhR-dependent manner and which could contribute to the liver steatosis present in very young AhR−/− mice (Matsubara et al., 2012), were also markedly upregulated in preweaning AhR−/− liver, concomitantly to an inhibition of CES3 expression (Figures 7F and 7G).
Lack of Polyploidy in Adult AhR-Null Liver May Be Related to Centrosome Amplification
In the liver, developmental polyploidy may arise after several rounds of genome duplication in the absence of cytokinesis (Pandit et al., 2013), with the appearance of supernumerary centrosomes (Conduit et al., 2015). Interestingly, continued expression of endogenous AhR was shown to promote centrosome amplification (e.g., increased number of centrosomes) in breast cancer cells (Korzeniewski et al., 2010). We considered the possibility that low-ploidy AhR−/− livers may have altered centrosome organization. Pericentrin (PCN), a prototypical component of the pericentriolar material (PCM) surrounding the centriole, was overexpressed in preweaning and adult AhR−/− livers with respect to AhR+/+ liver, as determined by immunoblotting (Figure 8A). Confocal immunofluorescence in liver sections stained for PCN showed that, in the absence of AhR, adult hepatocytes appeared to have larger and strongly stained centrosomes than AhR+/+ adult livers (Figure 8B). Image analyses were performed to quantify the area and volume of the centrosomes in AhR+/+ and AhR−/− livers. In agreement, centrosomes of AhR−/− livers had larger areas (Figure 8C) and volumes (Figure 8D) than centrosomes in AhR+/+ livers.
Figure 8.

Lower Ploidy in AhR−/− Liver Could Be Associated with Altered Centrosome Composition
(A) Total protein was obtained from preweaning and adult AhR+/+ and AhR−/− liver and analyzed for pericentrin (PCN) expression by immunoblotting using a specific antibody.
(B) Livers from adult AhR+/+ and AhR−/− mice were fixed, embedded in paraffin, sectioned, and analyzed for PCN expression by immunofluorescence using a specific primary antibody and an Alexa 633-labeled secondary antibody. Sections were visualized using an Olympus FV1000 confocal microscope (Olympus). DAPI was used to label cell nuclei. Details of centrosomes at higher magnification are shown within yellow boxes on the right panels.
(C and D) Centrosomal area (C) and PCN protein amount (D) were quantified using the ImageJ software, and the data show the different ranges in which centrosomes can be clustered for each mice genotype. β-Actin was used to confirm protein integrity and equal loading.
Two representative mice for each condition are shown. Six mice for each developmental time and genotype and four technical replicates were analyzed. Data are shown as mean ± SD. Scale bar, 15 μm. Antibodies used are indicated in Table S1. SD, standard deviation.
Pharmacological Inhibition of PI3K, ERK, and Wnt/β-Cat Signaling Partially Rescues Polyploidy in AhR−/− Liver
We next decided to investigate if inhibition of PI3K, ERK, and Wnt/β-Cat signaling could rescue polyploidy in AhR−/− liver (Figure 9). Primary hepatocytes were isolated from AhR+/+ and AhR−/− mice by collagenase perfusion and cultured in the presence of pharmacological inhibitors LY294002 (PI3K), PD98059 (ERK), and salinomycin (Wnt/β-Cat). Inhibition of these signaling pathways increased the number of hepatocytes with 4c DNA content in both genotypes and interestingly, stimulated the appearance of AhR−/− hepatocytes with >4c DNA content, including octaploid cells (Figures 9A and 9B).
Figure 9.

Pharmacological Inhibition of PI3K, ERK, and Wnt/β-Cat Signaling Partially Rescues Polyploidy in AhR−/− Liver
(A and B) Primary hepatocytes from AhR+/+ (A) and AhR−/− (B) mice were cultured for 48 hr in the presence of inhibitors for PI3K (LY294002), ERK (PD98059), and Wnt/β-Cat (salinomycin) at a low or high concentration (see Methods). Their DNA content was analyzed by flow cytometry and represented as 2c, 4c, and >4c containing cells.
(C) AhR−/− mice were treated in vivo with those inhibitors (see Methods), and the activation of PI3K signaling determined by the levels of p-AKTSer473 with respect to total AKT expression.
(D) mTOR expression was also determined in vivo under the same experimental conditions indicated above.
Two representative mice for each condition are shown. Six mice of each genotype were perfused to isolate primary hepatocytes for in vitro experiments and five mice were treated with inhibitors in vivo. Three technical replicates were analyzed. Data are shown as mean ± SD. Scale bar, 15 μm. Antibodies and oligonucleotides used are indicated in Tables S1 and S2, respectively. SD, standard deviation.
Treatment of AhR−/− mice with these inhibitors in vivo for 7 days reduced signaling through PI3K as determined by AKT phosphorylation at Ser473 (Figure 9C) and downregulated the expression of mTOR with respect to untreated AhR-null mice (Figure 9D). Overall, these in vitro and in vivo experiments support the involvement of PI3K, ERK, and Wnt/β-Cat signaling in liver polyploidization through a mechanism requiring AhR expression.
Discussion
Taking into account that AhR expression promotes differentiation in different cell types (Esser and Rannug, 2015, Mulero-Navarro and Fernandez-Salguero, 2016), and since polyploidization is in fact a differentiation process, we decided to investigate the role of AhR in the transition from a preweaning diploid liver to an adult polyploid liver and the signaling pathways that might be involved. The main conclusion from this work is that AhR maintains the activation of signaling pathways controlling proliferation, differentiation, and metabolism within physiological levels for proper liver polyploidization and maturation. The fact that several related pathways are modulated by AhR to control physiological liver polyploidy suggests that this receptor may be acting as a downstream hub in the signaling network. Future studies are guaranteed to determine the relative contribution of each of these pathways to the phenotype and the precise points for AhR interaction.
AhR deficiency increased the cellularity and reduced the average cell size in the preweaning proliferating liver and surprisingly, such hyperproliferative phenotype, rather than being blocked as in AhR wild-type mice, persisted in adult AhR−/− livers as demonstrated by Ki67 and PCNA immunodetection. These observations suggest that AhR is needed to maintain cell growth and proliferation within adequate physiological ranges in early stages of postnatal liver development. Even more, since the preweaning-to-adult transition involves a process of differentiation by which hepatocytes modify their cell cycle to inhibit division (Celton-Morizur et al., 2010, Sigal et al., 1999, Zielke et al., 2013), it appears likely that AhR is also required to switch from a proliferative to a differentiated adult liver. This hypothesis is supported by the fact that the liver differentiation marker albumin reached much higher expression levels in polyploid AhR+/+ livers than in mostly diploid AhR−/− livers. In addition, it is also known that AhR is able to prevent mitotic progression, induce differentiation, and inhibit pluripotency in different cell types (Contador-Troca et al., 2013, Esser and Rannug, 2015, Ko et al., 2016, Morales-Hernandez et al., 2016, Mulero-Navarro and Fernandez-Salguero, 2016).
Increased cell size, reduced proliferation, and terminal differentiation are common properties of polyploid cells including hepatocytes (Conlon and Raff, 1999, Raff, 1996). We therefore questioned whether AhR could be relevant for polyploidization of the adult liver. Indeed, AhR depletion maintained a predominantly diploid adult liver with a high content of mononuclear hepatocytes, suggestive of impaired polyploidy. To date, a sole single report has suggested that AhR may be needed for the differentiation and polyploidization of mouse megakaryocytes (Lindsey and Papoutsakis, 2011). The existence of this common function of AhR in different unrelated cell types emphasizes that ploidy maintenance may be an evolutionary conserved role for this receptor along phylogeny. The implication of AhR in inducing differentiation and polyploidy was further supported by the fact that, in the absence of AhR, compromised polyploidy in adult mice was accompanied by upregulation of cell cycle proteins promoting transition through G1/S and G2/M, Cyclin B1 and Cyclin E, and repression of cell cycle inhibitors p27Kip1 and p21Cip1. Thus, adult mice liver polyploidy could involve physiological control of the cell cycle by a process requiring AhR expression. Consistently, since AhR can bind to inactive hypophosphorylated retinoblastoma protein (pRB) and to displace p300 from E2F promoters, AhR deficiency may result in enhanced proliferation in adult hepatocytes. Interestingly, lack of E2F8 transcription factor impairs polyploidization in mouse liver through overexpression of E2F-dependent target genes (Pandit et al., 2012), thus producing a phenotype similar to that present in AhR-null mice. Since the E2F8-deficient phenotype was associated with the upregulation of pro-cytokinesis genes, it might be relevant to address to what extent cytokinesis affects ploidy in AhR−/− mouse liver. It is intriguing that preweaning AhR+/+ mice maintained higher levels of Cyclin B1 and Cyclin E despite their reduced proliferative potential with respect to AhR−/− mice. One possible explanation for these results is that wild-type livers overexpress positive regulators of the cell cycle to overcome their increased content in negative regulators p27Kip1 and p21Cip1.
Although the reduced polyploidy present in adult AhR-null liver could result from defective endoreplication (Zielke et al., 2013), it is also possible that the so-called ploidy reversal (Duncan et al., 2010) could generate cells of variable ploidy from a fraction of mature AhR−/− polyploid hepatocytes, ultimately restoring proliferative potential. Among the several molecular mechanisms driving polyploidy, failure to complete cytokinesis after mitosis appears relevant in mammalian liver (Zielke et al., 2013). We have found that the levels of PCN, a component of the PCM with relevant roles in spindle organization (Zimmerman et al., 2004), was restricted in centrosomes of AhR-expressing hepatocytes, suggesting that control of centrosomal activity by AhR may contribute to reduced proliferation and increased polyploidy. Since increased centrosome size appears related to a higher proliferative potential, adult diploid AhR−/− hepatocytes could enlarge their centrosomes to maintain proliferation in the absence of polyploidy. In addition, as PCN regulates mitotic responses in DNA damaged cells (Antonczak et al., 2016), AhR-null hepatocytes could widen their centrosomes to complete cell division under enforced non-polyploid conditions, whereas polyploid AhR wild-type hepatocytes would keep an increased number of smaller centrosomes. Interestingly, lack of the E2F8 transcription factor in mouse liver promotes overexpression of E2F-dependent target genes and enhanced cytokinesis with impaired polyploidization (Pandit et al., 2012). Since such phenotype was related to the upregulation of pro-cytokinesis genes, it could be relevant to address whether pro-cytokinesis genes are also deregulated in AhR−/− mouse liver. In addition, altered E2F transcriptional activity could also be involved in the mechanism since AhR inhibits cell proliferation by repressing E2F-dependent transcription through p300 displacement.
Insulin hormone signals through the INS-R to PI3K and Ras-ERK pathways to control cell survival, proliferation, and metabolism (Yu and Cui, 2016). A previous study has shown that AKT activation induced polyploidy in Wistar rat liver (Celton-Morizur et al., 2009, Celton-Morizur et al., 2010). Our characterization of the INS-R/PI3K/AKT/GSK3β and ERK axes indicated that it was persistently downregulated from preweaning-to-adult AhR+/+ liver when compared with AhR-deficient liver, suggesting that reduced signaling through those pathways may compromise proliferation and promote polyploid differentiation. In addition, diminished INS-R/PI3K/AKT/GSK3β and ERK signaling in polyploid AhR wild-type liver can be partly explained by the reduced insulin sensitivity of AhR+/+ with respect to AhR−/− mice (Wang et al., 2011). Remarkably, tumor suppressor p53, which is negatively regulated by PI3K signaling (Sabbatini and McCormick, 1999, Yamaguchi et al., 2001), was downregulated in preweaning AhR−/− liver but overexpressed in adults. Nonetheless, p53 target gene p21Cip1 was consistently suppressed in AhR-null livers at either developmental stage. These apparently contradictory results can be explained in the context of p53-regulating ploidy (Aylon and Oren, 2011, Kurinna et al., 2013) or in the proposed p53-dependent versus p53-independent regulation of p21Cip1 (Karimian et al., 2016, Macleod et al., 1995). Since previous work has indicated that p53 overexpression blocks polyploidy (Aylon and Oren, 2011, Kurinna et al., 2013), it can be suggested that high p53 expression impairs the diploid-to-polyploid transition in adult AhR-null liver, whereas a reduction in p53 levels could positively influence polyploidy in adult AhR+/+ liver. In addition, p21Cip1 could be downregulated independently on p53 in AhR-null liver, as previously reported in p53−/− mouse (Macleod et al., 1995).
AhR cross talks with Wnt/β-Cat signaling as AhR activation represses Wnt/β-Cat-related tissue regeneration in zebrafish (Mathew et al., 2009), differentiation of liver progenitor cells (Prochazkova et al., 2011), and early differentiation of mouse embryonic stem cells (Wang et al., 2016). On the other hand, Wnt/β-Cat signaling can increase the expression of AhR target genes in primary human hepatocytes (Gerbal-Chaloin et al., 2014). Wnt/β-Cat signaling was significantly reduced in adult primary AhR+/+ hepatocytes, suggesting in agreement with earlier data that inhibition of this pathway may contribute to polyploidization. An intriguing possibility is that AhR may be a component of the β-Cat repressive complex since both proteins co-immunoprecipitate under basal conditions in adult AhR+/+ liver. It is thus plausible that the presence of AhR in the β-Cat complex negatively affects Wnt/β-Cat-dependent functions, including inhibition of cell proliferation. Since active p-AKT co-immunoprecipitated with β-Cat and such effect was more noticeable and sustained in AhR−/− than in AhR+/+ liver, it is possible that cooperation between PI3K and Wnt/β-Cat supports liver proliferation and impairs polyploidy in the absence of AhR. Indeed, INS-R/PI3K can induce Wnt/β-Cat responses by inhibiting GSK3β (Yu and Cui, 2016), which was repressed in adult AhR−/− liver.
PI3K/AKT, ERK, and Wnt/β-Cat pathways converge to regulate mTORC1, which is a major complex in controlling proliferation, survival, and metabolism (Laplante and Sabatini, 2009, Laplante and Sabatini, 2012, Yu and Cui, 2016). Consistently, reduced activation of these pathways in preweaning and adult AhR+/+ livers coincided with lower mTOR expression and diminished activation of its canonical target S6K1. Altogether, we propose that AhR-expressing hepatocytes inhibit signaling from PI3K/AKT, ERK, and Wnt/β-Cat to mTORC1 to block proliferation, induce differentiation, and trigger polyploidy during the preweaning-to-adult transition. In fact, expression of a kinase-dead form of S6K1 in megakaryocytes increases polyploidy, whereas its rapamycin-resistant active form decreased polyploidy (Ma et al., 2009). mTORC1 also has relevant roles in cellular metabolism, and its activity can be increased by intracellular amino acids (Carroll et al., 2017). AhR expression reduced L-Leu and L-Gln levels in preweaning and L-Gln levels in adult liver. Since L-Leu and L-Gln are co-transported into the cell to activate mTORC1 (Boultwood et al., 2013, Laplante and Sabatini, 2009, Payne et al., 2012), it is possible that reduced L-Leu and L-Gln levels adversely affect mTORC1 in AhR+/+ polyploid liver. Furthermore, high L-Gln levels in adult AhR−/− liver may depend on Wnt/β-Cat since this pathway regulates L-Gln metabolism in the liver (Cadoret et al., 2002).
Polyploidization has been shown to favor glycolytic metabolism over mitochondrial oxidative phosphorylation in acute myeloid leukemia (Liu et al., 2013). Polyploid, low-proliferating AhR+/+ liver had reduced levels of succinate, considered the most energetic molecule of the mitochondrial aerobic metabolism (Chen et al., 2015). This result suggests that polyploid adult AhR wild-type liver may have a preferred glycolytic energy demand and a more differentiated status than the diploid proliferative adult AhR-null liver that would favor a more energetic mitochondrial metabolism. Finally, preweaning AhR+/+ livers had reduced levels of azelaic acid, likely due to overexpression of the AhR-regulated CES3 gene. Since high levels of azelaic acid have been associated with steatohepatitis (Matsubara et al., 2012), these data could help explain the steatosis described in postnatal AhR−/− mice (Schmidt et al., 1996).
We propose that AhR is needed to establish a physiological control of signaling pathways regulating cell proliferation, differentiation, metabolism, and polyploidy in mouse liver. This hypothesis is supported by the fact that inhibition of PI3K, ERK, and Wnt/β-Cat signaling rescues, at least partially, polyploidy in AhR−/− hepatocytes. Since polyploidization is a recurrent trait of human cancer (Zack et al., 2013), understanding how AhR expression influences ploidy may provide novel therapeutic opportunities using non-toxic AhR modulators. Because the pathways identified here are regulated within the same developmental window, it seems reasonable that AhR acts downstream of mTOR, PKB, ERK, and β-Cat possibly by acting as a common intermediate. This may lead to identify novel nuclear and non-nuclear functions of AhR, as previously found for the regulation of caveolin-1, β1-integrin, and c-Src (Enan and Matsumura, 1996, Rey-Barroso et al., 2013, Rey-Barroso et al., 2014). Elucidating whether ploidy reversal contributes to the reduced polyploidy of adult AhR−/− liver represents another relevant question that deserves further investigation.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by grants to P.M.F.-S. from the Spanish Ministry of Economy and Competitiveness (SAF2014-51813-R and SAF2017-82597-R) and from the Junta de Extremadura (GR15008 and IB160210). Research at the laboratory of P.M.F.-S. was also funded by the Red Temática de Investigación Cooperativa en Cáncer (RTICC), Carlos III Institute, and Spanish Ministry of Economy and Competitiveness (RD12/0036/0032). N.M.-M. was supported by the Ministerio de Economía y Competitividad. All Spanish funding is co-sponsored by the European Union FEDER Program. The authors acknowledge the support of the Servicio de Técnicas Aplicadas a las Biociencias (STAB-SAIUEX) of the Universidad de Extremadura.
Author Contributions
N.M.-M. design, performed, and discussed a major part of the experiments; J.M.M. helped in designing the study and organizing and discussing data; A.A.-B. contributed to cell analysis, flow cytometry, and confocal microscopy analysis; D.P.P. and S.T. assisted and helped with metabolomics experiments; J.M.G.-S. helped with in vitro assays for Wnt/β-Cat; P.G. helped to perform the analysis of pericentrin; R.M.R. contributed to the design and planning of centrosome experiments; A.M. contributed to the design and planning of Wnt/β-Cat experiments; F.J.G. contributed to the design and planning of metabolomic experiments; P.M.F.-S. designed, discussed, and coordinated the study and wrote the paper.
Declaration of Interests
The authors declare no competing interests.
Published: June 29, 2018
Footnotes
Supplemental Information includes Transparent Methods, one figure, and two tables and can be found with this article online at https://doi.org/10.1016/j.isci.2018.05.006.
Supplemental Information
References
- Antonczak A.K., Mullee L.I., Wang Y., Comartin D., Inoue T., Pelletier L., Morrison C.G. Opposing effects of pericentrin and microcephalin on the pericentriolar material regulate CHK1 activation in the DNA damage response. Oncogene. 2016;35:2003–2010. doi: 10.1038/onc.2015.257. [DOI] [PubMed] [Google Scholar]
- Aylon Y., Oren M. p53: guardian of ploidy. Mol. Oncol. 2011;5:315–323. doi: 10.1016/j.molonc.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhamouche S., Decaens T., Godard C., Chambrey R., Rickman D.S., Moinard C., Vasseur-Cognet M., Kuo C.J., Kahn A., Perret C. Apc tumor suppressor gene is the “zonation-keeper” of mouse liver. Dev. Cell. 2006;10:759–770. doi: 10.1016/j.devcel.2006.03.015. [DOI] [PubMed] [Google Scholar]
- Boultwood J., Yip B.H., Vuppusetty C., Pellagatti A., Wainscoat J.S. Activation of the mTOR pathway by the amino acid (L)-leucine in the 5q- syndrome and other ribosomopathies. Adv. Biol. Regul. 2013;53:8–17. doi: 10.1016/j.jbior.2012.09.002. [DOI] [PubMed] [Google Scholar]
- Bunney T.D., Katan M. Phosphoinositide signalling in cancer: beyond PI3K and PTEN. Nat. Rev. Cancer. 2010;10:342–352. doi: 10.1038/nrc2842. [DOI] [PubMed] [Google Scholar]
- Burke Z.D., Reed K.R., Phesse T.J., Sansom O.J., Clarke A.R., Tosh D. Liver zonation occurs through a beta-catenin-dependent, c-Myc-independent mechanism. Gastroenterology. 2009;136:2316–2324.e1-3. doi: 10.1053/j.gastro.2009.02.063. [DOI] [PubMed] [Google Scholar]
- Busca R., Pouyssegur J., Lenormand P. ERK1 and ERK2 map kinases: specific roles or functional redundancy? Front. Cell Dev. Biol. 2016;4:53. doi: 10.3389/fcell.2016.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadoret A., Ovejero C., Terris B., Souil E., Levy L., Lamers W.H., Kitajewski J., Kahn A., Perret C. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene. 2002;21:8293–8301. doi: 10.1038/sj.onc.1206118. [DOI] [PubMed] [Google Scholar]
- Carroll B., Nelson G., Rabanal-Ruiz Y., Kucheryavenko O., Dunhill-Turner N.A., Chesterman C.C., Zahari Q., Zhang T., Conduit S.E., Mitchell C.A. Persistent mTORC1 signaling in cell senescence results from defects in amino acid and growth factor sensing. J. Cell Biol. 2017;216:1949–1957. doi: 10.1083/jcb.201610113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celton-Morizur S., Merlen G., Couton D., Desdouets C. Polyploidy and liver proliferation: central role of insulin signaling. Cell Cycle. 2010;9:460–466. doi: 10.4161/cc.9.3.10542. [DOI] [PubMed] [Google Scholar]
- Celton-Morizur S., Merlen G., Couton D., Margall-Ducos G., Desdouets C. The insulin/Akt pathway controls a specific cell division program that leads to generation of binucleated tetraploid liver cells in rodents. J. Clin. Invest. 2009;119:1880–1887. doi: 10.1172/JCI38677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T.T., Maevsky E.I., Uchitel M.L. Maintenance of homeostasis in the aging hypothalamus: the central and peripheral roles of succinate. Front. Endocrinol. (Lausanne) 2015;6:7. doi: 10.3389/fendo.2015.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conduit P.T., Wainman A., Raff J.W. Centrosome function and assembly in animal cells. Nat. Rev. Mol. Cell Biol. 2015;16:611–624. doi: 10.1038/nrm4062. [DOI] [PubMed] [Google Scholar]
- Conlon I., Raff M. Size control in animal development. Cell. 1999;96:235–244. doi: 10.1016/s0092-8674(00)80563-2. [DOI] [PubMed] [Google Scholar]
- Contador-Troca M., Alvarez-Barrientos A., Barrasa E., Rico-Leo E.M., Catalina-Fernandez I., Menacho-Marquez M., Bustelo X.R., Garcia-Borron J.C., Gomez-Duran A., Saenz-Santamaria J. The dioxin receptor has tumor suppressor activity in melanoma growth and metastasis. Carcinogenesis. 2013;34:2683–2693. doi: 10.1093/carcin/bgt248. [DOI] [PubMed] [Google Scholar]
- Davoli T., de Lange T. The causes and consequences of polyploidy in normal development and cancer. Annu. Rev. Cell Dev. Biol. 2011;27:585–610. doi: 10.1146/annurev-cellbio-092910-154234. [DOI] [PubMed] [Google Scholar]
- Duncan A.W., Hanlon Newell A.E., Bi W., Finegold M.J., Olson S.B., Beaudet A.L., Grompe M. Aneuploidy as a mechanism for stress-induced liver adaptation. J. Clin. Invest. 2012;122:3307–3315. doi: 10.1172/JCI64026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan A.W., Hickey R.D., Paulk N.K., Culberson A.J., Olson S.B., Finegold M.J., Grompe M. Ploidy reductions in murine fusion-derived hepatocytes. PLoS Genet. 2009;5:e1000385. doi: 10.1371/journal.pgen.1000385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan A.W., Taylor M.H., Hickey R.D., Hanlon Newell A.E., Lenzi M.L., Olson S.B., Finegold M.J., Grompe M. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 2010;467:707–710. doi: 10.1038/nature09414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar B.A., Zielke N., Gutierrez C. Endocycles: a recurrent evolutionary innovation for post-mitotic cell growth. Nat. Rev. Mol. Cell Biol. 2014;15:197–210. doi: 10.1038/nrm3756. [DOI] [PubMed] [Google Scholar]
- Enan E., Matsumura F. Identification of c-Src as the integral component of the cytosolic Ah receptor complex, transducing the signal of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) through the protein phosphorylation pathway. Biochem. Pharmacol. 1996;52:1599–1612. doi: 10.1016/s0006-2952(96)00566-7. [DOI] [PubMed] [Google Scholar]
- Esser C., Rannug A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol. Rev. 2015;67:259–279. doi: 10.1124/pr.114.009001. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero P., Pineau T., Hilbert D.M., McPhail T., Lee S.S., Kimura S., Nebert D.W., Rudikoff S., Ward J.M., Gonzalez F.J. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science. 1995;268:722–726. doi: 10.1126/science.7732381. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero P., Ward J.M., Sundberg J.P., Gonzalez F.J. Lesions of aryl-hydrocarbon receptor-deficient mice. Vet. Pathol. 1997;34:605–614. doi: 10.1177/030098589703400609. [DOI] [PubMed] [Google Scholar]
- Fox D.T., Duronio R.J. Endoreplication and polyploidy: insights into development and disease. Development. 2013;140:3–12. doi: 10.1242/dev.080531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem N.J., Storchova Z., Pellman D. Tetraploidy, aneuploidy and cancer. Curr. Opin. Genet. Dev. 2007;17:157–162. doi: 10.1016/j.gde.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Gao Z., Bu Y., Zhang G., Liu X., Wang X., Ding S., Wang E., Shi R., Li Q., Fu J. Effect of TCDD on the fate of epithelial cells isolated from human fetal palatal shelves (hFPECs) Toxicol. Appl. Pharmacol. 2016;305:186–193. doi: 10.1016/j.taap.2016.06.016. [DOI] [PubMed] [Google Scholar]
- Gentric G., Celton-Morizur S., Desdouets C. Polyploidy and liver proliferation. Clin. Res. Hepatol. Gastroenterol. 2012;36:29–34. doi: 10.1016/j.clinre.2011.05.011. [DOI] [PubMed] [Google Scholar]
- Gentric G., Desdouets C. Polyploidization in liver tissue. Am. J. Pathol. 2014;184:322–331. doi: 10.1016/j.ajpath.2013.06.035. [DOI] [PubMed] [Google Scholar]
- Gerbal-Chaloin S., Dume A.S., Briolotti P., Klieber S., Raulet E., Duret C., Fabre J.M., Ramos J., Maurel P., Daujat-Chavanieu M. The WNT/beta-catenin pathway is a transcriptional regulator of CYP2E1, CYP1A2, and aryl hydrocarbon receptor gene expression in primary human hepatocytes. Mol. Pharmacol. 2014;86:624–634. doi: 10.1124/mol.114.094797. [DOI] [PubMed] [Google Scholar]
- Gerlyng P., Abyholm A., Grotmol T., Erikstein B., Huitfeldt H.S., Stokke T., Seglen P.O. Binucleation and polyploidization patterns in developmental and regenerative rat liver growth. Cell Prolif. 1993;26:557–565. doi: 10.1111/j.1365-2184.1993.tb00033.x. [DOI] [PubMed] [Google Scholar]
- Germain L., Blouin M.J., Marceau N. Biliary epithelial and hepatocytic cell lineage relationships in embryonic rat liver as determined by the differential expression of cytokeratins, alpha-fetoprotein, albumin, and cell surface-exposed components. Cancer Res. 1988;48:4909–4918. [PubMed] [Google Scholar]
- Guidotti J.E., Bregerie O., Robert A., Debey P., Brechot C., Desdouets C. Liver cell polyploidization: a pivotal role for binuclear hepatocytes. J. Biol. Chem. 2003;278:19095–19101. doi: 10.1074/jbc.M300982200. [DOI] [PubMed] [Google Scholar]
- Hannibal R.L., Chuong E.B., Rivera-Mulia J.C., Gilbert D.M., Valouev A., Baker J.C. Copy number variation is a fundamental aspect of the placental genome. PLoS Genet. 2014;10:e1004290. doi: 10.1371/journal.pgen.1004290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Y.S., Qian Y., Chen X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell. Signal. 2010;22:1003–1012. doi: 10.1016/j.cellsig.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimian A., Ahmadi Y., Yousefi B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair (Amst) 2016;42:63–71. doi: 10.1016/j.dnarep.2016.04.008. [DOI] [PubMed] [Google Scholar]
- Ko C.I., Fan Y., de Gannes M., Wang Q., Xia Y., Puga A. Repression of the aryl hydrocarbon receptor is required to maintain mitotic progression and prevent loss of pluripotency of embryonic stem cells. Stem Cells. 2016;34:2825–2839. doi: 10.1002/stem.2456. [DOI] [PubMed] [Google Scholar]
- Kolluri S.K., Weiss C., Koff A., Gottlicher M. p27(Kip1) induction and inhibition of proliferation by the intracellular Ah receptor in developing thymus and hepatoma cells. Genes Dev. 1999;13:1742–1753. doi: 10.1101/gad.13.13.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzeniewski N., Wheeler S., Chatterjee P., Duensing A., Duensing S. A novel role of the aryl hydrocarbon receptor (AhR) in centrosome amplification - implications for chemoprevention. Mol. Cancer. 2010;9:153. doi: 10.1186/1476-4598-9-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurinna S., Stratton S.A., Coban Z., Schumacher J.M., Grompe M., Duncan A.W., Barton M.C. p53 regulates a mitotic transcription program and determines ploidy in normal mouse liver. Hepatology. 2013;57:2004–2013. doi: 10.1002/hep.26233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahvis G.P., Lindell S.L., Thomas R.S., McCuskey R.S., Murphy C., Glover E., Bentz M., Southard J., Bradfield C.A. Portosystemic shunting and persistent fetal vascular structures in aryl hydrocarbon receptor-deficient mice. Proc. Natl. Acad. Sci. USA. 2000;97:10442–10447. doi: 10.1073/pnas.190256997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M., Sabatini D.M. mTOR signaling at a glance. J. Cell Sci. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M., Sabatini D.M. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey S., Papoutsakis E.T. The aryl hydrocarbon receptor (AHR) transcription factor regulates megakaryocytic polyploidization. Br. J. Haematol. 2011;152:469–484. doi: 10.1111/j.1365-2141.2010.08548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L.L., Long Z.J., Wang L.X., Zheng F.M., Fang Z.G., Yan M., Xu D.F., Chen J.J., Wang S.W., Lin D.J. Inhibition of mTOR pathway sensitizes acute myeloid leukemia cells to aurora inhibitors by suppression of glycolytic metabolism. Mol. Cancer Res. 2013;11:1326–1336. doi: 10.1158/1541-7786.MCR-13-0172. [DOI] [PubMed] [Google Scholar]
- Ma D., Yu H., Lin D., Sun Y., Liu L., Liu Y., Dai B., Chen W., Cao J. S6K1 is involved in polyploidization through its phosphorylation at Thr421/Ser424. J. Cell Physiol. 2009;219:31–44. doi: 10.1002/jcp.21647. [DOI] [PubMed] [Google Scholar]
- Macleod K.F., Sherry N., Hannon G., Beach D., Tokino T., Kinzler K., Vogelstein B., Jacks T. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev. 1995;9:935–944. doi: 10.1101/gad.9.8.935. [DOI] [PubMed] [Google Scholar]
- Marques J.M., Olsson I.A., Ogren S.O., Dahlborn K. Evaluation of exploration and risk assessment in pre-weaning mice using the novel cage test. Physiol. Behav. 2008;93:139–147. doi: 10.1016/j.physbeh.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Mathew L.K., Simonich M.T., Tanguay R.L. AHR-dependent misregulation of Wnt signaling disrupts tissue regeneration. Biochem. Pharmacol. 2009;77:498–507. doi: 10.1016/j.bcp.2008.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara T., Tanaka N., Krausz K.W., Manna S.K., Kang D.W., Anderson E.R., Luecke H., Patterson A.D., Shah Y.M., Gonzalez F.J. Metabolomics identifies an inflammatory cascade involved in dioxin- and diet-induced steatohepatitis. Cell Metab. 2012;16:634–644. doi: 10.1016/j.cmet.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen T.P., Pessa H.K., Caldez M.J., Fuhrer T., Diril M.K., Sauer U., Kaldis P., Bjorklund M. Identification of transcriptional and metabolic programs related to mammalian cell size. Curr. Biol. 2014;24:598–608. doi: 10.1016/j.cub.2014.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell K.A., Lockhart C.A., Huang G., Elferink C.J. Sustained aryl hydrocarbon receptor activity attenuates liver regeneration. Mol. Pharmacol. 2006;70:163–170. doi: 10.1124/mol.106.023465. [DOI] [PubMed] [Google Scholar]
- Morales-Hernandez A., Gonzalez-Rico F.J., Roman A.C., Rico-Leo E., Alvarez-Barrientos A., Sanchez L., Macia A., Heras S.R., Garcia-Perez J.L., Merino J.M. Alu retrotransposons promote differentiation of human carcinoma cells through the aryl hydrocarbon receptor. Nucleic Acids Res. 2016;44:4665–4683. doi: 10.1093/nar/gkw095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Marín N., Barrasa E., Morales-Hernández A., Paniagua B., Blanco-Fernández G., Merino J.M., Fernández-Salguero P.M. Dioxin receptor adjusts liver regeneration after acute toxic injury and protects against liver carcinogenesis. Sci. Rep. 2017;7:10420. doi: 10.1038/s41598-017-10984-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulero-Navarro S., Fernandez-Salguero P.M. New trends in aryl hydrocarbon receptor biology. Front. Cell Dev. Biol. 2016;4:45. doi: 10.3389/fcell.2016.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R., Clevers H. Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169:985–999. doi: 10.1016/j.cell.2017.05.016. [DOI] [PubMed] [Google Scholar]
- Pandit S.K., Westendorp B., de Bruin A. Physiological significance of polyploidization in mammalian cells. Trends Cell Biol. 2013;23:556–566. doi: 10.1016/j.tcb.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Pandit S.K., Westendorp B., Nantasanti S., van Liere E., Tooten P.C., Cornelissen P.W., Toussaint M.J., Lamers W.H., de Bruin A. E2F8 is essential for polyploidization in mammalian cells. Nat. Cell Biol. 2012;14:1181–1191. doi: 10.1038/ncb2585. [DOI] [PubMed] [Google Scholar]
- Payne E.M., Virgilio M., Narla A., Sun H., Levine M., Paw B.H., Berliner N., Look A.T., Ebert B.L., Khanna-Gupta A. L-Leucine improves the anemia and developmental defects associated with diamond-blackfan anemia and del(5q) MDS by activating the mTOR pathway. Blood. 2012;120:2214–2224. doi: 10.1182/blood-2011-10-382986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohjanvirta R. First Edition. John Wiley & Sons; 2012. The AH Receptor in Biology and Toxicology. [Google Scholar]
- Prochazkova J., Kabatkova M., Bryja V., Umannova L., Bernatik O., Kozubik A., Machala M., Vondracek J. The interplay of the aryl hydrocarbon receptor and beta-catenin alters both AhR-dependent transcription and Wnt/beta-catenin signaling in liver progenitors. Toxicol. Sci. 2011;122:349–360. doi: 10.1093/toxsci/kfr129. [DOI] [PubMed] [Google Scholar]
- Puga A., Ma C., Marlowe J.L. The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochem. Pharmacol. 2009;77:713–722. doi: 10.1016/j.bcp.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puga A., Marlowe J., Barnes S., Chang C.Y., Maier A., Tan Z., Kerzee J.K., Chang X., Strobeck M., Knudsen E.S. Role of the aryl hydrocarbon receptor in cell cycle regulation. Toxicology. 2002;181-182:171–177. doi: 10.1016/s0300-483x(02)00276-7. [DOI] [PubMed] [Google Scholar]
- Raff M.C. Size control: the regulation of cell numbers in animal development. Cell. 1996;86:173–175. doi: 10.1016/s0092-8674(00)80087-2. [DOI] [PubMed] [Google Scholar]
- Rey-Barroso J., Alvarez-Barrientos A., Rico-Leo E., Contador-Troca M., Carvajal-Gonzalez J.M., Echarri A., Del Pozo M.A., Fernandez-Salguero P.M. The dioxin receptor modulates caveolin-1 mobilization during directional migration: role of cholesterol. Cell Commun. Signal. 2014;12:57. doi: 10.1186/s12964-014-0057-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey-Barroso J., Colo G.P., Alvarez-Barrientos A., Redondo-Munoz J., Carvajal-Gonzalez J.M., Mulero-Navarro S., Garcia-Pardo A., Teixido J., Fernandez-Salguero P.M. The dioxin receptor controls beta 1 integrin activation in fibroblasts through a Cbp-Csk-Src pathway. Cell. Signal. 2013;25:848–859. doi: 10.1016/j.cellsig.2013.01.010. [DOI] [PubMed] [Google Scholar]
- Sabbatini P., McCormick F. Phosphoinositide 3-OH kinase (PI3K) and PKB/Akt delay the onset of p53-mediated, transcriptionally dependent apoptosis. J. Biol. Chem. 1999;274:24263–24269. doi: 10.1074/jbc.274.34.24263. [DOI] [PubMed] [Google Scholar]
- Saxton R.A., Sabatini D.M. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169:361–371. doi: 10.1016/j.cell.2017.03.035. [DOI] [PubMed] [Google Scholar]
- Schmidt J.V., Bradfield C.A. Ah receptor signaling pathways. Annu. Rev. Cell Dev. Biol. 1996;12:55–89. doi: 10.1146/annurev.cellbio.12.1.55. [DOI] [PubMed] [Google Scholar]
- Schmidt J.V., Su G.H.-T., Reddy J.K., Simon M.C., Bradfield C.A. Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc. Natl. Acad. Sci. USA. 1996;93:6731–6736. doi: 10.1073/pnas.93.13.6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfelder K.P., Fox D.T. The expanding implications of polyploidy. J. Cell Biol. 2015;209:485–491. doi: 10.1083/jcb.201502016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiojiri N., Lemire J.M., Fausto N. Cell lineages and oval cell progenitors in rat liver development. Cancer Res. 1991;51:2611–2620. [PubMed] [Google Scholar]
- Sigal S.H., Rajvanshi P., Gorla G.R., Sokhi R.P., Saxena R., Gebhard D.R., Jr., Reid L.M., Gupta S. Partial hepatectomy-induced polyploidy attenuates hepatocyte replication and activates cell aging events. Am. J. Physiol. 1999;276:G1260–G1272. doi: 10.1152/ajpgi.1999.276.5.G1260. [DOI] [PubMed] [Google Scholar]
- Siu K.T., Rosner M.R., Minella A.C. An integrated view of cyclin E function and regulation. Cell Cycle. 2012;11:57–64. doi: 10.4161/cc.11.1.18775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgill T.W. MAP kinase: it's been longer than fifteen minutes. Biochem. Biophys. Res. Commun. 2008;371:1–4. doi: 10.1016/j.bbrc.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Taub R. Liver regeneration: from myth to mechanism. Nat. Rev. Mol. Cell Biol. 2004;5:836–847. doi: 10.1038/nrm1489. [DOI] [PubMed] [Google Scholar]
- Ullah Z., Lee C.Y., Depamphilis M.L. Cip/Kip cyclin-dependent protein kinase inhibitors and the road to polyploidy. Cell Div. 2009;4:10. doi: 10.1186/1747-1028-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Xu C.X., Krager S.L., Bottum K.M., Liao D.F., Tischkau S.A. Aryl hydrocarbon receptor deficiency enhances insulin sensitivity and reduces PPAR-alpha pathway activity in mice. Environ. Health Perspect. 2011;119:1739–1744. doi: 10.1289/ehp.1103593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Kurita H., Carreira V., Ko C.I., Fan Y., Zhang X., Biesiada J., Medvedovic M., Puga A. Ah receptor activation by dioxin disrupts activin, BMP, and WNT signals during the early differentiation of mouse embryonic stem cells and inhibits cardiomyocyte functions. Toxicol. Sci. 2016;149:346–357. doi: 10.1093/toxsci/kfv246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Willenbring H., Akkari Y., Torimaru Y., Foster M., Al-Dhalimy M., Lagasse E., Finegold M., Olson S., Grompe M. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature. 2003;422:897–901. doi: 10.1038/nature01531. [DOI] [PubMed] [Google Scholar]
- Yamaguchi A., Tamatani M., Matsuzaki H., Namikawa K., Kiyama H., Vitek M.P., Mitsuda N., Tohyama M. Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J. Biol. Chem. 2001;276:5256–5264. doi: 10.1074/jbc.M008552200. [DOI] [PubMed] [Google Scholar]
- Yu J.S., Cui W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development. 2016;143:3050–3060. doi: 10.1242/dev.137075. [DOI] [PubMed] [Google Scholar]
- Zack T.I., Schumacher S.E., Carter S.L., Cherniack A.D., Saksena G., Tabak B., Lawrence M.S., Zhsng C.Z., Wala J., Mermel C.H. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielke N., Edgar B.A., DePamphilis M.L. Endoreplication. Cold Spring Harb. Perspect. Biol. 2013;5:a012948. doi: 10.1101/cshperspect.a012948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman W.C., Sillibourne J., Rosa J., Doxsey S.J. Mitosis-specific anchoring of gamma tubulin complexes by pericentrin controls spindle organization and mitotic entry. Mol. Biol. Cell. 2004;15:3642–3657. doi: 10.1091/mbc.E03-11-0796. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.