

Summary
Epithelial-to-mesenchymal transition (EMT) is integral to cancer progression, with considerable evidence that EMT has multiple intermediary stages. Understanding the mechanisms of this stepwise activation is of great interest. We recreated a genetically defined model in which primary cells were immortalized, resulting in migratory capacity, and subsequently H-Ras-transformed, causing malignancy and invasion. To determine the mechanisms coordinating stepwise malignancy, we quantified the changes in messenger RNA (mRNA) and protein abundance. During immortalization, we found dramatic changes in mRNA, consistent with EMT, which correlated with protein abundance. Many of these same proteins also changed following Ras transformation, suggesting that pre-malignant cells were primed for malignant conversion. Unexpectedly, changes in protein abundance did not correlate with changes in mRNA following transformation. Importantly, proteins involved in cellular adhesion and cytoskeletal structure decreased during immortalization and decreased further following Ras transformation, whereas their encoding mRNAs only changed during the immortalization step. Thus, Ras induced EMT-associated invasion via post-transcriptional mechanisms in primed pre-malignant cells.
Subject Areas: Molecular Network, Cancer Systems Biology, Transcriptomics
Graphical Abstract
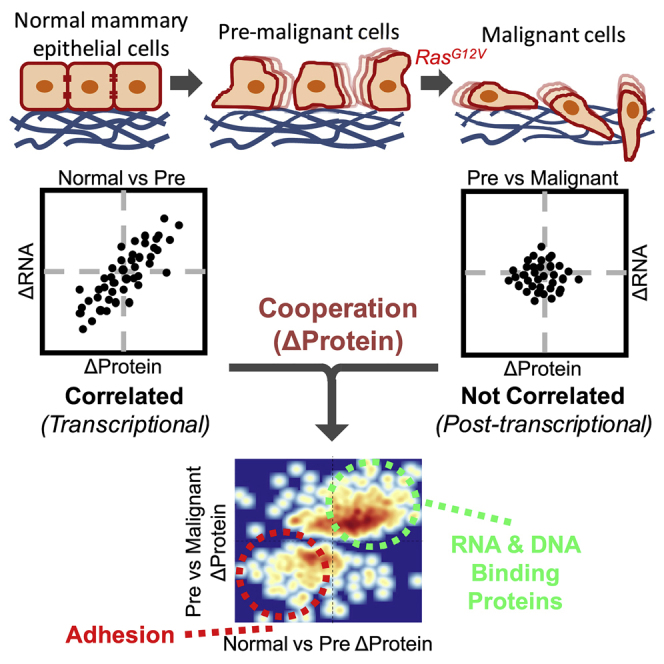
Highlights
-
•
Two-stage progressive cell culture model demonstrates partial EMT states
-
•
Pre-malignant immortalization alters RNA abundance to induce cell migration
-
•
Ras transformation alters protein abundance, but not RNA, to induce cell invasion
-
•
Both stages cooperate to regulate protein expression of adhesion molecules and RBPs
Molecular Network; Cancer Systems Biology; Transcriptomics
Introduction
The epithelial-to-mesenchymal transition (EMT), a process by which epithelial cells lose epithelial properties and gain mesenchymal characteristics, is centrally involved in metastasis (Lambert et al., 2017, Nieto et al., 2016, Shibue and Weinberg, 2017). This program involves reorganization of the cytoskeleton, loss of junctions and apical-basal polarity, and activation of signaling mechanisms that promote motility and invasion as well as interactions between the tumor cell and the surrounding microenvironment, all of which can promote metastatic dissemination. There is much evidence that EMT is not a binary process, but rather a dynamic program with multiple intermediary stages of partial, or incomplete, EMT wherein cells have a mixture of both epithelial and mesenchymal characteristics (Nieto et al., 2016). In fact, the EMT program is usually only activated partially in human carcinomas, and partial EMT cells are observed in circulating tumor cells from patients with breast cancer (Yu et al., 2013). Underlying these transitions are changes in gene expression, including a set of prevalent transcriptional and alternative splicing changes (Lamouille et al., 2014, Shapiro et al., 2011, Warzecha et al., 2010, Yang et al., 2016). However, it is unclear how, and by what mechanisms, cancer-associated genetic mutations cooperate to promote transitions through EMT states.
Cancers are derived from normal cells that evolve stepwise and progressively to a neoplastic state (Hanahan and Weinberg, 2011). Although historically treated as a late-stage event, several studies have demonstrated that the acquisition of EMT-associated traits can occur early on during multi-step tumorigenesis (Husemann et al., 2008, Mani et al., 2008, Podsypanina et al., 2008, Rhim et al., 2012). Cell culture models typically utilize cell lines that are already immortalized, and thus abnormal. Therefore, a more informative cell culture model with which to study the mechanisms regulating the earliest stages of tumorigenesis and EMT would be one that starts with normal human epithelial cells and perturbs only the pathways required for cell immortalization and subsequent tumorigenesis. Pioneering work in the Weinberg laboratory demonstrated that normal human epithelial and fibroblast cells can be converted to a tumorigenic state by expressing viral proteins SV40 T-Ag and t-Ag and mammalian proteins hTERT and HRasG12V(Hahn et al., 1999). Later work expanded these findings and identified a core set of non-viral proteins that together drive the processes of immortalization and transformation (Kendall et al., 2005, Kendall et al., 2006). With the modified system, the introduction of hTERT, p53DD, cyclin D1, CDK4R24C, and c-MYCT58A to primary epithelial cells immortalizes the cells, whereas subsequent expression of HRASG12V converts the immortalized cells to a fully tumorigenic state. Using this approach, well-defined genetic models of tumorigenesis can be established starting with normal primary epithelial cells. It was previously demonstrated that the Ras-transformed human mammary epithelial cells (HMECs), but not the pre-malignant immortalized line, form tumors in immunocompromised mice. The resulting tumors were surrounded by areas marked by local tissue invasion (Kendall et al., 2005), a characteristic associated with EMT.
We reconstructed this genetically defined cell culture system of stepwise tumorigenesis as a model with which to study global regulation of early transitions to both malignancy and EMT at multiple levels of gene expression. We demonstrate here that pre-malignant, immortalized cells have gained migratory capacity, whereas the Ras-transformed cells are significantly more invasive, indicating a stepwise gain of traits typically associated with EMT and metastasis. To determine the mechanisms of malignant progression, we quantified changes in messenger RNA (mRNA) expression using RNA sequencing and protein expression using ultraperformance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). Following immortalization, we found dramatic changes in mRNA expression and processing, which correlated with protein abundance. These changes were consistent with previously reported EMT mRNA and alternative splicing signatures. Surprisingly, during Ras transformation we observed enhanced changes of many of the same EMT-related proteins that previously changed during primary cell immortalization, whereas there were very few changes that were unique to the transformation step. These findings suggest that the pre-malignant cells were primed for malignant conversion, with immortalization and transformation cooperating to promote changes in protein abundance. Unlike the immortalization step, changes in protein abundance did not correlate with changes in mRNA following transformation, suggesting significant involvement of post-transcriptional regulation, rather than being solely driven by transcription. Thus, although the two sequential stages appear to cooperate, they use distinct mechanisms to promote EMT. For example, proteins involved in cellular adhesion and cytoskeletal structure, such as α-adducin (ADD1), decreased during immortalization and decreased further following Ras transformation, but their encoding mRNAs did not decrease during Ras transformation. Moreover, short hairpin RNA (shRNA) knockdown of ADD1 in pre-malignant cells resulted in a striking increase in invasive capacity, similar to what we observed in the Ras-transformed cells. Taken together, our data reveal that both malignancy and EMT-associated migration and invasion can be initiated in a two-stage process involving transcriptome priming of pre-malignant cells followed by Ras-triggered post-transcriptional regulation.
Results
Pre-malignant Immortalized Cells Have a Migratory Phenotype, whereas Ras-Transformed Cells Have Increased Invasive Capacity
To study cancer progression, we used a cell culture model in which we immortalized primary HMECs (PRIM) through the stable expression of hTERT, p53DD, cyclinD1, CDK4R24C, and c-MycT58A transgenes. This pre-malignant, immortalized cell line (IMO) was used to create a third, Ras-transformed cell line (TFO) through the expression of H-RasG12V (Figure 1A). We previously confirmed the expression of all transgenes in these cells with quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR), demonstrated that only the TFO cells have anchorage-independent growth using a standard soft agar assay, and confirmed that IMO and TFO cells can be passaged indefinitely, in contrast to the PRIM cells (Bisogno and Keene, 2017). Interestingly, PRIM cells grew in distinct clusters, whereas IMO and TFO cells had a scattered pattern (Figure S1), suggesting altered cell adhesion properties.
Figure 1.

Pre-malignant Immortalized Cells Have a Migratory Phenotype, whereas Ras-transformed Cells Have Increased Invasive Capacity
(A) Genetically defined system of breast cancer progression.
(B) Wound-healing scratch assay: Shown are representative photographs and quantification of scratch closure after 24 hr for four biological replicates. Data are represented as mean ± SEM. SEM, standard error of the mean.
(C) Transwell assay: Shown are representative images and the mean and standard deviation of three biological replicates, quantified as percentage that migrated through Matrigel versus through the control membrane without Matrigel. Data are represented as mean ± SEM.
See also Figure S1
To investigate the migratory properties of IMO and TFO cells, we measured wound closure using a scratch assay. Cells were grown to confluency, and a single scratch was made in the cell monolayer. Migration was quantified as the degree of wound closure after 24 hr, and both IMO and TFO cells were migratory with no significant difference in migration (Figure 1B). We quantified invasion of these cell lines using a Transwell Matrigel invasion assay, and, in contrast to migration, Ras transformation significantly increased invasiveness (Figure 1C). Given that migration and invasion are closely linked to EMT, these results suggest that this genetically defined, primary cell-derived system of tumorigenesis can serve as a stepwise model of gain for EMT-associated properties.
Changes in mRNA Are Consistent with an EMT and Correlate with Changes in Protein Abundance during Pre-malignant Immortalization
To understand the transition between distinct EMT phenotypic states, we quantified global mRNA and protein abundance in all three cell lines (PRIM, IMO, and TFO). We measured global transcriptomics using paired-end RNA sequencing and global proteomics using quantitative one-dimensional UPLC-MS/MS, both methods in biological triplicate (Table S1).
During the PRIM to IMO transition (also referred to as “immortalization”), we detected extensive changes in both mRNA and protein abundance (Figure 2A, left panel). Many of the mRNAs that changed during immortalization were expected based on the known pathways that were perturbed by the transgenes expressed in these cells (Figure S2A). For example, we saw enrichment for Gene Ontology (GO) categories including “telomere maintenance,” “cell proliferation,” “mitotic cell cycle process,” “chromosome organization,” “programmed cell death,” and “negative regulation of cell proliferation.” Importantly, we also observed enrichment of upregulated mRNAs involved in “epithelial cell migration” and enrichment of downregulated mRNAs involved in “cell adhesion,” consistent with our observation that IMO cells are migratory. For about 66% of the mRNAs that changed expression during immortalization, we observed a concordant change in corresponding protein abundance (Figure 2A, middle panel). Globally we observed a strong correlation between changes in mRNAs and corresponding proteins (Figure 2A, right panel). Therefore, changes in proteins that drive migration in these immortalized cells were, in large part, driven by dramatic changes in the transcriptome.
Figure 2.

Changes in mRNA and Protein Abundance Correlate during Immortalization, but Not Following Ras Transformation
(Left) Fraction of total expressed genes whose mRNA (red) or protein (purple) significantly increased (solid) or decreased (striped) during (A) immortalization (IMO) or (B) transformation (TFO).
(Middle) Venn diagram of mRNAs and proteins that increased or decreased significantly (p < 0.05) during (A) immortalization or (B) transformation. Overlapping area represents genes for which both the mRNA and corresponding proteins changed in the same direction during the indicated transition.
(Right) Log2 fold change (L2FC) of RNA (x axis) plotted against the L2FC of the corresponding proteins (y axis) during (A) immortalization and (B) transformation. Red dots denote significant (p < 0.05) changes in RNA only, blue is protein only, and yellow indicates that both RNA and corresponding protein changed significantly.
See also Figures S2 and S3.
To quantify the interplay between EMT progression and primary cell immortalization, we used a previously published method for EMT scoring based on global RNA abundance signatures (Tan et al., 2014). EMT signature scores were calculated using a two-sample Kolmogorov-Smirnov test based on multiple gene expression signatures and reported as a value between −1 and +1, with more positive scores indicating a more mesenchymal state. PRIM cells had a calculated score of 0.22905 (p = 0.05737), indicative of an intermediate or mixed epithelial-mesenchymal RNA signature, and IMO cells had a score of 0.71391 (p = 2.109 × 10−15), indicating a strong mesenchymal signature (Figure S2B).
Transcripts encoding proteins involved in RNA processing were also among the RNAs that increased during the transition from PRIM to IMO, and as a group, core splicing machinery components were significantly upregulated during immortalization (Figure S2C). This observation suggested that, in addition to changes in RNA abundance, immortalization may also cause changes in splicing. Indeed, we observed a large number of significant alternative splicing changes during immortalization (Figure S2D, blue). It has been previously reported that EMT drives an alternative splicing program associated with multiple tumor types (Shapiro et al., 2011, Yang et al., 2016). We therefore assessed the extent of EMT-specific alternative splicing by comparing previously reported EMT splicing signatures with changes observed during the PRIM to IMO transition. We observed close agreement between these datasets (Figures S2E and S2F), and we identified and validated many well-described mesenchymal isoforms, including those for CD44, ENAH (also known as MENA), and p120-catenin, in our system (Figure S3) (Prochazka et al., 2014, Rohan et al., 2014, Sarrio et al., 2004).
In addition to these isoforms that are known to be regulated during EMT, we identified several other splicing events that we would predict to have an impact on cell migration. For example, alternative splicing of Myo1b, which encodes unconventional myosin, yields proteins with lever arms of different lengths that regulate myosin step size and thus, are likely to increase motility rates (Laakso et al., 2010). Expression of this isoform of Myo1b has also been observed in other models of EMT (Yang et al., 2016). We also observed alternative splicing of Discs large homolog 1 (DLG1), a scaffolding protein, in a region that allows interactions with protein kinases, including Crk, which regulates cell adhesion, spreading, and migration (McLaughlin et al., 2002) (Figures S3 and S4). These alternative splicing events are novel in the context of EMT, and our data suggest that they may contribute to a migratory phenotype.
Changes in mRNA Abundance Do Not Correlate with Changes in Protein Abundance during Ras Transformation
In contrast to the transition from PRIM to IMO, very few significant changes in mRNA expression occurred during the IMO to TFO transition (Figure 2B, left panel). This was surprising in light of the drastic difference in anchorage-independent growth and invasiveness (Figure 1C) (Bisogno and Keene, 2017). When we applied the EMT signature scoring system, TFO cells were assigned a score of 0.53635 (p = 8.02 × 10−8) (Figure S2B), indicating that TFO cells have a gene expression signature typical of mesenchymal cells. However, this mRNA expression signature is not more mesenchymal when compared with IMO cells, despite their acquired invasiveness. In addition, the EMT-related alternative splicing events observed during the PRIM to IMO transition were maintained following Ras transformation, but we did not detect any additional changes in alternative splicing (Figures S2C–S2F). However, significantly more proteins were differentially expressed than mRNAs (Figure 2B, left panel). When comparing proteins that changed expression with their corresponding mRNA changes, there was very little overlap and poor correlation (Figure 2B, middle and right panels), suggesting that changes in protein abundance, but not RNA abundance or processing, may be driving changes in malignancy and invasiveness during transformation.
Ras Transformation Enhances the Changes in Protein Expression that Occur during Immortalization
We observed a progression of cellular behavior from IMO cells being more migratory than PRIM to TFO cells being more invasive than IMO, yet the EMT RNA signatures only showed a significant difference in immortalization and not during transformation. We reasoned that changes in protein abundance during transformation were driving the increase in invasion. To investigate how these changes of protein abundance induce migratory cells to become invasive, we compared protein changes during transformation with changes during immortalization. Interestingly, there was good correlation between the liquid chromatography (LC)-MS/MS-derived changes in protein abundance during both transitions, indicating that many proteins that changed during immortalization were altered in the same direction following transformation (Figure 3A, Groups 1 and 2). To see how changes in protein abundance during transformation relate to changes occurring in the preceding PRIM to IMO transition, we filtered the data for all proteins that changed significantly (p > 0.05) during Ras transformation and observed that 55.4% of these proteins increased during immortalization and further increased following transformation, whereas 24.9% of the proteins decreased during immortalization and further decreased during transformation (Figure 3B). Thus, less than 20% of all dynamically changing proteins showed opposite changes between immortalization and transformation. This indicates that Ras is primarily enhancing changes in protein abundance that had occurred during the preceding stage of immortalization.
Figure 3.

Ras Transformation Enhances the Changes in Protein Expression that Occur during Immortalization
(A) Log2 fold change (L2FC) protein abundance during immortalization (PRIM to IMO, x axis) versus transformation (IMO to TFO, y axis) for all expressed genes. Group 1 includes all proteins that increase, and Group 2 includes all proteins that decrease during both transitions.
(B) L2FC protein abundance during immortalization (PRIM to IMO, x axis) versus transformation (IMO to TFO, y axis) for all expressed genes, filtered for significantly changed proteins (p < 0.05) during the IMO to TFO transition (a total of 575 proteins met this criteria). Percent values indicate percentage of proteins contained within each quadrant.
(C) GO process categories for significantly changed proteins in “Group 1” (increased during both transitions) and “Group 2” (decreased during both transitions).
(D) Comparison of L2FC of RNA (left panel) and protein (right panel) during immortalization (x axis) and transformation (y axis) for the ncRNA processing GO category (Group 1).
(E) Comparison of L2FC of RNA (x axis) and protein (y axis) during immortalization (left panel) and transformation (right panel) for the ncRNA processing GO category (Group 1).
(F) Comparison of L2FC RNA (left panel) and protein (right panel) during immortalization (x axis) and transformation (y axis) for the regulation of actin filament-based processes GO category (Group 2).
(G) Comparison of L2FC of RNA (x axis) and protein (y axis) during immortalization (left panel) and transformation (right panel) for the regulation of actin filament-based processes GO category (Group 2).
See also Figure S4.
To investigate how these correlating changes relate to the observed phenotypic changes, we grouped proteins according to their changes during immortalization and transformation, with proteins increasing during both transitions belonging to Group 1 and those decreasing during both transitions belonging to Group 2. Among the proteins within each of these groups, we saw enrichment for numerous GO categories falling into eight broad groupings each. For Group 1, the categories fell into the following groupings: “post-transcriptional process,” “non-coding RNA processing,” “transcription/chromatin,” “DNA binding,” “DNA repair,” “nucleic acid metabolism,” “complex,” and “transport” (Figure 3C). For Group 2 proteins, the categories fell into the following groupings: “transport,” “vesicles/secretion,” “lipid metabolism,” “immune response,” “signaling,” “ion homeostasis,” and, importantly, “cytoskeletal” and “migration/adhesion” (Figure 3C).
Much like our observed global expression patterns, functional categories of mRNA and protein correlated during immortalization but were not correlated during transformation. For example, proteins involved in non-coding RNA processing (Group 1) increased from PRIM to IMO and then again from IMO to TFO (Figure 3D, right panel), whereas the mRNAs encoding these proteins did not follow the same pattern (Figure 3D, left panel). Both RNA and protein increased during the PRIM to IMO transition (Figure 3E, left panel), whereas only proteins increased during the IMO to TFO transition (Figure 3E, right panel). Similarly, proteins involved in the regulation of actin filament-based processes (Group 2) decreased in abundance from PRIM to IMO and then decreased again from IMO to TFO, whereas the mRNAs encoding these proteins had decreased expression from PRIM to IMO but subsequently did not change, as a group, from IMO to TFO (Figures 3F and 3G). We validated the UPLC-MS/MS data by western blotting and RNA-seq data via qRT-PCR for several Group 1 and Group 2 proteins (Figure S5). Indeed, individual protein levels increased (Figure S5A) or decreased (Figure S5B) during Ras transformation without significant corresponding changes in mRNA. Therefore, although immortalization and transformation appear to cooperate to promote EMT, they do so by distinct mechanisms. Changes in protein expression correlate with changes in mRNA expression during immortalization, suggesting regulation of transcription and/or RNA stability, while oncogenic Ras further enhances these changes in protein abundance with no corresponding changes in RNA expression, suggesting regulation via post-transcriptional mechanisms, such as translation and/or protein stability, during transformation.
Knockdown of α-Adducin Enhances Invasion of Immortalized Cells
Since the enriched GO categories for Group 2 proteins contained many categories related to “cytoskeletal” and “adhesion/migration,” suggesting that Ras-driven changes in protein abundance may result in increased invasion, we sought to test whether mimicking these changes in IMO cells could induce the transformation-specific invasive phenotype. As a proof of principle, we examined ADD1, a Group 2 protein. ADD1 is a membrane skeletal protein that plays a role in the formation and stabilization of the membrane cytoskeleton by binding and capping actin filaments and promoting actin association with spectrin (Li et al., 1998). ADD1 has a demonstrated role in maintaining epithelial junction and barrier integrity, and knockdown of ADD1 decreases adherens junctions and tight junction assembly through diminished spectrin recruitment and a decrease in F-actin abundance, resulting in the impaired assembly of actin bundles (Naydenov and Ivanov, 2010).
Expression of oncogenic Ras resulted in a decrease in ADD1 protein abundance with no significant change in mRNA expression (Figures 4A and 4B). We reduced the levels of ADD1 in IMO cells using three shRNA constructs (Figure 4A). ADD1 reduction in IMO cells drastically increased cell invasion while having no effect on cell survival, similar to what we observed during Ras transformation (Figure 4C). In summary, ADD1 protein expression levels decreased during immortalization, yet did not lead to an invasive phenotype. ADD1 protein, but not its encoding mRNA, was further reduced in response to oncogenic Ras transformation, and both oncogenic Ras expression and direct knockdown of ADD1 resulted in increased cell invasion. Therefore, our data suggest that oncogenic Ras induces EMT-associated invasion, at least in part, by directly decreasing the levels of ADD1 protein.
Figure 4.

Knockdown of α-Adducin Enhances Invasion of Immortalized Cells
(A) Western blot validation of the LC-MS/MS detected decrease of ADD1 in TFO versus IMO cells and shRNA knockdown of ADD1 in IMO cells.
(B) qRT-PCR of ADD1-encoding mRNA in IMO and TFO cells, shown as relative expression to PRIM cells, normalized to Glyceraldehyde 3-phosphate dehydrogenase (GapDH). Data are represented as mean ± standard deviation, n = 4.
(C) Transwell assay: Shown are representative images and the mean and standard deviation of two biological replicates of each shRNA, six biological replicates of IMO, and four biological replicates of TFO, quantified as percentage that migrated through Matrigel versus through the membrane without Matrigel. Data are represented as the mean ± SEM.
Discussion
EMT Induction and Synergistic Cooperation with Ras
Just as normal cells evolve to a neoplastic state during malignant progression, epithelial cells transition through a series of intermediate stages during the process of EMT. During an EMT, epithelial cells pass through intermediate and reversible states with a mixture of epithelial and mesenchymal properties, and cells in a partial EMT state may simultaneously express epithelial and mesenchymal markers and acquire some EMT-related phenotypes (Nieto et al., 2016). Unlike traditional one-step models of EMT, our genetically defined, stepwise system enabled us to gain novel mechanistic insights into different EMT-intermediate states. In the first stage of primary cell immortalization, we observed a repression of the epithelial gene expression signature, activation of a mesenchymal program, and a dramatic increase in cell migration. Ras transformation of these immortalized cells enhanced the changes laid out during the first stage by enhancing the expression levels of a subset of proteins, resulting in an invasive phenotype. In fact, over 80% of the Ras-induced changes were changed in the same direction up or down during immortalization, suggesting a synergy between the two stages where pre-malignant cells were primed for Ras-triggered activation.
Although the acquisition of most cancer hallmarks can be encompassed into a model of Darwinian evolution in which genetic mutations confer a selective advantage, activation of migration and invasion does not provide an obvious survival advantage to the primary tumor (Gerlinger et al., 2012, Gupta et al., 2005, Hanahan and Weinberg, 2011). Therefore, it has been postulated that mutations that directly contribute to metastatic competence may be harbored within genetic lesions for another trait that renders a selective advantage within the primary tumor (Gupta et al., 2005). Given the cooperation between stages that promote invasion and malignancy in our model (Figures 1 and S2), we speculate that invasive traits may be harbored within multiple mutations, and in some instances, these mutations may be able to act synergistically.
Ras Synergy with Multiple EMT-Promoting Pathways
Our results suggest that oncogenic Ras works cooperatively with the genetic alterations induced during primary cell immortalization to promote EMT-associated invasion. Interestingly, a previous study showed that KRAS suppression in pancreatic cancer cells attenuated transforming growth factor β (TGF-β)-induced upregulation of Snail, a transcription factor involved in EMT (Horiguchi et al., 2009). In addition, expression of the H-RasG12V transgene in HeLa cells boosted the upregulation of Snail in response to TGF-β, suggesting that TGF-β signaling and Ras cooperate to induce Snail (Horiguchi et al., 2009). Notably, this TGF-β-Ras synergism induced EMT through a distinct mechanism from immortalization in our system, as we did not observe changes to Snail expression. This suggests that Ras can cooperate with multiple, distinct mechanisms to promote EMT.
We observed synergistic regulation by immortalization and Ras transformation for many proteins involved in EMT-related processes, including the canonical mesenchymal markers vimentin and N-cadherin, as well as many cell adhesion and cytoskeleton organization proteins (Figure S5). Our data suggest that Ras may cooperate with the genetic mutations in the immortalized cells to regulate many EMT-related proteins to promote malignancy and invasion. One example is RIO kinase 1 (RIOK1), which increased protein expression following both immortalization and Ras transformation. Interestingly, a recent study revealed that RIOK1 knockdown reduced invasion in Ras-transformed breast, colon, and lung cancer cells, but it did not have the same effect on cells with wild-type Ras (Weinberg et al., 2017). Moreover, knockdown of RIOK1 in immortalized breast cells reduced KRAS transformation efficiency, suggesting a requirement for RIOK1 in the context of oncogenic Ras signaling (Weinberg et al., 2017). Another example of an EMT-related change was Drebrin-like (DBNL) actin-binding protein, which was reduced following both immortalization and Ras transformation. DBNL is necessary for the formation of podosome rosettes (Boateng et al., 2012). Podosomes are dynamic structures that regulate extracellular matrix adhesion and degradation, and knockdown of DBNL in Src-transformed fibroblasts greatly increased invasive cell migration (Boateng et al., 2012). In breast cancer, DBNL has been characterized as a metastatic suppressor, and it is stabilized by the RNA-binding protein MBNL1 (Fish et al., 2016).
Post-transcriptional Mechanisms Triggered by Oncogenic Ras
Although many studies have relied on steady-state mRNA levels as a proxy for gene expression, it has become increasingly clear that mRNA levels alone do not provide a complete picture of the complex regulatory processes occurring during cancer cell progression, as evidenced by the fact that steady-state protein levels often do not correlate with steady-state mRNA levels (Griffin et al., 2002, Ideker et al., 2001, Keene, 2001, Mansfield and Keene, 2009). Our global datasets suggest that Ras likely controls invasion through post-transcriptional mechanisms, such as translation and/or protein stability (Figure 2).
Ras signaling has previously been shown to regulate translation. In glioblastoma, Ras and Akt signaling have minimal effects on RNA abundance, whereas they significantly alter mRNA translation (Rajasekhar et al., 2003). Ras signaling is also required for phosphorylation of RPS6 and is thereby involved in stimulating cap-dependent translation (Roux et al., 2007). Furthermore, Ras is required for mitogen-induced phosphorylation of the cap-binding protein eIF4E, thus regulating translation initiation (Frederickson et al., 1992). eIF4E is also known to be phosphorylated during TGF-β-induced EMT, which in turn regulates translation of Snail and Mmp-3 (Robichaud et al., 2015).
In addition to directly regulating the translational machinery, Ras can regulate the activity of sequence-specific RNA-binding proteins, which bind to elements located in regulatory regions of mRNA, such as 3′ or 5′ untranslated regions (UTRs), and regulate the translation of multiple functionally related genes, or RNA regulons. For example, the CELF6 RNA-binding protein has been demonstrated to rescue KRAS suppression in oncogene-addicted cells (Shao et al., 2014). Furthermore, a recent study established a role for CELF1, a related family member of CELF6, in regulating EMT-associated translational regulation (Chaudhury et al., 2016). Several other studies have demonstrated roles for translational RNA regulons in the acquisition of cancer-related phenotypes downstream of major oncogenic signaling pathways (Jung et al., 2014, Romeo et al., 2013, Shahbazian et al., 2010, Truitt and Ruggero, 2016, Tsukumo et al., 2016, Wurth et al., 2016).
Regulation of protein localization and turnover also greatly determines cellular processes. EMT is regulated by post-translational modifications that can influence protein localization and degradation. For example, Snail is phosphorylated by glycogen synthase kinase (GSK)-3β, leading to nuclear export and subsequent ubiquitin-mediated proteasome degradation, whereas dephosphorylation by SCP inhibits Snail degradation (Wu et al., 2009, Zhou et al., 2004). Ras signaling can lead to the activation of mitogen-activated protein kinases (MAPK), which phosphorylates downstream targets involved in cancer progression and EMT. MAPK-mediated phosphorylation of the transcription factor TWIST1 increased protein levels without affecting mRNA levels, leading to breast cancer cell invasiveness (Hong et al., 2011). Furthermore, the Ras-ERK MAPK signaling pathway has been demonstrated to inhibit the ubiquitin-proteasome pathway in both developing T cells and during mitogen activation of fibroblasts (Sears et al., 1999, Yamashita et al., 2005). Interestingly, our data showed upregulation of components of the anaphase-promoting complex/cyclosome (APC/C), including Bub1B, CDC20, UBE2C, and UBE2S (Table S1). The APC/C is a ubiquitin ligase complex that regulates a variety of cellular processes, including invasion, by targeting different substrates for ubiquitination (Zhou et al., 2016). It is therefore possible that a number of the proteins that are decreased in response to oncogenic Ras could be targeted for ubiquitination by the APC/C complex.
In summary, we have identified a novel and synergistic transition in gene expression whereby mutated oncogenic Ras post-transcriptionally enhances changes primed via genetic mutations in pre-malignant cells, resulting in a significant increase in malignancy and cell invasiveness. This work provides important insights into the mechanisms by which cancer-associated genetic mutations cooperate to promote transitions through partial EMT states.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Jeff Blackinton, Kyle Mansfield, and Cindo Nicholson for helpful discussions and critical insights. We also thank Duke University's Center for Genomic and Computational Biology, as well as Matt Foster and Will Thompson at Duke University's Proteomics Core Facility. This work was funded by grants from the NIH: R01CA157268 (J.D.K.) and F31CA185892 (L.S.B.).
Author Contributions
Conceptualization, J.D.K., L.S.B., and M.B.F.; Methodology, L.S.B., M.B.F., and J.D.K.; Formal Analysis, L.S.B. and M.B.F.; Investigation, L.S.B.; Resources, J.D.K.; Data Curation, M.B.F.; Writing – Original Draft, Review, & Editing, L.S.B., M.B.F., and J.D.K.; Visualization, L.S.B. and M.B.F.; Funding Acquisition, J.D.K. and L.S.B.
Declaration of Interests
The authors declare no conflict of interest.
Published: June 29, 2018
Footnotes
Supplemental Information includes Transparent Methods, five figures, and six tables and can be found with this article online at https://doi.org/10.1016/j.isci.2018.05.011.
Supplemental Information
References
- Bisogno L.S., Keene J.D. Analysis of post-transcriptional regulation during cancer progression using a donor-derived isogenic model of tumorigenesis. Methods. 2017;126:193–200. doi: 10.1016/j.ymeth.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boateng L.R., Cortesio C.L., Huttenlocher A. Src-mediated phosphorylation of mammalian Abp1 (DBNL) regulates podosome rosette formation in transformed fibroblasts. J. Cell Sci. 2012;125:1329–1341. doi: 10.1242/jcs.096529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury A., Cheema S., Fachini J.M., Kongchan N., Lu G., Simon L.M., Wang T., Mao S., Rosen D.G., Ittmann M.M. CELF1 is a central node in post-transcriptional regulatory programmes underlying EMT. Nat. Commun. 2016;7:13362. doi: 10.1038/ncomms13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish L., Pencheva N., Goodarzi H., Tran H., Yoshida M., Tavazoie S.F. Muscleblind-like 1 suppresses breast cancer metastatic colonization and stabilizes metastasis suppressor transcripts. Genes Dev. 2016;30:386–398. doi: 10.1101/gad.270645.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederickson R.M., Mushynski W.E., Sonenberg N. Phosphorylation of translation initiation factor eIF-4E is induced in a ras-dependent manner during nerve growth factor-mediated PC12 cell differentiation. Mol. Cell Biol. 1992;12:1239–1247. doi: 10.1128/mcb.12.3.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M., Rowan A.J., Horswell S., Larkin J., Endesfelder D., Gronroos E., Martinez P., Matthews N., Stewart A., Tarpey P. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin T.J., Gygi S.P., Ideker T., Rist B., Eng J., Hood L., Aebersold R. Complementary profiling of gene expression at the transcriptome and proteome levels in Saccharomyces cerevisiae. Mol. Cell Proteomics. 2002;1:323–333. doi: 10.1074/mcp.m200001-mcp200. [DOI] [PubMed] [Google Scholar]
- Gupta P.B., Mani S., Yang J., Hartwell K., Weinberg R.A. The evolving portrait of cancer metastasis. Cold Spring Harb. Symp. Quant. Biol. 2005;70:291–297. doi: 10.1101/sqb.2005.70.033. [DOI] [PubMed] [Google Scholar]
- Hahn W.C., Counter C.M., Lundberg A.S., Beijersbergen R.L., Brooks M.W., Weinberg R.A. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hong J., Zhou J., Fu J., He T., Qin J., Wang L., Liao L., Xu J. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011;71:3980–3990. doi: 10.1158/0008-5472.CAN-10-2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiguchi K., Shirakihara T., Nakano A., Imamura T., Miyazono K., Saitoh M. Role of Ras signaling in the induction of snail by transforming growth factor-beta. J. Biol. Chem. 2009;284:245–253. doi: 10.1074/jbc.M804777200. [DOI] [PubMed] [Google Scholar]
- Husemann Y., Geigl J.B., Schubert F., Musiani P., Meyer M., Burghart E., Forni G., Eils R., Fehm T., Riethmuller G. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13:58–68. doi: 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Ideker T., Thorsson V., Ranish J.A., Christmas R., Buhler J., Eng J.K., Bumgarner R., Goodlett D.R., Aebersold R., Hood L. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science. 2001;292:929–934. doi: 10.1126/science.292.5518.929. [DOI] [PubMed] [Google Scholar]
- Jung H., Gkogkas C.G., Sonenberg N., Holt C.E. Remote control of gene function by local translation. Cell. 2014;157:26–40. doi: 10.1016/j.cell.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene J.D. Ribonucleoprotein infrastructure regulating the flow of genetic information between the genome and the proteome. Proc. Natl. Acad. Sci. USA. 2001;98:7018–7024. doi: 10.1073/pnas.111145598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall S.D., Adam S.J., Counter C.M. Genetically engineered human cancer models utilizing mammalian transgene expression. Cell Cycle. 2006;5:1074–1079. doi: 10.4161/cc.5.10.2734. [DOI] [PubMed] [Google Scholar]
- Kendall S.D., Linardic C.M., Adam S.J., Counter C.M. A network of genetic events sufficient to convert normal human cells to a tumorigenic state. Cancer Res. 2005;65:9824–9828. doi: 10.1158/0008-5472.CAN-05-1543. [DOI] [PubMed] [Google Scholar]
- Laakso J.M., Lewis J.H., Shuman H., Ostap E.M. Control of myosin-I force sensing by alternative splicing. Proc. Natl. Acad. Sci. USA. 2010;107:698–702. doi: 10.1073/pnas.0911426107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert A.W., Pattabiraman D.R., Weinberg R.A. Emerging biological principles of metastasis. Cell. 2017;168:670–691. doi: 10.1016/j.cell.2016.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S., Xu J., Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Matsuoka Y., Bennett V. Adducin preferentially recruits spectrin to the fast growing ends of actin filaments in a complex requiring the MARCKS-related domain and a newly defined oligomerization domain. J. Biol. Chem. 1998;273:19329–19338. doi: 10.1074/jbc.273.30.19329. [DOI] [PubMed] [Google Scholar]
- Mani S.A., Guo W., Liao M.J., Eaton E.N., Ayyanan A., Zhou A.Y., Brooks M., Reinhard F., Zhang C.C., Shipitsin M. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield K.D., Keene J.D. The ribonome: a dominant force in co-ordinating gene expression. Biol. Cell. 2009;101:169–181. doi: 10.1042/BC20080055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin M., Hale R., Ellston D., Gaudet S., Lue R.A., Viel A. The distribution and function of alternatively spliced insertions in hDlg. J. Biol. Chem. 2002;277:6406–6412. doi: 10.1074/jbc.M108724200. [DOI] [PubMed] [Google Scholar]
- Naydenov N.G., Ivanov A.I. Adducins regulate remodeling of apical junctions in human epithelial cells. Mol. Biol. Cell. 2010;21:3506–3517. doi: 10.1091/mbc.E10-03-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto M.A., Huang R.Y., Jackson R.A., Thiery J.P. Emt: 2016. Cell. 2016;166:21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- Podsypanina K., Du Y.C., Jechlinger M., Beverly L.J., Hambardzumyan D., Varmus H. Seeding and propagation of untransformed mouse mammary cells in the lung. Science. 2008;321:1841–1844. doi: 10.1126/science.1161621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochazka L., Tesarik R., Turanek J. Regulation of alternative splicing of CD44 in cancer. Cell. Signal. 2014;26:2234–2239. doi: 10.1016/j.cellsig.2014.07.011. [DOI] [PubMed] [Google Scholar]
- Rajasekhar V.K., Viale A., Socci N.D., Wiedmann M., Hu X., Holland E.C. Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol. Cell. 2003;12:889–901. doi: 10.1016/s1097-2765(03)00395-2. [DOI] [PubMed] [Google Scholar]
- Rhim A.D., Mirek E.T., Aiello N.M., Maitra A., Bailey J.M., McAllister F., Reichert M., Beatty G.L., Rustgi A.K., Vonderheide R.H. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaud N., del Rincon S.V., Huor B., Alain T., Petruccelli L.A., Hearnden J., Goncalves C., Grotegut S., Spruck C.H., Furic L. Phosphorylation of eIF4E promotes EMT and metastasis via translational control of SNAIL and MMP-3. Oncogene. 2015;34:2032–2042. doi: 10.1038/onc.2014.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohan T.E., Xue X., Lin H.M., D'Alfonso T.M., Ginter P.S., Oktay M.H., Robinson B.D., Ginsberg M., Gertler F.B., Glass A.G. Tumor microenvironment of metastasis and risk of distant metastasis of breast cancer. J. Natl. Cancer Inst. 2014;106 doi: 10.1093/jnci/dju136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo Y., Moreau J., Zindy P.J., Saba-El-Leil M., Lavoie G., Dandachi F., Baptissart M., Borden K.L., Meloche S., Roux P.P. RSK regulates activated BRAF signalling to mTORC1 and promotes melanoma growth. Oncogene. 2013;32:2917–2926. doi: 10.1038/onc.2012.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux P.P., Shahbazian D., Vu H., Holz M.K., Cohen M.S., Taunton J., Sonenberg N., Blenis J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 2007;282:14056–14064. doi: 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrio D., Perez-Mies B., Hardisson D., Moreno-Bueno G., Suarez A., Cano A., Martinez-Perez J., Gamallo C., Palacios J. Cytoplasmic localization of p120ctn and E-cadherin loss characterize lobular breast carcinoma from preinvasive to metastatic lesions. Oncogene. 2004;23:3272–3283. doi: 10.1038/sj.onc.1207439. [DOI] [PubMed] [Google Scholar]
- Sears R., Leone G., DeGregori J., Nevins J.R. Ras enhances Myc protein stability. Mol. Cell. 1999;3:169–179. doi: 10.1016/s1097-2765(00)80308-1. [DOI] [PubMed] [Google Scholar]
- Shahbazian D., Parsyan A., Petroulakis E., Hershey J., Sonenberg N. eIF4B controls survival and proliferation and is regulated by proto-oncogenic signaling pathways. Cell Cycle. 2010;9:4106–4109. doi: 10.4161/cc.9.20.13630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao D.D., Xue W., Krall E.B., Bhutkar A., Piccioni F., Wang X., Schinzel A.C., Sood S., Rosenbluh J., Kim J.W. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell. 2014;158:171–184. doi: 10.1016/j.cell.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro I.M., Cheng A.W., Flytzanis N.C., Balsamo M., Condeelis J.S., Oktay M.H., Burge C.B., Gertler F.B. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011;7:e1002218. doi: 10.1371/journal.pgen.1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibue T., Weinberg R.A. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017;14:611–629. doi: 10.1038/nrclinonc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan T.Z., Miow Q.H., Miki Y., Noda T., Mori S., Huang R.Y., Thiery J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014;6:1279–1293. doi: 10.15252/emmm.201404208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truitt M.L., Ruggero D. New frontiers in translational control of the cancer genome. Nat. Rev. Cancer. 2016;16:288–304. doi: 10.1038/nrc.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukumo Y., Alain T., Fonseca B.D., Nadon R., Sonenberg N. Translation control during prolonged mTORC1 inhibition mediated by 4E-BP3. Nat. Commun. 2016;7:11776. doi: 10.1038/ncomms11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha C.C., Jiang P., Amirikian K., Dittmar K.A., Lu H., Shen S., Guo W., Xing Y., Carstens R.P. An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. EMBO J. 2010;29:3286–3300. doi: 10.1038/emboj.2010.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg F., Reischmann N., Fauth L., Taromi S., Mastroianni J., Kohler M., Halbach S., Becker A.C., Deng N., Schmitz T. The atypical kinase RIOK1 promotes tumor growth and invasive behavior. EBioMedicine. 2017;20:79–97. doi: 10.1016/j.ebiom.2017.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Evers B.M., Zhou B.P. Small C-terminal domain phosphatase enhances snail activity through dephosphorylation. J. Biol. Chem. 2009;284:640–648. doi: 10.1074/jbc.M806916200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurth L., Papasaikas P., Olmeda D., Bley N., Calvo G.T., Guerrero S., Cerezo-Wallis D., Martinez-Useros J., Garcia-Fernandez M., Huttelmaier S. UNR/CSDE1 drives a post-transcriptional program to promote melanoma invasion and metastasis. Cancer Cell. 2016;30:694–707. doi: 10.1016/j.ccell.2016.10.004. [DOI] [PubMed] [Google Scholar]
- Yamashita M., Shinnakasu R., Asou H., Kimura M., Hasegawa A., Hashimoto K., Hatano N., Ogata M., Nakayama T. Ras-ERK MAPK cascade regulates GATA3 stability and Th2 differentiation through ubiquitin-proteasome pathway. J. Biol. Chem. 2005;280:29409–29419. doi: 10.1074/jbc.M502333200. [DOI] [PubMed] [Google Scholar]
- Yang Y., Park J.W., Bebee T.W., Warzecha C.C., Guo Y., Shang X., Xing Y., Carstens R.P. Determination of a comprehensive alternative splicing regulatory network and combinatorial regulation by key factors during the epithelial-to-mesenchymal transition. Mol. Cell Biol. 2016;36:1704–1719. doi: 10.1128/MCB.00019-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M., Bardia A., Wittner B.S., Stott S.L., Smas M.E., Ting D.T., Isakoff S.J., Ciciliano J.C., Wells M.N., Shah A.M. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339:580–584. doi: 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.P., Deng J., Xia W., Xu J., Li Y.M., Gunduz M., Hung M.C. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 2004;6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- Zhou Z., He M., Shah A.A., Wan Y. Insights into APC/C: from cellular function to diseases and therapeutics. Cell Div. 2016;11:9. doi: 10.1186/s13008-016-0021-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.