

Abstract
Ischaemia-induced tissue injury has wide-ranging clinical implications including myocardial infarction, stroke, compartment syndrome, ischaemic renal failure and replantation and revascularization. However, the restoration of blood flow produces a 'second hit' phenomenon, the effect of which is greater than the initial ischaemic event and characterizes ischaemiareperfusion (IR) injury. Some examples of potential settings of IR injury include: following thrombolytic therapy for stroke, invasive cardiovascular procedures, solid organ transplantation, and major trauma resuscitation. Pathophysiological events of IR injury are the result of reactive oxygen species (ROS) production, microvascular vasoconstriction, and ultimately endothelial cell-neutrophil adhesion with subsequent neutrophil infiltration of the affected tissue. Initially thought to increase the amount of free radical oxygen in the system, hyperbaric oxygen (HBO) has demonstrated a protective effect on tissues by influencing the same mechanisms responsible for IR injury. Consequently, HBO has tremendous therapeutic value. We review the biochemical mechanisms of ischaemia-reperfusion injury and the effects of HBO following ischaemia-reperfusion.
Keywords: Hypoxia, Hyperoxia, Reperfusion injury, Free radicals, Nitric oxide, Ischaemic preconditioning, Review article
Introduction
Tissue ischaemia represents the final common pathway of various disease states that include myocardial infarction, stroke, amputations, compartment syndromes and failing tissue flaps and grafts. In these scenarios, emergent interventions are undertaken to restore blood flow to the affected areas, which in some cases may be life- and limb-saving on a global scale. However, this reperfusion is not without consequence; despite restoration of flow, further tissue and microcirculation injury still occur, even to a greater extent than the initial ischaemic insult. Tissue necrosis and microcirculatory collapse that occur because of reperfusion following prolonged ischaemia is referred to as ischaemia-reperfusion (IR) injury. Examples of IR injury can be seen in many settings: thrombolytic therapy for stroke, any cardiovascular invasive procedure (e.g., angioplasty of the popliteal artery to coronary artery bypass with assisted circulation), organ transplantation, and major trauma resuscitation. Reactive oxygen species (ROS) have been shown to be the principal mediators of this phenomenon. During IR injury, the blood-endothelial cell interface shows increased microcirculatory neutrophil adhesion that incites tissue necrosis and starts a feedback loop that results in further ROS production and injury. Given the potentially devastating clinical outcomes of IR injury, much investigation has been undertaken to better understand the molecular signals and changes that occur in the microcirculation. As our understanding of the mechanisms of IR injury has evolved, so too has interest in therapeutic interventions to reverse or prevent it, particularly with hyperbaric oxygen (HBO). The purpose of this article is to discuss the biochemical mechanisms of ischaemia-reperfusion injury and review the effects of HBO treatment.
Ischaemia-reperfusion injury (Figure 1)
Figure 1.
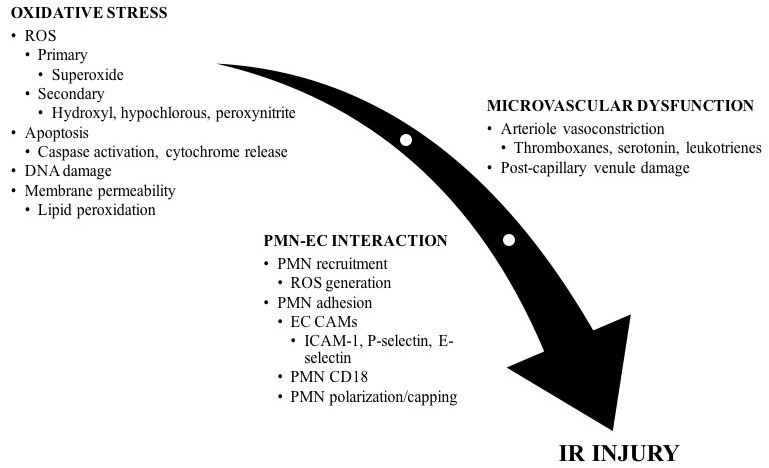
Mediators of ischaemia-reperfusion injury; oxidative stress, microvascular dysfunction, and the neutrophil-endothelial cell interaction produce changes in cellular physiology that increase cell damage and tissue death; CAM – cellular adhesion molecule; EC – endothelial cell; PMN – polymorphonuclear neutrophil; ROS – reactive oxygen species
OXIDATIVE STRESS
ROS, chiefly oxygen free radicals, serve a cell-signalling role and are formed during cellular metabolism.[ 1] While their role in normal homeostasis continues to be investigated, their involvement in mediating oxidative damage seen during IR injury has been documented extensively.[ 2] The predominant ROS in the cell are superoxide and hydroxyl radicals. Their cellular toxicity results from lipid peroxidation and its associated membrane damage, direct DNA damage, and production of other free radical and reactive species.[ 3] Due to the instability of free radical species, free radical scavengers have been utilized to indirectly prove the detrimental effects of ROS in IR injury. Using a mouse hind limb model, improvement in inflammatory cell infiltration in skeletal muscle after IR was shown when pretreating with edavarone, a synthetic free radical scavenger.[ 4]
In the normal physiologic state, multiple antioxidant mechanisms exist to counteract the effect of these ROS: superoxide dismutase (SOD), glutathione, and catalase.[ 5] Once these systems are overwhelmed, as occurs during IR injury, excess ROS is produced and incites tissue damage. These antioxidant systems have also been utilized as physiologic free radical scavengers that provide improvements in skeletal muscle function after undergoing IR injury. As a model for limb replantation, rabbit tibialis anterior muscle was subjected to IR at five-and eight-hour intervals and muscle function was examined after administration of the hydroxyl free radical scavenger dimethylsulfoxide (DMSO) and superoxide free radical scavenger SOD prior to reperfusion.[ 6]
During IR injury, two sources for ROS generation are xanthine oxidase and neutrophils. Xanthine oxidase, the conversion product of oxidative damage to xanthine dehydrogenase found in skeletal muscle endothelial cells, produces superoxide and hydrogen peroxide (H₂O₂) during purine metabolism.[ 3] These ROS recruit neutrophils to the blood-endothelial cell interface, thereby initiating migration into the surrounding tissues. Neutrophils then produce a greater amount of ROS, further precipitating the effects of IR injury. Consequently, much research into IR injury has focused on the interaction between neutrophils at the blood-endothelial cell interface. Neutrophils generate a large amount of extracellular superoxide owing to the presence of membrane-bound nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. Following dismutation to H₂O₂, subsequent reactions result in the formation of other toxic molecules: hydroxyl radical (reaction with ferritin) and hypochlorous acid (reaction with chloride via neutrophil myeloperoxidase). It appears that, in IR injury, xanthine oxidase may produce the initial liberation of ROS species, with further propagation coming from neutrophils, finally culminating in tissue injury.[ 7 , 8]
Programmed cell death (apoptosis) has also been implicated in IR injury although the mechanisms remain unclear. ROS accumulation has been shown to induce apoptosis.[ 9] More recently, nitric oxide (NO) has been suggested as a mediator of IR injury. NO is produced by nitric oxide synthase (NOS), which has three isoforms: neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS).[ 10] The former two isoforms are expressed constitutively, whereas the latter requires protein synthesis. It should be noted that NO competes with oxygen in binding to terminal cytochrome c oxidase, which has a higher affinity for NO than for oxygen. At higher oxygen concentrations, NO is consumed by cytochrome c oxidase and thus simulates a hypoxic environment. Conversely in lower oxygen tension, NO is not consumed and is available to mediate its physiologic effects.[ 11]
High levels of NO produced by iNOS may interact with superoxide to produce peroxynitrite, resulting in mitochondrial cytochrome c release and caspase activation, and ultimately apoptosis.[ 12 , 13] Rat intestinal mucosa subjected to IR injury demonstrated iNOS, NO, and apoptosis.[ 14] The data regarding NO and apoptosis is conflicting as other studies have demonstrated a protective role for NO against apoptosis.[ 15 , 16] Further investigation is required to clarify this relationship. Therefore, it appears that the secondary products of superoxide radical interactions (i.e., hydroxyl radical, hypochlorous acid, peroxynitrite) are the primary mediators of the toxic effects of IR injury rather than superoxide itself. Moreover, there is a growing body of evidence demonstrating a cellular signalling role for superoxide and other oxidative species.[ 17]
MICROCIRCULATION DYNAMICS
In studies investigating reperfusion following myocardial infarction in dogs, reperfusion results in improved flow through the large vessels. However, the microcirculation demonstrates obstruction, circulatory collapse, and myocardial function still suffers.[ 18] These findings suggest that IR injury involves changes in the microcirculation. An early study found significant increases in vasoconstricting thromboxanes compared to vasodilating prostaglandins in a rat hind-limb model of the no-reflow state, a phenomenon describing complete microcirculatory failure, compared to ischaemic limbs with reflow.[ 19] This study also found decreased venous outflow with absent vascular thrombosis, suggesting a mechanism of excess thromboxane release producing microcirculatory vasoconstriction in the impending no-reflow state.[ 19] Similar findings regarding vasoconstriction in IR injury were observed in a rat skeletal muscle utilizing intravital microscopy to examine arteriolar and venule diameters.[ 20]
Another study demonstrated that vasoactive substances exert a greater effect on arteriolar smooth muscle than on the endothelium in the microcirculation.[ 21] It was concluded that there is a barrier to diffusion of water-soluble vasoactive substances between the smooth muscle and endothelium. During IR injury, the post-capillary venule endothelial cells are damaged by neutrophils. This might provide a putative explanation as to why the vasoconstrictive effect of IR injury is confined to the immediately adjacent microarterioles. Other vasoactive substances have been implicated in modulating microarteriolar vasoconstriction, including serotonin and leukotrienes.[ 22 , 23] While much of the focus has been on neutrophil-induced injury following reperfusion, other cell lines may be involved. Skeletal muscle IR injury results in adenosine-regulated mast cell degranulation that initiates arteriolar vasoconstriction.[ 24] Therefore, it is likely that the dynamic microvascular changes occurring during IR injury reflect the complex interplay between multiple cell lines and vasoactive molecules. Additional research is necessary to better understand these interactions.
NEUTROPHIL-ENDOTHELIAL CELL ADHESION
It is interesting to note that tissue damage from ischaemia differs microscopically from that from IR injury. With ischaemia, tissue architecture is preserved and there is a relative acellularity. In contrast, IR injury is characterized by tissue necrosis and leukocyte infiltration, especially neutrophils. Neutrophils are recruited to the ischaemic site following reperfusion and, prior to extravasation, adhere to the endothelium. Given the role of neutrophils in mediating IR injury and ROS production, investigation of the neutrophil-endothelial cell interface has increased for its potential therapeutic targetting.
Neutrophil adhesion to the microvascular endothelium is dependent on interactions between receptors and ligands on the surface of the neutrophil and endothelium called cell adhesion molecules (CAMs). Examples include P-selectin, E-selectin, and ICAM-1. Expression of these molecules varies depending on the local tissue conditions and on cytokine release. P-selectin is expressed on the surface of endothelial cells within 15 minutes of middle cerebral artery occlusion, where it can bind to its respective neutrophil ligand (P-selectin glycoprotein ligand-1).[ 25] P-selectin is contained within Weibel-Palade bodies and is secreted onto the endothelial cell surface after these storage granules are exocytosed.[ 26]
Whereas P-selectin expression occurs acutely following IR injury, other CAM molecules (e.g., E-selectin and ICAM-1) expression occurs in a delayed fashion, likely because of increased CAM molecule expression via transcription and translation.[ 27] E-selectin and ICAM-1 then bind to neutrophil ligands to produce the neutrophil-endothelial cell adhesion. Using monoclonal antibody against CD18 to block neutrophil-endothelial cell adherence, a study of rat skeletal muscle showed that ICAM-1 interacts with neutrophil cell-surface CD18 molecules during IR injury.[ 28] Microarteriolar vasoconstriction was also blocked, suggesting that neutrophil CD18 plays a key role in altering the microcirculation during IR injury. Using phorbol-12 myristate 13-acetate (PMA) to simulate endothelial cell injury, it has been shown that ischaemia-reperfusion also upregulates neutrophil CD18.[ 29] In another in vitro study of neutrophil adherence, confocal microscopy demonstrated neutrophil capping, which refers to the change in polarity of surface CD18 molecules by concentrating them in one area, thereby increasing the likelihood of adhesion to endothelial ICAM-1 after IR injury.[ 30]
Protection against IR injury has been shown by blocking neutrophil-endothelial cell adhesion in vivo. Use of anti-CD18 monoclonal antibody in baboons decreased cerebral infarction.[ 31] Additionally, a knock-out mice model for ICAM-1 exhibited a six-fold decrease in infarction size compared to wild-type.[ 32] Interestingly, neutrophil recruitment was similar between the groups, suggesting that the extent of inflammatory cell response is less crucial than the interaction between neutrophils and the endothelium.[ 32]
Hyperbaric oxygen (Figure 2)
Figure 2.
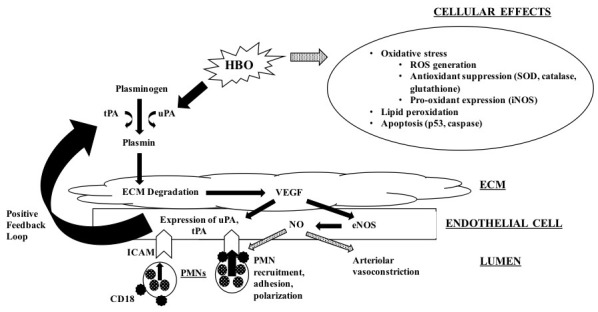
Physiologic effects following hyperbaric oxygen therapy for ischaemia-reperfusion; hyperbaric oxygen results in interference with neutrophil-endothelial cell interactions, promotes arteriolar vasodilation, and ameliorates cellular damage; solid arrows represent stimulatory pathways; dotted arrows represent inhibitory pathways; ECM − extracellular matrix; eNOS − endothelial nitric oxide synthase; HBO − hyperbaric oxygen; ICAM − intercellular adhesion molecule; iNOS − inducible nitric oxide synthase; NO − nitric oxide; PMN − polymorphonuclear neutrophil; ROS − reactive oxygen species; SOD − superoxide dismutase;tPA − tissue plasminogen activator; uPA − urinokinase-like plasminogen activator; VEGF− vascular endothelial growth factor
HALLMARK STUDIES
The initial ischaemic insult deprives tissues of oxygen and results in cellular injury, so it follows that restoration of oxygenation through the microcirculation would halt or perhaps reverse the tissue necrosis. IR therefore represents a paradoxical process because reperfusion produces a greater degree of tissue damage, dependent on ROS. In addition, it was thought that a hyperoxic tissue environment provided by administration of HBO following IR injury would increase ROS production and worsen the extent of tissue necrosis.
Unexpectedly, after subjecting an axial skin flap model to eight hours of ischaemia to determine the effects of HBO during reperfusion, skin-flap survival was significantly improved following HBO.[ 33] It had been anticipated that HBO would increase free radical liberation, worsen IR injury and decrease skin-flap survival. Subsequent findings of increased perfusion of ischaemic skin flaps treated with HBO using laser Doppler analysis confirmed these results.[ 34] Similarly, HBO provided a three- to six-fold improvement in free skin-flap survival in rats after microvascular anastomosis.[ 35] The authors posited that 24 hours was the threshold for irreversible ischaemic damage.[ 35] Consequently, we now understand that HBO ameliorates the detrimental effects of IR injury by improving tissue microcirculation.
IMPROVEMENT IN OXIDATIVE DAMAGE AND CELL DEATH
Since ROS are a key mediator in IR injury, studies have investigated the effects of HBO on the generation of these toxic molecules. Carbon monoxide (CO) simulates ischaemic conditions, a concept that was utilized in a rat model of brain injury.[ 36] Lipid peroxidation mediated by CO was inhibited by preventing xanthine oxidase formation, presumably decreasing superoxide radical and hydrogen peroxide levels.[ 36] Free radical scavenging systems, specifically SOD, may also be upregulated following HBO treatment.[ 35] HBO has also been shown to upregulate antioxidant gene expression in human endothelial cells which may protect against oxidative damage seen in IR injury.[ 37] As a result, HBO appears to produce its beneficial effects on ischaemic tissue by both decreasing production of ROS and increasing their degradation. Rat studies of renal IR injury treated with HBO prior to ischaemia demonstrated decreased oxygen radical-induced lipid peroxidation.[ 38] A more recent study demonstrated HBO-induced inhibition of apoptosis and improvement in cellular proliferation following renal IR injury.[ 39]
Additionally, utilizing a validated ischaemic flap model in rats, it was demonstrated that HBO improves ischaemic wound healing compared to untreated and N-acetylcysteine-treated (a non-specific free radical scavenger) groups. The mechanisms for this appeared to be by down-regulation of hypoxia-inducible factor-1 alpha (HIF-1 alpha) and p53- and caspase-3-mediated apoptosis.[ 40] In addition, the inflammatory response, as demonstrated by VEGF, cyclooxygenase-2, and neutrophil counts, were reduced in the HBO group.[ 40] Subsequently it was demonstrated that HBO increased antioxidant enzyme expression (SOD, catalase, and glutathione peroxidase) and decreased prooxidant enzyme expression (iNOS and gp91-phox) in ischaemic wounds.[ 41] These findings show that HBO does not exacerbate ROS-mediated tissue injury.
INTERFERENCE WITH NEUTROPHIL-ENDOTHELIAL CELL ADHESION
The potential mechanisms of HBO on neutrophil adhesion have also been of great interest. In a rat gracilis model investigating neutrophil adherence following HBO, adherence was significantly decreased during and after four hours of ischaemia. When HBO was initiated one hour after reperfusion, however, leukocyte adhesion was reduced to a lesser degree.[ 20] In the absence of ischaemia, HBO had no observable effect on the neutrophil-endothelial cell interaction. The same study also reviewed the effects on microvascular vasoconstriction. Vasoconstriction was inhibited when HBO was initiated during ischaemia, immediately after reperfusion, and one hour after reperfusion, with maximal effect during ischaemia.[ 20] These findings suggest that the maximal beneficial response to HBO occurs during the ischaemic phase and may be time-dependent. A rodent model of renal IR injury demonstrated decreased neutrophil infiltration following HBO and associated improvements in blood urea nitrogen and creatinine levels.[ 42]
A study at 284 and 304 kPa reported on the inhibition of human neutrophil adherence to injured endothelium via beta-2-integrin (CD18) function.[ 43] CD18-mediated neutrophil adhesion was inhibited but expression was not affected. Moreover, this study demonstrated that this function is cyclic GMP (cGMP)-regulated. HBO inhibited the function of guanylate cyclase and neutrophil adhesion was restored directly by cGMP incubation and also by increasing guanylate cyclase activity with N-formyl-Met-Leu-Phe (FMLP).[ 43] cGMP production was altered by inhibition of the membrane-bound guanylate cyclase, but free intracellular guanylate cyclase was unaffected. Therefore, it appears that HBO inhibits CD18 activity via impaired cGMP production. The finding that CD18 expression is not decreased by HBO treatment has been confirmed by others in a skeletal muscle rat model.[ 29] Neutrophil capping and CD18 surface polarization were inhibited by HBO, providing another plausible mechanism for the reversal of IR injury.[ 44]
The role of the endothelial cell cannot be overlooked in IR injury and other investigators have sought to focus on endothelial cell CAM (E-selectin and ICAM-1) expression after HBO. Ischaemia-reperfusion was simulated with hypoxia and hypoglycaemia exposure and demonstrated increased adhesion of neutrophils to endothelium.[ 45] HBO significantly reduced ICAM-1 expression and neutrophil adhesion.[ 45] Similar results were noted in in vivo experiments of rat skeletal muscle flaps.[ 46] HBO was administered at 253 kPa for 90 minutes and down regulated ICAM-1 expression with a resultant improvement in flap survival.
THE ROLE OF NITRIC OXIDE
NO has been shown to regulate many processes in IR injury including the microcirculation through its vasodilatory properties and reversal of leukocyte adhesion. Loss of the protective effect of NO resulted in increased CAM expression.[ 47] Indirect evidence has been found for increased survival and decreased neutrophil-endothelial adhesion after infusion of L-Arginine, a NO precursor, into ischaemic muscle.[ 48] eNOS inhibition promotes neutrophil adherence in the endothelium. In a previously mentioned study, HBO induced expression of eNOS; additionally, inhibition of NOS attenuated inhibition of ICAM-1 after HBO.[ 45] Another study of isolated rat neutrophils showed that NO inhibited CD18 activity by decreasing guanylate cyclase activity, corroborating the finding that NO decreases neutrophil adhesion.[ 49] Rat studies have demonstrated a favorable effect of HBO following IR injury on intestinal mucosa and hepatic cell apoptosis which may be mediated through decreased iNOS activity with a resultant decrease in peroxynitrite.[ 50 , 51]
The NO-dependent effect of HBO on CD18 polarization also has been examined.[ 52] Following administration of a NO scavenger, CD18 polarization and adherent neutrophils increased significantly compared to untreated controls. Furthermore, NOS inhibitors given before HBO restored neutrophil adhesion and capping via CD18. Together these findings represent a NO-regulated mechanism underlying the beneficial effects of HBO after IR injury and its associated CD18 polarization.[ 52] More recently, two important findings on the effect of HBO on NOS activity and expression in IR injury in rats have been demonstrated. First, eNOS was increased in pulmonary tissues, which supports the theory that the beneficial effects of HBO therapy occur systemically, not locally. Additionally, a temporal relationship of HBO-mediated NOS effects exists with an early phase increase in eNOS enzymatic activity and a subsequent late-phase increase in protein expression, the delay accounted for by transcription and translation.[ 53] Unpublished data from our laboratory suggests that the NO-dependent effect of HBO on CD18 polarization occurs through the plasmin-mediated release of membrane-bound vascular endothelial growth factor (VEGF).
The role of VEGF has recently been an area of investigative focus. HBO improves wound angiogenesis by increasing VEGF transcription and protein production.[ 54] In contrast to the acute changes seen after HBO following IR injury, the effects on VEGF represent long-term effects. VEGF can be bound to the extracellular matrix and released by the activity of various proteases, including plasmin.[ 55] Plasminogen is activated to yield plasmin by tissue or urokinase-like (tPA or uPA) plasminogen activators, the expression of which is increased with HBO.[ 56] The enhanced plasmin activation results in release of VEGF from the extracellular matrix and increased NO production.[ 57] IR injury increases alpha2-antiplasmin, thereby decreasing the amount of plasmin available to release stored VEGF.[ 58] Part of the beneficial effect of HBO after IR injury may be to increase levels of tPA and uPA, increasing plasmin beyond the inactivating capability of alpha2-antiplasmin present from the initial ischaemic event.
HYPERBARIC OXYGEN PRECONDITIONING
Preconditioning refers to the administration of HBO to limit the effects of subsequent ischaemia. The putative mechanism appears to be from the induction of antioxidant intracellular systems including catalase and SOD.[ 59 , 60] It has been suggested that the cardio-protective effects of HBO therapy in a rat model were NOS-regulated.[ 61] Much of the recent literature regarding HBO and IR injury has focused on this phenomenon. There is evidence that preconditioning rat skin flaps with HBO prior to IR injury resulted in improved survival and microcirculatory perfusion.[ 62] This beneficial response was the result of increased expression of anti-apoptotic factor B-cell lymphoma-2 (Bcl-2) and inhibition of apoptotic factors phosphorylated apoptosis signal-regulating kinase 1 (pASK-1), phosphorylated c-Jun N-terminal kinase (pJNK) and Bcl2-associated K protein (Bax). A diminution of the inflammatory cytokine cascade has also been advocated.[ 63]
The preconditioning effect of HBO was confirmed in an hepatic IR study in rats. HBO not only resulted in improvements in serum alanine aminotransferase and aspartate aminotransferase levels, but also demonstrated improvements in mitochondrial respiration and swelling.[ 64] The relationship between mitochondrial injury, cytochrome c release, and apoptosis suggests that the improvement in mitochondrial function revealed by this study may result in inhibition of apoptosis. HBO preconditioning provides systemic and local tissue benefits; however, the response appears to be dependent on the timing of HBO exposure with the exact window remaining unknown.[ 65] HBO preconditioning may be useful for limiting the well-documented neurological complications following elective cardiac surgery and carotid endarterectomy. It may also prove useful in complex reconstructive procedures requiring composite tissue allotransplantation, such as face and hand transplants, where ischaemia times can be extremely prolonged and multiple tissue types are involved including muscle and bone.
Limitations
The studies mentioned above, and others, have provided a great deal of clarity in this challenging area. However, it should be noted that much of our understanding of IR injury and HBO derives from experimental studies in animal models or cell culture. While these experimental findings are promising, it is unclear whether the physiologic outcomes correlate to clinically relevant findings. The clinical data regarding this topic are mostly from retrospective studies and are insufficiently powered with small sample sizes, and thus, the clinical impact remains debatable.
Conclusions
Studies of the microcirculation, neutrophil adhesion, and ROS reflect the complex interactions between various cell signalling molecules, intracellular pathways, and cell types in IR injury. HBO can ameliorate the cytotoxic effects of reperfusion injury in a dose-dependent manner. However, the critical issue at the centre of IR injury is the duration of ischaemia as the primary factor in determining the outcome from injury. The adage 'time is muscle/nerve' emphasizes the well-established fact that irreversible damage is observed in ischaemia-sensitive tissues such as muscle and nerve tissue after six hours of ischaemia. The window of reversible changes presents a valuable opportunity not only for promptly initiating the appropriate interventions to reverse the ischaemic aetiology, but also to institute adjunctive therapies such as HBO to limit the extent of tissue damage after IR injury.
Our understanding of IR and the protective effects of HBO continues to evolve. The evidence from rat skeletal muscle and skin flap studies provides insight into the potential for HBO in the treatment of IR injury following crush injuries, and failing grafts and flaps. Other studies of animal heart, brain, kidney, intestine and liver demonstrate the positive effects of HBO in the context of myocardial infarction, stroke, ischaemic renal failure and solid organ transplantation. HBO preconditioning has promising therapeutic value and its applications are under investigation. Additionally, standardized protocols for HBO have not been defined. It is apparent that questions remain for these processes and therefore more basic-science and clinical research is required to elucidate them.
Contributor Information
A Francis, Division of Plastic Surgery, Department of Surgery, University of Nevada School of Medicine, Las Vegas, USA.
R Baynosa, Division of Plastic Surgery, Department of Surgery, University of Nevada School of Medicine, Las Vegas, USA.
References
- Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology . Cardiovasc Res. 2004;61:461–470. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- Russell RC, Roth AC, Kucan JO, Zook EG. Reperfusion injury and oxygen free radicals: a review . J Reconstr Micro. 1989;5:79. doi: 10.1055/s-2007-1006855. [DOI] [PubMed] [Google Scholar]
- Zweier JL, Talukder MA. The role of oxidants and free radicals in reperfusion injury . Cardiovasc Res. 2006;70:181–190. doi: 10.1016/j.cardiores.2006.02.025. [DOI] [PubMed] [Google Scholar]
- Hori K, Tsujii M, Iino T, Satonaka H, Uemura T, Akeda K, et al. Protective effect of edaravone for tourniquet-induced ischaemia-reperfusion injury on skeletal muscle in murine hindlimb . BMC Musculoskelet Disord. 2013;14:113. doi: 10.1186/1471-2474-14-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AA, Aziz FA, Hall JC. Reperfusion injury . Plast Reconstr Surg. 2006;117:1024–1033. doi: 10.1097/01.prs.0000204766.17127.54. [DOI] [PubMed] [Google Scholar]
- Feller Am, Roth AC, Russell RC, Eagleton B, Suchy H, Debs N. Experimental evaluation of oxygen free radical scavengers in the prevention of reperfusion injury to skeletal muscle . Ann Plast Surg. 1989;22:321–331. doi: 10.1097/00000637-198904000-00007. [DOI] [PubMed] [Google Scholar]
- Inauen W, Suzuki M, Granger DN. Mechanisms of cellular injury: potential sources of oxygen free radicals in ischaemia/reperfusion . Microcirc Endothelium Lymphatics. 1989;5:143–155. [PubMed] [Google Scholar]
- Pang CY. Ischaemia-induced reperfusion injury in muscle flaps: pathogenesis and major source of free radicals . Reconstr Microsurg. 1990;6:77–83. doi: 10.1055/s-2007-1006807. [DOI] [PubMed] [Google Scholar]
- Steller H. Mechanisms and genes of cellular suicide . Science. 1995;267:1445–1449. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- Nathan C, Xie QW. Nitric oxide synthasis: role, tolls, and controls . Cell. 1994;78:915–918. doi: 10.1016/0092-8674(94)90266-6. [DOI] [PubMed] [Google Scholar]
- Taylor CT, Moncada S. Nitric oxide, cytochrome C oxidase, and the cellular response to hypoxia . Arterioscler Thromb Vasc Biol. 2010;30:643–647. doi: 10.1161/ATVBAHA.108.181628. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly . Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- Ghavami S, Hashemi M, Kadkhoda K, Alavian SM, Bay GH, Los M. Apoptosis in liver disease – detection and therapeutic applications . Med Sci Monit. 2005;11:RA337–RA345. [PubMed] [Google Scholar]
- Wu B, Iwakiri R, Tsunada S, Utsumi H, Kojima M, Fujise T, Ootani A, Fujimoto K. iNOS enhances rat intestinal apoptosis after ischaemia-reperfusion . Free Radic Biol Med. 2002;5:549–555. doi: 10.1016/s0891-5849(02)00917-6. [DOI] [PubMed] [Google Scholar]
- Kim YM, Kim TH, Seo DW, Talanian RV, Billiar TR. Nitric oxide suppression of apoptosis occurs in association with an inhibition of Bcl-2 cleavage and cytochrome c release . J Biol Chem. 1998;273:31437–31441. doi: 10.1074/jbc.273.47.31437. [DOI] [PubMed] [Google Scholar]
- Mannick KE, Miao XQ, Stamler JS. Nitric oxide inhibits Fasinduced apoptosis . J Biol Chem. 1997;272:24125–24128. doi: 10.1074/jbc.272.39.24125. [DOI] [PubMed] [Google Scholar]
- Weidinger A, Kozlov AV. Biological activities of reactive oxygen and nitrogen species: oxidative stress versus signal transduction . Biomolecules. 2015;5:472–484. doi: 10.3390/biom5020472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KC, Kim RJ, Bluemke DA, Rochitte CE, Zerhouni EA, Becker LC, Lima JA. Quantification and time course of microvascular obstruction by contrast-enhanced echocardiography and magnetic resonance imaging following acute myocardial infarction and reperfusion . J Am Coll Cardiol. 1998;32:1756–1764. doi: 10.1016/s0735-1097(98)00429-x. [DOI] [PubMed] [Google Scholar]
- Feng LJ, Berger BE, Lysz TW, Shaw WW. Vasoactive prostaglandins in the impending no-reflow state: evidence for a primary disturbance in microvascular tone . Plast Reconstr Surg. 1988;81:755–767. doi: 10.1097/00006534-198805000-00018. [DOI] [PubMed] [Google Scholar]
- Zamboni WA, Roth AC, Russell RC, Graham B, Suchy H, Kucan JO. Morphologic analysis of the microcirculation during reperfusion of ischaemic skeletal muscle and the effect of hyperbaric oxygen . Plast Reconstr Surg. 1993;91:1110–1123. doi: 10.1097/00006534-199305000-00022. [DOI] [PubMed] [Google Scholar]
- Lew MJ, Rivers RJ, Duling BR. Arteriolar smooth muscle responses are modulated by an intramural diffusion barrier . Am J Physiol. 1989;257:H10–H16. doi: 10.1152/ajpheart.1989.257.1.H10. [DOI] [PubMed] [Google Scholar]
- Eidt JF, Ashton J, Golino p, McNatt J, Buja LM, Willerson JT. Thromboxane A2 and serotonin mediate coronary blood flow reductions in unsedated dogs . Am J Physiol. 1989;257:H873–H882. doi: 10.1152/ajpheart.1989.257.3.H873. [DOI] [PubMed] [Google Scholar]
- Schumacher WA, Heran CL, Allen GT, Ogletree ML. Leukotrienes cause mesenteric vasoconstriction and hemoconcentration in rats without activating thromboxane receptors . Prostaglandins. 1989;38:335–344. doi: 10.1016/0090-6980(89)90137-8. [DOI] [PubMed] [Google Scholar]
- Keller MW. Arteriolar constriction in skeletal muscle during vascular stunning: role of mast cells . Am J Physiol. 1997;272:H2 154–H2 163. doi: 10.1152/ajpheart.1997.272.5.H2154. [DOI] [PubMed] [Google Scholar]
- Zhang R, Chopp M, Zhang Z, Jiang N, Powers C. The expression of P- and E-selectins in three models of middle cerebral artery occlusion . Brain Res. 1998;785:207–214. doi: 10.1016/s0006-8993(97)01343-7. [DOI] [PubMed] [Google Scholar]
- Valentijn KM, Sadler JE, Valentijn JA, Voorberg J, Eikenboom J. Functional architecture of Weibel-Palade bodies . Blood. 2011;117:5033–5043. doi: 10.1182/blood-2010-09-267492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokura S, Yoshida N, Yoshikawa T. Anoxia/reoxygenationinduced leukocyte-endothelial cell interactions . Free Radic Biol Med. 2002;33:427–432. doi: 10.1016/s0891-5849(02)00852-3. [DOI] [PubMed] [Google Scholar]
- Zamboni WA, Stephenson LL, Roth AC, Suchy H, Russell RC. Ischaemia-reperfusion injury in skeletal muscle: CD 18-dependent neutrophil-endothelial adhesion and arteriolar vasoconstriction . Plast Reconstr Surg. 1997;99:2002–2007. doi: 10.1097/00006534-199706000-00028. [DOI] [PubMed] [Google Scholar]
- Larson JL, Stephenson LL, Zamboni WA. Effect of hyperbaric oxygen on neutrophil CD18 expression . Plast Reconstr Surg. 2000;105:1375–1381. doi: 10.1097/00006534-200004040-00017. [DOI] [PubMed] [Google Scholar]
- Khiabani KT, Stephenson LL, Gabriel A, Nataraj C, Wang WZ, Zamboni WA. A quantitative method for determining polarization of neutrophil adhesion molecules associated with ischaemia reperfusion . Plast Reconstr Surg. 2004;114:1846–1850. doi: 10.1097/01.prs.0000143580.45631.dd. [DOI] [PubMed] [Google Scholar]
- Mori E, del Zoppo GJ, Chambers JD, Copeland BR, Arfors KE. Inhibition of polymorphonuclear leukocyte adherence suppresses no-reflow after focal cerebral ischaemia in baboons . Stroke. 1992;23:712–718. doi: 10.1161/01.str.23.5.712. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Mabuchi T, Yagita Y, Ohtsuki T, Hori M, Yangihara T. Deficiency of intercellular adhesion molecule 1 attenuates microcirculatory disturbance and infarction size in focal cerebral ischaemia . J Cereb Blood Flow Metab. 1998;18:1336–1345. doi: 10.1097/00004647-199812000-00008. [DOI] [PubMed] [Google Scholar]
- Zamboni WA, Doweck I, Roth AC, Russell RC, Nemiroff PM, Casssas L, Smoot EC. The effect of acute hyperbaric oxygen therapy on axial pattern skin flap survival when administered during and after total ischaemia . J Reconstr Micro. 1989;5:343. doi: 10.1055/s-2007-1006884. [DOI] [PubMed] [Google Scholar]
- Zamboni WA, Roth AC, Russell RC, Smoot EC. Effect of hyperbaric oxygen on reperfusion of ischaemic axial skin flaps: a laser doppler analysis . Ann Plast Surg. 1992;23:339. doi: 10.1097/00000637-199204000-00008. [DOI] [PubMed] [Google Scholar]
- Kaelin CM, Im MJ, Myers RA, Manson PN, Hoopes JE. The effects of hyperbaric oxygen on free flaps in rats . Arch Surg. 1990;125:607–609. doi: 10.1001/archsurg.1990.01410170053011. [DOI] [PubMed] [Google Scholar]
- Thom SR. Functional inhibition of leukocyte B2 integrins by hyperbaric oxygen in carbon monoxide-mediated brain injury in rats . Toxicol Appl Pharmacol. 1993;123:248–256. doi: 10.1006/taap.1993.1243. [DOI] [PubMed] [Google Scholar]
- Godman CA, Joshi R, Giardina C, Perdrizet G, Hightower LE. Hyperbaric oxygen treatment induces antioxidant gene expression . Ann N Y Acad Sci. 2010;1197:178–183. doi: 10.1111/j.1749-6632.2009.05393.x. [DOI] [PubMed] [Google Scholar]
- Gurer A, Ozdogan I, Gomceli I, Demirag A, Gulbahar O, Arikok T, et al. Hyperbaric oxygenation attenuates renal ischaemia-reperfusion injury in rats . Transplant Proc. 2006;38:3337–3340. doi: 10.1016/j.transproceed.2006.10.184. [DOI] [PubMed] [Google Scholar]
- Migita H, Yoshitake S, Tange Y, Choijookhuu N, Hishikawa Y. Hyperbaric oxygen therapy suppresses apoptosis and promotes renal tubular regeneration after renal ischaemia/reperfusion injury in rats . Nephrourol Mon. 2016;8:e34421. doi: 10.5812/numonthly.34421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Chang Q, Cox RA, Gong X, Gould LJ. Hyperbaric oxygen attenuates apoptosis and decreases inflammation in an ischaemic wound model . J Invest Dermatol. 2008;128:2102–2112. doi: 10.1038/jid.2008.53. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Gould LJ. Hyperbaric oxygen reduces matrix metalloproteinases in ischaemia wounds through a redoxdependent mechanism . J Invest Dermatol. 2014;134:237–246. doi: 10.1038/jid.2013.301. [DOI] [PubMed] [Google Scholar]
- Solmazgul E, Uzun G, Cermik H, Atasoyu EM, Aydinoz S, Yildiz S. Hyperbaric oxygen therapy attenuates renal ischaemia/perfusion injury in rats . Urol Int. 2007;78:82–85. doi: 10.1159/000096941. [DOI] [PubMed] [Google Scholar]
- Thom SR, Mendiguren I, Hardy K, Bolotin T, Fisher D, Nebolon M, Kilpatrick L. Inhibition of human neutrophil beta2-integrin-dependent adherence by hyperbaric O2 . Am J Physiol. 1997;272:C770–C777. doi: 10.1152/ajpcell.1997.272.3.C770. [DOI] [PubMed] [Google Scholar]
- Khiabani KT, Bellister SR, Skaggs SS, Stephenson LL, Wang WZ, Zamboni WA. Reperfusion induced neutrophil CD18 polarization: effect of hyperbaric oxygen . J Surg Res. 2008;150:11–16. doi: 10.1016/j.jss.2007.12.780. [DOI] [PubMed] [Google Scholar]
- Buras JA, Stahl GI, Svoboda KK, Reenstra WR. Hyperbaric oxygen downregulates ICAM-1 expression induced by hypoxia and hypoglycemia: the role of NOS . Am J Physiol Cell Physiol. 2000;278:C292–C302. doi: 10.1152/ajpcell.2000.278.2.C292. [DOI] [PubMed] [Google Scholar]
- Hong JP, Kwon H, Chung YK, Jung SH. The effect of hyperbaric oxygen on ischaemia-reperfusion injury: An experimental study in a rat musculocutaneous flap . Ann Plast Surg. 2003;51:478–487. doi: 10.1097/01.sap.0000095651.05156.0f. [DOI] [PubMed] [Google Scholar]
- Lefer AM, Lefer DJ. The role of nitric oxide and cell adhesion molecules on the microcirculation in ischaemia-reperfusion . Cardiovasc Res. 1996;32:743–751. [PubMed] [Google Scholar]
- Meldrum DG, Stephenson LL, Zamboni WA. Effects of L-NAME and L-Arginine on ischaemia-reperfusion injury in rat skeletal muscle . Plast Reconstr Surg. 1999;103:935–940. doi: 10.1097/00006534-199903000-00025. [DOI] [PubMed] [Google Scholar]
- Banick PD, Chen Q, Xu YA, Thom SR. Nitric oxide inhibits neutrophil beta 2 integrin function by inhibiting membrane associated cyclic GMP synthesis . J Cell Physiol. 1997;172:12–24. doi: 10.1002/(SICI)1097-4652(199707)172:1<12::AID-JCP2>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Bertoletto PR, Fagundes DJ, Simoes MJ, Oshima CT, Montero EF, Simoes RS, Fagundes AT. Effects of hyperbaric oxygen therapy on the rat intestinal mucosa apoptosis caused by ischaemia-reperfusion injury . Microsurgery. 2007;27:224–227. doi: 10.1002/micr.20349. [DOI] [PubMed] [Google Scholar]
- Chaves JC, Fagundes DJ, Simoes MJ, Bertoletto PR, Oshima CT, Taha MO, et al. Hyperbaric oxygen therapy protects the liver from apoptosis caused by ischaemia-reperfusion injury in rats . Microsurgery. 2009;29:578–583. doi: 10.1002/micr.20664. [DOI] [PubMed] [Google Scholar]
- Jones SR, Carpin KM, Woodward SM, Khiabani KT, Stephenson LL, Wang WZ, Zamboni WA. Hyperbaric oxygen inhibits ischaemia-reperfusion-induced neutrophil CD18 polarization by a nitric oxide mechanism. . Plast Reconstr Surg. 2010;126:403–411. doi: 10.1097/PRS.0b013e3181df64a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baynosa RC, Naig AL, Murphy PS, Fang FH, Stephenson LL, Khiabani KT, et al. The effect of hyperbaric oxygen on nitric oxide synthase activity and expression in ischaemia reperfusion injury . J Surg Res. 2013;183:355–361. doi: 10.1016/j.jss.2013.01.004. [DOI] [PubMed] [Google Scholar]
- Sheikh AY, Gibson JJ, Rollins MD, Hopf HW, Hussain Z, Hunt TK. Effect of hyperoxia on vascular endothelial growth factor levels in a wound model.factor levels in a wound model . Arch Surg. 2000;135:1293, 1297. doi: 10.1001/archsurg.135.11.1293. [DOI] [PubMed] [Google Scholar]
- Keyt BA, Berleau LT, Nguyen HV, Chen H, Heinsohn H, Vandlen R, Ferrara N. The carboxyl-terminal domain (111-165) of vascular endothelial growth factor is critical for its mitogenic potency . J Biol Chem. 1996;271:7788–7795. doi: 10.1074/jbc.271.13.7788. [DOI] [PubMed] [Google Scholar]
- Tjarnstrom J, Holmdahl L, Falk P, Falkenberg M, Arnell P, Risberg B. Effects of hyperbaric oxygen on expression of fibrinolytic factors of human endothelium in a stimulated ischaemia/reperfusion situation . Scand J Clin Lab Invest. 2001;61:539–546. doi: 10.1080/003655101753218300. [DOI] [PubMed] [Google Scholar]
- Hood JD, Meininger CJ, Ziche M, Granger HJ. VEGF upregulates ecNOS message, protein, and NO production in human endothelial cells . Am J Physiol. 1998. pp. H1054–H1058. [DOI] [PubMed]
- Bayes-Genis A, Guindo J, Oliver A, Badimon L, Fiol M, Mateo J, et al. Elevated levels of plasmin-alpha2 antiplasmin complexes in unstable angina . Thromb Haemost. 1999;81:865–868. [PubMed] [Google Scholar]
- Kim CH, Choi H, Chun YS, Kim GT, Park JW, Kim MS. Hyperbaric oxygenation pretreatment induces catalase and reduces infarct size in ischaemic rat myocardium . Pflugers Arch. 2001;442:519–525. doi: 10.1007/s004240100571. [DOI] [PubMed] [Google Scholar]
- Nie H, Xiong L, Lao N, Chen S, Xu N, Zhu Z. Hyperbaric oxygen preconditioning induces tolerance against spinal cord ischaemia by upregulation of antioxidant enzymes in rabbits . J Cereb Blood Flow Metab. 2006;26:666–674. doi: 10.1038/sj.jcbfm.9600221. [DOI] [PubMed] [Google Scholar]
- Cabigas BP, Su J, Hutchins W, Shi RB, Recinos RF, et al. Hyperoxic and hyperbaric-induced cardioprotection: role of nitric oxide synthase 3 . Cardiovasc Res. 2006;72:143–151. doi: 10.1016/j.cardiores.2006.06.031. [DOI] [PubMed] [Google Scholar]
- Xiao YD, Liu YQ, Li JL, Ma XM, Wang YB, Liu YF, et al. Hyperbaric oxygen preconditioning inhibits skin flap apoptosis in a rat ischaemia-reperfusion model . J Surg Res. 2015;199:732–739. doi: 10.1016/j.jss.2015.06.038. [DOI] [PubMed] [Google Scholar]
- Kang N, Hai Y, Liang F, Gao CJ, Liu XH. Preconditioned hyperbaric oxygenation protects skin flap grafts in rats against ischaemia/reperfusion injury . Mol Med Rep. 2014;9:2124–2130. doi: 10.3892/mmr.2014.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losada DM, Chies AB, Feres O, Chaib E, D'Albuquerque LA, Castro-e-Silva O. Effects of hyperbaric oxygen therapy as hepatic preconditioning in rats submitted to hepatic ischaemia/reperfusion injury . Acta Cir Bras. 2014;29:61–66. doi: 10.1590/s0102-8650201400140012. [DOI] [PubMed] [Google Scholar]
- Losada DM, Jordani ME, Jordani MC, Piccinato MA, Fina CF, Feres O, et al. Should preconditioning hyperbaric oxygenation protect the liver against ischaemia-reperfusion injury? An experimental study in a rat model . Transplant Proc. 2014;46:56–62. doi: 10.1016/j.transproceed.2013.10.044. [DOI] [PubMed] [Google Scholar]