

Abstract
2,3-Dichloroquinoxaline and some of its derivatives have been reacted with malononitrile and ethyl cyanoacetate to yield a variety of 3-chloro-2-(cyanomethylene)-1,2-dihydroquinoxaline derivatives. The reaction of 3-chloro-2-(dicyanomethylene)-1,2-dihydroquinoxaline (2e) with pyridine and its methyl derivatives led to the zwitterionic structures 6a-6c. The structures of the newly synthesized compounds were assigned by spectroscopic data and elemental analyses.
Keywords: 2,3-Dichloroquinoxaline; ethoxycarbonylcyanomethylene- and dicyano-methylene-3-chloro-1,2-dihydroquinoxalines; betaine structures; spectroscopic data
Introduction
The two reactive chlorine atoms in 2,3-dichloroquionoxaline (1a) are prone to nucleophilic displacement reactions by a wide variety of nucleophiles which react in a stepwise manner [1,2]:
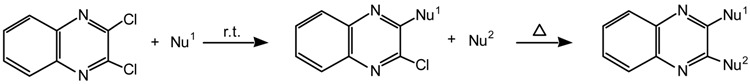 |
The reaction of 2,3–dichloroquinoxaline (1a) with carbanions generated from active methylene compounds has not been fully investigated. Pratt and Keresztesy [3] have reported the synthesis of indolizino – and dihydroindolizinoquinoxalines from either the reaction of 1a with ethyl cyanoacetate and isoquinoline in a one-step process, or the isolation of the monosubstituted intermediates derived from the reaction with ethyl cyanoacetate or malononitrile, followed by the treatment of the reaction products with isoquinoline or pyridine. Novel colored compounds containing the dicyano-methylidene groups conjugated with other chromophores have also been reported [4,5,6,7]. In the present paper, detailed synthesis, analysis and spectroscopic properties of some colored quinoxaline compounds containing the ethoxycarbonylcyanomethylene and dicyanomethylene groups are reported.
Results and Discussion
2,3-Dichloroquinoxaline and its derivatives 1a–d were prepared following the procedure of Komin and Carmack [8], by treatment of 1,2,3,4-tetrahydroquinoxaline-2,3-diones with thionyl chloride in the presence of dimethylformamide (DMF). The reaction of dichloroquinoxalines 1a–d and the appropriate active methylene compounds (ethyl cyanoacetate or malononitrile) in dimethoxyethane, with sodium hydride as a base, gave good yields of colored crystals of a variety of 3-chloro-2-(cyanomethylene)-1,2-dihydroquinoxaline derivatives as isomeric mixtures of 2 and 3 (Scheme 1). The main reaction products 2 were isolated in pure form, whereas the isomers 3 have been detected only by their NMR-spectra. The structures of the newly synthesized compounds were assigned by their IR-spectra,1H- and 13C-NMR spectra (see Experimental section).
Scheme 1.

The products can occur as tautomers 2 and 2', however, according to their 1H-NMR spectra the compounds exist mainly as the 2-(cyanomethylene)-1,2-dihydroquinoxaline derivatives 2 or 3 (in CDCl3 or DMSO-d6), with the NH protons (exchangeable with D2O) showing broad singlets in the downfield region (in the range δ 10.99–14.49 ppm). In the 13C-NMR spectra, the central carbon in the =C(CN)R grouping resonates as a quartenary carbon in the δ 41.9–69.3 ppm region, according to Attached Proton Test (APT) experiments. This is in the region reported for the 13C- chemical shifts of the central carbon of the =C(CN)2 in some compounds containing the dicyanomethylene group [9,10,11,12].
The 2-position of 6-substituted-2,3-dichloroquinoxalines is presumed to be preferentially attacked by nucleophiles, especially when the substituent at the 6-position is electron-withdrawing, based on greater stability of the intermediate σ-complex and molecular orbital calculations [13]. Inspection of the 1H-NMR spectra of the reaction products shows that they are mixtures of 6- and 7-substituted isomers 2 and 3, of which the 6-isomer 2 was isolated as the main product. We have failed to observe the replacement of the second chlorine atom of 1 by the active methylene compounds even when large excesses of the carbanions were employed. However, nucleophilic replacement of the second chlorine atom occurred with sodium azide and hydrazine. For example, both 3-chloro-2-(ethoxycarbonylcyano-methylene)-1,2-dihydroquinoxaline (2a) and 3-chloro-2-(dicyanomethylene)-1,2-dihydroquinoxaline (2e) reacted with sodium azide in dimethylformamide (DMF) to afford the tetrazolo[1,5-a]quinoxaline derivatives 4a (86 % yield) and 4b (85 % yield), respectively. Compound 2a also reacted with hydrazine in DMF to give the crystalline derivative 5 in 53 % yield.
The infrared spectra of 2a – 2d, 4a and 5 further show that in the solid state, these compounds exist in the methylidene tautomeric form, most probably with an intramolecular hydrogen bond, as shown by the presence, in each infrared spectrum, of carbonyl absorptions at around 1650–1630 cm-1, instead of the normal absorption expected at > 1700 cm-1 for unconjugated esters. In addition, all the quinoxaline derivatives showed IR absorption bands at around 2200 cm-1, characteristic of the cyano group. The predominant existence of 4a and 4b in the tetrazolo forms was supported by the absence of absorption bands at around 2140 cm-1 in their IR spectra, as expected for the azido (N3) group.
As mentioned earlier, excess hydrazine converted 2a into 5 at room temperature. The ester group (COOEt) does not react under these mild conditions, showing that it is deactivated, most probably due to conjugation of the carbonyl function of the COOEt with the NH group in the HN-C=C-C=O(OR) system [14], and the -NH…O=C-hydrogen bonding.
3-Chloro-2-(dicyanomethylene)-1,2-dihydroquinoxaline (2e) was reported to react with pyridine to give 12,12-dicyano-12,12a-dihydroindolizino[2,3-b]quinoxaline (9) [3]. We have found that the reaction of 2e with pyridine and its methyl derivatives did not produce 9, but rather gave red or orange crystals of dicyano(3-pyridinium-1-ylquinoxalin-2-yl)methanides 6a-c (Scheme 1), whose structural assignments were based on their spectra and analytical data.
The yields, infrared, 1H- and 13C-NMR spectral and analytical data for the dicyanomethanides, 6, are given in the Experimental section. In the 13 C-NMR spectra, the cyano groups resonate at around δ 120.7 ppm, compared to δ 116.5 ppm observed for the cyano groups in the starting material and related structures 2.
The formation of the dicyanoquinoxalinemethanides 6, instead of the reported dicyanoindolizino-quinoxalines 9, may account for the observation of Pratt and Keresztesy [3] that hydrogen cyanide could not be eliminated from their products. Also in the same report, they suggested structure 10 (based on analysis only) for the product obtained upon heating 3-chloro-2-(ethoxycarbonylcyano-methylene)-1,2-dihydroquinoxaline (2a) with quinoxaline. However, in the present studies, the main product obtained upon reacting 2a with quinoxaline is 2-chloro-3-cyanomethylquinoxaline (8), as proven by analysis, 1H-NMR and mass spectral data.
Experimental
General:
Melting points were recorded on a Gallenkamp (variable heater) melting point apparatus and are uncorrected. UV spectra were run in MeOH solution on a Lambda-15-Perkin Elmer spectrometer (λmax in nm (log ε), sh = shoulder). Infrared spectra were recorded on a Buck spectrometer as potassium bromide pellets. 1H- and 13C-NMR were recorded as CDCl3 or DMSO-d6 solutions on a Bruker- AC- 250 or JEOL-JNM-GX 400-MHz spectrometers (δ in ppm relative to Me4Si and H3PO4). Mass spectra (E1-MS) were recorded on a Finnigan MAT 312 machine [in m/z (rel. %)].
General Preparation of 3-Chloro-2-(cyanomethylene)-1,2-dihydroquinoxalines (2a-g).
The 3-chloro-2-(cyanomethylene)-1,2-dihydroquinoxalines were prepared by reacting 2,3-dichloro-quinoxaline derivatives 1a-d with the appropriate active methylene compound in the presence of sodium hydride. This is exemplified by the preparation of compound 2a as follows: ethyl cyanoacetate (1.1 mL, 10 mmoles) was added dropwise with stirring to a suspension of sodium hydride (0.25 g, 10.4 mmoles) in dimethoxyethane (20 mL). After the addition, the stirring was continued for 30 min. and then 2,3-dichloroquinoxaline (1.0 g, 5 mmoles) was added. The reaction mixture was stirred at room temperature for 3 h and then heated under reflux for 1 h. The dimethoxyethane was removed on a rotatory evaporator in vacuo and the resulting residue was treated with cold aqueous hydrochloric acid to give a yellow product. This was filtered, washed with cold water, dried and then recrystallized from ethanol to give yellow crystals of 3-chloro-2-(ethoxycarbonylcyanomethylene)-1,2-dihydroquinoxaline (2a, yield: 85%); m.p. 175–177°C (lit. [3] 174–175°C); IR (cm-1) (KBr): 2200 (νCN), 1630 (sh) (νC=O); 1H-NMR: 14.49 (br s, NH, D2O exchangeable), 7.78 (d, IH, ArH), 7.63 (t, IH, ArH), 7.37-7.50 (m, 2H, ArH), 4.37 (q, 2H, CH2), 1.41 (t, 3H, CH3 ); 13C-NMR: 170.7 (C=O), 146.0, 143.6, 134.2, 132.3, 128.6, 126.6, 116.5 (CN), 116.4, 69.3 (=C(CN)2), 62.1 (OCH2), 14.2 (CH3); MS: 275 (19.4, M+), 240 (2.2, [M–Cl]+), 231 (7.3, [M-CO2]+), 212 (10.5, [M-Cl–C2H4]+), 203 (100, [M–CO2–C2H4]+), 167 (62.4, [M–CO2C2H5–Cl]+), 114 (14.2), 102 (53.5), 76 (23.9), 75 (21.0).
Compounds 2b-2g were obtained similarly:
3,6-Dichloro-2-(ethoxycarbonylcyanomethylene)-1,2-dihydroquinoxaline (2b): golden yellow crystals (yield: 82%); m.p. 162-164oC (dec.); IR (cm-1): 2200 (νCN), 1640 (νCO); 1H-NMR: 13.84 (br s, NH, D2O exchangeable), 8.01 (s, 1H, H-5), 7.72 (d, 1H, H-8), 7.46 (d, 1H, H-7), 4.28 (q, 2H, CH2), 1.30 (t, 3H, CH3); Analysis: Calc. For C13H9Cl2N3O2: C, 50.34; H, 2.93; N, 13.55. Found: C, 50.15; H, 2.85; N, 13.39.
3,7-Dichloro-2-(ethoxycarbonylcyanomethylene)-1,2-dihydroquinoxaline (3b): 1H-NMR: 13.85 (br s, NH, D2O exchangeable), 8.10 (s, 1H, H-8), 7.80 (d, 1H, H-5), 7.66 (d, 1H, H-6), 4.28 (q, 2H, CH2), 1.21 (t, 3H, CH3).
3-Chloro-6-methyl-2-(ethoxycarbonylcyanomethylene)-1,2-dihydroquinoxaline (2c): golden yellow crystals (yield: 79%); m.p. 159-161oC (dec.); IR (cm-1): 2190 (ν CN), 1640 (ν C=O); 1H‑NMR: 14.46 (br s, NH, D2O exchangeable), 7.51 (s, 1H, H-5), 7.45 (d, 1H, H-7), 7.31 (d, 1H, H-8), 4.35 (q, 2H, CH2), 2.48 (s, 3H, CH3), 1.40 (t, 3H, CH3); 13C-NMR: 170.8 (C=O), 145.5, 143.2, 137.2, 134.2, 133.8, 127.9, 125.9, 116.5 (CN), 68,3 (=C–(CN)2), 62.5 (OCH2) 21.0 (CH3), 14.0 (CH3); Analysis: Calc. For C14H12ClN3O2: C, 58.04; H, 4.17; N, 14.5. Found: C, 58.00; H, 4.10; N, 14.26.
3-Chloro-7-methyl-2-(ethoxycarbonylcyanomethylene)-1,2-dihydroquinoxaline (3c): 1H-NMR: 14.39 (br s, NH, D2O exchangeable), 7.59 (d, 1H, H-5), 7.26 (d, 1H, H-6), 7.15 (s, 1H, H-8), 4.35 (q,2H, CH2), 2.51 (s, 3H, CH3). 1.35 (t, 3H, CH3); 13C-NMR: 170.7 (C=O), 145.8, 143.8, 133.8, 132.5, 128.3, 128.1, 128.0, 116.0 (CN), 68.6 (=C(CN)2), 61.9 (OCH2), 21.8 (CH3), 14.1 (CH3).
3-chloro-2-(ethoxycarbonylcyanomethylene)-6-nitro-1,2-dihydroquinoxaline (2d): golden yellow crystals (yield: 88%); m.p 191-1993oC (dec.); IR (cm-1): 2210 (υCN), 1650 (υCO); 1H‑NMR: 11.42 (br s, NH, D2O exchangeable), 8.48 (s, 1H, H-5), 8.27 (d, 1H, H-7), 7.48 (d, 1H, H-8), 4.37 (q, 2H, CH2) 1.39 (t, 3H, CH3); Analysis: Calc. for C13H9ClN4O4: C 48.69, H 2.83, N 17.47. Found C, 48.51; H. 2.80; N, 17.28.
3-Chloro-2-(dicyanomethylene)-1,2-dihydroquiinoxaline (2e): golden brown crystals (yield: 78%); m.p. 240-243o C (dec.) (lit. [3] 217o C); IR (cm-1): 2210 (υCN); 1H-NMR: 10.99 (br s, NH, D2O exchangeable), 7.85 (d, 1H, Ar-H), 7.63-7.75 (m, 2H, Ar-H), 7.46 (t, 1H, Ar-H). 13C-NMR: 147.3, 142.3, 134.1, 132.0, 130.8, 127.7, 126.1, 118.0, 116.7 (CN), 47.3 (=C(CN)2); UV (MeOH): 206 (4.18), 222 (4.43), 290 (4.32), 315(sh) (3.95), 435 (4.03); MS:228 (79.8, M+), 192 (20.7, [M–HCl]+), 163 (100, [M–CH (CN)2]+ ), 114 (8.3), 102 (83.8), 76 (37.7), 75 (38.3).
3,6-Dichloro-2-(dicyanomethylene)-1,2-dihydroquinoxaline (2f): brown crystals (yield: 75%); m.p. 190-191o C (dec.); IR (cm-1): 2200 (υCN); 1H-NMR: 10.14 (br s, NH, D2O exchangeable), 8.27 (s, 1H, H-5), 7.63 (d, 1H, H-8), 7.30 (d, 1H, H-7); 13C-NMR: 150.0, 142.3, 135.3, 133.6, 129.1, 125.5, 119.7, 118.5 (CN), 45.6 (=C(CN)2). MS: 264 (54.8, [M+2]+), 262 (79.5, M+), 226 (23.4, [M–HCl]+), 197 (100, [M–CH(CN) 2]+), 162 (12.4, [M–CH(CN)2–Cl]+), 136(52.5), 110 (9.1), 100 (42.4), 75 (28.9). Analysis : Calc. for C11H4Cl2N4: C, 50.22; H, 1.53; N, 21.30. Found: C, 50.41; H, 1.50; N, 21.18.
3-Chloro-2-(dicyanomethylene)-6-nitro-1,2-dihdroquinoxaline (2g): brown crystals (yield: 80%); m.p 255-257oC (dec.); IR (cm-1): 2208 (υCN); 1H-NMR: 8.33(s, 1H, H-5), 8.19 (d, 1H, H-7), 7.50 (d, 1H, H-8); 13C-NMR: 155.5, 145.6, 144.5, 143.4, 142.0, 134.3, 125.8, 123.9, 123.3 (CN?), 44.5 (-C(CN)2); MS: 273 (41.9, M+), 243 (27.6, [M–NO]+), 237 (10.8, [M–HCl]+), 227 (25.2 [M–NO2]+), 215 (24.21), 208 (12.7, [M–CH(CN)2]+), 162 (16.0), 117 (13.0), 101 (30.0), 75 (48.5); Analysis: Calc. for C11H4ClN5O2: C, 48.28; H, 1.47; N, 25.59. Found: C, 48.20; H, 1.44; N, 25.40.
The Reaction of 2a and 2e with Sodium Azide.
A solution of 2a (0.5 g, 1.8 mmoles) in DMF (20 mL) was stirred with sodium azide (0.3 g, 4.6 mmoles) at room temperature for 12 h. The reaction mixture was poured into water and the resulting solid filtered, washed with water and dried in the oven. It was then recrystallized from ethanol to give tetrazolo[1,5-a]quinoxalin-4(5H)-ylidenecyanoacetic ester (4a) (0.44 g, yield: 86%); m.p 315-316oC (dec.); IR (cm-1): 2200 (υCN), 1635 (υCO); MS: 282 (27.8, M+), 254 (2.0, [M–N2]+), 226 (9.4, [M–N2–C2H4]+), 210 (17.4, [M–CO2–C2H4]+), 208 (17.6), 182 (67.7, [M–CO2–C2H4–N2]+), 181 (100, [M–CO2 –C2H5–N2]+), 155 (46.5), 128 (26.7), 102 (40.8), 90 (48.7), 77 (22.2); Analysis: Calc. for C13H10N6O2: C, 55.32; H, 3.57; N, 29.78. Found: C, 55.02; H, 3.38; N, 29.49.
In a similar manner tetrazolo[1,5-a]quinoxalin-4(5H)-ylidenemalononitrile (4b) was synthesized: yellow crystals (yield: 85%); m.p > 340o C; IR (cm-1): 2200 (υCN); 1H-NMR: 8.20 (d, 1H, Ar-H), 7.49-7.65 (m, 2H, Ar-H), 7.36 (t, 1H, Ar-H); 13C-NMR: 148.9, 140.5 138.0, 129.3, 125.0, 123.6, 121.5, 120.7, 115.5 (CN), 41.8 (=C(CN)2); MS: 235 (30.3, M+], 207 (36.4, [M–N2]+), 181 (28.5, [M–N2– CN]+), 180 (33.9, [M–N2–HCN]+), 155 (100), 128 (26.1), 102 (52.1), 90 (71.5), 77 (24.8); Analysis: Calc. for C11H5N7: C, 56.17; H, 2..14; N, 41.69. Found: C, 56.26; H, 2.00; N, 41.57.
Reaction of 2a with Hydrazine
A solution of 2a (0.5 g, 1.8 mmoles) and hydrazine hydrate (0.9 ml, 18.5 mmoles) in DMF (20 mL) was stirred at room temperature for 15 h, after which the solvent was removed on a rotatory evaporator under reduced pressure. The resulting residue was recrystallized from ethanol to give light yellow crystals (0.26 g) of 3-hydrazino-2-(ethoxycarbonylcyanomethylene)-1,2-dihydroquinoxaline (5) (yield: 53%); m.p. 222-225oC (dec.); IR (cm-1): 2200 (υCN), 1590 (υCO); 1H-NMR: 8.14 (br s, 1H, NH, D2O exchangeable),7.86-7.96 [m, 3H, 2Ar-H+NH (D2O exchangeable)], 7.49-7.60 (m, 2H, Ar-H), 5.81 (br, s, NH2, D2O exchangeable), 4.32 (q, 2H, CH2), 1.34 (t, 3H, CH3); MS: 271 (70.7, M+), 227 (19.4 [M– CO2]+), 225 (76.5, [M–C2H5–NH3]+), 199 (100, [M–C2H5–NH3-CN]+), 169 (21.0), 102 (24.0), 90 (18.7); Analysis: Calc. for C13H13N5O2:: C,57.56; H, 4.83; N, 25.82. Found: C, 57.50; H, 4.81; N, 25.71.
Typical Procedure for the Synthesis of Dicyano(3-pyridinium-1-ylquinoxalin-2-yl)methanides 6a-c from 2e
To a solution of 2e (0.5 g, 2.2. mmoles) in absolute EtOH (40 mL) was added pyridine (3.5 mL, 43 mmoles) and the resulting solution refluxed for 6 h. The reaction mixture was cooled and the colored solid was filtered off, washed with EtOH and then dried in vacuo to give 0.56 g of dicyano(3-pyridinium-1-ylquinoxalin-2-yl)methanide (6a) as orange crystals (yield: 95%); m.p 342-345oC; IR (cm-1): 2200, 2160 (υCN); 1H-NMR: 9.53 (d, 2H, J2’3’ = 5.45 , 2’-PyH), 8.89 (t, 1H, J4’3’= 7.89 4’-Py H), 8.42 (t, 2H, J = 7.24, 3’-PyH), 7.72 – 7..79 (m, 3H, quinoxalinyl-H), 7.42-7.49 (m, 1H quinoxalinyl-H); 13C-NMR: 120.7 (CN) 144.0 (C-2) 150.2 (C-3) 128.3 (C-5), 125.4 (C-6), 132.0 (C-7), 125.6 (C-8), 141.5 (C-9), 134.5 (C-10), 145.5 (C-2’), 128.5 (C-3’), 148.5 (C-4’), 128.5 (C-5’), 145.4 (C-6’), 37.1 (=C(CN)2); UV (MeOH): 220 (4.52), 277 (4.28), 311 (4.30), 415 (3.68), 460 (sh) (3.64); MS: 271 (100, M+), 245 (11.2, [M–CN]+), 192 (22.6, [M–C5H5N]+), 167 (74.3), 102 (67.9), 79 (19.6); Analysis: Calc. for C16H9N5: C, 70.84; H, 3.34, N, 25.82. Found: C, 70.65; H, 3.11; N, 25.80.
In a similar manner, 6b and 6c were synthesized:
Dicyano(3-(3’-methylpyridinium)-1-ylquinoxalin-2-yl)methanide (6b): red crystals (yield: 94%); m.p. 297-300oC ; IR (cm-1): 2210, 2160 (υCN); 1H-NMR: 9.43 (s, 1H, 2’-pyH), 9.35 (d, 1H, J6’5’ = 6.01. 6’-PyH), 8.73 (d, 1H, J4’5’ = 8.11, 4’-PyH), 8.29 (t, 1H, J = 7.06, 5’-PyH), 7.69-7.78 (m, 3H, quinoxalinyl-H), 7.41-7.48 (m, 1H quinoxalinyl-H), 2.59 (s, 3H, CH3); 13C-NMR: 120.7 (CN), 144.0 (C-2), 150.2 (C-3), 127.8 (C-5), 125.4 (C-6), 132.0 (C-7), 125.6 (C-8), 141.3 (C-9), 134.5 (C-10), 142.7 (C-2’), 139.4 (-3), 148.6 (C-4’), 128.3 (C-5’), 144.5 (C-6’) 37.2 (=C(CN)2), 17.9 CH3). MS: 285 (100, M+), 270 (5.0, [M–CH3]+), 259 (45.1, [M–CN]+), 192 (11.1, [M–CH3–C5H4N]+), 167 (40.3), 129 (8.9), 102 (53.5), 93 (26.7); Analysis: Calc for C17H11N5: C, 71.56; H, 3.89; N, 24.55. Found: C, 71.27; H, 3.68; N, 24.50.
Dicyano(3-(4’-methylpyridinium)-1-ylquinoxalin-2-yl)methanide (6c): as red crystals (yield: 96%); m.p. 283-285oC; IR (cm-1): 2210, 2170 (υCN); 1H-NMR: 9.33 (d, 1H, J = 6.77, 2’-PyH), 8.23 (d, 2H, J = 6.47, 3’-PyH), 7.68-7.77 (m, 3H, quinoxalinyl-H), 7.40-7.47 (m, 1H, quinoxalinyl-H), 2.72 (s, 3H, CH3); 13C-NMR: 120.8 (CN), 144.0 (C-2), 150.5 (C-3), 128.4 (C-5), 125.3 (C-6), 132.0 (C-7), 125.7 (C-8), 141.3 (C-9), 144.2 (C-2’), 128.7 (C-3’), 162.4 (C-4’), 128.7 (C-5’), 144.2 (C-6’), 37.2 (=C(CN)2), 22.0 (CH3); MS: 285 (100, M+), 270 (2.0, [M–CH3]+), 259 (14.3, [M–CN]+), 192 (8.5, [M –CH3–C5H4N]+), 167 (47.0), 142 (9.4), 102 (40.2), 93 (15.6); Analysis: Cal. For C17H11N5: C, 71.56; H, 3.89; N, 24.55. Found: C, 71.33; H, 3.71; N, 24.46.
Reaction of 2a with Quinoxaline
A mixture of 2a (1.0 g, 3.6 mmoles) and quinoxaline (1.1 g, 8.5 mmoles) in dimethoxyethane (50 mL) was heated under reflux for 20 h. TLC showed the presence of two main spots, corresponding to the starting material 2a and the main reaction product, 2-chloro-3-cyanomethylquinoxaline (8). Separation was achieved by column chromatography (silica gel) to give 8 as orange crystals (0.2 g, yield: 27%); m.p 140-143oC (dec.); IR (cm-1): 2240 (υCN); 1H-NMR: 8.13-8.19 (m, 1H, Ar-H), 8.02-8.09 (m, 1H, ArH), 7.81 – 7.89 (m, 2H, Ar-H), 4.29 (s, 2H, CH2); 13C-NMR (CDCl3): 145.4, 144.3, 141.7, 140.6, 131.7, 131.0, 129.1, 128.2 (quinoxalinyl group), 114.7 (CN), 25.8 (CH2); MS: 205 (31.9, [M+2]+), 203 (100, M+), 168 (74.9, [M–Cl]+), 163 (8.9, [M–CH2CN]+), 102 (38.4), 76 (12.9); Analysis: Calc. for C10H6ClN5 : C, 58.98; H, 2.97; N, 20.64. Found: C, 58.90; H, 2.80; N, 20.54.
Acknowledgements
We thank the Alexander von Humboldt Foundation for a post-doctoral fellowship and Professor Dr J.C. Jochims for valuable discussions on the NMR spectra.
Footnotes
Sample Availability: Available from the authors.
References
- 1.Krishnan H. V. S, Chowdary S. K, Dubey P. K, Naidu A, Sunkara V. Heterocycl. Commun. 2000;6:333. [Google Scholar]
- 2.Goncharova I. N, Postovskii I. Ya. J. Gen. Chem. USSR. 1962;32:3271. [Google Scholar]
- 3.Pratt E. F, Keresztesy J. C. J. Org. Chem. 1967;32:49. [Google Scholar]
- 4.Hertler W. R, Hartzler H. D, Acker D. S, Benson R.E . J. Amer. Chem. Soc. 1962;84:3387. [Google Scholar]
- 5.Maxfield M, Bloch A. N, Cowan D. O. J. Org. Chem. 1985;50:3789. [Google Scholar]
- 6.Katritzky A. R, Fan W.-Q, Liang D_S, Li Q.-L. J. Heterocyclic Chem. 1989;26:1541. [Google Scholar]
- 7.Hans J, Albin H, Andre B. M. Monatsh Chem. 1975;106:715. [Google Scholar]
- 8.Komin A. P, Carmack M. J. Heterocyclic Chem. 1976;13:13. [Google Scholar]
- 9.Kim J.H., Lee J.K. Bull. Korean Chem. Soc. 2001;22:999. [Google Scholar]
- 10.Kay A.J., Woolhouse A.D, Gainsford G.J, Haskell T.G, Wyss C.P, Giffin S.M, McKinnie I.T, Barnes T.H. J. Mater. Chem. 2001;11:2271. [Google Scholar]
- 11.Geurn N, Gong M.S. Bull. Korean Chem. Soc. 2000;21:1111. [Google Scholar]
- 12.Shin G.I, Lee J.I, Kim J.H. Bull. Korean Chem. Soc. 1996;17:29. [Google Scholar]
- 13.Tanaka K, Takahashi H, Takimoto K, Sugita M, Mitsuhashi J. J. Heterocyclic Chem. 1992;29:771. [Google Scholar]
- 14.Chekavichus B. S, Sausin A. E, Dubur G. Ya. Khim. Geterotsikl. Soedin. 1982;1072 [Google Scholar]