

Abstract
Colorectal carcinogenesis (CRC) imposes a major health burden in developing countries. It is the third major cause of cancer deaths. Despite several treatment strategies, novel drugs are warranted to reduce the severity of this disease. Adenomatous polyps in the colon are the major culprits in CRC and found in 45% of cancers, especially in patients 60 years of age. Inflammatory polyps are currently gaining attention in CRC, and a growing body of evidence denotes the role of inflammation in CRC. Several experimental models are being employed to investigate CRC in animals, which include the APCmin/+ mouse model, Azoxymethane, Dimethyl hydrazine, and a combination of Dextran sodium sulphate and dimethyl hydrazine. During CRC progression, several signal transduction pathways are activated. Among the major signal transduction pathways are p53, Transforming growth factor beta, Wnt/β-catenin, Delta Notch, Hippo signalling, nuclear factor erythroid 2-related factor 2 and Kelch-like ECH-associated protein 1 pathways. These signalling pathways collaborate with cell death mechanisms, which include apoptosis, necroptosis and autophagy, to determine cell fate. Extensive research has been carried out in our laboratory to investigate these signal transduction and cell death mechanistic pathways in CRC. This review summarizes CRC pathogenesis and the related cell death and signal transduction pathways.
Keywords: Colorectal cancer, Cell death, Apoptosis, Autophagy, Inflammation, Hippo signalling, Nuclear factor erythroid 2-related factor 2, Wnt signaling
Core tip: Colorectal carcinogenesis (CRC) imposes a major health burden. This review addresses the cell death mechanisms and major signal transduction pathways involved in CRC. Regulated cell death is important for maintaining normal homeostasis, and the dysregulation of cell death processes leads to a spectrum of diseases including cancer. It is interesting to note that cell death pathways collaborate with each other, so understanding the various cell death mechanisms are therefore essential. CRC is orchestrated by various signal transduction pathways, which are used as drug targets. This review highlights the key concepts concerning cell death mechanisms and signal transduction in CRC.
INTRODUCTION
Cancer is a dreadful disease caused to an anomalous growth of cells, which leads to an irregular balance of cell proliferation and death. Cell death is a physiological process where normal cells are regulated by “touch contact-inhibition”. However, proliferating tumor cells metastasize to distant sites and invade other tissues, often causing morbidity[1,2]. In recent years, colorectal carcinogenesis (CRC) has imposed a major health burden in developing countries[3,4]. CRC is the second highest cause of cancer deaths in women, and the third highest cause of cancer deaths in men[5]. Due to environmental factors, a sedentary lifestyle and diet, the risk of CRC has been growing over the past few years. In many cases, the symptoms are not recognized by the individual. Although awareness via cancer screenings and the knowledge of therapy modalities has increased, the burden of CRC is much more pronounced in developing countries. The mortality rate of CRC is particularly high in Asian and African populations. Recently, mortality rates are declining in Western countries because of early screening and better treatment procedures[6]. An increase in mortality has been reported in several Latin American countries, the Caribbean and Asia, likely due to inadequate health infrastructure and the lack of awareness about cancer screenings[7]. It is well-known that dietary factors influence the incidences of CRC[8]. Diets that are rich in fiber and that have low fat content tend to prevent CRC. The food stuffs we intake determine our quality of health. Fried foods, red meat, and processed foods all increase CRC risk[9,10].
ROLE OF POLYPS IN COLORECTAL CANCER
The cells in the lining of the colon change morphologically and proliferate uncontrollably. Benign (non-cancerous) polyps are often found lining the bowels. They occur in several areas of the gastrointestinal tract, but predominantly arise in the colon. They appear as small protrusions in the lumen. As aging progresses, the number of polyps increases. Malignant polyps indicate an adenoma that appears benign. Adenomas are precursor lesions in CRC that arise through the adenoma-carcinoma sequence. CRC develops due to the formation of malignant neoplasms within the lining of the large intestine[11]. Malignancy risk has been linked to the site, size, and histological characteristics of polyps. Polyps < 5 mm in diameter are harmless and pose an insignificant risk of malignancy, whereas those with a diameter > 25 mm pose a significant risk[12]. Colonic polyps are aberrant growths that appears in the colon. Polyps, in principle, can be diagnosed by screening the colon via endoscopy or colonoscopy. Three types of colonic polyps include hyperplastic polyps, adenomatous polyps and malignant polyps[13]. These small colorectal polyps vary in size, ranging from small (< 10 mm) to diminutive (< 6 mm), and develop into cancer in 3%-5% of cases[14]. The larger polyps have a greater chance of developing into a tumor. Among polyps, the most common ones are adenomas, which have the potential to become cancerous and can be removed during screening tests. Hyperplastic polyps must be differentiated from adenomatous polyps, as they have less cancerous potential unless localized in the proximal colon[15]. Inflammatory polyps are gaining attention and often contribute to ulcerative colitis. Ulcerative colitis therefore increases the overall risk of CRC[16,17]. A recent article highlights the importance of both managing these complex polyps and resecting colonic tumors[18]. It is known that 5% of all CRC cases are attributed to two specific inherited syndromes, which include hereditary nonpolyposis colorectal cancer and familial adenomatous polyposis[19,20].
SYMPTOMS AND RISK FACTORS OF COLORECTAL CANCER
Common symptoms of CRC are rectal bleeding, significant changes in the colour of stool (especially dark or black-colored stools), irregular bowel habits, pain or discomfort in the lower abdomen, weakness or fatigue, and certain types of anemias[21]. Several risk factors are thought to cause CRC. Age is a major risk factor. About 90% of CRC patients are above the age of 50. The median age of CRC diagnosis is 68 in men and 72 in women. CRC risk also increases due to environmental factors, which include consuming a diet rich in red meat and fat, poor intake of dietary fiber, sedimentary life style, obesity, diabetes mellitus, smoking and consumption of alcohol[22,23]. One possible mechanism of diet-associated CRC is the production of heterocyclic amines during the cooking of meat, as well as higher levels of fecal bile acids[24]. Conversely, the consumption of fish oil rich in omega 3 - fatty acids (Omega 3 PUFA) reduces CRC risk. Personal history of sporadic tumours is also known to increase the risk of CRC. A previous history of colonic polyps, small bowel, endometrial, breast or ovarian cancers are additional factors that contribute to CRC[25,26]. In recent years, there has been an increasing interest in evaluating the genetic pathways that contribute to CRC. The current research trend has been diverted towards chromosome instability pathways, which correlate with sporadic CRC via mutations arising in K-ras, p53 and adenomatous polysposis coli (APC). The microsatellite instability pathway describes hereditary non-polyposis through frequent mutations in DNA mismatch repair pathway genes[27,28].
STAGES OF COLORECTAL CANCER
CRC is a horrendous disease that progresses gradually through three precisely-connected stages: Initiation - a process that alters the molecular message of the normal cell, promotion - aberrant signal transduction cascades and progression - phenotypically-altered, transformed cells. CRC can be divided into five stages, stage 0 to IV (Figure 1). Disease severity and the corresponding therapeutic options depend on the stage[29]. Stage 0 can be characterized by a tumour at the region of the mucosa or inner lining of the colon. CRC stage I is when cancer cells grow in the mucosa, yet their invasive capacity is restricted to the muscular region and not present in the neighbouring tissues of the colon)[30]. Stage II can be subcategorized into three types based on invasive growth into: the walls of the colon, the muscular layer of abdomen lining, and nearby tissues[31]. Depending upon the growth of the cancer, stage III can be further divided into three types. During this stage, the cancer grows into the inner lining of the colonic muscular layer and forms lymph nodules in surrounding tissues. Based on the number of nodule formations, this stage can be named IIIA, IIIB or IIIC. Stage IV describes the worst stage of the disease where the cancer has spread to distant parts of the body, such as the liver, lungs, etc.[32].
Figure 1.

Different stages during the progression of colorectal carcinogenesis. Stage 0: The cancerous cells grow within the inner lining of the mucosa; Stage I: The cancerous cells grow throughout the mucosa and submucosa. The cancerous growth invades into the muscular layer of the colon; Stage II: The cancerous growth penetrates through the wall of the colon without spreading to neighbouring tissues or lymph nodes; Stage III: The cancer penetrates through layers of muscle into the serosa, the layer of visceral peritoneum. The cancer begins to spread to the lymph nodes; Stage IV: A tumour nodule forms in the tissue surrounding the colon, cancer cells appears within the lymph nodes, and the cancer begins to metastasize.
MURINE MODELS OF COLORECTAL CANCER
Basic CRC research using animal models has grown[33,34]. Especially in recent times, animal models have contributed towards our understanding of CRC pathogenesis and yielded insights into the development of novel chemotherapeutic drugs. In spite of this, murine models have become a key tool in understanding the effects of genetic modifications that occur during the process of CRC formation[35,36]. Researchers have developed and modified murine models of CRC, which is a resource with immense potential. Murine models have been segregated into three different classifications, namely genetically-modified, western diet-induced, and chemically-induced models[37].
APCmin/+ mouse model
Studies over the past few decades involving preclinical CRC research utilize the APCmin/+ mouse[38]. The APCmin/+ mouse is a genetically-engineered model of mouse colon carcinogenesis. When these mice reach the age of 4 wk old, they spontaneously develop tumors in the intestine and colon. It is a well-known phenomenon that about 80% of CRCs arise due to mutations in the APC gene. Researchers removed one allele of the APC gene, thus creating the APCmin/+mouse model. The APCmin/+model of intestinal/colorectal cancer has been extensively studied in the context of developing novel chemotherapeutic drugs[39,40].
Dimethyl hydrazine and azoxymethane
Azoxymethane (AOM) and 1,2 dimethyl hydrazine (DMH) are the two notorious chemical carcinogens used to induce and study CRC in rat and mouse models[41,42]. AOM and DMH are alkylating agents that produce free radicals that bind to DNA and cause mutations. These accumulating mutations will develop into tumours. These agents are injected either intraperitoneally (i.p.) or subcutaneously (s.c.) into animals for several weeks to induce colonic tumors[43]. Detailed analysis of colonic tumours from these chemically- induced rodents harbour mutations in the β-catenin gene, which is quite similar to Human Non Polyposis Colorectal Cancer [44]. In our laboratory, we extensively use this model to develop many natural chemotherapeutic agents[45].
DSS/DMH model of ulcerative colitis-induced CRC
Chronic inflammatory bowel disease, where the colon is extensively injured over a prolonged period of time due to inflammation, increases the risk of CRC. The most common forms of inflammatory bowel disease are ulcerative colitis and Crohn’s disease[46]. A combination of Dextran sodium sulfate (DSS) and DMH are now used to induce CRC in Fisher rats[47]. A single dose of AOM and three cycles of 2% DSS in drinking water for seven days results in tumor formation within 8 wk. These AOM/DSS or DMH/DSS mouse models are largely used by researchers to screen drugs.
N-methyl-N-nitro-N-nitrosoguanidine and N-methyl-N-nitrosourea
Chemically-induced N-methyl N-nitro-N-nitrosoguanidine and N-methyl-N-nitrosourea are non-specific colon cancer models. These carcinogens induce neoplasia in multiple organs when administered to rodents [48-51]. N-methyl-N-nitrosourea injection into rodents also induces prostate and breast cancer[52]. When N-methyl-N-nitrosourea is administered through the rectum, it not only causes a greater incidence of CRC, but also induces thymic lymphoma and lung cancers[53]. Since this is considered to be a non-specific colon cancer model, it is not frequently used to study colorectal cancer.
Western diet-induced rodent CRC model
Epidemiologic studies indicate that diet plays a vital role in the development of colorectal cancer risk in humans[54]. Many studies have examined the influence of typical western diets on the incidence of colorectal cancer. About 12 wk of feeding these western diets to rats and mice promotes hyperplasia in colonic crypts[55,56]. Approximately 70% of the mice fed with this Western diet exhibited nuclear atypia in their colonic epithelia, and 40% of the mice showed features of dysplastic crypts at the end of two years[57,58]. These reports suggest the involvement of a Western diet in eliciting CRC.
EPITHELIAL-MESENCHYMAL TRANSITION IN COLORECTAL CANCER
Epithelial cells: targets of colorectal cancer
CRC is believed to originate in epithelial cells that line the colon and rectum. The epithelium is highly vulnerable to mutation and carcinogenesis, as the replication rate of cells in the epithelium of the colon and rectum is relatively high, with a replication rate of 1010 cells every day[59]. The abnormal accumulation of epithelial cells can cause mutations in oncogenes and tumour suppressor genes, which may lead to neoplastic growth. Thus, these abnormal changes in cells of the colon and rectum, which form benign lesions, have the potential to further develop into cancer and metastasize to other organs[60].
Epithelial-to-mesenchymal transition: a complex mechanism in cancer metastasis
Epithelial-to-mesenchymal transition (EMT) represents a well-organized mechanism in which epithelial cells alter their cellular characteristics and behaviour, and reform into a mesenchymal-like phenotype[61]. Polarized epithelial cells are tightly-packed through tight junction molecules such as claudin, occludin, and zonula occludens; adherens junction molecules such as E-cadherin and desmosomes form a sheet-like structure in the normal epithelium[62]. In contrast to epithelial cells, mesenchymal cells do not possess cell-cell adhesion molecules, which give mesenchymal cells migratory capacity and invasiveness. The dissolution of cell adhesion molecules results in loss of apical-basolateral cell polarity in mesenchymal cells. Another important feature of these mesenchymal cells is resistance to cellular senescence and apoptosis. Mesenchymal cells are characterized by the enhanced expression of extracellular proteases and transcription factors, such as snail, slug and twist, which activate the cells to produce collagen, fibronectin, vimentin, α-smooth muscle actin (α-SMA), etc[63]. Interestingly, the shift from an epithelial to mesenchymal state is complex. Upon triggering by mediators, these events begin with the dissolution of cell-cell adhesion, which results in the loss of microvilli and cilia at the apical surface of epithelial cells. At this stage, cytoskeletal reorganization takes place, which releases α-smooth muscle actin and matrix metalloproteinases. These secreted matrix metalloproteinases degrade the extracellular matrix, which facilitates the dissolution of cells from the basement membrane and allows cells to move along the matrix[64].
EMT plays a key role in the spreading of cancer throughout distant parts of the body. Newly-produced cells by EMT display several properties associated with cancer metastasis. Reports suggest that EMT cells can avoid cellular senescence by inhibiting tumour-suppressor proteins[65]. Furthermore, research evidence shows that high levels of vimentin in EMT cells makes these cells more resistant to chemotherapeutic drugs[66]. The mechanism of EMT in colorectal cancer metastasis is depicted in Figure 2. The mechanism of EMT is considered to be complex because of the heterogeneity within this population. Interestingly, not all epithelial cells in a mutated epithelium undergo EMT. Moreover, not all EMT cells facilitate metastasis. Several environmental factors as well as signalling cascades regulate these mechanisms of EMT[67]. A successful metastasis is achieved through the involvement of another mechanism called mesenchymal-to-epithelial transition. The invasive mesenchymal cells produced by EMT travel through systemic circulation and anchor themselves in other distant parts. For this, cells must regain their epithelial features and thereby undergo a mesenchymal-to-epithelial transition. The modulation of cells between EMT and mesenchymal-to-epithelial transition states facilitates cancer metastasis[68].
Figure 2.
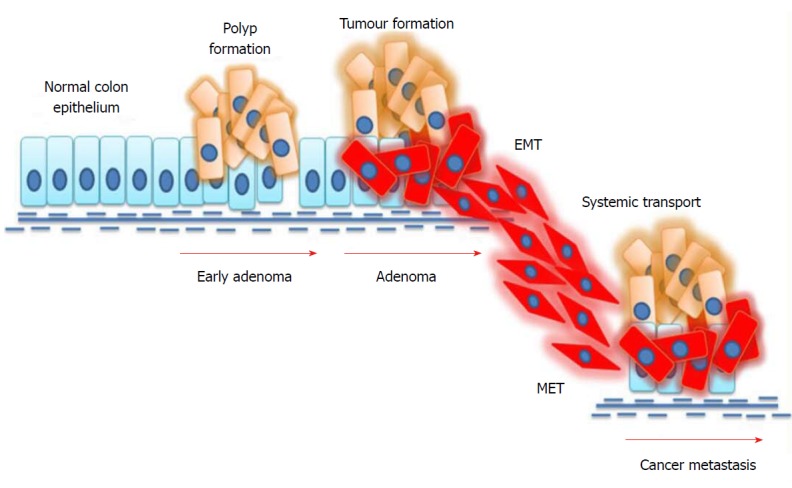
Mechanism of epithelial-to-mesenchymal transition in colorectal cancer. External stimuli or mutations in cancer cells induce epithelial-to-mesenchymal transition (EMT), where epithelial cells undergo phenotypic changes and transit into an invasive mesenchymal cell state. Mesenchymal cells invade the systemic circulation and undergo a mesenchymal-to-epithelial transition (MET) in distant organs, thus facilitating metastasis.
Interestingly, E-cadherin, a hallmark for EMT, is reported as a biomarker for colorectal cancer[69]. Recently, a research group reported that the silencing of ubiquitin-specific protease 47, a deubiquitinating enzyme, augments the proteasomal degradation of Snail, the transcription factor involved in EMT, to prevent the progression of colorectal cancer[70]. A molecular genetic approach towards the involvement of EMT in colorectal cancer revealed that the epithelial nature of colon cancer cells might be sensitive to several drugs, including Src, Notch, and epidermal growth factor receptor inhibitors[71]. Further studies are warranted to identify novel regulators of EMT in order to find novel cellular targets of colorectal cancer.
CELL DEATH IN COLORECTAL CANCER: “CUTS TWO WAYS” PROCESS FROM WOMB TO TOMB
Although Carl Vogt reported the incidence of cell death in metamorphic toads in 1842, the mechanisms of cell death was recognized in the middle of the 19th century[72]. However, research attempts have yet to come out with a clear picture of the phenomena of cell death, and confusions still remain between the alternative forms of cell death. As an essential physiological process required to maintain tissue homeostasis, the different modalities of cell death are intensively studied[73]. The decision of a cell to live or die is important and can be the determining factor in the progression of cancers[74]. Chemotherapies targeting cell death mechanisms are highly encouraged in order to prevent cancer progression and metastasis[75,76]. Dysregulated cell death signalling cascades are considered to be fundamental to the progression and worsening of CRC. Considering this notion, a conceptual understanding of the involvement of different modes of cell death in colon carcinogenesis and its progression would shed light on novel cellular targets against colorectal cancer.
Death-triggering environmental cues in the colorectum
The urogenital system and hindgut, which include the colon and rectum, begin to divide in the 4th week of human gestation and become separate units by the 7th week[77]. Cell death, particularly apoptosis in the mesenchyme, plays a predominant role in this process. Research evidence shows that apoptotic cells are concentrated in the mesenchyme of the terminal rectum during the formation of the anal canal in the 7th week of gestation[78]. A number of developmental regulatory signalling molecules such as Wnt 5a, Cdx1, Hoxd-13, Tcf4 and Shh actively participate in the up- and downregulation of apoptotic cell death during the formation of the colorectum[79,80]. Interestingly, researchers have reported the decisive role of autophagy in the activation of cellular signals that are required for the phagocytic engulfment of apoptotic cells during embryonic development[81]. Yet another research group has reported that alternative cell death mechanisms such as autophagy, cornification, entosis, and necroptosis exist when apoptosis machinery fails during embryogenesis[82]. Previous reports clearly point out that cell death mechanisms are not only important in shaping the embryo, but also for maintaining adult tissue homeostasis, and can therefore be considered as key machinery from womb to tomb.
TYPES AND CHARACTERIZATION OF CELL DEATH
According to the 2018 nomenclature committee on cell death, all cell death processes taken together can be classified into fourteen or more subgroups based on their morphological characteristics, enzymological criteria, functional phases, and immunological reactions. These subgroups include apoptosis, necrosis, necroptosis[83], ferroptosis[84], pyroptosis[85], parthanatos[86], entosis[87], NETosis[88], autophagy[89], and mitotic catastrophe[90]. Genetically-programmed mechanisms for the targeted eradication of permanently-damaged and destructive cells or organs are collectively termed as regulated cell death mechanisms. The major classifications of different cell death modalities with each of stheir functional aspects are depicted in Figure 3.
Figure 3.

Different modalities of cell death. Unpredictable perturbations in the extracellular or intracellular microenvironments of a cell can activate several signal transduction cascades that ultimately lead to various forms of cell death. Type I cell death apoptosis: the extrinsic pathway of apoptosis is mediated by Fas-associated death domain protein (FADD). Caspase 8, in turn, triggers caspase 3 and 7, which then activates caspase 9. Both intrinsic and mitochondrial pathways of apoptosis are mediated through the inhibition of anti-apoptotic Bcl2, which in turn activates Bax/Bak and induces release of cytochrome c from the mitochondria. The activation of caspase 9 in the apoptosome induces apoptotic cell death. Type II cell death autophagy: autophagy is an active lysosomal degradative flux, which can be divided into three distinct types; macroautophagy, microautophagy and chaperone-mediated autophagy. Macroautophagy involves four different steps: initiation, autophagosome nucleation, phagosome expansion and completion, and autolysosome docking. The tightly-regulated autophagy machinery is mediated through several autophagy-related (Atg) molecules. Regulated necrosis/necroptosis: regulated necrosis is mediated through the interaction of receptor interacting protein 1 (RIP1) with RIP3 upon caspase 8 inhibition. RIP3 and mixed-lineage kinase domain-like (MLKL) are phosphorylated and assembled into complex IIb, which is then translocated to the plasma membrane to mediate membrane permeabilization. Ferroptosis: this regulated form of cell death is driven by the loss of glutathione peroxidase 4 (GPX4) activity, a lipid repair enzyme, followed by the accumulation of lipid hydroperoxides. Autosis: a plasma membrane Na+/K+-dependent autophagy form of cell death. Entosis: internalized cells undergo entotic cell death through the formation of entotic vacuoles, which is mediated by autophagy proteins like Vps34, etc. Pyroptosis: a caspase-dependent cell death mechanism that is an intermediary variation of apoptosis and necrosis. Caspase 1 is activated by the NLRP3 inflammasome, which activates the inflammatory cytokines interleukin 1β and interleukin 18, which in turn mediate the lytic cycle.
Targeting cell death in colorectal cancer: implications for therapy
An interesting finding about cancer is that several genes that are responsible for cancer development are very much active during embryogenesis and fetal development, particularly regulating embryonic growth and organ formation. These genetic programs remain silent throughout the rest of the life of an organism; however, they are turned on in cells during cancer formation[91]. The genetic paradigm of colorectal cancer reveals that APC or β-catenin is responsible for the initial changes in normal mucosa that form dysplastic aberrant crypt foci. COX-2 mainly regulates the formation of early adenomas, and K-RAS regulates the formation of intermediate adenomas. CPC4/SMAD4 is responsible for late adenomas and p53 is majorly responsible for carcinomas[92]. During these sequential events from benign polyp formation to adenomas and finally carcinomas, cell death plays an essential role.
A low rate of apoptosis in the base of the crypt, where stem cells are expected to reside, is fundamental to the function of the normal intestine. It is interesting to note that epithelial cells residing in the villi of the small intestine and colon are resistant to apoptosis[93]. Changes in the expression patterns of several apoptotic proteins during the transformation of adenomas into carcinomas reveal the importance of apoptosis during colon cancer progression[94]. Since 70% of reported CRCs are associated with mutations in the tumour suppressor p53 gene, the transition from adenomas to carcinomas in the colorectal region is considered to involve a mechanism whereby apoptosis machinery fails[95]. Therefore, chemotherapies intended to stimulate apoptosis in colon cells would be central to controlling disease progression[96]. With this notion, our laboratory is interested in elucidating the apoptosis-inducing effect of certain phytochemicals in order to eradicate cancer cells and provide protection against CRC progression. We have provided evidence that the bioflavonoid luteolin has strong anti-proliferative activity. Luteolin inhibits the Wnt/β-catenin signalling cascade, which induces apoptosis and cell cycle growth arrest in the G2/M phase in HCT-15 colon cancer cells[97]. In addition, azoxymethane induces colon carcinogenesis in BALB/c mice[98]. Our reports suggest that apoptosis is an efficient parameter in preventing malignant transformation since it eradicates harmful cells. On the contrary, apoptosis can also promote cancer growth by preventing the removal of certain genetic variants that have a high potential to induce malignancy. Yet another interesting hypothesis about cancer is that tumour tissues possess a higher apoptotic index than normal tissues. Notably, a higher apoptotic index in tissue indicates more a malignant tumour[99]. Therefore, apoptosis can be considered as a double-edged sword in cancer progression. However, the mechanism linking a high apoptotic rate with increased cancer cell proliferation and metastasis needs to be further elucidated.
Apart from apoptosis, other cell death modes are also gaining attention in cancer research in order to find better therapeutic targets. From this point of view, the pro-and anti-metastatic effects of autophagy have been studied in several cancers including brain, liver, pancreas, colon etc. Several signalling cascades are known to regulate autophagy. Among these, PI3K/Akt/mammalian target of rapamycin (mTOR) is an important signalling pathway that acts as a checkpoint in autophagy and promotes cancer progression. Interestingly, PI3K/Akt hyperactivation, PIK3CA mutations, and both PTEN mutations and deletions have been reported in the incidence of CRC[100]. Autophagy is reported as an anti-metastatic mechanism in the early stages of cancer metastasis by preventing both the infiltration of inflammatory cells as well as tumour cell necrosis, thus helps reduce cancer cell invasion and metastasis. However, autophagy may act as a promoter of metastasis during advanced cancer stages by enhancing EMT, cell survival and metastasis[101]. Moreover, high expression of LC3I/II, which is a key regulator of autophagosome nucleation and is known to downregulate Beclin 1, has been reported in the advanced stages of CRC[102]. This research evidence points out that autophagy machinery influences all stages of cancer progression, including initiation, proliferation and metastasis, while its effect on inhibiting or promoting cancer metastasis seems to be context-dependant.
Targeted therapies for necroptosis, a caspase-independent, receptor-interacting protein kinase-mediated form of regulated cell death, has recently been postulated as a novel strategy for cancer prevention. Very few reports are available concerning the role of necroptosis in regulating CRC progression. Moriwaki and colleagues have shown a significant downregulation in RIPK1 and RIPK3 expression in colon cancer tissues when compared with normal colon tissues[103]. Interestingly, dimethyl fumarate, an approved drug for the treatment of multiple sclerosis, is reported to induce necroptosis through the depletion of glutathione in colon cancer cells[104]. Colon cancer cell resistance against the 5-fluorouracil drug is sensitized by the usage of pan-caspase inhibitors, which facilitate 5-fluorouracil-induced necroptosis in CRC cells[105]. However, more research should be conducted to identify the possible regulatory role of necroptosis in the prevention of CRC. Altogether, these reports shed a limelight on colon cancer research by revealing a promising therapeutic target against cancer progression.
SIGNALLING PATHWAYS IN COLORECTAL CANCER
The development of colorectal involves various signalling pathways that regulate cellular proliferation, differentiation and immortalization. Signalling activation of Wnt/β-catenin, inactivation of transforming growth factor β (TGFβ) and epidermal growth factor receptor, and mutation in k-ras signalling all play a vital role in the progression of CRC[106,107].
Wnt/β-catenin signalling in colon cancer
Wnt signaling plays divergent biological roles, such as regulating cellular homeostasis and maintaining cell self-renewal throughout embryogenesis and adulthood. This pathway particularly promotes intestinal epithelial proliferation and differentiation of the intestinal crypt[108]. In the presence of Wnt ligand, the receptor frizzled inhibits the phosphorylation of Glycogen synthase kinase-3 beta, thus impeding the degradation of β-catenin by ubiquitins. Accumulated cytoplasmic β-catenin translocates to the nucleus and transcribes target genes (Figure 4). The activity of this signalling pathway depends on the cellular localization of β-catenin. Among 90% of colonic tumours have a mutation in the APC and β-catenin genes[109]. Mutations in the cluster region of APC leads to the generation of truncated protein, which fails to prevent complex formation. This mutational dysregulation in Wnt signalling stabilizes cytoplasmic β-catenin, and its nuclear translocation promotes β-catenin-dependent transcription of Wnt target genes, which therein contributes to CRC progression[110]. Nuclear β-catenin favours peripheral cellular changes that impact cell adhesion and migration. Interestingly, Wnt signaling is necessary for the initial activation of intestinal stem cells. This plays a crucial role not only for stem cell maintenance but also for crypt homeostasis. Research evidence shows that experimental abolition of Wnt signalling in cells leads to the specific loss of proliferative crypts[111,112].
Figure 4.

Wnt/β-catenin pathways. In the absence of Wnt, cytoplasmic β-catenin forms a complex with Axin (yellow), APC (blue), GSK3 (red), and CK1 (purple). Phosphorylated β-catenin undergoes ubiquitin-mediated proteosomal degradation. In the presence of Wnt, Wnt binds to the frizzled receptor, which in turn recruits the Axin complex. This disrupts Axin-mediated proteosomal degradation of β-catenin. Cytoplasmic β-catenin then travels to the nucleus and functions as a co-activator with TCF to activate Wnt-regulated gene expression. GSK: Glycogen synthase kinase; APC: Adenomatous polyposis coli; CK1: Casein kinase 1.
PI3K/Akt/mTOR signalling in colorectal cancer
PI3K/Akt/mTOR is the second most frequently mutated oncogenic signalling network in human cancers. The dysregulation of PI3K is observed in almost 30% of human cancers, making this signalling cascade an important therapeutic target in controlling cancer progression[113]. The involvement of PI3K/Akt /mTOR signalling in colon carcinogenesis has been intensively studied. Overexpression of p-Akt and impaired expression of PTEN, a tumor suppressor negative regulator of Akt, have been reported in 70% of colorectal cancer patients[114]. The carotenoid Lycopene has been reported to supress leptin-mediated cell invasion in CRC HT-29 cells through the inhibition of Akt phosphorylation[115]. Yet another research group has reported that aspirin, an inhibitor of mTOR and activator of AMP-activated protein kinase, induces autophagy and protects against the progression of colorectal cancer[116].
TGFβ / Smad signalling in colorectal cancer
TGFβ and related bone morphogenetic proteins belong to the family of cytokines involved in the governing of various cellular processes, including proliferation, differentiation, and apoptosis[117]. The TGFβ superfamily of cytokines contains many proteins, including TGFβ1, TGFβ2, TGFβ3, and activins. TGFβ conducts its signals via numerous intracellular signalling molecules, including the Smad family of proteins, mainly Smads 2 and 3[118,119]. TGFβ enhances the expression of several fibrogenic and pro-inflammatory cytokines, such as platelet-derived growth factor, tumor necrosis factor α or interleukin 1β, and promotes the development and progression of the fibrotic reaction[120]. Three major isoforms of TGFβ have been identified in mammals, namely TGFβ1, TGFβ2, and TGFβ3. In general, TGFβ is secreted in an inactivated form through its attachment to a latent TGFβ-binding protein[121]. The downstream regulation of TGFβ signalling is activated upon ligand binding to type II receptors, which phosphorylates the type I receptor, which then further phosphorylates Smads 2 and 3. The phosphorylated Smads heterodimerize with Smad4 and translocate into the nucleus to promote gene transcription (Figure 5)[122]. TGFβ plays a dual role in early cancer progression. TGFβ can perform as a tumor-suppressor pathway in normal colon epithelial cells by regulating cell proliferation and apoptosis. In later stages of cancer, however, TGFβ promotes cell migration by increasing EMT events and supressing the immune response[123,124]. The involvement of TGFβ signalling in CRC was described earlier[125-127].
Figure 5.

TGF/Smad pathway. TGFβ signalling is initiated via the binding of the TGFβ1 ligand to receptor II, which promotes dimerization between receptor II and receptor I and the subsequent transphosphorylation of TGFβRI. Activated TGFβRI therein phosphorylates and activates receptor-regulated Smads (Smad2 and Smad3). Phosphorylated Smads, along with co-Smads, form a trimeric complex and translocate to the nucleus to induce the transcription of target genes and promote cell growth and survival. TGF: Transforming growth factor.
Epidermal growth factor receptor and Ras-Raf-MEK-ERK signalling
Epidermal growth factor receptor, a membrane-bound receptor tyrosine kinase, plays a vibrant role in the development and progression of many cancers. Ligand-activated receptors form homo and heterodimers with the other ErbB family members and autophosphorylate their tyrosine residues[128]. Once ligand binds to the receptor, it triggers the activation of downstream signalling such as Ras, MAPK, ERK, NFκB and PI3K/Akt. These pathways are very critical to CRC development. The overexpression of epidermal growth factor receptor and its ligands correlates with the development of human cancer and its poor prognosis[129].
P53 AND COLORECTAL CANCER
p53 is well known gene for its tumor suppressor role and is one of the most mutated genes in all forms of human cancer. Activation of the p53 DNA damage stress response induces DNA repair and regulates the cell cycle to prevent oncogenic mutation[130]. Alteration of p53 signalling in colon cancer, which results in the loss of apoptosis and cellular checkpoints while altering genetic integrity, ultimately leads to malignancy. Accumulation of mutations in cancer-related genes, such as K-ras, p53 and APC, instigates the transition from normal epithelium to adenomatous to colorectal cancer[131].
NOTCH SIGNALLING IN COLORECTAL CANCER
In mammals, the major components of Notch signalling include five ligands (Delta like ligands 1, 3 and 4, and Jagged 1 and 2 (Sterrate-like ligands)), four Notch receptors (Notch 1-4), and several downstream target genes[132]. Signal-transduction is initiated by the interaction of a notch ligand that is present on one cell with the transmembrane Notch receptor that is present on a neighbouring cell. This binding interaction activates metalloproteases that cleave the transmembrane domain of the Notch receptor, resulting in the release of the constitutively-active Notch intracellular domain. Translocation of this domain to the nucleus regulates transcriptional complexes to induce expression of target genes (Figure 6)[133]. Alhough currently available reports provide little information about cell-specific functions of Notch signalling in CRC when compared with other solid tumours, aberrant activation of Notch signalling has been reported in CRC. In a recent study, the superior therapeutic effect of targeting both Notch and MAPK signalling on colon cancer growth, as well as its role in regulating tumor cell plasticity, has been reported[134]. Notch signalling has been reported to induce cellular resistance to chemotherapeutic drugs. It was demonstrated that Notch signalling is significantly upregulated in SW480 cells that are resistant to the experimentally-generated Regorafenib drug, a multi-kinase inhibitor. Interestingly, the inhibition of Notch signalling in resistant cells restored their sensitivity to Regorafenib, thus suggesting the important role of Notch in promoting resistance to chemotherapeutic drugs[135]. The dysregulation of Notch signalling in colon cancer metastasis has been studied in detail[136]. These reports strongly suggest the importance of Notch signalling in the pathogenesis and progression of CRC.
Figure 6.
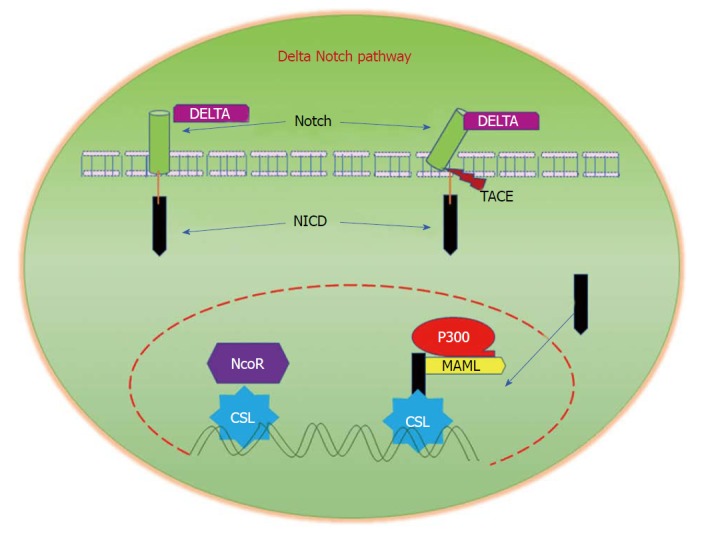
Delta/Notch Signalling. Notch signalling is initiated by the binding of Notch on one cell to the transmembrane ligands Delta or Jagged on a neighbouring cell. This binding interaction promotes cleavage of the Notch receptor and releases the Notch intracellular domain (NICD), which travels to the nucleus and controls the transcription of Notch responsive genes. In the nucleus, NICD binds to the transcriptional repressor CBF1, which recruits Mastermind-like (MAML) and other co-activators to initiate the transcription of downstream Notch-regulated genes.
Nrf2/Keap signalling in colorectal cancer
Oxidative stress is denoted as an imbalance between oxidant production and antioxidant defences, where oxidants dominate and lead to cellular dysfunction and tissue damage. Oxidative stress caused by harmful reactive oxygen species are involved in colorectal cancer. Reactive oxygen species cause cellular damage, leading to the progression of many diseases such as cancers, fibrosis, neurodegenerative disorders etc. In turn, cells possess detoxification genes (Phase II) and antioxidant genes to counterbalance the lethal effects of reactive oxygen species[137]. In many disease settings, NF-E2-related factor 2 (Nrf2), which is a basic leucine zipper transcription factor, plays a crucial role in protecting tissues against free radical-mediated insults including carcinogens, drugs, inflammation, etc[138]. Nrf2 is a member of the Cap-N-collar transcription factor family. It recognizes the antioxidant response element in the promoter of target genes[139,140]. Under basal conditions, Nrf2 is restricted to the cytoplasm by Kelch like ECH associated protein 1. Kelch like ECH associated protein 1 is very critical, as it serves as a linker protein substrate between the Cul3-based E3-ubiquitin ligase complex and Nrf2, leading to the ubiquitination and proteosomal degradation of Nrf2[141]. Certain conditions, such as the induction of the antioxidant response element, promote the detachment of Nrf2 from its partner Kelch like ECH associated protein 1, thereby facilitating the translocation of Nrf2 to the nucleus. Inside the nucleus, Nrf2 dimerizes and associates with small Maf proteins, leading to the binding of Nrf2 to antioxidant response elements, which therein promotes transcriptional activation of these genes. In colorectal cancer, the chemopreventive effect of many drugs greatly depends on this signalling[142-144].
Hippo signalling and colorectal cancer
The origin of the hippo pathway began with observations in Drosophila melanogaster flies with concomitant mutations that led to tissue overgrowth[145]. Hippo signalling has gained attention in cancer biology because of its crosstalk with oncogenic signalling pathways[146]. Yes associated protein 1 is the key transcriptional regulator of the Hippo pathway. This protein, along with its partner PDZ-binding domain taffazin, orchestrate the Hippo pathway[147]. In principle, hippo signalling plays an important role in the regulation of tissue homeostasis, development, regeneration, and cancer[148]. Three protein components in mammals are depicted: Mammalian Ste 2 like kinase 1 and 2, and large tumor suppressor kinase 1 and 2. These kinases phosphorylate Yes associated protein 1 and PDZ-binding domain taffazin, which leads to their nuclear exclusion and ubiquitin-mediated proteosomal degradation in the cytoplasm, thus promoting the suppression of Yes associated protein 1/ PDZ-binding domain taffazin-targeted genes[149,150]. Recently, a huge body of evidence suggests the critical role of Hippo signalling in CRC[151,152]. The Hippo signalling pathway has been reported to crosstalk with other signalling pathways [153,154].
MiRNAs AND COLORECTAL CANCER
Over the years, several molecular mechanisms have been identified to be involved in CRC[155]. In recent years, the discovery of microRNAs (miRNAs) has attracted considerable attention in different disease conditions. An understanding of the roles of miRNAs in development and disease, especially in cancer, have made miRNAs both an attractive tool and novel therapeutic target[156]. Generally, miRNAs are non-coding RNAs that are 20-24 nucleotides in length and were classified into Oncomirs, including the tumor-suppressor miRNAs that are related to cancer. According to recent research relating miRNAs and cancer, miRNAs impact several vital processes such as the cell cycle, proliferation, differentiation, metabolism and apoptosis[157]. It was reported that miRNAs such as miR-21, miR-181b1, miR-101, the let7 family, miR-133b, and miR-126 were dysregulated in CRC[158,159]. Recently, miR-760 was found to suppress human colorectal cancer growth by targeting BATF3/AP-1/cyclinD1 signalling[160]. MiR-422a acts as a tumor-suppressor in colorectal cancer, and its expression is limited to CRC tumours. Increasing the expression of miR-422a could inhibit CRC cell growth and promote cell apoptosis in colorectal cancer cells. It was also reported that miR-422a restricts colorectal cancer by inhibiting the p38/MAPK pathway[161]. Therefore, miRNAs are emerging as potential targets in CRC.
CONCLUSION
Research attempts to develop more effective therapies against CRC progression are of outstanding importance, as the effectiveness of mono-therapeutic approaches in CRC treatment are very limited. However, combinational therapies are gaining attention due to their ability to manipulate certain signalling cascades and induce different modalities of cell death to prevent cancer metastasis. The regulation of both cell signalling pathways and cell death represents a promising tool to improve patient responses to chemotherapy. When the normal orchestra of cellular signalling is dysregulated, cells become pathological and ultimately decide whether to die or survive. A subset of novel signalling pathways, and their association with colorectal cancer progression and metastasis, was discussed in this review. A better understanding of anticancer agents that target these cellular pathways and induce cell death modalities will hopefully provide more insights into the complicated molecular mechanisms that underlie colorectal cancer, thus facilitating the development of more effective treatments.
ACKNOWLEDGEMENT
GS acknowledges Council of Scientific and Industrial research (CSIR), New Delhi for funding Colon cancer project [37 (1364) /09/EMR-II].
Footnotes
Conflict-of-interest statement: We, the authors declare no conflict of interest.
Manuscript source: Invited manuscript
Peer-review started: April 16, 2018
First decision: April 27, 2018
Article in press: June 28, 2018
Specialty type: Oncology
Country of origin: India
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C, C, C
Grade D (Fair): D
Grade E (Poor): 0
P- Reviewer: Chmiela M, Gassler N, Perse M, Tanaka T S- Editor: Ji FF L- Editor: Filipodia E- Editor: Tan WW
Contributor Information
Ashok kumar Pandurangan, Cell Biology Laboratory, Department of Biochemistry, University of Madras, Guindy Campus, Chennai 600025, India; School of Life sciences, B.S. Abdur Rahman Crescent Institute of Science and Technology, Chennai 600048, India.
Thomas Divya, Cell Biology Laboratory, Department of Biochemistry, University of Madras, Guindy Campus, Chennai 600025, India.
Kalaivani Kumar, School of Life sciences, B.S. Abdur Rahman Crescent Institute of Science and Technology, Chennai 600048, India.
Vadivel Dineshbabu, Cell Biology Laboratory, Department of Biochemistry, University of Madras, Guindy Campus, Chennai 600025, India.
Bakthavatchalam Velavan, Cell Biology Laboratory, Department of Biochemistry, University of Madras, Guindy Campus, Chennai 600025, India.
Ganapasam Sudhandiran, Cell Biology Laboratory, Department of Biochemistry, University of Madras, Guindy Campus, Chennai 600025, India. sudhandiran@unom.ac.in.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Gryfe R, Swallow C, Bapat B, Redston M, Gallinger S, Couture J. Molecular biology of colorectal cancer. Curr Probl Cancer. 1997;21:233–300. doi: 10.1016/s0147-0272(97)80003-7. [DOI] [PubMed] [Google Scholar]
- 3.Tamas K, Walenkamp AM, de Vries EG, van Vugt MA, Beets-Tan RG, van Etten B, de Groot DJ, Hospers GA. Rectal and colon cancer: Not just a different anatomic site. Cancer Treat Rev. 2015;41:671–679. doi: 10.1016/j.ctrv.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 4.Tariq H, Kamal MU, Mehershahi S, Saad M, Azam S, Kumar K, Niazi M, Makker J, Daniel M. A Rare Case of Colonic Metastases From Tonsillar Carcinoma: Case Report and Review of Literature. World J Oncol. 2018;9:35–37. doi: 10.14740/wjon1073w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Center MM, Jemal A, Smith RA, Ward E. Worldwide variations in colorectal cancer. CA Cancer J Clin. 2009;59:366–378. doi: 10.3322/caac.20038. [DOI] [PubMed] [Google Scholar]
- 6.Chatenoud L, Bertuccio P, Bosetti C, Malvezzi M, Levi F, Negri E, La Vecchia C. Trends in mortality from major cancers in the Americas: 1980-2010. Ann Oncol. 2014;25:1843–1853. doi: 10.1093/annonc/mdu206. [DOI] [PubMed] [Google Scholar]
- 7.Deng Y. Rectal Cancer in Asian vs. Western Countries: Why the Variation in Incidence? Curr Treat Options Oncol. 2017;18:64. doi: 10.1007/s11864-017-0500-2. [DOI] [PubMed] [Google Scholar]
- 8.Pan P, Yu J, Wang LS. Colon Cancer: What We Eat. Surg Oncol Clin N Am. 2018;27:243–267. doi: 10.1016/j.soc.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marques-Vidal P, Ravasco P, Ermelinda Camilo M. Foodstuffs and colorectal cancer risk: a review. Clin Nutr. 2006;25:14–36. doi: 10.1016/j.clnu.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 10.Schwingshackl L, Schwedhelm C, Hoffmann G, Knüppel S, Laure Preterre A, Iqbal K, Bechthold A, De Henauw S, Michels N, Devleesschauwer B, et al. Food groups and risk of colorectal cancer. Int J Cancer. 2018;142:1748–1758. doi: 10.1002/ijc.31198. [DOI] [PubMed] [Google Scholar]
- 11.Calvert PM, Frucht H. The genetics of colorectal cancer. Ann Intern Med. 2002;137:603–612. doi: 10.7326/0003-4819-137-7-200210010-00012. [DOI] [PubMed] [Google Scholar]
- 12.Williams JG, Pullan RD, Hill J, Horgan PG, Salmo E, Buchanan GN, Rasheed S, McGee SG, Haboubi N; Association of Coloproctology of Great Britain and Ireland. Management of the malignant colorectal polyp: ACPGBI position statement. Colorectal Dis. 2013;15 Suppl 2:1–38. doi: 10.1111/codi.12262. [DOI] [PubMed] [Google Scholar]
- 13.Oving IM, Clevers HC. Molecular causes of colon cancer. Eur J Clin Invest. 2002;32:448–457. doi: 10.1046/j.1365-2362.2002.01004.x. [DOI] [PubMed] [Google Scholar]
- 14.Schoefl R, Ziachehabi A, Wewalka F. Small colorectal polyps. Dig Dis. 2015;33:38–41. doi: 10.1159/000366034. [DOI] [PubMed] [Google Scholar]
- 15.Angarita FA, Feinberg AE, Feinberg SM, Riddell RH, McCart JA. Management of complex polyps of the colon and rectum. Int J Colorectal Dis. 2018;33:115–129. doi: 10.1007/s00384-017-2950-1. [DOI] [PubMed] [Google Scholar]
- 16.Kim EC, Lance P. Colorectal polyps and their relationship to cancer. Gastroenterol Clin North Am. 1997;26:1–17. doi: 10.1016/s0889-8553(05)70280-6. [DOI] [PubMed] [Google Scholar]
- 17.Ponz de Leon M, Di Gregorio C. Pathology of colorectal cancer. Dig Liver Dis. 2001;33:372–388. doi: 10.1016/s1590-8658(01)80095-5. [DOI] [PubMed] [Google Scholar]
- 18.Lord R, Burr NE, Mohammed N, Subramanian V. Colonic lesion characterization in inflammatory bowel disease: A systematic review and meta-analysis. World J Gastroenterol. 2018;24:1167–1180. doi: 10.3748/wjg.v24.i10.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Witold K, Anna K, Maciej T, Jakub J. Adenomas - Genetic factors in colorectal cancer prevention. Rep Pract Oncol Radiother. 2018;23:75–83. doi: 10.1016/j.rpor.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boland PM, Yurgelun MB, Boland CR. Recent progress in Lynch syndrome and other familial colorectal cancer syndromes. CA Cancer J Clin. 2018;68:217–231. doi: 10.3322/caac.21448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maida M, Macaluso FS, Ianiro G, Mangiola F, Sinagra E, Hold G, Maida C, Cammarota G, Gasbarrini A, Scarpulla G. Screening of colorectal cancer: present and future. Expert Rev Anticancer Ther. 2017;17:1131–1146. doi: 10.1080/14737140.2017.1392243. [DOI] [PubMed] [Google Scholar]
- 22.Yantiss RK, Goodarzi M, Zhou XK, Rennert H, Pirog EC, Banner BF, Chen YT. Clinical, pathologic, and molecular features of early-onset colorectal carcinoma. Am J Surg Pathol. 2009;33:572–582. doi: 10.1097/PAS.0b013e31818afd6b. [DOI] [PubMed] [Google Scholar]
- 23.Carr PR, Jansen L, Bienert S, Roth W, Herpel E, Kloor M, Bläker H, Chang-Claude J, Brenner H, Hoffmeister M. Associations of red and processed meat intake with major molecular pathological features of colorectal cancer. Eur J Epidemiol. 2017;32:409–418. doi: 10.1007/s10654-017-0275-6. [DOI] [PubMed] [Google Scholar]
- 24.Murff HJ, Shrubsole MJ, Cai Q, Smalley WE, Dai Q, Milne GL, Ness RM, Zheng W. Dietary intake of PUFAs and colorectal polyp risk. Am J Clin Nutr. 2012;95:703–712. doi: 10.3945/ajcn.111.024000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kudryavtseva AV, Lipatova AV, Zaretsky AR, Moskalev AA, Fedorova MS, Rasskazova AS, Shibukhova GA, Snezhkina AV, Kaprin AD, Alekseev BY, et al. Important molecular genetic markers of colorectal cancer. Oncotarget. 2016;7:53959–53983. doi: 10.18632/oncotarget.9796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raskov H, Pommergaard HC, Burcharth J, Rosenberg J. Colorectal carcinogenesis--update and perspectives. World J Gastroenterol. 2014;20:18151–18164. doi: 10.3748/wjg.v20.i48.18151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burghel GJ, Lin WY, Whitehouse H, Brock I, Hammond D, Bury J, Stephenson Y, George R, Cox A. Identification of candidate driver genes in common focal chromosomal aberrations of microsatellite stable colorectal cancer. PLoS One. 2013;8:e83859. doi: 10.1371/journal.pone.0083859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iacopetta B, Grieu F, Amanuel B. Microsatellite instability in colorectal cancer. Asia Pac J Clin Oncol. 2010;6:260–269. doi: 10.1111/j.1743-7563.2010.01335.x. [DOI] [PubMed] [Google Scholar]
- 29.Slattery ML, Edwards SL, Samowitz W. Stage of colon cancer at diagnosis: implications for risk factor associations? Int J Epidemiol. 1998;27:382–387. doi: 10.1093/ije/27.3.382. [DOI] [PubMed] [Google Scholar]
- 30.Freeman HJ. Early stage colon cancer. World J Gastroenterol. 2013;19:8468–8473. doi: 10.3748/wjg.v19.i46.8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hatano S, Ishida H, Ishibashi K, Kumamoto K, Haga N, Miura I. Identification of risk factors for recurrence in high-risk stage II colon cancer. Int Surg. 2013;98:114–121. doi: 10.9738/CC131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robinson JR, Newcomb PA, Hardikar S, Cohen SA, Phipps AI. Stage IV colorectal cancer primary site and patterns of distant metastasis. Cancer Epidemiol. 2017;48:92–95. doi: 10.1016/j.canep.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson RL, Fleet JC. Animal models of colorectal cancer. Cancer Metastasis Rev. 2013;32:39–61. doi: 10.1007/s10555-012-9404-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosenberg DW, Giardina C, Tanaka T. Mouse models for the study of colon carcinogenesis. Carcinogenesis. 2009;30:183–196. doi: 10.1093/carcin/bgn267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edelmann L, Edelmann W. Loss of DNA mismatch repair function and cancer predisposition in the mouse: animal models for human hereditary nonpolyposis colorectal cancer. Am J Med Genet C Semin Med Genet. 2004;129C:91–99. doi: 10.1002/ajmg.c.30021. [DOI] [PubMed] [Google Scholar]
- 36.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 37.Heyer J, Yang K, Lipkin M, Edelmann W, Kucherlapati R. Mouse models for colorectal cancer. Oncogene. 1999;18:5325–5333. doi: 10.1038/sj.onc.1203036. [DOI] [PubMed] [Google Scholar]
- 38.Reichling T, Goss KH, Carson DJ, Holdcraft RW, Ley-Ebert C, Witte D, Aronow BJ, Groden J. Transcriptional profiles of intestinal tumors in Apc(Min) mice are unique from those of embryonic intestine and identify novel gene targets dysregulated in human colorectal tumors. Cancer Res. 2005;65:166–176. [PubMed] [Google Scholar]
- 39.Jin D, Liu T, Dong W, Zhang Y, Wang S, Xie R, Wang B, Cao H. Dietary feeding of freeze-dried whole cranberry inhibits intestinal tumor development in Apcmin/+ mice. Oncotarget. 2017;8:97787–97800. doi: 10.18632/oncotarget.22081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang G, Khan I, Li X, Chen L, Leong W, Ho LT, Hsiao WLW. Ginsenosides Rb3 and Rd reduce polyps formation while reinstate the dysbiotic gut microbiota and the intestinal microenvironment in ApcMin/+ mice. Sci Rep. 2017;7:12552. doi: 10.1038/s41598-017-12644-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bissahoyo A, Pearsall RS, Hanlon K, Amann V, Hicks D, Godfrey VL, Threadgill DW. Azoxymethane is a genetic background-dependent colorectal tumor initiator and promoter in mice: effects of dose, route, and diet. Toxicol Sci. 2005;88:340–345. doi: 10.1093/toxsci/kfi313. [DOI] [PubMed] [Google Scholar]
- 42.Perše M, Cerar A. Morphological and molecular alterations in 1,2 dimethylhydrazine and azoxymethane induced colon carcinogenesis in rats. J Biomed Biotechnol. 2011;2011:473964. doi: 10.1155/2011/473964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ashokkumar P, Sudhandiran G. Luteolin inhibits cell proliferation during Azoxymethane-induced experimental colon carcinogenesis via Wnt/β-catenin pathway. Invest New Drugs. 2011;29:273–284. doi: 10.1007/s10637-009-9359-9. [DOI] [PubMed] [Google Scholar]
- 44.Miyaki M, Iijima T, Kimura J, Yasuno M, Mori T, Hayashi Y, Koike M, Shitara N, Iwama T, Kuroki T. Frequent mutation of beta-catenin and APC genes in primary colorectal tumors from patients with hereditary nonpolyposis colorectal cancer. Cancer Res. 1999;59:4506–4509. [PubMed] [Google Scholar]
- 45.Umesalma S, Sudhandiran G. Differential inhibitory effects of the polyphenol ellagic acid on inflammatory mediators NF-kappaB, iNOS, COX-2, TNF-alpha, and IL-6 in 1,2-dimethylhydrazine-induced rat colon carcinogenesis. Basic Clin Pharmacol Toxicol. 2010;107:650–655. doi: 10.1111/j.1742-7843.2010.00565.x. [DOI] [PubMed] [Google Scholar]
- 46.Ordás I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet. 2012;380:1606–1619. doi: 10.1016/S0140-6736(12)60150-0. [DOI] [PubMed] [Google Scholar]
- 47.Kohno H, Suzuki R, Sugie S, Tanaka T. Beta-Catenin mutations in a mouse model of inflammation-related colon carcinogenesis induced by 1,2-dimethylhydrazine and dextran sodium sulfate. Cancer Sci. 2005;96:69–76. doi: 10.1111/j.1349-7006.2005.00020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jobst K. Teratogenous changes and tumors in rats following treatment with methylnitroso-urea (MNU) Neoplasma. 1967;14:435–436. [PubMed] [Google Scholar]
- 49.Koestner AW, Ruecker FA, Koestner A. Morphology and pathogenesis of tumors of the thymus and stomach in Sprague-Dawley rats following intragastric administration of methyl nitrosourea (MNU) Int J Cancer. 1977;20:418–426. doi: 10.1002/ijc.2910200314. [DOI] [PubMed] [Google Scholar]
- 50.Maekawa A, Onodera H, Kanno J, Furuta K, Nagaoka T, Todate A, Matsushima Y, Oh-hara T, Kawazoe Y. Carcinogenicity and organ specificity of N-trimethylsilylmethyl-N-nitrosourea (TMS-MNU), N-neopentyl-N-nitrosourea (neoPNU), and N-methyl-N-nitrosourea (MNU) in rats. J Cancer Res Clin Oncol. 1988;114:473–476. doi: 10.1007/BF00391494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Almora-Pinedo Y, Arroyo-Acevedo J, Herrera-Calderon O, Chumpitaz-Cerrate V, Hañari-Quispe R, Tinco-Jayo A, Franco-Quino C, Figueroa-Salvador L. Preventive effect of Oenothera rosea on N-methyl-N-nitrosourea-(NMU) induced gastric cancer in rats. Clin Exp Gastroenterol. 2017;10:327–332. doi: 10.2147/CEG.S142515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pollard M, Luckert PH. Autochthonous prostate adenocarcinomas in Lobund-Wistar rats: a model system. Prostate. 1987;11:219–227. doi: 10.1002/pros.2990110303. [DOI] [PubMed] [Google Scholar]
- 53.Narisawa T, Wong CQ, Maronpot RR, Weisburger JH. Large bowel carcinogenesis in mice and rats by several intrarectal doses of methylnitrosourea and negative effect of nitrite plus methylurea. Cancer Res. 1976;36:505–510. [PubMed] [Google Scholar]
- 54.Magalhães B, Peleteiro B, Lunet N. Dietary patterns and colorectal cancer: systematic review and meta-analysis. Eur J Cancer Prev. 2012;21:15–23. doi: 10.1097/CEJ.0b013e3283472241. [DOI] [PubMed] [Google Scholar]
- 55.Newmark HL, Yang K, Kurihara N, Fan K, Augenlicht LH, Lipkin M. Western-style diet-induced colonic tumors and their modulation by calcium and vitamin D in C57Bl/6 mice: a preclinical model for human sporadic colon cancer. Carcinogenesis. 2009;30:88–92. doi: 10.1093/carcin/bgn229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Newmark HL, Lipkin M, Maheshwari N. Colonic hyperplasia and hyperproliferation induced by a nutritional stress diet with four components of Western-style diet. J Natl Cancer Inst. 1990;82:491–496. doi: 10.1093/jnci/82.6.491. [DOI] [PubMed] [Google Scholar]
- 57.Risio M, Lipkin M, Newmark H, Yang K, Rossini FP, Steele VE, Boone CW, Kelloff GJ. Apoptosis, cell replication, and Western-style diet-induced tumorigenesis in mouse colon. Cancer Res. 1996;56:4910–4916. [PubMed] [Google Scholar]
- 58.Wang D, Peregrina K, Dhima E, Lin EY, Mariadason JM, Augenlicht LH. Paneth cell marker expression in intestinal villi and colon crypts characterizes dietary induced risk for mouse sporadic intestinal cancer. Proc Natl Acad Sci USA. 2011;108:10272–10277. doi: 10.1073/pnas.1017668108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Muto T, Bussey HJ, Morson BC. The evolution of cancer of the colon and rectum. Cancer. 1975;36:2251–2270. doi: 10.1002/cncr.2820360944. [DOI] [PubMed] [Google Scholar]
- 60.Subramaniam R, Mizoguchi A, Mizoguchi E. Mechanistic roles of epithelial and immune cell signaling during the development of colitis-associated cancer. Cancer Res Front. 2016;2:1–21. doi: 10.17980/2016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Varga J, Greten FR. Cell plasticity in epithelial homeostasis and tumorigenesis. Nat Cell Biol. 2017;19:1133–1141. doi: 10.1038/ncb3611. [DOI] [PubMed] [Google Scholar]
- 63.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 64.Škovierová H, Okajčeková T, Strnádel J, Vidomanová E, Halašová E. Molecular regulation of epithelial-to-mesenchymal transition in tumorigenesis (Review) Int J Mol Med. 2018;41:1187–1200. doi: 10.3892/ijmm.2017.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S, et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell. 2008;14:79–89. doi: 10.1016/j.ccr.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 66.Lazarova DL, Bordonaro M. Vimentin, colon cancer progression and resistance to butyrate and other HDACis. J Cell Mol Med. 2016;20:989–993. doi: 10.1111/jcmm.12850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328–337. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yao D, Dai C, Peng S. Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumor formation. Mol Cancer Res. 2011;9:1608–1620. doi: 10.1158/1541-7786.MCR-10-0568. [DOI] [PubMed] [Google Scholar]
- 69.Christou N, Perraud A, Blondy S, Jauberteau MO, Battu S, Mathonnet M. E-cadherin: A potential biomarker of colorectal cancer prognosis. Oncol Lett. 2017;13:4571–4576. doi: 10.3892/ol.2017.6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Choi BJ, Park SA, Lee SY, Cha YN, Surh YJ. Hypoxia induces epithelial-mesenchymal transition in colorectal cancer cells through ubiquitin-specific protease 47-mediated stabilization of Snail: A potential role of Sox9. Sci Rep. 2017;7:15918. doi: 10.1038/s41598-017-15139-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Loboda A, Nebozhyn MV, Watters JW, Buser CA, Shaw PM, Huang PS, Van't Veer L, Tollenaar RA, Jackson DB, Agrawal D, et al. EMT is the dominant program in human colon cancer. BMC Med Genomics. 2011;4:9. doi: 10.1186/1755-8794-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Clarke PG, Clarke S. Nineteenth century research on naturally occurring cell death and related phenomena. Anat Embryol (Berl) 1996;193:81–99. doi: 10.1007/BF00214700. [DOI] [PubMed] [Google Scholar]
- 73.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fulda S. Regulation of cell death in cancer-possible implications for immunotherapy. Front Oncol. 2013;3:29. doi: 10.3389/fonc.2013.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Letai A. Cell Death and Cancer Therapy: Don't Forget to Kill the Cancer Cell! Clin Cancer Res. 2015;21:5015–5020. doi: 10.1158/1078-0432.CCR-15-1204. [DOI] [PubMed] [Google Scholar]
- 76.Ricci MS, Zong WX. Chemotherapeutic approaches for targeting cell death pathways. Oncologist. 2006;11:342–357. doi: 10.1634/theoncologist.11-4-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nievelstein RA, van der Werff JF, Verbeek FJ, Valk J, Vermeij-Keers C. Normal and abnormal embryonic development of the anorectum in human embryos. Teratology. 1998;57:70–78. doi: 10.1002/(SICI)1096-9926(199802)57:2<70::AID-TERA5>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 78.Zhang T, Zhang HL, Wang da J, Tang XB, Jia HM, Bai YZ, Yuan ZW, Wang WL. Normal development of hindgut and anorectum in human embryo. Int J Colorectal Dis. 2011;26:109–116. doi: 10.1007/s00384-010-1034-2. [DOI] [PubMed] [Google Scholar]
- 79.Nakata M, Takada Y, Hishiki T, Saito T, Terui K, Sato Y, Koseki H, Yoshida H. Induction of Wnt5a-expressing mesenchymal cells adjacent to the cloacal plate is an essential process for its proximodistal elongation and subsequent anorectal development. Pediatr Res. 2009;66:149–154. doi: 10.1203/PDR.0b013e3181aa304a. [DOI] [PubMed] [Google Scholar]
- 80.Zhang T, Tang XB, Wang LL, Bai YZ, Qiu GR, Yuan ZW, Wang WL. Mutations and down-regulation of CDX1 in children with anorectal malformations. Int J Med Sci. 2013;10:191–197. doi: 10.7150/ijms.4929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, Gilpin C, Levine B. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–946. doi: 10.1016/j.cell.2006.12.044. [DOI] [PubMed] [Google Scholar]
- 82.Yuan J, Kroemer G. Alternative cell death mechanisms in development and beyond. Genes Dev. 2010;24:2592–2602. doi: 10.1101/gad.1984410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhou W, Yuan J. Necroptosis in health and diseases. Semin Cell Dev Biol. 2014;35:14–23. doi: 10.1016/j.semcdb.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 84.Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–379. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vande Walle L, Lamkanfi M. Pyroptosis. Curr Biol. 2016;26:R568–R572. doi: 10.1016/j.cub.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 86.Fatokun AA, Dawson VL, Dawson TM. Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br J Pharmacol. 2014;171:2000–2016. doi: 10.1111/bph.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kroemer G, Perfettini JL. Entosis, a key player in cancer cell competition. Cell Res. 2014;24:1280–1281. doi: 10.1038/cr.2014.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mesa MA, Vasquez G. NETosis. Autoimmune Dis. 2013;2013:651497. doi: 10.1155/2013/651497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim KH, Lee MS. Autophagy--a key player in cellular and body metabolism. Nat Rev Endocrinol. 2014;10:322–337. doi: 10.1038/nrendo.2014.35. [DOI] [PubMed] [Google Scholar]
- 90.Galluzzi L, Bravo-San Pedro JM, Kepp O, Kroemer G. Regulated cell death and adaptive stress responses. Cell Mol Life Sci. 2016;73:2405–2410. doi: 10.1007/s00018-016-2209-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008;2:333–344. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 93.Marshman E, Ottewell PD, Potten CS, Watson AJ. Caspase activation during spontaneous and radiation-induced apoptosis in the murine intestine. J Pathol. 2001;195:285–292. doi: 10.1002/path.967. [DOI] [PubMed] [Google Scholar]
- 94.Manne U, Shanmugam C, Katkoori VR, Bumpers HL, Grizzle WE. Development and progression of colorectal neoplasia. Cancer Biomark. 2010;9:235–265. doi: 10.3233/CBM-2011-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li XL, Zhou J, Chen ZR, Chng WJ. P53 mutations in colorectal cancer - molecular pathogenesis and pharmacological reactivation. World J Gastroenterol. 2015;21:84–93. doi: 10.3748/wjg.v21.i1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kumar M, Kaur V, Kumar S, Kaur S. Phytoconstituents as apoptosis inducing agents: strategy to combat cancer. Cytotechnology. 2016;68:531–563. doi: 10.1007/s10616-015-9897-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pandurangan AK, Dharmalingam P, Sadagopan SK, Ramar M, Munusamy A, Ganapasam S. Luteolin induces growth arrest in colon cancer cells through involvement of Wnt/β-catenin/GSK-3β signaling. J Environ Pathol Toxicol Oncol. 2013;32:131–139. doi: 10.1615/jenvironpatholtoxicoloncol.2013007522. [DOI] [PubMed] [Google Scholar]
- 98.Gerola S, Nittka S, Kähler G, Tao S, Brenner H, Binelli G, Eils R, Brors B, Neumaier M. Genetic variants in apoptosis-related genes associated with colorectal hyperplasia. Genes Chromosomes Cancer. 2014;53:769–778. doi: 10.1002/gcc.22185. [DOI] [PubMed] [Google Scholar]
- 99.Sinicrope FA, Hart J, Hsu HA, Lemoine M, Michelassi F, Stephens LC. Apoptotic and mitotic indices predict survival rates in lymph node-negative colon carcinomas. Clin Cancer Res. 1999;5:1793–1804. [PubMed] [Google Scholar]
- 100.Danielsen SA, Eide PW, Nesbakken A, Guren T, Leithe E, Lothe RA. Portrait of the PI3K/AKT pathway in colorectal cancer. Biochim Biophys Acta. 2015;1855:104–121. doi: 10.1016/j.bbcan.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 101.Kenific CM, Thorburn A, Debnath J. Autophagy and metastasis: another double-edged sword. Curr Opin Cell Biol. 2010;22:241–245. doi: 10.1016/j.ceb.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen Z, Li Y, Zhang C, Yi H, Wu C, Wang J, Liu Y, Tan J, Wen J. Downregulation of Beclin 1 and impairment of autophagy in a small population of colorectal cancer. Dig Dis Sci. 2013;58:2887–2894. doi: 10.1007/s10620-013-2732-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Moriwaki K, Bertin J, Gough PJ, Orlowski GM, Chan FK. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis. 2015;6:e1636. doi: 10.1038/cddis.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xie X, Zhao Y, Ma CY, Xu XM, Zhang YQ, Wang CG, Jin J, Shen X, Gao JL, Li N, et al. Dimethyl fumarate induces necroptosis in colon cancer cells through GSH depletion/ROS increase/MAPKs activation pathway. Br J Pharmacol. 2015;172:3929–3943. doi: 10.1111/bph.13184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Oliver Metzig M, Fuchs D, Tagscherer KE, Gröne HJ, Schirmacher P, Roth W. Inhibition of caspases primes colon cancer cells for 5-fluorouracil-induced TNF-α-dependent necroptosis driven by RIP1 kinase and NF-κB. Oncogene. 2016;35:3399–3409. doi: 10.1038/onc.2015.398. [DOI] [PubMed] [Google Scholar]
- 106.Katz LH, Li Y, Chen JS, Muñoz NM, Majumdar A, Chen J, Mishra L. Targeting TGF-β signaling in cancer. Expert Opin Ther Targets. 2013;17:743–760. doi: 10.1517/14728222.2013.782287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Goel S, Huang J, Klampfer L. K-Ras, intestinal homeostasis and colon cancer. Curr Clin Pharmacol. 2015;10:73–81. doi: 10.2174/1574884708666131111204440. [DOI] [PubMed] [Google Scholar]
- 108.Giles RH, van Es JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta. 2003;1653:1–24. doi: 10.1016/s0304-419x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 109.Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene. 2006;25:7531–7537. doi: 10.1038/sj.onc.1210059. [DOI] [PubMed] [Google Scholar]
- 110.Rosin-Arbesfeld R, Townsley F, Bienz M. The APC tumour suppressor has a nuclear export function. Nature. 2000;406:1009–1012. doi: 10.1038/35023016. [DOI] [PubMed] [Google Scholar]
- 111.Fevr T, Robine S, Louvard D, Huelsken J. Wnt/beta-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol Cell Biol. 2007;27:7551–7559. doi: 10.1128/MCB.01034-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 113.Fortin J, Mak TW. Targeting PI3K Signaling in Cancer: A Cautionary Tale of Two AKTs. Cancer Cell. 2016;29:429–431. doi: 10.1016/j.ccell.2016.03.020. [DOI] [PubMed] [Google Scholar]
- 114.Colakoglu T, Yildirim S, Kayaselcuk F, Nursal TZ, Ezer A, Noyan T, Karakayali H, Haberal M. Clinicopathological significance of PTEN loss and the phosphoinositide 3-kinase/Akt pathway in sporadic colorectal neoplasms: is PTEN loss predictor of local recurrence? Am J Surg. 2008;195:719–725. doi: 10.1016/j.amjsurg.2007.05.061. [DOI] [PubMed] [Google Scholar]
- 115.Lin MC, Wang FY, Kuo YH, Tang FY. Cancer chemopreventive effects of lycopene: suppression of MMP-7 expression and cell invasion in human colon cancer cells. J Agric Food Chem. 2011;59:11304–11318. doi: 10.1021/jf202433f. [DOI] [PubMed] [Google Scholar]
- 116.Din FV, Valanciute A, Houde VP, Zibrova D, Green KA, Sakamoto K, Alessi DR, Dunlop MG. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology. 2012;142:1504–1515.e3. doi: 10.1053/j.gastro.2012.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Massagué J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 118.Kaklamani VG, Pasche B. Role of TGF-beta in cancer and the potential for therapy and prevention. Expert Rev Anticancer Ther. 2004;4:649–661. doi: 10.1586/14737140.4.4.649. [DOI] [PubMed] [Google Scholar]
- 119.Xu Y, Pasche B. TGF-beta signaling alterations and susceptibility to colorectal cancer. Hum Mol Genet. 2007;16 Spec No 1:R14–R20. doi: 10.1093/hmg/ddl486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 121.Javelaud D, Mauviel A. Mammalian transforming growth factor-betas: Smad signaling and physio-pathological roles. Int J Biochem Cell Biol. 2004;36:1161–1165. doi: 10.1016/S1357-2725(03)00255-3. [DOI] [PubMed] [Google Scholar]
- 122.Clark DA, Coker R. Transforming growth factor-beta (TGF-beta) Int J Biochem Cell Biol. 1998;30:293–298. doi: 10.1016/s1357-2725(97)00128-3. [DOI] [PubMed] [Google Scholar]
- 123.Bellam N, Pasche B. Tgf-beta signaling alterations and colon cancer. Cancer Treat Res. 2010;155:85–103. doi: 10.1007/978-1-4419-6033-7_5. [DOI] [PubMed] [Google Scholar]
- 124.Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- 125.Geng L, Chaudhuri A, Talmon G, Wisecarver JL, Wang J. TGF-Beta suppresses VEGFA-mediated angiogenesis in colon cancer metastasis. PLoS One. 2013;8:e59918. doi: 10.1371/journal.pone.0059918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gatza CE, Holtzhausen A, Kirkbride KC, Morton A, Gatza ML, Datto MB, Blobe GC. Type III TGF-β receptor enhances colon cancer cell migration and anchorage-independent growth. Neoplasia. 2011;13:758–770. doi: 10.1593/neo.11528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Oshima H, Nakayama M, Han TS, Naoi K, Ju X, Maeda Y, Robine S, Tsuchiya K, Sato T, Sato H, et al. Suppressing TGFβ signaling in regenerating epithelia in an inflammatory microenvironment is sufficient to cause invasive intestinal cancer. Cancer Res. 2015;75:766–776. doi: 10.1158/0008-5472.CAN-14-2036. [DOI] [PubMed] [Google Scholar]
- 128.Ruzzo A, Graziano F, Canestrari E, Magnani M. Molecular predictors of efficacy to anti-EGFR agents in colorectal cancer patients. Curr Cancer Drug Targets. 2010;10:68–79. doi: 10.2174/156800910790980205. [DOI] [PubMed] [Google Scholar]
- 129.Mendelsohn J. The epidermal growth factor receptor as a target for cancer therapy. Endocr Relat Cancer. 2001;8:3–9. doi: 10.1677/erc.0.0080003. [DOI] [PubMed] [Google Scholar]
- 130.Rotter V, Prokocimer M. p53 and human malignancies. Adv Cancer Res. 1991;57:257–272. doi: 10.1016/s0065-230x(08)61001-5. [DOI] [PubMed] [Google Scholar]
- 131.Hsieh JS, Lin SR, Chang MY, Chen FM, Lu CY, Huang TJ, Huang YS, Huang CJ, Wang JY. APC, K-ras, and p53 gene mutations in colorectal cancer patients: correlation to clinicopathologic features and postoperative surveillance. Am Surg. 2005;71:336–343. [PubMed] [Google Scholar]
- 132.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 133.Bray SJ. Notch signalling in context. Nat Rev Mol Cell Biol. 2016;17:722–735. doi: 10.1038/nrm.2016.94. [DOI] [PubMed] [Google Scholar]
- 134.Schmidt EM, Lamprecht S, Blaj C, Schaaf C, Krebs S, Blum H, Hermeking H, Jung A, Kirchner T, Horst D. Targeting tumor cell plasticity by combined inhibition of NOTCH and MAPK signaling in colon cancer. J Exp Med. 2018;215:1693–1708. doi: 10.1084/jem.20171455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mirone G, Perna S, Shukla A, Marfe G. Involvement of Notch-1 in Resistance to Regorafenib in Colon Cancer Cells. J Cell Physiol. 2016;231:1097–1105. doi: 10.1002/jcp.25206. [DOI] [PubMed] [Google Scholar]
- 136.Kranenburg O. Prometastatic NOTCH Signaling in Colon Cancer. Cancer Discov. 2015;5:115–117. doi: 10.1158/2159-8290.CD-14-1456. [DOI] [PubMed] [Google Scholar]
- 137.Rajendran P, Dashwood WM, Li L, Kang Y, Kim E, Johnson G, Fischer KA, Löhr CV, Williams DE, Ho E, et al. Nrf2 status affects tumor growth, HDAC3 gene promoter associations, and the response to sulforaphane in the colon. Clin Epigenetics. 2015;7:102. doi: 10.1186/s13148-015-0132-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Copple IM. The Keap1-Nrf2 cell defense pathway--a promising therapeutic target? Adv Pharmacol. 2012;63:43–79. doi: 10.1016/B978-0-12-398339-8.00002-1. [DOI] [PubMed] [Google Scholar]
- 139.Manigandan K, Manimaran D, Jayaraj RL, Elangovan N, Dhivya V, Kaphle A. Taxifolin curbs NF-κB-mediated Wnt/β-catenin signaling via up-regulating Nrf2 pathway in experimental colon carcinogenesis. Biochimie. 2015;119:103–112. doi: 10.1016/j.biochi.2015.10.014. [DOI] [PubMed] [Google Scholar]
- 140.Niture SK, Khatri R, Jaiswal AK. Regulation of Nrf2-an update. Free Radic Biol Med. 2014;66:36–44. doi: 10.1016/j.freeradbiomed.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27:2179–2191. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yang Y, Cai X, Yang J, Sun X, Hu C, Yan Z, Xu X, Lu W, Wang X, Cao P. Chemoprevention of dietary digitoflavone on colitis-associated colon tumorigenesis through inducing Nrf2 signaling pathway and inhibition of inflammation. Mol Cancer. 2014;13:48. doi: 10.1186/1476-4598-13-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yao J, Zhao L, Zhao Q, Zhao Y, Sun Y, Zhang Y, Miao H, You QD, Hu R, Guo QL. NF-κB and Nrf2 signaling pathways contribute to wogonin-mediated inhibition of inflammation-associated colorectal carcinogenesis. Cell Death Dis. 2014;5:e1283. doi: 10.1038/cddis.2014.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wondrak GT, Villeneuve NF, Lamore SD, Bause AS, Jiang T, Zhang DD. The cinnamon-derived dietary factor cinnamic aldehyde activates the Nrf2-dependent antioxidant response in human epithelial colon cells. Molecules. 2010;15:3338–3355. doi: 10.3390/molecules15053338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Irvine KD, Harvey KF. Control of organ growth by patterning and hippo signaling in Drosophila. Cold Spring Harb Perspect Biol. 2015;7:pii: a019224. doi: 10.1101/cshperspect.a019224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kosik KS, Kowall NW, McKee A. Along the way to a neurofibrillary tangle: a look at the structure of tau. Ann Med. 1989;21:109–112. doi: 10.3109/07853898909149195. [DOI] [PubMed] [Google Scholar]
- 147.Santucci M, Vignudelli T, Ferrari S, Mor M, Scalvini L, Bolognesi ML, Uliassi E, Costi MP. The Hippo Pathway and YAP/TAZ-TEAD Protein-Protein Interaction as Targets for Regenerative Medicine and Cancer Treatment. J Med Chem. 2015;58:4857–4873. doi: 10.1021/jm501615v. [DOI] [PubMed] [Google Scholar]
- 148.Nagashima S, Bao Y, Hata Y. The Hippo Pathway as Drug Targets in Cancer Therapy and Regenerative Medicine. Curr Drug Targets. 2017;18:447–454. doi: 10.2174/1389450117666160112115641. [DOI] [PubMed] [Google Scholar]
- 149.Guo L, Teng L. YAP/TAZ for cancer therapy: opportunities and challenges (review) Int J Oncol. 2015;46:1444–1452. doi: 10.3892/ijo.2015.2877. [DOI] [PubMed] [Google Scholar]
- 150.Hansen CG, Moroishi T, Guan KL. YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol. 2015;25:499–513. doi: 10.1016/j.tcb.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Liang K, Zhou G, Zhang Q, Li J, Zhang C. Expression of hippo pathway in colorectal cancer. Saudi J Gastroenterol. 2014;20:188–194. doi: 10.4103/1319-3767.133025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cho SY, Gwak JW, Shin YC, Moon D, Ahn J, Sol HW, Kim S, Kim G, Shin HM, Lee KH, et al. Expression of Hippo pathway genes and their clinical significance in colon adenocarcinoma. Oncol Lett. 2018;15:4926–4936. doi: 10.3892/ol.2018.7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Konsavage WM Jr, Kyler SL, Rennoll SA, Jin G, Yochum GS. Wnt/β-catenin signaling regulates Yes-associated protein (YAP) gene expression in colorectal carcinoma cells. J Biol Chem. 2012;287:11730–11739. doi: 10.1074/jbc.M111.327767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kim M, Jho EH. Cross-talk between Wnt/β-catenin and Hippo signaling pathways: a brief review. BMB Rep. 2014;47:540–545. doi: 10.5483/BMBRep.2014.47.10.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tanaka T. Colorectal carcinogenesis: Review of human and experimental animal studies. J Carcinog. 2009;8:5. doi: 10.4103/1477-3163.49014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203–222. doi: 10.1038/nrd.2016.246. [DOI] [PubMed] [Google Scholar]
- 157.Jansson MD, Lund AH. MicroRNA and cancer. Mol Oncol. 2012;6:590–610. doi: 10.1016/j.molonc.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Masuda T, Hayashi N, Kuroda Y, Ito S, Eguchi H, Mimori K. MicroRNAs as Biomarkers in Colorectal Cancer. Cancers (Basel) 2017;9:pii: E124. doi: 10.3390/cancers9090124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Degagné E, Pandurangan A, Bandhuvula P, Kumar A, Eltanawy A, Zhang M, Yoshinaga Y, Nefedov M, de Jong PJ, Fong LG, et al. Sphingosine-1-phosphate lyase downregulation promotes colon carcinogenesis through STAT3-activated microRNAs. J Clin Invest. 2014;124:5368–5384. doi: 10.1172/JCI74188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cao L, Liu Y, Wang D, Huang L, Li F, Liu J, Zhang C, Shen Z, Gao Q, Yuan W, et al. MiR-760 suppresses human colorectal cancer growth by targeting BATF3/AP-1/cyclinD1 signaling. J Exp Clin Cancer Res. 2018;37:83. doi: 10.1186/s13046-018-0757-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhang W, Sun Z, Su L, Wang F, Jiang Y, Yu D, Zhang F, Sun Z, Liang W. miRNA-185 serves as a prognostic factor and suppresses migration and invasion through Wnt1 in colon cancer. Eur J Pharmacol. 2018;825:75–84. doi: 10.1016/j.ejphar.2018.02.019. [DOI] [PubMed] [Google Scholar]