

Abstract
We localized and characterized a new regulatory element with promoter activity in the human c-ets-2 intron 1. This promoter governs the expression of 5′ divergent c-ets-2 transcripts through multiple start sites dispersed within 300 bp. Among the multiple start sites detected, three are major transcriptional initiation points. We detected transcripts initiated from this new promoter in various cell lines such as COLO 320, NBE, or HepG2 cells. This promoter exhibits transcriptional activity when linked to the CAT gene, and deletion constructs reveal that it contains activating and repressing elements. The sequence of the promoter reveals putative binding sites for ETS, MYB, GATA, and Oct factors. In addition, we show that this promoter is functionally conserved in the chicken.
Keywords: Oncogene, c-ets-2, Gene expression, Promoters
THE ets gene family includes a number of related transcription factors that are conserved throughout evolution from early metazoans to humans (10,13,16,18). This family is defined by the presence of a DNA binding domain, the ETS domain, most often located at the carboxy-terminus of the protein. The ets family members act as transcription factors by interacting in a sequence-specific manner with purine-rich motifs containing the core sequence GGAA/T (32,33). ETS family members binding sites are found in a wide variety of cellular or viral promoters and enhancers, including ets gene members themselves (9,10). Interestingly, whereas ETS proteins recognize DNA as monomers, a dual function for ETS family members has come to light from the observation that some of them can form ternary complexes, in association with unrelated transcription factors such as SRF (12,15), AP-1 (7,33), or PEBP2/CBF (14,31).
The human c-ets-2 gene is one of the best characterized members of the ets family and is localized on human chromosome 21 in position q22. It has been proposed that the c-ets-2 gene may be implicated in some aspects of Down syndrome (11,30). c-ets-2 is expressed in a large variety of tissues but its level of expression varies greatly from one tissue to another (1,21). The expression of c-ets-2 is regulated by growth factors. For example, in chicken a rapid and transient expression of c-ets-2 can be observed in macrophages following stimulation with chicken myelomonocytic growth factor (cMGF) or LPS (5,6). In addition c-ets-2 has been shown to be expressed during T-lymphocyte differentiation (1,2) and activation (3). During oocyte maturation in Xenopus, c-ets-2 was clearly shown to be required for meiotic progression as demonstrated by antisense experiments (8). Overexpression of the c-ets-2 gene in rodent fibroblasts such as NIH 3T3 cells leads to cell transformation and tumorigenesis in nude mice (26). A promoter has been described for the human c-ets-2 gene. It displays no TATA or CAAT box but contains Sp1 sites as well as a dinucleotide repeat tract (22,23).
A better understanding of the positive and negative controls that underlie a cell-specific pattern of gene expression requires careful examination of the mechanism regulating the expression of the regulators themselves. The arrangement of multiple control elements in the regulatory region of a gene enables and facilitates the coordinate functioning of multiple transcription factors in potentiating transcription. The ability of distinct factors to recognize common control elements in the regulatory region of a gene allows precise transcriptional regulation via the interplay of the transcription factors present within a given cell at any given time. Furthermore, the presence of several independent regulatory units such as distinct promoters or enhancers in a given gene allows a refined tuning of its regulation. For example, out of the two promoters present in the chicken c-ets-1 gene one is regulated by its own product whereas the other is probably not (9). Thus, in several situations or in particular tissues, the c-ets-1 gene may be autoregulated whereas in others it may not.
In this article we reveal the existence of a second promoter governing the expression of the human c-ets-2 gene. This promoter is located in the first intron of c-ets-2 and controls the expression of transcripts containing different 5′ ends. We found several transcripts expressed from this promoter in human cell lines. We have mapped multiple start sites among which three appear to be of major importance. This promoter contains no clear TATA box sequences or GC-rich region but includes several putative EBS/PEA3 sites (ETS binding sites), MYB and GATA sites, and a perfect octamer sequence. Furthermore, this regulatory element is functionally conserved in the chicken.
MATERIALS AND METHODS
Molecular Probes, cDNA Isolation, and Sequencing Analysis
For the isolation of the 5′ end of the human c-ets-2 gene, a K562 cells genomic DNA library (partial EcoRI digestion inserted in the CHARON 4A vector) was screened with a 365-bp fragment from human c-ets-2 corresponding to positions 135–500 (35). From one recombinant we subcloned into M13 mpl8 a 2.1-kb EcoRI fragment [localized in the c-ets-2 intron 1 (23,36)], directly or after digestion by AluI or HaeIII. Cloning, phage and plasmid growth, and isolation were performed using standard procedures (20). The nucleotide sequence was determined on both directions with an Applied Biosystem 370 A automatic sequencer using protocols and reagents recommended by the supplier. To isolate the AL1 cDNA, a human adult liver cDNA library constructed in XNM1149 kindly provided by C. de Taisne was screened with both the 2.1-kb EcoRI intronic fragment mentioned above, and a 382-bp XmnI-PstI fragment from the human c-ets-2 cDNA [positions 298-680 (35)]. The AG3 cDNA was isolated as described in the Results section from a fetal liver cDNA library.
Northern Blots
RNAs were prepared by the guanidine isothiocyanate method. Polyadenylated RNAs were selected by oligo-dT cellulose chromatography (20). Poly(A)+ RNA (15 μg) was run on denaturing formaldehyde gel, stained with ethidium bromide, and transferred overnight to nitrocellulose membrane (Schleicher and Schuell). Hybridizations were carried out at 42°C during 48 h according to standard procedures (20). Probes used were: (i) a 381-bp Exo-SstI fragment (positions 1012–1393) generated by 5′ end ExoIII deletion of the intronic 2.1-kb EcoRI fragment, followed by SstI digestion. This deleted fragment was inserted in the 5′-3′ orientation into M13 mp19; (ii) a 382-bp XmnI-PstI fragment from the human c-ets-2 cDNA (positions 298–680) inserted in the 5′-3′ orientation into M13 mp18. Uniformly 32P-labeled single-strand DNA probes corresponding to the complementary sense transcripts were synthetized with Klenow DNA polymerase as described in Maniatis et al. (20).
RNAase Protection
To perform RNAase protection experiments two probes were used: (i) a 859-bp BamHI-SstI fragment (positions 534–1393) of the intronic region of c-ets-2 (sequence in Fig. 6) cloned into pSP65 plasmid, and (ii) the already described 381 Exo-SstI fragment cloned into pSP65. Antisense RNA probes were transcribed in vitro using the Riboprobe kit of Promega from linearized plasmids using 25 units of SP6 polymerase (Stratagene) in the presence of 80 μCi [32P]UTP or 80 μCi [32P]CTP. Protection experiments were performed according to Dozier et al. (13a). Briefly, 105 cpm of labeled antisense RNA probe and 20 μg of total RNA were hybridized overnight at 45°C. RNaseA (Boehringer) (40 μg/ml) and 2000 units/ml RNseT1 (BRL) were added and the reaction mixture was incubated 45 min at 32°C. After phenolchloroform extraction and ethanol precipitation the samples were analyzed in a denaturing 6% acrylamide gel containing 8 M urea.
FIG. 6.

Sequence of the c-ets-2 P2 human promoter. Restriction sites mentioned in the text are indicated. Putative sites for transcription factors are boxed and the name of the factor is indicated. The direct or inverted repeats observed in the sequence are underlined. The sequence of the GT repeat is italicized. The 5′ boundaries of the various deletion constructs used to measure the activity of the promoter are indicated with arrows. The three start sites S1, S2, and S3 are indicated by black dots below the sequence. Other additional minor start sites observed either in RNAase protection or in primer extension experiments are indicated by stars below the sequence.
Primer Extension
Oligonucleotides (500 ng) were labeled in the 5′ end using phage T4 polynucleotide kinase (Amersham). Oligonucleotides were incubated in a total volume of 20 pi for 45 min at 37°C in 50 mM Tris-HCl (pH 7.6), 10 mM MgCl2, 5 mM DTT, 100 mM spermine with 20 μCi of [γ-32P]ATP, and the reaction was stopped by addition of 200 μl of 20 mM EDTA (pH 8.0). The mixture was then ethanol precipited using 10 μg of tRNA as a carrier. For the annealing of the labeled primer, 106 cpm of oligonucleotide and 10 μg of total RNA were mixed in a standard reverse transcriptase buffer from BRL (50 mM Tris-HCl, pH 7.5; 75 mM KC1; 3 mM MgCl2; 10 mM DTT) and incubated for 3 min at 80°C. The temperature was then slowly decreased to the T m of the oligonucleotides (70°C for primer 1, 52°C for primer 2), incubated for 30 min at T m, and then finally cooled on ice. An aliquot of the annealing mixture corresponding to 2.5 μg of RNA was then extended for 30 min at 45°C in 10 mM DTT, 100 μM dATP, 100 μM dGTP, 100 μM TTP, 100 μM dCTP, and 100 units of Moloney Murine Leukemia Virus (Mu-MLV) reverse transcriptase (BRL) in the described buffer. The reaction was stopped by addition of water. Products were extracted by phenolchloroform, ethanol precipitated, and analyzed on a 6% acrylamide gel containing 8.3 M urea.
The oligonucleotides used are depicted in Fig. 1A and were as follows: PE 1 (5′-TCA CCT CTA GGG GGA GGC CTT GCC CTC CGA GGG TCA TGG C-3′), which corresponds to positions 1234–1274 is a 40-mer oligonucleotide localized 119 bp upstream of the SstI site in the intron 1 of c-ets-2; PE 2 (5′ -TTT ACA ACT GAG AAC TTT TTA AGT-3′), which is a 24-mer located at positions 1142–1166, 227 bp upstream the same SstI site.
FIG. 1.
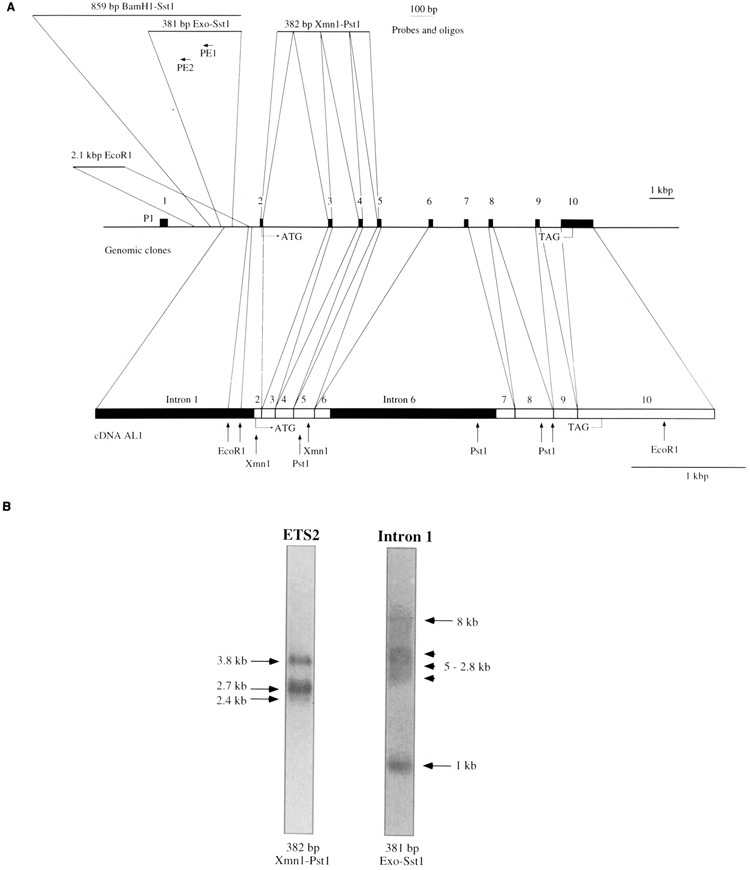
(A) Map of the human c-ets-2 gene. In the middle of the panel the genomic organization of c-ets-2 is depicted from exon 1 to exon 10 containing the stop codon. Exons are represented by black boxes. In the lower part of the panel the AL1 cDNA is represented at a different scale and limits between exons are indicated by vertical bars. Representative restriction sites are indicated as well as initiation and stop codons. The two intronic sequences contained in AL1 are indicated (intron 1 and intron 6) In the upper part of the panel the various probes used in this study are indicated together with their location in the c-ets-2 gene. The two primers used for primer extension experiments are also indicated. (B) Northern blot performed with 15 Mg of poly(A)+ RNAs extracted from COLO 320 cells and hybridized with a common c-ets-2 probe (ETS2: 382 bp XmnI-PstI) or an intron 1-specific probe (Intron 1:381 bp Exo-SstI). The exposure times of the two lanes are different.
In Vitro Promoter Assay
The 1062-bp XbaI-SstI fragment (positions 331–1393 in Fig. 5) was first cloned in plasmid pSP64. A series of nested 5′ deletions was produced from the internal BamHI site at position 534 using an exonuclease111/mung bean nuclease system (Stratagene). The full-length and the deleted fragments were subsequently cloned, after T4 DNA polymerase treatment to yield blunt-ended DNA fragments of the 3′ SstI site, and digestion of the 5′ part by HindIII into the pBL-CAT3 vector digested by HindIII, and XbaI (after filling of that site). The endpoints of the deletions were determined by dideoxy sequencing using an oligonucleotide complementary to the vector sequence ACG ACG TTG TAA AAC GAC GG. The longer XbaI-XbaI fragment (positions 331–1928 in Fig. 6) was also inserted in both orientations into pBLCAT3. The 549-bp EcoRI-SstI fragment of the human c-ets-2 promoter [positions –495, +54 (22)] was also inserted in the pBL-CAT3. Five micrograms of plasmids DNA were cotransfected with 0.5 mg of β-galactosidase expression plasmid pRSVbgal into NBE cells by the calcium phosphate method. The CAT activity was measured using 14C acetyl-coenzyme A (New England Nuclear) with the company-supplied protocol. The transfection efficiency was normalized using the amount of cellular proteins measured by Coomassie assay (Biorad) and by β-galactosidase assay. The experiments were done at least four times (two different DNA preparations, two times).
FIG. 5.
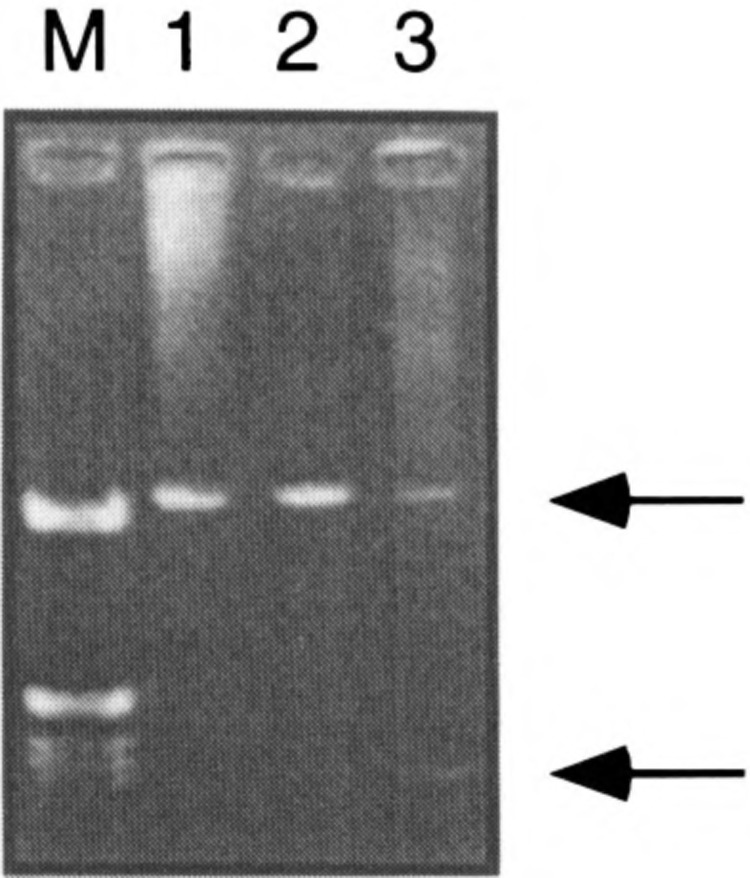
RT-PCR detection of transcripts initiated at the P2 promoter. RNAs from COLO 320 (lane 1), HepG2 (lane 2), and NBE (lane 3) cells were retrotranscribed using the oligo dT primer and amplified using RT1 and RT2 primers. (M) Molecular size marker. Arrows point to the two fragments discussed in the text.
RT-PCR
Poly(A)+ RNA (5 μg) was reverse-transcribed using the first strand synthesis kit of GIBCO-BRL. PCR was performed to generate amplified fragments of c-ets-2 transcripts starting at P2. The primers were as follows: RT1: 5′-ACC AGC CTT CAG TTC CAT-3′ (positions 1215–1232 in Fig. 6), and RT2: AGG CAA TTC ACA GTT GGC GGA (exon 4 of c-ets-2). The PCR reaction mixture contained cDNA (2 μl), 250 μM each dNTP, 10 × PCR buffer with Mg2+, 20 pmol RT2 primers, and 2.5 units of Taq polymerase from Eurogentec (Sart Tillman, Belgium). Reactions were carried out in a PCR apparatus from Perkin Elmer Cetus. One PCR cycle consisted of 1 min 30 s denaturation (94°C), 1 min annealing (60°C), and 3 min extension (72°C). Each PCR reaction consisted of 30 cycles.
RESULTS
Isolation of an Alternative c-ets-2 cDNA
The human c-ets-2 gene directs the synthesis of three different mRNAs of 3.8, 2.7, and 2.4 kb (34), which encode a single 56-kDa nuclear protein. These mRNAs were shown to be generated through different polyadenylation sites (36). In order to search for other possible mRNA species generated by this locus, we attempted to isolate new cDNAs. A cDNA library prepared from human adult liver cloned in λXNM1149 was screened with a 382-bp XmnI-PstI fragment from the 5′ part of a bona fide c-ets-2 cDNA isolated from a fetal liver cDNA library [A. Gegonne, unpublished data; (35); Fig. 1A). This screen yielded an unusual c-ets-2 cDNA that contained 5′ sequences homologous to intron 1 of the gene. We thus further screened the library with two probes: first a 2.1-kb EcoRI fragment from intron 1 of the human c-ets-2 gene, corresponding to the third EcoRI fragment of the phage λD8 (22), isolated by us from a K562 genomic DNA library (see Materials and Methods), and second, the 382-bp XmnI-PstI fragment. Three double-positive clones with inserts ranging from 2.7 to 5 kbp were obtained. The two shortest clones of 2.7 and 3 kbp exhibited a common 5′ end that begins 1.7 kbp upstream of the 3′ EcoRI site of the intronic probe. Mapping and partial sequence data of the 3′ part of these clones revealed that they corresponded to partially unspliced precursor transcripts. The third clone (named AL1, 5 kbp long, Fig. 1A), which ends at the level of the second polyadenylation signal determined by Watson et al. (36), seems to be an interesting candidate and contained, as the two others, intron 1 sequences. We characterized this cDNA clone by fine mapping and partial sequencing analysis. This analysis showed that the clone begins inside the 2.1-kbp EcoRI fragment (position 1088 in Fig. 6) and remains colinear to the genomic DNA up to exon 2. At the end of exon 2 splicing occurs normally (like in the classical c-ets-2 transcripts) except for the 1.3 kbp of intron 6, which is not excised (Fig. 1A). Using a probe corresponding to the c-ets-2 coding region, we also isolated a cDNA starting in a much more 3′ position (nt 1679 in Fig. 6) but that cannot be attributed to the activity of the classical c-ets-2 promoter. The isolation of this cDNA in a different library suggests that the clones previously isolated do not result from a library artefact.
The presence of a portion of the first c-ets-2 intron in these cDNA clones could be explained by several hypotheses: (i) the cloning of unspliced precursor transcripts, (ii) the use of alternative splice sites, or (iii) an initiation of transcription within this intronic region. To confirm the existence of intron 1-containing transcripts in living cells and to determine the size of an mRNA corresponding to the AL1 cDNA we performed Northern blots using poly(A)+ RNA isolated from the colon cancer cell line COLO 320, which expresses high levels of c-ets-2 transcripts (22). This Northern blot was hybridized with a single-strand probe corresponding to the complementary strand of a 381-bp Exo-SstI DNA fragment (Fig. 1A; see Materials and Methods, position 1012–1393 in Fig. 6), which is strictly specific to the intron 1 region contained in AL1 (Fig. 1A). Three types of products were observed: a large mRNA of about 8 kb, several unresolved mRNAs with sizes ranging from 5 to 2.8 kb, and a small product of about 1 kb (Fig. 1B). In contrast, the labeled 382-bp XmnI-PstI fragment corresponding to the coding region of the c-ets-2 cDNA (Fig. 1A) detects the three bona fide c-ets-2 mRNAs (3.8, 2.7, and 2.4 kb). Nevertheless, the 1.0-kb band could be detected at a low level using both the 381-bp Exo-SStI and the 382-bp XmnI-PstI probes in HepG2 cells (data not shown). This suggest that this 1.0-band contains at least part of the sequences present in the regular c-ets-2 transcripts. Interestingly, some of the transcripts recognized by the intron 1-specific probe seem not to be recognized by the ETS2 probe. Nevertheless, it has to be noted that the intensity of the hybridization of the intronic probe was much lower than that observed with the cDNA c-ets-2 probe.
Mapping of Intronic c-ets-2 Initiation Sites
To determine if transcription really initiated within c-ets-2 intron 1, and to eliminate the possibility of an alternative splicing of products initiated in the upstream c-ets-2 promoter, we performed RNAase protection and primer extension. For RNAase protection experiments, we first used as a probe a 859-bp BamHI-SstI DNA fragment (positions 534–1393 in Fig. 6) from the intronic region. This antisense probe was hybridized to 20 μg of total RNA from various cell lines (Fig. 2A). Surprisingly, a large number of bands ranging from 65 to 500 nucleotides were observed in RNAs from COLO 320, NBE, and HepG2 cell lines. No signal was detected in a yeast tRNA negative control (Fig. 2A). In all the human cell lines tested the pattern obtained was identical, suggesting that these bands were indeed specific. Repetition of this experiment with other RNA preparations extracted from HeLa, K562, or MRC5 cells yielded identical results (data not shown). Of note, the intensity was much higher in COLO 320 cells in which the intron 1-specific probe led to a high signal in Northern blot. In order to confirm these results, a shorter RNA probe of 381 bp (Exo-SstI, Fig. 1A and Materials and Methods) was hybridized to 20 μg of total RNA. Again yeast tRNA was used as a negative control and yielded no signal (Fig. 2B). Using this probe we also observed multiple bands (310, 250, 185, 125, and 65 nt) that were common with those observed with the first probe (see Figs. 1A and 2). Here again, the intensity of the protected band was higher in COLO 320 than in other cell lines. These RNAase protection experiments were reproduced using poly(A)+ mRNA and in that case we obtained a greatly enriched signal. This clearly shows that the mRNAs starting at P2 are correctly polyadenylated and spliced. As a negative control we also performed RNAase protection experiments using the same 381-bp probe in the opposite orientation. As expected, we did not observe protected bands in the three cell lines tested (data not shown). From these experiments we concluded that the observation of multiple bands in the RNAase protection assays reflects the heterogeneity of the transcripts generated in intron 1.
FIG. 2.
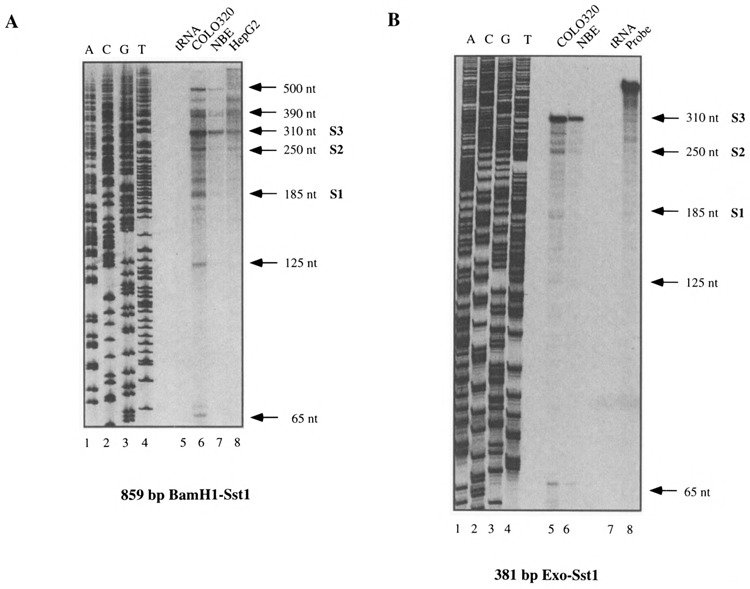
RNAase protection experiments performed with the 859-bp BamHI-SstI probe (A) or the 381-bp Exo-SstI probe (B). Total RNAs (20 μg) from various cell lines indicated above each panel were hybridized with the corresponding probe. In each case a protection experiment was also performed with tRNAs as a negative control. The sizes of the protected fragments are indicated. The three major start sites S1, S2, and S3 are also depicted.
To confirm these observations, we performed primer extension using COLO 320 and NBE RNAs, and two primers PEI and PE2 (Figs. 1A and 3). Again, numerous products extending upstream of the primers were observed (Fig. 3A and B). This pattern is consistent with the RNAase protection assays and suggests a multiplicity of transcriptional start sites. The plausible hypothesis of a series of reverse transcriptase-dependent artefactual arrests was eliminated by the absence of any similar bands in the control of extension performed with an in vitro-synthetized RNA from the 381-bp DNA fragment (lane CTRL in Fig. 3A and B). From the RNAase protection and primer extension experiments we concluded that there may be more than 10 transcription initiation sites within a 300-bp region. Three of these sites (positions 1056, 1117, and 1208 in Fig. 6) appeared to be major start sites and are subsequently referred to as S3, S2, and S1. Only those bands that are common to the two sets of RNAase protection and primer extension experiments will be discussed in the rest of the article.
FIG. 3.
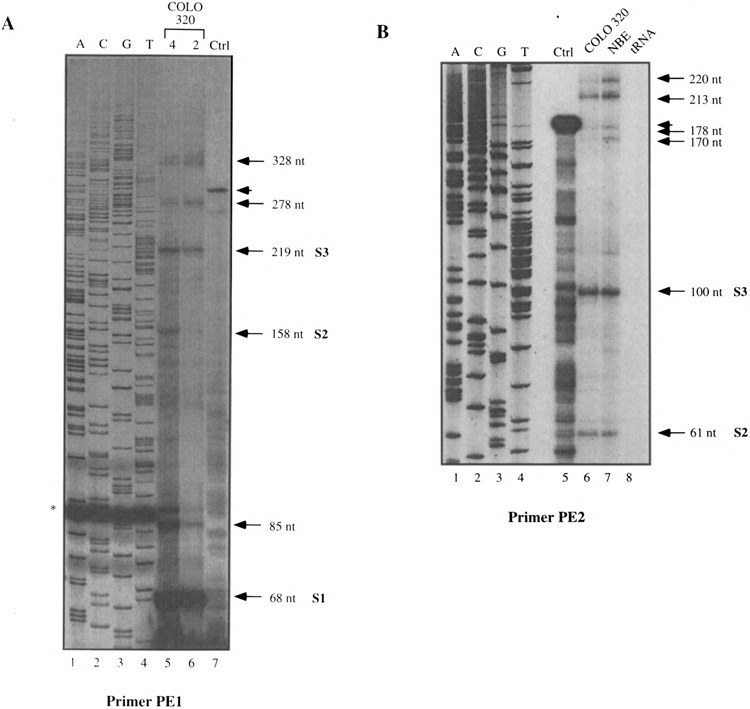
Primer extension experiments performed with the PE1 (A) or PE2 (B) oligonucleotides. For (A) two different amounts of COLO 320 (4 and 2 μg) total RNAs were used whereas for (B) COLO 320 and NBE total RNAs were used. Ctrl: Control experiment with in vitro synthetized cold RNA as explained in the text. The sizes of the extended fragments as the three major start sites S1, S2, and S3 are indicated. In (A) the star indicates an artefactual band in the sequencing reaction and the arrowhead indicates the extended product in the control lane.
Functional Activity of the Intron 1 Initiation Region
In order to demonstrate that we had isolated an efficient promoter region in the first intron of c-ets-2, we inserted a 1.6-kbp XbaI fragment (positions 330–1928; Figs. 4A and 6) upstream of the bacterial CAT gene. This fragment was cloned in both orientations in the pBLCAT3 vector and tested for CAT activity following transfection into NBE cells. The NBE cells were used because they are easily transfectable and because the intronic promoter we described is highly expressed in these cells (Figs. 2 and 3). As a negative control, we included a pBLCAT3 without any insert. The full-length 1.6-kbp XbaI fragment clearly promoted CAT activity when transfected into NBE cells whereas the same fragment cloned in the reverse orientation as well as the pBLCAT3 vector alone did not induce any detectable CAT activity (Fig. 4B). When the same amount of vector was transfected we noticed that the 1.6-kbp XbaI fragment induced a CAT activity threefold lower than the already described c-ets-2 promoter (not shown). The initiation sites used in the transfected construct are identical to the ones used in the endogenous gene mapped by RNase protection and primer extension experiments (data not shown). Thus, we had isolated a new c-ets-2 promoter that we named the P2 promoter.
FIG. 4.
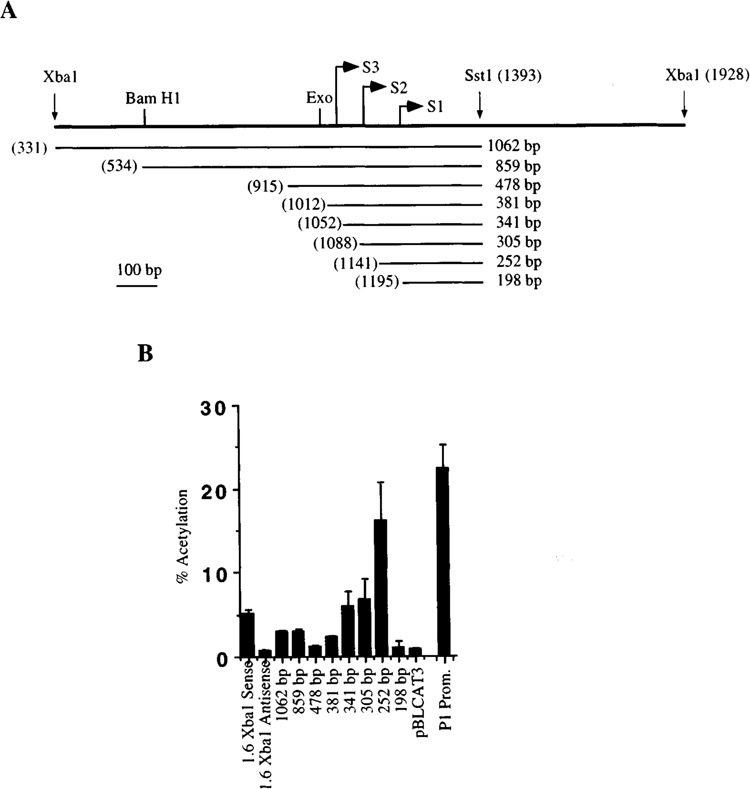
(A) Map of the deletion constructs used to study the activity of the P2 human c-ets-2 promoter. The different constructs are named following the size of their respective inserted fragments. 5′ end of each construct is numbered according to its position in the sequence presented in Fig. 6. The size of the fragment is indicated at the right. The first 5′ base of each deletion is indicated with the numbering system used for Fig. 6. The three major start sites S1, S2, and S3 are also depicted together with representative restriction sites. (B) Activity of the c-ets-2 P2 human promoter and deletion constructs cloned in the pBLCAT3 vector and their transcriptional activity assayed in NBE cells. The averaged result of three experiments is presented with standard deviation. The activity mediated by the 1.6-kbp XbaI fragment cloned in sense or antisense orientation is shown together with the activity of the wild-type vector (pBLCAT3).
To identify regions required for initiation of transcription, a series of deletion fragments of the P2 promoter were ligated 5′ to the bacterial CAT gene and tested for CAT activity. The structure of the various deletion mutants and their relative promoter activities are shown in Fig. 4A and B. The results of this analysis show strong variations of the CAT activity. Deletion of 500 bp 3′ of the 1.6-kbp XbaI fragment (1062-bp fragment) induces an activity slightly lower than the full-length fragment. 5′ deletions constructed from this 1062-bp XbaI-SstI fragment generate two deletion mutants (859 and 478 bp) with equal or lower activity than the 1062-bp fragment. The case of the 478-bp deletion (Fig. 4B) is particularly interesting because in our conditions this mutant is devoid of CAT activity when compared to the pBLCAT3 vector alone. This result strongly suggests that the deletion has unraveled a strong repression sequence located in the promoter that may be bypassed by activating elements present in the 5′ region. This is confirmed by the shorter deletions that effectively induce an increase of CAT activity, suggesting that the repressive element was deleted. This is obvious for the 252-bp deletion, which shows the highest CAT activity, even threefold higher than the full-length 1.6-kbp XbaI fragment. The CAT activity generated by this fragment is nearly identical to the activity induced by the classical c-ets-2 promoter (not shown). Thus, we have mapped a strong repressing activity in the 331 to 1012 region of the intronic c-ets-2 promoter. Of note, the last deletion 198 bp does not induce CAT activity when transfected into NBE cells. This is fully consistent with the initiation site mapping experiments because this deletion removes nearly all of the numerous start sites we have mapped except S1.
We compared the activity of the P2 promoter with that of PI by transient transfection assays performed in NBE cells. We observed that the 550-bp EcoRI-SacI fragment of P1 (22,23) induced a fourfold higher CAT activity than the 1.6-kb XbaI fragment or than the 1062-bp fragment linked to CAT. This suggests that, in these experimental conditions, P2 is less active than P1.
We next decided to test whether the P2 promoter exhibits an enhancer or silencer activity. We cloned the 1.6-kb XbaI fragment in both orientations downstream of the minimal thymidine kinase promoter of vector pBLCAT5S (4) or downstream of the PI promoter [region –495 to +54 (23)] in a promotorless CAT vector (28). We tested the resulting recombinants for CAT activity and we noticed that the basal activities induced by the tk or the PI promoters were not modified by the presence of the 1.6-kbp intronic fragment (not shown). Thus, we conclude that the P2 promoter exhibits no enhancer or silencer activity and does not modulate the activity of the P1 promoter.
Detection of Transcripts Starting at the P2 Promoter
In order to detect in cultured cells transcripts that originate in the P2 promoter we designed an RT-PCR experiment. RNAs from COLO 320, HepG2, and NBE cells were reverse transcribed using oligo dT. The reverse transcribed products were then amplified using RT2 (a primer located in exon 4) and RT1 (a primer corresponding to the P2 promoter; positions 1215 to 1232 in Fig. 5). The PCR products were cloned and their sequence compared to the one of the P2 promoter and of the c-ets-2 cDNA. Interestingly, two PCR products were obtained (see Figs. 5 and 6): (i) a transcript starting in P2 and continuing until exon 2, then regularly spliced until the RT2 oligonucleotide in exon 4, and (ii) a transcript generated also in P2 but spliced at position 1293 (see Fig. 6) where a classical splice site is located. The transcript then reaches directly exon 2 and is then regularly spliced until exon 4. Noteworthy, the coding regions encompassed in these two transcripts are indistinguishable to the classical c-ets-2 transcripts starting at P1. Thus, they are likely to be translated into the classical c-ets-2 product. These transcripts were apparently detected at the higher level in COLO 320 and HepG2 cells than in NBE cells. The small transcript seems to be detected at a much lower level than the larger one. This experiment clearly proves that the P2 promoter is able to control the expression of bona fide transcripts.
Sequence of the c-ets-2 Intronic Promoter
We sequenced the 2.1-kb fragment containing this intronic promoter region (Figs. 1A and 6). The DNA sequence immediately upstream from the major start sites (either S1, S2, or S3; Figs. 1 and 4) does not contain the typical TATA box or CAAT box elements found in many characterized eukaryotic genes. From the sequence data it appears that this promoter does not present any bias toward G and C bases and may thus not be considered as a GC-rich promoter. We did not notice the presence of consensus initiator sequences (Inr) often present in some TATA-less promoters acting in an Sp1 site context (17,27). Because this intronic promoter is not GC rich and does not contain the Sp1 site, this observation is not surprising. Some sequences upstream to the major start sites (indicated by black dots in Fig. 6) may possibly be viewed as highly degenerate TATA signals. It is known that in such cases of degenerated TATA boxes the affinity and selectivity of the RNA polymerase is decreased and that numerous start sites are observed, as in the new promoter we described (38).
We have noted the presence of several direct or inverted repeats in the intronic promoter. These repeats do not correspond to any known binding sites for transcription factors but it is conceivable that unknown molecules are able to regulate c-ets-2 expression through these repeated sites. The sequence of Fig. 6 also contains a GT dinucleotide repeat that is located in the region containing a strong repressive activity. Whether this GT repeat is implicated in the observed repression is not known but the presence of such sequences acting in other promoters or enhancers suggests that it may be the case (see Discussion).
A systematic search for known binding sites of trans-acting factors was achieved using the transcription factor data base. It is interesting to note the presence of an octamer sequence ATG-CAAAT (position 1064), which could be recognized by OCT trans-acting factors and which is located near the start site S1. Other interesting putative binding sites for transcription factors have been observed such as three PEA3 sites, which may be recognized by ETS family members, five MYB consensus sequences, and three GATA sites. As shown in Fig. 6, the promoter also contains possible binding sites for CREB and MYOD1. It is interesting to note that, near the start site S2, there is an intriguing overlap between a GATA site and a MYB consensus sequence. Whether this accounts for a mutually exclusive binding of these factors remains to be established. Interestingly, a large number of putative sites are located in the 5′ region of the promoter in which our deletion experiments have located an activating region (see Figs. 4B and 6).
Functional Conservation of the Human c-ets-2 Intronic Promoter in the Chicken
We next decided to test whether this new promoter element was evolutionary conserved. We thus cloned the intron 1 region from the chicken ets-2 gene. A 2.3-kbp Pst fragment was cloned in both orientations upstream of the CAT gene (see Fig. 7A). The promoter activity of this fragment was tested in the macrophage HD1 cells by transient transfection assays. The result of a typical experiment is shown in Fig. 7B and revealed that this fragment exhibits a clear promoter activity. This strongly suggested that the new intronic promoter that we defined in humans is functionally conserved in chickens.
FIG. 7.
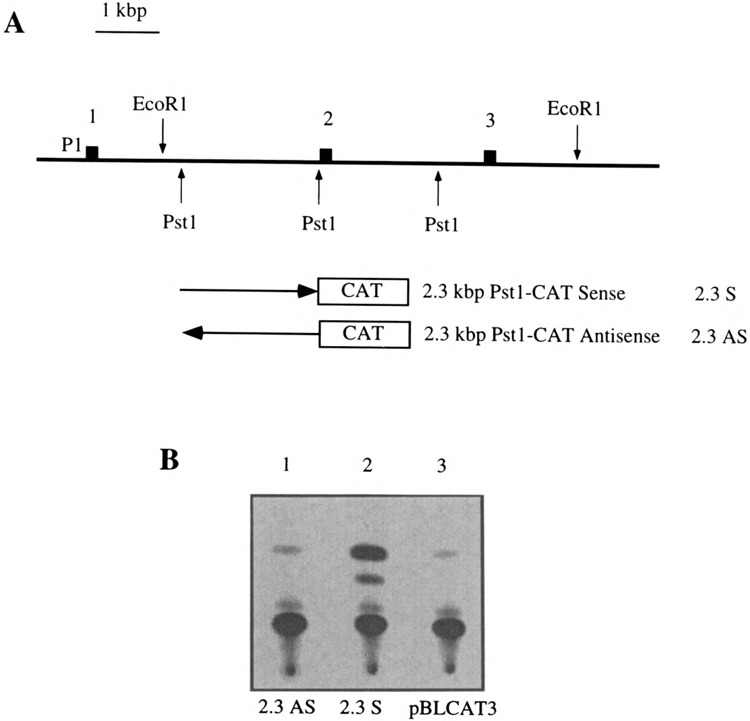
(A) Map of the 5′ part of the chicken c-ets-2 gene. The first three exons are indicated by black boxes as well as representative restriction sites. The fragments used to measure the transcriptional activity mediated by intron 1 are indicated. The P1 promoter is indicated. These fragments were cloned in sense and antisense orientation in the pBLCAT3 vector. (B) Typical result of a CAT assay performed in the HD11 macrophagic cells. The 2.3AS and the 2.3S constructs were used together with the pBLCAT3 vector as a negative control.
We have sequenced the 2.3-kbp Pst fragment (data not shown). Of note, the chicken intronic promoter contains no clear TATA signal and no GC-rich stretches, which may be reminiscent of classical promoters, and it harbors PEA3, MYB, and GATA sites as the human sequence. Outside of these features, this fragment does not harbor convincing homologies with the human intronic promoter. This observation is not surprising because the sequence of promoters is generally not conserved between divergent species.
DISCUSSION
Features of the Human c-ets-2 P2 Promoter
In this article, we report the isolation and characterization of a second functional promoter for the human c-ets-2 gene. The intronic P2 promoter we described displays several interesting features, (i) It does not harbor clear initiation signals such as TATA box or Inr sequences and is not particularly GC rich. Nevertheless, when linked to a pro-motorless CAT reporter vector it promotes a clear transcriptional activity. Furthermore, we detected transcripts specifically initiated from this promoter in several human cell lines (see below). (ii) It contains a number of start sites dispersed in 300 bp among which three could be considered as major initiation sites. Deletion of these sites greatly reduced the CAT activity, showing that they are indeed necessary for the normal activity of the promoter, (iii) It contains a GT dinucleotide repeat of 21 elements interrupted once by an adenosine that is located in a region where we noticed a strong activity of cis-transcriptional repression. Whether this sequence is actively engaged in this repressing activity remains to be determined. Other promoters containing stretches of repeated nucleotides have been described. Most often such types of repeats are homopurine–homopyrimidine repeats, which are present and active in mouse c-Ki-ras (25), human c-ets-2 PI promoter (22), human EGFR, IR and c-myc promoters, mouse TGFb3, and Drosophila hsp26. In the case of human c-Ki-ras promoter this homopurine–homopyrimidine tract has been shown to positively regulate transcription. This structure adopts an unusual DNA structure called H-DNA, responsible for nuclease hypersensitivity sites in vitro. But it has been shown by mutational analysis that it is the sequence integrity rather than the H-DNA forming potential that is responsible for the positive effect in transcription. In the case of the dinucleotide GT repeats in promoters the data are much more rare. It is clear that H-DNA structure cannot be adopted by a CA dinucleotide repeat but it is not known whether this sequence can be specifically recognized by DNA binding proteins, as is the case for the homopurine–homopyrimidine tracts.
Several types of transcripts were detected by Northern blot experiments in COLO 320 cells: some are located in the 2.8- and 3.8-kb range and are also recognized by a c-ets-2 probe containing the sequences of exons 2 to 5. These transcripts were also detected in HepG2 cells. Other transcripts migrate as 8.0 kb and 5.0 kb, where several unresolved mRNAs are visible. These transcripts are not detected with an exonic c-ets-2 probe. Finally, we also observed a 1.0-kb mRNA with the 381-bp Exo-SstI probe. This mRNA was also detected with the exonic probe in HepG2 cells, suggesting that it contains sequences corresponding to exons 2 to 5. These results are in accordance with the RT-PCR experiment in which we observed transcripts starting in P2 that encompass bona fide c-ets-2 coding sequences. Taken together, these results suggest that the P2 promoter may, as P1, govern the synthesis of the c-ets-2 proteins. The identity of the large size transcripts (8.0 and 5.0 kb) remains unresolved. One possible explanation may be that the intensity of the transcripts detected with the intron 1-specific probe is low, suggesting that we may have missed some of these with the c-ets-2 probe. Interestingly, the 5.0-kb band was observed on Northern blots hybridized with a probe containing exons 8 to 10 of c-ets-2 gene. This suggests that this transcript effectively contains c-ets-2 coding sequences.
The activity of the P2 promoter appears in NBE cells to be fourfold lower than that of P1 (Fig. 4B). This weaker activity is confirmed by the different intensity of the mRNA visualized on Northern blots. Indeed, the transcript initiated at P1 appears much more intense than that of P2 (Fig. 1B). Although we cannot exclude a differential maturation or stability of these types of transcripts, these observations suggest that P2 is a weaker promoter than P1. Nevertheless, we observed that some deletion constructs of P2 (252 bp, Fig. 4B) exhibit in transient transfection an activity in the same range as that of P1. This suggests that in some situations the activity of P2 may reach the level of P1. This is further supported by RNAase protection experiments, which show that the activity of P2 appears to be higher in certain cell types such as COLO 320 when compared to NBE or HeLa cells (data not shown).
The high number of observed start sites in the P2 promoter revealed by the RNAase protection and primer extension experiments renders difficult the precise mapping of all of them because the intensity of some of the start sites such as S1 may vary when both types of experiments are compared. Thus, there is still a relative uncertainty on the precise number of major start sites. This structure of the P2 promoter with a high number of possible start sites, in addition to the three major ones, is reminiscent to the situation that is found in some immunoglobulin genes. It has been demonstrated that within the human immunoglobulin μ heavy chain enhancer region there is initiation of mRNA transcripts that are extremely heterogeneous at the 5′ end (19,29). More than 50 initiation sites mapping near an octamer sequence ATTTGCGT have been identified (24). This is highly similar to what we observed in the c-ets-2 P2 promoter, which indeed contains an OCT site in the reverse orientation around the start sites. The Igμ heavy chain enhancer region seems to produce sterile transcription because mRNAs starting in this region contains nontranslatable exons. The in vivo significance of such bizarre promoters is still unknown.
Putative Binding Sites of the P2 Promoter
The human c-ets-2 promoter we described contains several sequences similar to cis-acting elements shown to bind transcription factors. Of course, the most noticeable are the three EBS/PEA3 sites, which may be recognized by ets family members and thus by the c-ets-2 gene product itself. Whether c-ets-2 gene may be autoregulated as is the case for its closest related gene c-ets-1 is not known. It has to be emphasized that the P1 promoter already described for c-ets-2 does not seem to be autoregulated. It contains a EBS/ PEA3 site 3′ of the start sites, but this site is recognized by a factor that is not ETS-1 nor ETS-2 (21). One could hypothetize that the new regulatory element we described allows a fine ETS family member-dependent tuning of c-ets-2 expression whereas the PI promoter remains insensitive to ETS products. Such a model could explain some of the in vivo data regarding c-ets-2 expression. For example, during T-lymphocyte activation, an autoregulation of c-ets-2 has been proposed (3). The P2 promoter also contains MYB and GATA sites. Both myb and Gata proteins are expressed in hematopoietic organs and their action on the P2 promoter may account for the c-ets-2 expression in these cells. Here again, it has to be emphasized that the P1 c-ets-2 promoter does not contain MYB or GATA sites (23). Near the major start site S2 a MYB site and a GATA site overlap. Although the effective binding of these factors on such a site has to be demonstrated, such an overlap between putative sites is provocative. It is tempting to imagine a mutually exclusive occupancy of the site by MYB and GATA factors. The close proximity of this MYB/GATA site to a transcriptional start site leads us to propose that the influence of the binding of transcription factors on the site may have a profound effect on transcription. Whether this effectively occurs remains to be experimentally addressed. Interestingly, we also noticed the presence of an octamer site. This finding is particularly interesting because it has been known from a long time that c-ets-2 is expressed during early stages of T-cell development (2). Indeed, the octamer sequence is known to be sufficient to confer lymphoid-specific promoter activity (37). In the c-ets-2 P2 promoter this site is just downstream of the SI major start site. When this OCT site is deleted there is no clear modification of the CAT activity in the cells tested (compare the 381- and 3045-bp deletions in Fig. 4), but this site could be used for regulation of the promoter activity in a cell-specific fashion. Given the number of apparently important putative transcription factor sites on the P2 promoter that are located in close proximity to the major start sites (OCT for S1 and Myb/Gata for S2), we can hypothesize that the occupancy of these sites may regulate the usage of a given start site. For example, occupancy of the octamer site may decrease transcription from SI and subsequently increase transcription efficiency from S2 and/or S3. The combinatorial interaction of several transcription factors and of the transcriptional machinery on this promoter may give rise to an intricate specific control of the P2 expression independently of the more generalist housekeeping like the P1 promoter.
ACKNOWLEDGEMENTS
We thank Serge Plaza, Fabienne Denhez, and Patrick Martin for help and advice during the course of this work; Rachid Safi for help in sequencing; and Jean Coll, Jean-Marc Vanacker, and Christine Dozier for critical reading of the manuscript. This work was financed by CNRS, Institut Pasteur de Lille and ARC.
REFERENCES
- 1. Bhat N. K.; Fisher R. J.; Fujiwara S.; Ascione R.; Papas T. S. Temporal and tissue-specific expression of mouse ets genes. Proc. Natl. Acad. Sci. USA 84:3161–3165; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bhat N. K.; Komschlies K. L.; Fujiwara S.; Fisher R. J.; Mathieson B. J.; Gregorio T. A.; Young H. A.; Kasik J. W.; Ozato K.; Papas T. S. Expression of ets genes in mouse thymocyte subsets T cells. J. Immunol. 142:672–678; 1989. [PubMed] [Google Scholar]
- 3. Bhat N. K.; Thompson C. B.; Lindsten T.; June C. H.; Fujiwara S.; Koizum S.; Fisher R. J.; Papas T. S. Reciprocal expression of human ETS1 and ETS2 genes during T-cell activation: Regulatory role for the protooncogene ETS1. Proc. Natl. Acad. Sci. USA 87:3723–3727; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boshart M.; Kluppel M.; Schmidt A.; Schutz C.; Luckow B. Reporter constructs with low background activity utilizing the cat gene. Gene 110:129–130; 1992. [DOI] [PubMed] [Google Scholar]
- 5. Boulukos K. E.; Pognonec P.; Bègue A.; Galibert F.; Gesquière J. C.; Stéhelin D.; Ghysdael J. Identification in chickens of an evolutionary conserved cellular ets-2 gene (c-ets-2) encoding nuclear proteins related to the products of the c-ets proto-oncogene. EMBO J. 7:697–705; 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boulukos K. E.; Pognonec P.; Sariban E.; Bailly M.; Lagrou C.; Ghysdael J. Rapid and transient expression of Ets2 in mature macrophages following stimulation with CMGF, LPS and PKC activators. Genes Dev. 4:401–409; 1990. [DOI] [PubMed] [Google Scholar]
- 7. Butticè G.; Duterque-Coquillaud M.; Basuyaux J. P.; Carrère S.; Kurkinen M.; Stéhelin D. Erg, an Ets-family member, differentially regulates collagenasel and stromelysin1 gene expression by physically interacting with the Fos/Jun complex. Oncogene 13:2297–2306; 1996. [PubMed] [Google Scholar]
- 8. Chen Z. Q.; Burdett L. A.; Seth A. K.; Lautenberger J. A.; Papas T. S. Requirement of ets-2 expression for Xenopus oocyte maturation. Science 250:1416–1418; 1990. [DOI] [PubMed] [Google Scholar]
- 9. Crépieux P.; Leprince D.; Flourens A.; Albagli O.; Stéhelin D. The two functionally distinct N termini of chicken c-ets-1 products arise from alternative promoters usage. Gene Expr. 3:215–225; 1993. [PMC free article] [PubMed] [Google Scholar]
- 10. Crépieux P.; Coll J.; Stéhelin D. The Ets family of proteins: Weak modulators of gene expression in quest for transcriptional partners. Crit. Rev. Oncog. 5:615–638; 1995. [PubMed] [Google Scholar]
- 11. Crété N.; Delabar J. M.; Rahmani Z.; Yaspo M. L.; Kraus J.; Marks A.; Sinet P. M.; Creau-Goldberg N. Partial physical map of human chromosome 21 from fibroblast and lymphocyte DNA. Hum. Genet. 91:245–253; 1993. [DOI] [PubMed] [Google Scholar]
- 12. Dalton S.; Treisman R. Characterization of SAP-1, a protein recruited by serum response factor to the c-fos serum response element. Cell 68:597–612; 1992. [DOI] [PubMed] [Google Scholar]
- 13. Degnan B. M.; Degnan S. M.; Naganuma T.; Morse D. E. The ets multigene family is conserved throughout the Metazoa. Nucleic Acids Res. 21:3479–3484; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13a. Dozier C.; Ansieu S.; Ferreira E.; Coll J.; Stéhelin D. An alternatively spliced c-mil/raf mRNA is predominantly expressed in chicken muscular tissues and conserved among vertebrate species. Oncogene 6:1307–1311; 1991. [PubMed] [Google Scholar]
- 14. Giese K.; Kingsley C.; Kirshner J. R.; Grosschedl R. Assembly and function of a TCR alpha enhancer complex is dependent on LEF-1-induced DNA bending and multiple protein-protein interactions. Gene Dev. 9:995–1008; 1995. [DOI] [PubMed] [Google Scholar]
- 15. Hipskind R. A.; Rao V. N.; Mueller C. G. G.; Reddy E. S. P.; Nordheim A. Ets-related protein Elk-1 is homologous to the c-fos regulatory factor p62Ets . Nature 354:531–534; 1991. [DOI] [PubMed] [Google Scholar]
- 16. Laudet V.; Niel C.; Duterque-Coquillaud M.; Leprince D.; Stéhelin D. Evolution of the ets gene family. Biochem. Biophys. Res. Commun. 190:8–14; 1993. [DOI] [PubMed] [Google Scholar]
- 17. Laudet V.; Vanacker J. M.; Adelmant G.; Bègue A.; Stéhelin D. Characterization of a functional promoter for the human thyroid hormone receptor alpha c-erbA-1 gene. Oncogene 8:975–982; 1993. [PubMed] [Google Scholar]
- 18. Lautenberger J. A.; Burdett L. A.; Gunnell M. A.; Qi S.; Watson D. K.; O’Brien S. J.; Papas T. S. Genomic dispersal of the ets gene family during metazoan evolution. Oncogene 7:1713–1719; 1992. [PubMed] [Google Scholar]
- 19. Lennon G. G.; Perry R. P. Cμ-containing transcripts initiate heterogeneously within the IgH enhancer region and contain a novel 5′-nontranslatable exon. Nature 318:475–478; 1985. [DOI] [PubMed] [Google Scholar]
- 20. Maniatis T.; Fritsch E. F.; Sambrook J. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1989. [Google Scholar]
- 21. Mavrothalassitis G. J.; Papas T. S. Positive and negative factors regulate the transcription of the ETS2 gene via an oncogene-responsive-like unit within the ETS2 promoter region. Cell Growth Differ. 2:215–224; 1991. [PubMed] [Google Scholar]
- 22. Mavrothalassitis G. J.; Watson D. K.; Papas T. S. Molecular and functional characterization of the promoter of TS2, the human c-ets-2 gene. Proc. Natl. Acad. Sci. USA 87:1047–1051; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mavrothalassitis G. J.; Watson D. K.; Papas T. S. The human ETS-2 gene promoter: Molecular dissection and nuclease hypersensitivity. Oncogene 5:1337–1342; 1990. [PubMed] [Google Scholar]
- 24. Neale G. A. M.; Kitchingman G. R. mRNA transcripts initiating within the human immunoglobulin mu heavy chain enhancer region contain a non-translatable exon and are extremely heterogeneous at the 5′ end. Nucleic Acids Res. 19:2427–2433; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Raghu G.; Tevosian S.; Anant S.; Subramanian K. N.; George D. L.; Mirkin S. M. Transcriptional activity of the homopurine-homopyrimidine repeat of the c-Ki-ras promoter is independent of its H-forming potential. Nucleic Acids Res. 22:3271–3279; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Seth A.; Watson D. K.; Blair D. G.; Papas T. S. c-ets-2 protooncogene has mitogenic and oncogenic activity. Proc. Natl. Acad. Sci. USA 86:7833–7837; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Smale S. T.; Schmidt M. C.; Berk A. J.; Baltimore D. Transcriptional activation by Sp1 as directed through TATA or initiator: Specific requirement for mammalian transcription factor IID. Proc. Natl. Acad. Sci. USA 87:4509–4513; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stein B.; Rahmsdorf H. J.; Steffen A.; Litfin M.; Herrlich P. UV-induced DNA damage is an intermediate step in UV-induced expression of human immunodeficiency virus type 1, collagenase, c-fos, and metallothionein. Mol. Cell. Biol. 9:5169–5181; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Su L. K.; Kadesch T. The immunoglobulin heavy-chain enhancer functions as the promoter for Iμ sterile transcription. Mol. Cell. Biol. 10:2619–2624; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sumarsono S. H.; Wilson T. J.; Tymms M. J.; Venter D. J.; Corrick C. M.; Kola R.; Lahoud M. H.; Papas T. S.; Seth A.; Kola I. Down’s syndrome-like skeletal abnormalities in Ets2 transgenic mice. Nature 379:534–537; 1996. [DOI] [PubMed] [Google Scholar]
- 31. Sun W.; Graves B. J.; Speck N. A. Transactivation of the Moloney murine leukemia virus and T-cell receptor beta-chain enhancers by cbf and ets requires intact binding sites for both protein. J. Virol. 69:4941–4949; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang C. Y.; Petryniak B.; Ho I. C.; Thompson C. B.; Leiden J. M. Evolutionary conserved Ets family members display distinct DNA binding specificities. J. Exp. Med. 175:1391–1399; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wasylyk B.; Wasylyk C.; Flores P.; Bègue A.; Leprince D.; Stéhelin D. The c-ets protooncogenes encode transcription factors that cooperate with c-Fos and c-Jun for transcriptional activation. Nature 346:191–193; 1990. [DOI] [PubMed] [Google Scholar]
- 34. Watson D. K.; McWilliam-Smith M. J.; Nunn M. F.; Duesberg P. H.; O’Brien S. J.; Papas T. S. The ets sequence from the transforming gene of avianerythroblastosis virus, E26, has unique domain on human chromosomes 11 and 21: Both loci are transcriptionally active. Proc. Natl. Acad. Sci. USA 82:7294–7298; 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Watson D. K.; McWilliams M. J.; Lapis P.; Lautenberger J. A.; Schweintest C. W.; Papas T. S. Mammalian ets-1 and ets-2 genes encode highly conserved proteins. Proc. Natl. Acad. Sci. USA 85:7862–7866; 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Watson D. K.; Ascione R.; Papas T. S. Molecular analysis of the Ets genes and their products. Crit. Rev. Oncog. 1:409–436; 1990. [PubMed] [Google Scholar]
- 37. Wirth T.; Staudt L.; Baltimore D. An octamer oligonucleotide upstream of a TATA motif is sufficient for lymphoid-specific promoter activity. Nature 329:174–178; 1987. [DOI] [PubMed] [Google Scholar]
- 38. Zenzie-Gregory B.; Khachi A.; Garraway I. P.; Smale S. T. Mechanism of initiator-mediated transcription: Evidence for a functional interaction between the TATA-binding protein and DNA in the absence of a specific recognition sequence. Mol. Cell. Biol. 13:3841–3849; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]