

Abstract
The product of the REF2 gene is required for optimal levels of endonucleolytic cleavage at the 3′ ends of yeast mRNA, prior to the addition of a poly(A) tail. To test the role of the previously demonstrated nonspecific affinity of REF2 for RNA in this process, we have identified RNA binding mutants in vitro and tested them for function within the cell. One REF2 variant, with an internal deletion of 82 amino acids (269–350), displays a 10-fold reduction in RNA binding, yet still retains full levels of processing activity in vivo. Conversely, a series of carboxyl-terminal deletions that maintain full RNA binding capability have progressively decreasing activity. These results rule out a major role for the central RNA binding domain of REF2 in mRNA 3′ end processing and demonstrate the importance of the carboxyl-terminal region. To ask if the stimulatory role of REF2 depends on interactions with other proteins, we used a two-hybrid screen to identify a new protein termed FIR1 (Factor Interacting with REF) encoded on chromosome V. FIR1 interacts with two independent regions of REF2, one of which (amino acids 268–345) overlaps the RNA binding domain and is dispensible for REF2 function, whereas the other (amino acids 391–533) is located within the critical carboxyl-terminus. As with REF2, FIR1 has a small but detectable role in influencing the efficiency of poly(A) site use. Yeast strains containing a disrupted FIR1 gene are slightly less efficient in the use of cryptic poly(A) sites located within the lacZ portion of an ACTl-lacZ reporter construct. Likewise, a double Δref2, Δfir1 mutant is more defective in processing of a reporter CYC1 poly(A) site than Δref2 alone. This synergistic response provides additional support for the interaction of FIR1 with REF2 in vivo, and suggests that a number of gene products may be involved in regulating the cleavage reaction in yeast.
Keywords: Yeast mRNA processing, 3′ End cleavage factors, RNA binding, Protein-protein interactions
Beginning with the gene encoding yeast poly(A) polymerase (PAP1) (26,39) a remarkable number of new S. cerevisiae genes whose products are involved in mRNA 3′ end processing have been discovered by combining a variety of biochemical and genetic strategies [for reviews, see (20,52)]. The development of a reconstituted in vitro system using whole-cell extracts (5,6) has played a central role, leading to the separation and characterization of four cellular fractions involved in the cleavage and polyadenylation reactions (9,21a,55).
The first of these, designated cleavage factor Iy (CF Iy), is essential for both sequence-specific 3′ endonucleolytic cleavage [when combined with cleavage factor IIy (CF IIy)], as well as the attachment of a poly(A) tract by poly(A) polymerase (encoded by the PAP1 gene) to the same site, and thus is thought to be a specificity factor involved in recognizing yeast poly(A) signals (9). In functional terms, this factor resembles mammalian cleavage and polyadenylation specificity factor (CPSF), which interacts with the conserved metazoan element AAUAAA and is also needed for both the cleavage and polyadenylation reactions. Three components of CF Iy are encoded by PCF11 (la), RNA14, and RNA15. The latter two were initially isolated as temperature-sensitive mutations that displayed a shortening of poly(A) tails and a rapid decrease in the steady-state concentration of polyadenylated RNAs at the nonpermissive temperature (33). In addition, strains rna14-1 and rna15-1 are synergistically lethal with a pap1-5 mutation at the permissive temperature, and extracts made from these strains are defective for both cleavage and polyadenylation reactions in vitro (34). The direct role of these two proteins in 3′ end processing was confirmed by the restoration of cleavage and polyadenylation activity to RNA14- and RNA15-depleted extracts with purified CF Iy, as well as the cofractionation of both proteins with CF Iy. RNA14 shares significant homology with the suppressor-of-forked protein that regulates gene expression in Drosophila (35), and to the human 77-kd subunit of cleavage stimulation factor (CstF) (51). RNA15 has an amino-terminal RNA recognition motif (RRM) that resembles both the Drosophila and human 64-kd RNA-interactive component of CstF (51) and has been demonstrated to bind poly(U) RNA in vitro (33).
Using a sensitive assay to monitor the use of multiple poly(A) sites located within the 3′ untranslated region of ACT1, Mandart and Parker (30) observed the preferential use of more distal poly(A) sites in rna14-3, rna15-2, and pap1-1 strains, at the restrictive temperature. This provides evidence that mutations in these genes can alter poly(A) site selection in vivo and strengthens previous suggestions from in vitro work that poly(A) polymerase can enhance the efficiency of the cleavage reaction (9,34). In addition to FIP1 (see below), there is some genetic evidence that RNA14 may interact with the product of the nonessential SSM4 gene because mutations or deletions in SSM4 suppress the thermosensitivity of the rna14-1 allele but not a deletion of RNA14 or mutations in RNA15 (29). Interestingly, a deletion of SSM4 can also correct the poly(A) site alteration observed in the rna14-3 strain (30).
Of the other cleavage and polyadenylation factors, CF IIy is only recently characterized (55). Whereas polyadenylation factor I (PF I), in concert with CF Iy and poly(A) polymerase, is required for the polyadenylation reaction in vitro. One component of PF I, encoded by the essential FIP1 gene, was identified in a two-hybrid screen using PAP1 as the bait (41). Extracts made from cells carrying a mutant fip1-1 gene could cleave but not polyadenylate pre-mRNA substrates unless purified PF I fractions, which contain FIP1, were added back to the reactions. Coimmunoprecipitation studies showed that FIP1 also interacts with RNA14 and immunodepletion of FIP1 from extracts led to a two- to threefold decrease in cleavage activity. All of the above observations support a model wherein FIP1 forms a bridge between the RNA14 component of CF Iy and poly(A) polymerase (41). More recently, the CFT1 protein has been implicated as another component of CF IIy (49) and the product of the BRR5/YSH1 gene is a component of PF I and the homologue of the 73-kd subunit of mammalian CPSF (7,19).
An additional component of this system is the nonessential yeast gene REF2, identified genetically by a mutation that reduced processing of an intronic poly(A) site, thus allowing expression of a downstream reporter gene (44,45). Recombinant REF2 stimulated cleavage of pre-mRNA substrates carrying weak, or suboptimal, processing signals in extracts made from Δref2 cells. Despite lacking any consensus RNA-interactive motifs, REF2 also bound RNA in a standard band-retention assay, but in a nonspecific fashion. This binding was not competed by either single-stranded or double-stranded DNA and, in fact, REF2 displayed a preferential affinity for pyrimidine bases, especially poly(U) RNA. The stimulatory effect of REF2 both in vivo and in vitro suggested that it participated in the stabilization of a precleavage complex as an integral component of the assembled ribonucleoprotein particle (45), possibly through increased nonspecific contacts with the underlying RNA substrate. Indeed, a number of studies in mammalian systems have shown that poly(A) site use is a function of the stability with which 3′ end processing factors assemble upon it (1,16,53).
In this study we set out to examine the biological importance of the RNA binding capability of REF2 and to define its novel mode of RNA recognition, because REF2 shows no discernible homology to other known RNA binding proteins. A combination of REF2 deletions was tested for their ability to both bind RNA in vitro and stimulate poly(A) site use in vivo. The results indicate that the RNA binding activity of REF2 resides in multiple, partially redundant elements but, most importantly, is dispensible for its role in 3′ end processing. To address the possibilty that REF2 may be stabilizing the precleavage complex through an association with other proteins, we carried out a two-hybrid screen and identified a single interacting protein, FIR1. Disruption of FIR1 in a Δref2 background resulted in further decreased use of our reporter poly(A) site, suggesting that FIR1 is a novel component involved in regulating the efficiency of 3′ end processing.
MATERIALS AND METHODS
General Procedures
Table 1 lists the genotypes of the S. cerevisiae haploid strains used in this study. Cells were maintained in either YPD medium or supplemented SD medium (47). Histidinol-selective medium replaced histidine with 642 mg/l of histidinol. Transformation of yeast was carried out according to Chen et al. (8) using sheared calf thymus DNA as carrier. For maintenance of plasmids in E. coli, strain DH5α was used.
TABLE 1.
S. CEREVISIAE STRAINS USED IN THIS STUDY
| Strain | Genotype | Reference |
|---|---|---|
| DC35-15D | MATa ura3-52 leu2-3, 112 HOL1 ade2 ade5 gal80 his4-401:: ACT1-CYC1-HIS4 | 41 |
| RR4 | MATa ref2::LEU2 ura3-52 trp1 leu2-3, 112 HOL1 ade2 ade5 gal80 his4-401::ACT1-CYC1-HIS4 | 41 |
| RR9 | MATa fir1::LEU2 ura3-52 trp1 leu2-3, 112 HOL1 ade2 ade5 gal80 his4-401::ACT1-CYC1-HIS4 | this study |
| RR10 | MATa ref2::LEU2 fir1::TRP1 ura3-52 trp1 leu2-3, 112 HOL1 ade2 ade5 ga180 his4-401:: ACT1-CYC1-HIS4 | this study |
| CYT10-5d | MATa ade2 trp1-901 leu2-3, 112 his3-200 Δgal4 Δgal80 URA::lexA op-lacZ | 3 |
Plasmids Used for Bacterial Expression
To create pTrcF533, an NcoI-EcoRV fragment was purified from pFLAGRF2 and cloned into pTrcFRF2 (45), which had been EcoRI digested, end-repaired with Klenow, and further digested with NcoI. The protein expressed from pTrcF484 (FgREF2484) is the same as Ref2pF described in Russnak et al. (45) and contained the first 484 amino acids of REF2 followed by 17 residues encoded by vector sequence (see text). The pTrcF345 construct was created by cloning an XbaI-EcoRI fragment from pGEM3Zf(+)ref2-l (45) into XbaI-EcoRI cut pTrcFRF2. In turn, pFLAG345 was constructed by exchanging the NcoI-StyI fragment of pTrcF345 with that of pFLAGRF2. All of the other pTrc derivatives were created by exchanging an NcoI-EcoRI fragment from the appropriate pFLAG vectors (see below) with the corresponding wild-type region in pTrcFRF2.
For pFLAG406, a partial digestion of pFLAGRF2 with XmnI yielded linear plasmid that was religated with an SpeI linker carrying an amber codon in all three reading frames. The construct pFLAG268 resulted from AccI digestion, Klenow repair, and religation of pFLAGRF2. In pFLAGP1/P2, two unique PstI sites were created by PCR-directed mutagenesis using pFLAGRF2 as a template. The oligomers used were: 1) oligo P1, nt 872-GCGTCATCCTTCTG CAGATATTTAGAA-846, 2) oligo P2, nt 1061-TTAGAGTCC TGCA GGGATATTTTGGTTTT-1033 (underlined sequences represent the PstI sites whereas bold indicates a change from the wild-type sequence; numbering is based on designating the A within the initiation codon as 1), 3) oligo 2.2, nt 653-GGATAAATCAAAAGTCAAG-671, and 4) the SP6 promoter primer (USB), which annealed to vector sequences downstream of the cloned REF2 gene in pFLAGRF2.
A typical PCR reaction was made up to 0.1 ml containing 20 mM Tris-Cl (pH 8.7), 10 mM KCl, 10 mM (NH4)2SO4, 2 mM MgSO4, 0.1% Triton X-100, 0.1 mg/ml BSA, 0.2 mM dNTPs, 200 ng template DNA, 50 pmol of each primer, and 2.5 units of Pfu Polymerase. The reactions were layered with 0.05 ml mineral oil and thermocycled 30 times at 94°C for 1 min, 48°C for 1 min, and 72°C for 3 min. The two PstI sites were introduced sequentially. First, a PCR reaction using oligo 2.2 and PI primers yielded a 220-bp product that was gel-purified and used in a second round of PCR as the upstream primer, together with the downstream SP6 promoter oligomer. This 1.25-kb PCR product was digested with AccI-NsiI, cloned into AccI-NsiI-digested pFLAGRF2, and sequenced to ensure that the only mutation present in the cloned sequence was the desired PstI site at position 860 (amino acid 287, an asparagine to glutamine change). This plasmid, referred to as pFLAGP1, was used as a template along with oligos 2.2 and P2, and the resultant PCR product (408 bp) was used as the upstream primer in a second round of PCR. The final 1.25-kb product was cloned into pFLAGRF2 to produce pFLAGP1/P2 and sequencing again confirmed the introduction of a second PstI site at amino acid 350 (a valine to isoleucine change). An NcoI-EcoRI fragment from this vector was cloned into the pTrcFRF2 vector to produce pTrcFP1/P2. To create either pFLAGΔ287-350 or pTrcFΔ287-350, an internal collapse at the PstI sites was carried out to produce an in-frame deletion of 63 amino acids. The vector pFLAG406Δ287-350 was created from pFLAGΔ287-350 as described for pFLAG406.
To create pFLAGΔ243-327, pFLAGP1 was linearized with PstI, treated with Bal31 at 37°C for 7 min, end-repaired with Klenow and T4 DNA polymerase, and self-ligated. Plasmids that demonstrated a loss of 250 bp were sequenced and one carrying an in-frame deletion of 84 amino acids from 243 to 327 was identified. To create pFLAGΔ287-406, pFLAGΔ287-350 was partially digested with XmnI, followed by complete digestion with PstI, after which the ends were repaired with T4 DNA polymerase and ligated, leading to an in-frame deletion of 120 amino acids from 287 to 406. The pFLAG268-484 vector was created by digestion of pFLAGP1/P2 with NdeI-AccI, end-repair with Klenow, and religation. To create pFLAGΔRBD, pFLAGP1/P2 was digested with AccI-PstI, and religated with BamHI linkers to produce an 82-amino acid in-frame deletion from residues 269 to 350.
Expression and Purification of FLAG Epitope-Tagged REF2 Proteins
Affinity purification was carried out according to Chiang and Roeder (10), with the following modifications. Briefly, 100-ml cultures of DH5α cells harboring the appropriate pTrcF plasmids were grown in 2xYT at 37°C to an optical density of 1.0 at 600 nm. The pTrc promoter was induced by the addition of IPTG (isopropyl-d-thiogalacto-pyranoside) to 1 mM at 30°C for 4 h. Cells were harvested, resuspended in 3 ml 1 × PBS (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4), 10% glycerol, 0.2 mM DTT, 0.5 mM PMSF, and lysed by sonication. The cell lysates were incubated with 50–60 μl of anti-FLAG M2 agarose (IBI) at 4°C for 4 h. The resin was separated from the supernatant by centrifugation, washed six times with 1 × PBS, 5% glycerol, 0.2 mM DTT, 0.5 mM PMSF, 0.1% Nonidet P-40, twice more with the buffer lacking the detergent, and transferred to a microfuge tube. The FgREF2 fusion proteins were eluted from the resin in two 20-min washes of 0.2 ml FLAG peptide (IBI) at a concentration of 0.2 mg/ml. The eluants were pooled and dialysed twice against 500 ml of REF2 storage buffer (10 mM HEPES, pH 7.0, 0.2 mM EDTA, 50 mM KAc, 50% glycerol) at 4°C. The concentration of the affinity-purified proteins was estimated by Coomassie blue staining against a known amount of protein, and preparations were estimated to be routinely 80–90% pure. The integrity of the proteins was also determined by Western analysis using an anti-FLAG M5 antibody (IBI) at a dilution of 1:500 and the antibody–FgREF2 complexes were detected using chemiluminescence and a horseradish peroxidase-coupled secondary antibody at 1:5000 (Amersham).
RNA Binding Assays
For the band-shift analysis, 100 nM (approximately 50 ng) of the FgREF2 proteins was incubated in a 0.01-ml reaction volume with 1 nM [32P]GTP-labeled RNA, 10 mM HEPES (pH 7.0), 0.2 mM EDTA, 50 mM KAc, 1 mM DTT, and 2 mg/ml heparin. The reaction mixes were electrophoresed at room temperature in a 4% acrylamide:bis (30:1), 10% glycerol, 0.5X TBE gel after the addition of 2 μl DNA loading dye. The gels were dried onto Whatman paper and visualized by autoradiography. Estimations of the percent RNA shifted were carried out on a Phosphor-Imager. For the protein titration experiments, the indicated amounts of protein were added to the reaction mixes as described.
The 110-nt C/S RNA used in the RNA binding studies was synthesized by SP6 RNA polymerase from pC/X(-PL), linearized at a unique SspI site within the U3 region of Ty and located 112 nt upstream of the site of polyadenylation. The pC/X(-PL) vector is a derivative of pU3RU5 (18) in which a XhoI-SstI fragment had been excised, deleting the RU5 region and part of the downstream polylinker region. Further removal of sequences between the BamHI and HindIII sites resulted in the deletion of the polylinker region found upstream of U3.
Complementation Analysis
Full-length REF2533 was expressed in yeast cells from the low-copy plasmid YCpREF2. For its construction, a 2.6-kb Klenow-repaired SalI-BglII fragment was excised from a derivative of pRF2.3Z (45) in which the XbaI site within the polylinker was destroyed by partial digestion with XbaI followed by filling-in and religation. This fragment was cloned into YCplac33 (13), which had been digested with HindIII and EcoRI and end-repaired. YCp484 was created by digestion, Klenow repair, and religation of the unique EcoRI site in YCpP1/P2, resulting in a frameshift at amino acid 484 and the addition of 19 residues before the new stop codon. To create YCpP1/P2, YCpΔ287-350, YCpΔRBD, YCp406, YCp345, and YCp268, an XbaI-EcoRI fragment purified from the appropriate pFLAG.RF2 derivative (see above) was exchanged with the corresponding region in YCpREF2. For YCp458, the XbaI-EcoRI fragment was purified from pTrcF458. The described expression plasmids, which carry a URA3 gene, were introduced into the Δref2 strain RR4 (Table 1) and Ura+ transformants were tested for growth at 30°C on histidinol plates or at 37°C on YPD.
Two-Hybrid Interactive Screen
The entire coding region of REF2 was expressed as a LexA fusion protein from the plasmid pLBDRF2, created by the insertion of a 1.9-kb BamHI-BglII fragment purified from pRF2.3Z-(Nde) (45) into BamHI-digested pBTM116 (3). The yeast strain CYT10-5d (3) was cotransformed with both pLBDRF2 and either one of two pGAD.F-derived libraries (11) containing yeast genomic DNA fragments fused in different translation frames to the GAL4 transcriptional activation domain. After 3–4 days growth at 30°C, Trp + Leu+ transformants were replica-plated to medium containing 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) and incubated further. Activation domain plasmids were recovered from blue colonies and retransformed into the original indicator strain with either pBTM116 or pLBDRF2 to identify those that resulted in REF2-specific transcriptional activation. Two positive clones, designated pGAD24-1 and pGAD24-4, were further characterized by restriction mapping and sequencing and found to contain the GAL4 open reading frame fused to the carboxy-terminal 847 or 801 amino acids, respectively, of the same gene (FIR1; see text). In pFR1.3Z(E), a 2.5-kb EcoRI fragment from pGAD24-l harboring GAL4 sequences and nucleotides 235–2706 of FIR1 (nucleotide numbering is relative to the initiation codon; see below) was cloned into EcoRI-digested pGEM3Zf(+).
Isolation of the 5′ End of FIR1 and Disruption of the Chromosomal Copy
To obtain the 5′ portion of FIR1, genomic DNA, isolated from wild-type strain DC35-15D (see Table 1) by the method of Philippsen et al. (40), was used to construct a library of size-fractionated (∼4 kb) PstI fragments cloned into pGEM3Zf(+). Colony hybridization was carried out as described (46), using an RNA probe (incubation at 50°C), which corresponded to FIR1 sequences between NcoI (nt 477) and HindIII (nt 1078). A positive clone [designated pFR1.3Z(P)] was characterized further and found to contain ∼1.6 kb of sequence upstream of the putative initiation codon and extending to an internal PstI site at 2570.
To make pFR1LEU2, a 3.0-kb BglII fragment harboring the LEU2 gene was purified from CV13, end-repaired with Klenow enzyme, and cloned into pFR1.3Z(P), which had been digested with BglII (nt 124) and ClaI (nt 1977), recessed ends being filled in prior to ligation. For pFR1TRP1, the TRP1 gene, contained within a 1.5-kb SspI-NdeI fragment purified from YIplac204 (13) and made blunt-ended, was cloned into EcoRV (nt 833)-ClaI (nt 1977)-digested pFR1.3Z(E) (see above), which had been treated with Klenow. Yeast strains carrying disrupted FIR1 alleles were created by transforming either strain DC35-15D with pFR1LEU2 digested with PstI or strain RR4 with NcoI-PstI-digested pFR1TRP1 and selecting on the appropriate medium. In all cases, proper integration was determined by Southern analysis of EcoRI-digested genomic DNA using the ∼600 nt riboprobe described for the colony hybridization above.
Monitoring Poly(A) Site Use In Vivo
Analysis of transcripts derived from the integrated ACT1-CYC1-HIS4 reporter gene (Fig. 5B) was carried out as described in Russnak et al. (45). To assay the efficiency of use of cryptic poly(A) site(s) located within the lacZ gene when expressed in yeast, the various strains to be tested were transformed with pSB20, in which the CYC1 poly(A) site located within the intron of the ACT1-HIS4 gene in pSB17 (45) was missing. β-Galactosidase activity was quantified by liquid assay as described by Guarente (17). Briefly, individual colonies were grown to mid-log phase in 2.5 ml of SC-Trp medium and the optical density at 600 nm (A600) was determined. Cells were resuspended in 0.5 ml of Z buffer (0.1 M NaPO4, pH 7.0, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol, 0.01% SDS), 30 ml chloroform was added, and the cells permeabilized by vortexing for 10 s. After a preincubation at 28°C for 5 min, reactions were started by adding 200 μl of 4 mg/ml o-nitrophenyl-β-d-galactoside (ONPG) and, upon pale color development, were stopped with the addition of 500 μl of 1 M Na2CO3. The optical density of the o-nitrophenol product was measured at 420 nm (A420) and units of β-galactosidase activity expressed as A420 × 1000/A600 × volume assayed (in ml) × time (in min).
FIG. 5.
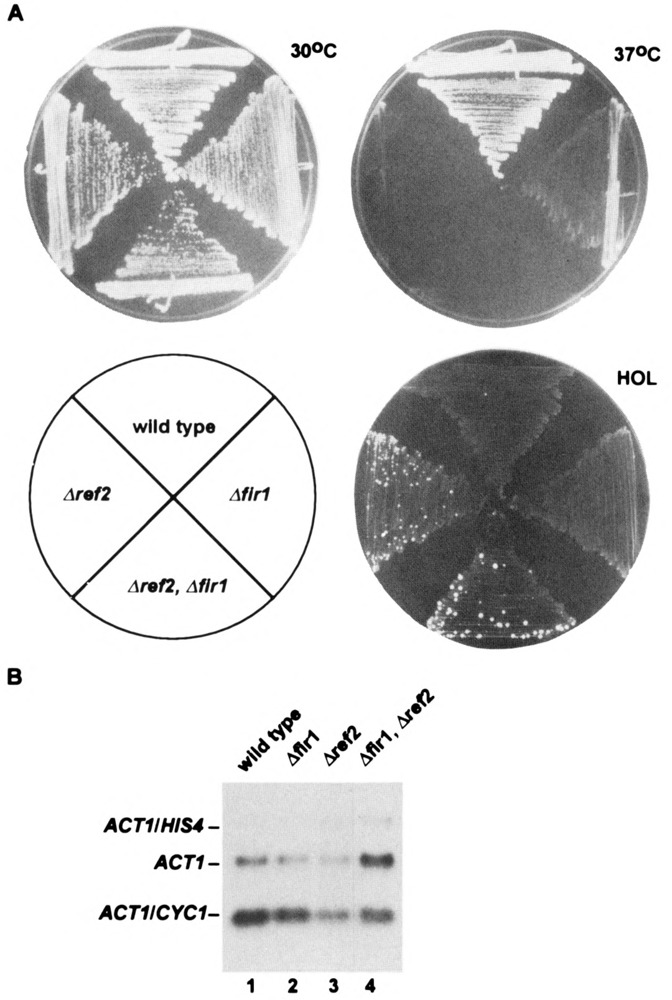
(A) Growth characteristics on YPD and synthetic medium containing histidinol (HOL) of strains carrying the indicated single-gene and double-gene disruptions. (B) Processing efficiency of the reporter CYC1 poly(A) site found within the same cells. Approximately 5–10 μg of total RNA, isolated from the indicated strains that all carry an integrated ACT1- CYC1-HIS4 gene (see text), was subjected to Northern analysis. Shown are the positions of the 300-nt ACT1/CYC1 transcript [processed at the up-stream CYC1 poly(A) site inserted within the ACT1 intron], the extended 2.8-kb ACT1/HIS4 transcript [processed at the downstream poly(A) site], and the endogenous 1.35-kb ACT1 message.
Plasmid Construction and Expression of LexA Fusion Proteins
To create pLBD484, the EcoRI site at amino acid 484 within pLBDREF2 was destroyed by partial digestion, Klenow repair, and religation, resulting in the same frameshift that was described for YCp484 above. Insertion of a SpeI-amber codon linker at the unique StyI site (made blunt-ended) within pLBDREF2 resulted in pLBD391. For the construction of pLBD458 and pLBD345, a Klenow-repaired 1.5-kb NdeI-EcoRI fragment was purified from either pTrcF458 or pFLAG345, respectively, and cloned into pBTM116 digested with SmaI. For pLBD268, a 1.9-kb NdeI-NsiI fragment purified from pFLAG268 and end-repaired with T4 DNA polymerase was cloned in the same manner. Removal of sequences between the BamHI and StyI sites of pLBDREF2 by digestion, filling-in, and religation resulted in pLBD391-533. The remaining carboxy-terminal constructs pLBD350-533, pLBD350-484, and pLBD350-458 were made by cloning a 620-bp, T4-repaired PstI-EcoRV fragment purified from YCpP1/P2, YCp484, or YCp458, respectively, into SmaI-digested pBTM116. Strain CYT10-5d was cotransformed with the above plasmids along with pGAD24-1 and, in each case, four Trp+ Leu+ strains were established and assayed multiple times for blue-color development on X-gal plates. For each of the LxREF2 proteins described, at least two sets of transformations were tested.
RESULTS
Correct Sequence of the REF2 Gene
The product of the REF2 gene was reported to be 429 amino acids (45), but we discovered an error in our initial sequence determination that resulted in a frameshift at amino acid 425. The corrected sequence results in an extended open reading frame of 533 amino acids on chromosome IV (Fig. 1), which encodes a lysine-rich (15.6%) protein with a molecular weight of 59,781. The new carboxyl-terminal extension contains no notable homologies to other proteins but carries a canonical amino transferase class II pyridoxal phosphate attachment site at lys498, as defined by the MOTIFS program (Genetics Computer Group, Inc.). The significance of this site in REF2 is not known, but it is probably not required for function because REF2484, which is lacking this site, is fully functional in 3′ end processing in vivo (see below). As a result of the cloning strategy employed by Russnak et al. (45), the described bacterial expression plasmid pTrcFRF2 (renamed here pTrcF484) produced a protein with the first 484 amino acids of REF2 fused to 17 amino acids encoded by vector sequences. Therefore, the in vitro experiments described therein, which demonstrated the participation of REF2 in the 3′ end cleavage reaction as well as describing the RNA binding properties of REF2, were carried out with this truncated product. We have now purified from E. coli the full-length FLAG-derivatized protein and shown it to bind RNA with the same affinity (Fig. 2) and with the same lack of specificity (data not shown) as FgREF2484.
FIG. 1.

Corrected sequence (see text) of the 533-amino acid REF2 gene product (GenBank accession number U20261; see also Z48784 for localization to chromosome IV). Arrows denote the endpoints of the various analyzed carboxyl-terminal truncations. REF2484 is analogous to the protein expressed from pTrcFRF2 (45), which was found to stimulate 3′ end processing in vitro. Numbered are the lysine (K)-rich sequences or clusters that are implicated in RNA binding (see text) and that are denoted schematically by thick lines in subsequent figures. Also indicated by underlining is the FXXXLNK sequence, which is found in both of the determined FIR1-interactive domains.
FIG. 2.
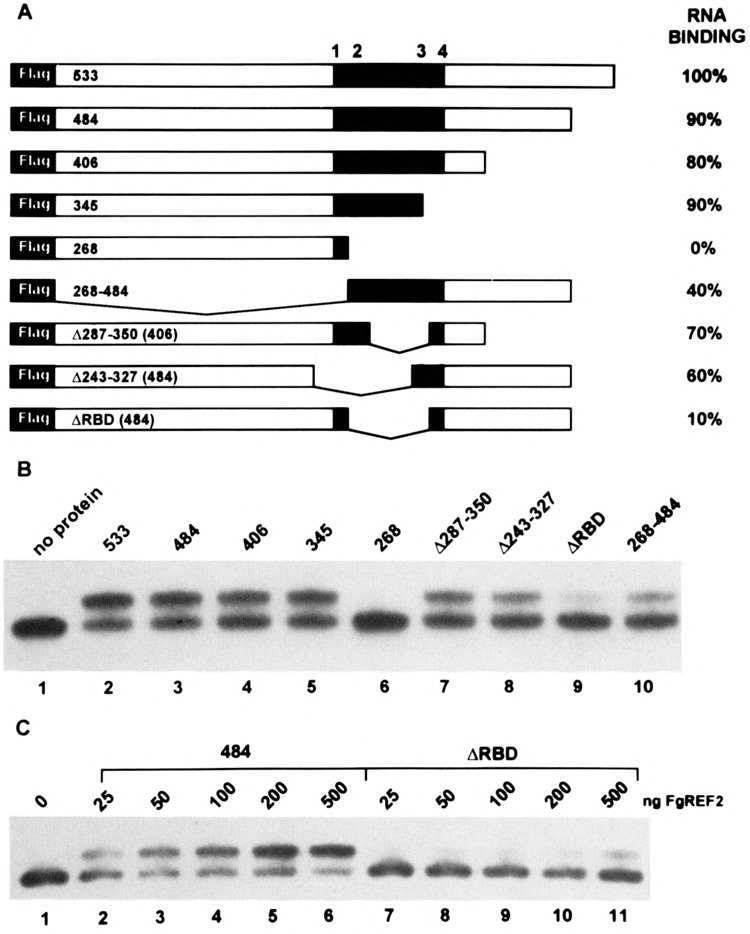
RNA binding analysis. The 110-nt AU-rich RNA substrate used in the band-retention experiments was derived from the U3 region found within the yeast retrotransposable element Ty but lacks the functional poly(A) site as well as elements TS1 and TS2 (18). (A) Diagram of the FLAG epitope-tagged REF2 proteins that were expressed in E. coli and purified by affinity chromatography. The shaded region from amino acid 250 to 375 represents the approximate boundary of the putative RNA binding domain, which is distinquished by multiple K-clusters as shown (see also Fig. 1). The relative binding affinities are expressed to the right as the percent RNA shifted by each protein at 100 nM compared to FgREF2533 (wild-type) and was obtained by Phosphorlmager quantification of band-shift data similar to that shown in (B) and represents an average value obtained from multiple protein isolates. (C) Titration of the FgREF2484 and FgREF2ΔRBD proteins using the indicated amounts (see Materials and Methods for details).
Delineation of the RNA Binding Domain
To identify those regions that were required for RNA recognition, we extended our previous in vitro analysis by testing a variety of truncated forms of REF2. The REF2-derived proteins used in the RNA binding experiments are shown in Fig. 2A and contained at their amino-terminus a 16 amino acid extension that harbors the FLAG epitope DYDDDDK, allowing for their affinity purification with an antibody-containing resin (see Materials and Methods). Coomassie staining and Western blot analysis using an anti-FLAG antibody revealed that the wild-type protein migrated with an apparent MW of 80 kDa (compared to a calculated value of 60 kDa), and that all of the FgREF2 mutants migrated in accordance with the extent of their predicted deletions (data not shown). The affinity purification procedure yielded, in most cases, largely intact full-length protein. The preparation of FgREF2533, FgREF2406, and FgREF2268, however, routinely resulted in the appearance of distinct smaller peptides that were determined to be carboxyl-terminal degradation products and thus these proteins appeared to be more labile than the rest.
The analysis of a series of carboxyl-terminal deletions showed that the removal of 188 amino acids did not affect RNA binding, as FgREF2345 still retained 80–90% of the wild-type binding activity (Fig. 2B, lane 5) whereas further deletion to amino acid 268 completely abolished it (Fig. 2B, lane 6). This suggested that the RNA binding domain lay within amino acids 268–345 and, indeed, removal of this region in FgREF2ΔRBD resulted in a protein that was severely defective for RNA binding (Fig. 2B, lane 9). Internal deletions that removed either the amino-terminal (FgREF2Δ243–327, lane 8) or carboxyl-terminal (FgREF2Δ287–350, lane 7) portions of the putative RNA binding domain retained significant binding activity, suggesting that multiple determinants were involved. Interestingly, inspection of REF2 from amino acids 250–375 revealed four clusters of lysine residues (K-clusters), the positions of which are indicated in Fig. 2A (sequences are shown in Fig. 1). The most compelling argument, perhaps, that K-clusters may be involved in RNA binding in vitro comes from the observation that the only difference between the internal deletion in FgREF2Δ287–350 (70% binding) versus FgREF2ΔRBD (10% binding) is the removal of 20 amino acids harboring the second K-cluster. Also, FgREF2268, which does not bind RNA in vitro, maintains the same high content of basic amino acids (16% lys and arg) but they are (excluding the first K-cluster) evenly distributed or, in the case of the unusual KEK motif from amino acids 208–220, alternating with negatively charged residues. The presence of K-clusters does not, by itself, determine RNA binding, which must also depend on their sequence content because both FgREF2Δ243–327 and FgREF2ΔRBD each contain two K-clusters, but bind RNA with much different affinity.
Amino acids 268–484 (harboring K-clusters 2, 3, and 4) were sufficient for significant, albeit partial, RNA binding activity (Fig. 2B, lane 10). This indicated that the region from approximately 250 to 375 represented the actual RNA-interactive domain and not sequences that modulated the activity of a hypothetical amino-terminal one. The reduced binding of FgREF2268–484 compared with FgREF2Δ243–327, however, may indicate that amino-terminal sequences play a role in stabilizing the RNA-protein interaction.
The band-shift experiments shown in Fig. 2B were all carried out with a constant concentration of FgREF2 protein. To determine whether higher amounts of the severely defective FgREF2ΔRBD protein could promote efficient RNA binding, we carried out the titration analysis shown in Fig. 2C using FgREF2484 as control. A comparison of the binding efficiencies using varying amounts of protein demonstrated that, whereas only 25 ng (50 nM) of FgREF2484 was required to produce a discernable shifted species (apparent K d ∼ 100 nM), FgREF2ΔRBD required 200 ng (400 nM) to achieve a similar result (Fig. 2C, compare lanes 2 and 10). Thus, although there was a linear increase in the binding of FgREF2ΔRBD, it always remained eightfold weaker than FgREF2484.
Together, the RNA binding results indicate that the interactive region of REF2 is not restricted to a simple set of sequence elements, but rather, appears to be made up of multiple, redundant elements spread over a region of approximatley 125 amino acids. Nonetheless, we have identified two internal deletion mutants, FgREF2Δ287–350 and FgREF2ΔRBD, which differ by only 20 amino acids but have drastically altered RNA binding behavior. These proteins were further tested, along with the carboxyl deletions, for in vivo function.
The RNA Binding Domain Is Dispensible for REF2 Function In Vivo
In our complementation analysis, we have scored the various mutant REF2 proteins for their ability to restore normal levels of 3′ end processing and to relieve the associated growth defect in a Δref2 yeast strain. The first involves the use of a reporter CYC1 poly(A) site located within the intron of an integrated ACT1-HIS4 fusion gene that determines whether cells can grow on histidinol (44,45). For example, wild-type cells failed to grow on histidinol (are Hol–) due to the efficient use of this internal CYC1 poly(A) site, which resulted in the accumulation of truncated RNAs approximately 300 nt in length (Fig. 5B, lane 1). In Δref2 cells, increased histidinol dehydrogenase activity (encoded by HIS4) allowed for the growth of a subset of cells on selective medium (Hol+) and is associated with an increase in normal ACT1-HIS4 RNA (after splicing) due to a 50% reduction in the use of the CYC1 poly(A) site (Fig. 5B, lane 3). In addition, Δref2 strains are slightly sick at 30 °C and grow very poorly at 37 °C (see below and Fig. 5A).
To test the activity of the REF2 proteins shown in Fig. 3A, we expressed them in a Δref2 strain carrying an ACT1-CYC1-HIS4 reporter gene and transformants were scored for growth on histidinol at 30°C or growth on YPD plates at 37°C (Fig. 3B). Like the wild-type REF2533 protein and REF2484, we observed that cells expressing the RNA binding mutation REF2ΔRBD (10% activity) reverted to a Hol– phenotype and grew very well at 37°C, indicating full 3′ end processing activity. As expected, the same phenotype was observed for the smaller internal deletion, REF2Δ287–350, which affected RNA binding to a lesser degree (70% activity). Unlike REF2484, however, the carboxyl-terminal deletions REF2406 and REF2345, which also retained high levels of RNA binding in vitro, remained Hol+ as they were not able to fully restore CYC1 poly(A) site use. Interestingly, these proteins may have some function because they were observed to partially rescue growth at 37°C. This is in contrast to REF2268, which also remained Hol+ and grew as poorly at 37°C as Δref2 cells transformed with YCplac33 alone.
FIG. 3.
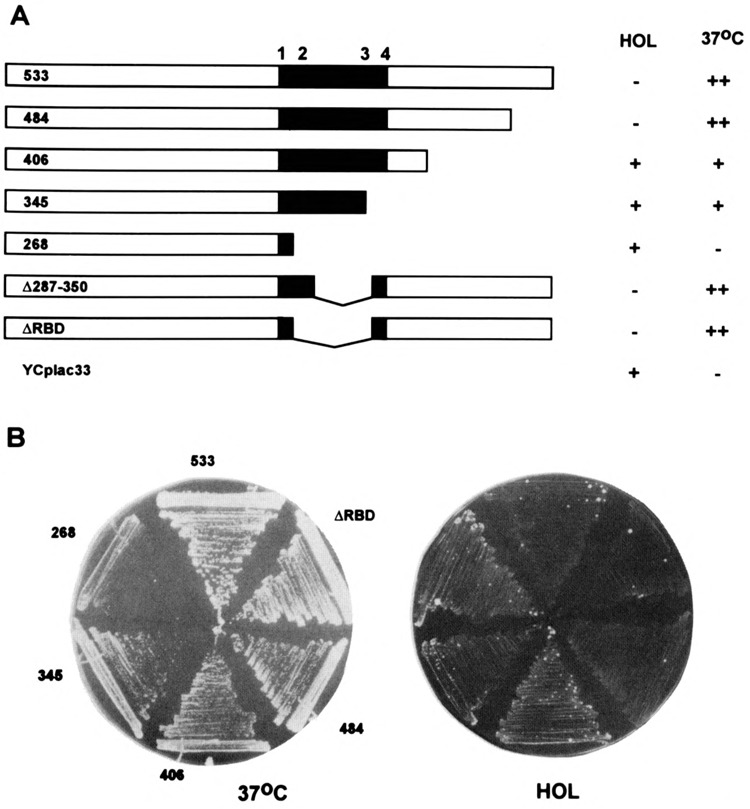
Complementation analysis. The unmodified REF2 proteins shown in (A) were expressed in a Δref2 strain from a low-copy plasmid under the control of the natural REF2 promoter. YCplac33 represents the parental vector into which the various mutated REF2 genes were inserted. The truncated protein REF2345 represents the gene product of the ref2-1 allele, which was originally identified in a selection for Hol+ phenotypes (45). Transformed strains were tested (B) for their ability to grow at 37°C or on medium containing histidinol in place of histidine (see text) and results are depicted on the right. In the case of growth at 37 °C, + + denotes wild-type growth, – indicates slight growth as observed for the Δref2 strain, whereas + represents an intermediate phenotype.
Together these results indicate that RNA binding plays only a minor role in REF2 function, at best, and points to an alternative explanation for the essential nature of the carboxyl-terminal half. We cannot rule out the possibility that the carboxyl-terminal deletions in REF2406 and REF2345 are destabilizing when expressed in yeast cells (although they are not in bacteria or as part of the LexA-REF2 fusion proteins described below) or that they disrupt nuclear localization signals (which are usually found at the amino-terminus in yeast). The lack of an antibody to REF2 coupled with our inability to fully complement Δref2 with an epitope-tagged version of REF2 has prevented the resolution of these issues to date.
Isolation and Characterization of the FIR1 Gene
To identify proteins that interact with REF2 we employed a two-hybrid strategy. The complete coding region of the REF2 gene was fused in-frame to the 202-amino acid bacterial DNA binding protein LexA that had, attached to its amino end, an SV40 T antigen nuclear localization signal. This LxREF2 protein was expressed in the reporter strain CYT10-5d, which carried an integrated GAL1-lacZ reporter gene under the control of eight LexA dimer binding sites. These cells were then transformed with genomic DNA libraries fused, in separate frames, to sequences encoding the SV40 T antigen nuclear localization signal and the GAL4 transcriptional activation domain (GAD; amino acids 768–881). Approximately 140,000 colonies were screened for β-galactosidase activity (see Materials and Methods for details), which would arise from the binding of a GAD-fusion protein to LxREF2 localized at the GAL1-lacZ promoter. Two dark blue colonies were identified against a uniform white background on X-gal plates. The GAD-fusion plasmids were rescued from these cells and further shown to cause blue color in the presence of LxREF2 but not LexA alone. Sequencing of plasmids pGAD24-1 and pGAD24-4 revealed that the GAL4 activation domains were fused in-frame to 847 or 801 amino acids, respectively, of the same gene that we have named FIR1 for Factor interacting with REF. As a further test of the specificity of the interaction between FIR1 and REF2, we have analyzed the effects of a number of deletions of the REF2 portion in LxREF2 and have identified those that disrupt binding (see below). In addition to interacting with REF2, FIR1 contains sequences which can activate transcription, because the expression of a LexA-FIR1 fusion protein alone was also positive (data not shown).
Because pGAD24-1 and pGAD24-4 contained incomplete genes, we isolated an overlapping 5′ clone of FIR1 by screening a library containing genomic PstI fragments with a FIR1-specific probe (see Materials and Methods). This led to the determination of the complete 925-amino acid sequence of FIR1 (presented in Fig. 4A), which is serine rich (13.6%) and predicted to have a molecular weight of 104,592. Like REF2, FIR1 has no closely related genes within the available data bases and, furthermore, carries no known sequence elements or motifs. A homology search with nucleotide sequences taken from the 5′ untranslated region revealed that the putative initiation ATG of FIR1 was separated by 168 bp from the ATG of the essential divergently transcribed YPT8 gene on chromosome V (22,48). Northern analysis of the FIR1 transcript revealed a single transcript of approximately 3.0 kb as expected (data not shown).
FIG. 4.
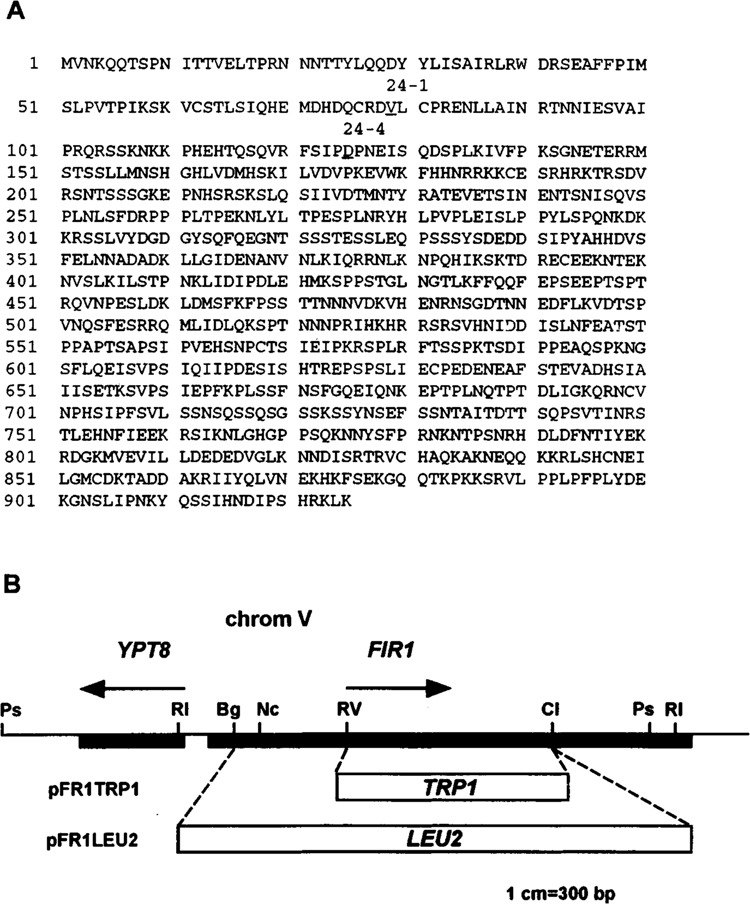
(A) Complete sequence of the 925-amino acid FIR1 protein that is expressed from the YPT8-GLN3 intergenic region on chromosome V (GenBank accession number U18778; locus SCE9537; ORF YER032W). Underlined are amino acids 79 and 125, which are fused to the GAL4 activation domain in pGAD24-1 and pGAD24-4, respectively. (B) Map of the F1R1 locus as well as the gene disruption constructs, pFRlLEU2 and pFRITRPI, which were used for generating the Δfir1 and Δref2, Δfir1 strains used in this study (see Table 1 and Materials and Methods for details).
Synergistic Action of REF2 and FIR1 in 3′ End Processing
Our discovery that FIR1 is encoded by a single nonessential gene has allowed us to compare the growth characteristics and 3′ end processing activities of isogenic haploid strains carrying inactivated alleles. To create Δfir1, amino acids 43–660 of the normal gene in strain DC35-15D were replaced with a functional LEU2 gene after transformation with pFR1LEU2 (Fig. 4B). As shown in Fig. 5A, and in stark contrast to Δref2 cells, Δfir1 had a wild-type Hol– phenotype on histidinol medium. As described above, this resulted from the relatively efficient use of a CYC1 poly(A) site situated within the ACT1-HIS4 reporter gene located on chromosome III (Fig. 5B, compare lanes 1, 2, and 3). The Δfir1 strain did, however, display reduced growth at 37°C compared to wild-type, but it was not as severe as that observed for Δref2; this is in accord with findings on Ty inactivation of chromosome V ORFs (48). The temperature sensitivity displayed by processing mutants such as Δref2 is not understood but must be a secondary effect of general suboptimal poly(A) site use because processing of the CYC1 site does not become further reduced at higher temperatures (data not shown). This suggested to us that Δfir1 may have a slightly weakened capacity for processing but that it was not severe enough to detect, in a consistent manner, by RNA analysis.
To determine if the disruption of FIR1 had any effect on poly(A) site use, we employed a sensitive assay, which measures β-galactosidase levels that result from proper expression of an ACT1-lacZ fusion gene. Previous results with both the ref2-1 and Δref2 strains [see Figs. 1 and 2 of Russnak et al. (45), for description and analysis of RNA transcribed from pSB17] demonstrated that increased blue color development on X-gal plates correlated with the appearance of extended transcripts resulting from transcriptional readthrough of cryptic poly(A) sites located within the body of the lacZ gene. In our present analysis, the low-copy plasmid pSB20, which is identical to pSB17 but does not contain an intronic CYC1 poly(A) site, was introduced into the strains indicated in Table 2 and β-galactosidase levels were quantified by liquid assay. This experiment revealed a small yet reproducible increase (∼twofold) in β-galactosidase activity in Δfir1 compared to wild-type but which was still significantly lower than the ∼fivefold effect observed for Δref2.
TABLE 2.
STEADY-STATE β-GALACTOSIDASE ACTIVITY IN STRAINS CARRYING A LOW-COPY ACTI-lacZ GENE (pSB20)
| Strain | Experiment | |||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| DC35-15D (wild-type) | 0.6 ± 0.1 | 0.8 ± 0.01 | 0.7 ± 0.05 | 0.3 ± 0.03 |
| RR9 (Δfir1) | 2.9 ± 0.4 | 1.8 ± 0.3 | 1.2 ± 0.1 | 0.66 ± 0.05 |
| (4.8 ×) | (2.25 ×) | (1.8 ×) | (2.2 ×) | |
| RR4 (Δref2) | 5.0 ± 0.5 | 3.6 ± 0.3 | 2.5 ± 0.5w | 1.8 ± 0.4 |
| (8.3 ×) | (4.5 ×) | (3.8 ×) | (6.0 ×) | |
Each number represents an average value obtained from at least three colonies (see Materials and Methods for details). Experiments 1 and 2 were carried out with the same set of pSB20 transformants whereas for Experiments 3 and 4, a second independent set was generated. Numbers in parentheses indicate the fold increase relative to wild-type.
To further test the possibilty that FIR1 and REF2 were involved in the same process and, therefore, may function synergistically, we created a double mutant by transforming strain RR4 (Δref2::LEU2) with pFRITRP1, effectively replacing amino acids 279–660 of FIR1 with a second selectable marker. This strain remained viable and did not display any growth characteristics that were distinct from the Δref2 mutant alone, either on glucose as shown in Fig. 5A or on alternative carbon sources such as galactose, acetate, lactate, glycerol, and ethanol (data not shown). RNA analysis, however, revealed that the double mutant (lane 4) had an approximate fourfold reduction in the level of the truncated RNA (relative to ACT1 message) compared to the twofold decrease that is normally observed in Δref2 (lane 3). It should be pointed out that, like Δref2 cells, the double mutant still did not result in any changes in the appearance or levels of normal endogenous mRNAs such as that transcribed from the HTB2 locus (data not shown). Regardless, removal of both REF2 and FIR1 from the cell resulted in lower 3′ end processing at the intronic CYC1 poly(A) site than was observed with the loss of either protein by itself. Because REF2 is known to influence the efficiency of 3′ end cleavage, these results implicate FIR1 in the same process, although we cannot yet formally rule out that it may instead be an inhibitor of splicing, for example.
FIR1 Interacts With the Essential Carboxyl-Terminal Domain of REF2
To further investigate the interaction between REF2 and FIR1, we asked whether the successive loss of activity observed with the carboxyl-terminal deletions described above correlated at all with changes in FIR1 binding. Thus, various segments of REF2 were expressed as LexA fusion proteins in cells also expressing the GADFIR1 protein identified in the original screen. The relative strengths of the various protein–protein interactions were estimated from the kinetics of blue color development on X-gal medium and are shown in Fig. 6.
FIG. 6.
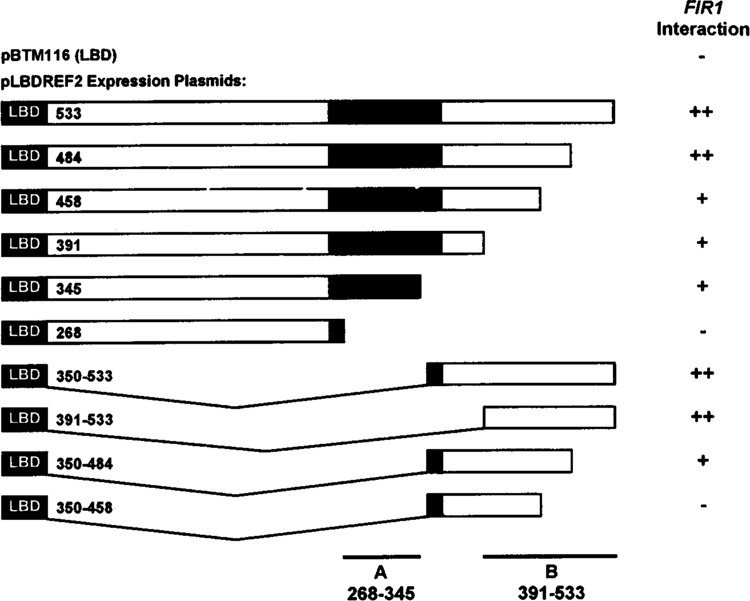
Identification of REF2 sequences that interact specifically with FIR1. Strain CYT10-5d, harboring a lacZ gene with a LexA-responsive operator, was cotransformed with pGAD24-1 (containing FIR1 amino acids 79–925) and each of the plasmids shown, which express either the LexA protein alone (pBTM116) or fusions to various regions of REF2. Transformants were streaked heavily onto X-gal indicator plates and incubated at 30°C for several days. The strength of the interaction was estimated and is shown to the right whereby + + represents blue color development within 24 h, + indicates blue color development in 48–72 h, and in the case of a negative, no blue color was observed for at least a week. Although we have not confirmed (e.g., by Western blotting) that all of these constructs are expressed at equivalent levels, the positive results are internally consistent, and the negative results are in qualitative agreement with our overall conclusions.
Although LxREF2484 was indistinguishable from wild-type in its association with FIR1, a further deletion of only 26 amino acids in LxREF2458 resulted in a discernible decrease in FIR1 binding to a level that was also observed for LxREF2391 and LxREF2345. This activity was completely removed by a deletion to amino acid 268 (LxREF2268) and so we have tentatively assigned a FIR1-interactive region (Fig. 6, site A) to sequences that, surprisingly, overlap those involved in RNA binding in vitro. Analysis of the amino-terminal deletions LxREF2350–533 and LxREF2391–533 (in which site A has been removed) demonstrated that the carboxyl-terminus of REF2 from amino acids 391–533 was sufficent for strong FIR1 binding and so we have designated this domain as site B. Binding to site B was completely disrupted by a carboxyl-terminal deletion to amino acid 458 (LxREF2350–458) consistent with the partial phenotype observed with the same deletion in LxREF2458, which still carried an intact A site. Interestingly, a comparison of sequences found between amino acids 458 and 484, whose removal abolished FIR1 binding to site B, and those found in the loosely defined A site revealed a conserved leucineasparagine-lysine triplet which was preceded, in each case, by a phenylalanine spaced three residues away (see Fig. 1).
These results suggested that the interaction between REF2 and FIR1 is complex and that the carboxyl-terminal half of REF2 has the potential of forming multiple protein–protein contacts. In particular, sequences near the tail of REF2 constitute a distinct, separable domain that is capable of tight binding to at least one protein, namely FIR1. This is important as it provides a plausible protein-interactive role for sequences that are necessary for REF2 function and again implicates FIR1 in poly(A) site function.
DISCUSSION
A growing number of genes have been identified that are required for precise and efficient endonucleolytic cleavage at the 3′ ends of eucaryotic mRNA and this process is much more complex than initially envisioned [see (31,42)]. In yeast, proteins that participate directly in this reaction and are known components of the basal cleavage apparatus (CF Iy) include RNA14 and RNA15. Cell-free extracts prepared from strains harboring mutations in either of these essential genes lack the ability to cleave pre-mRNA substrates in vitro (34) and in vivo; these strains display transcriptional readthrough of normal poly(A) sites (30). REF2, on the other hand, is a nonessential accessory factor that has been shown to stimulate cleavage of weak poly(A) sites in vitro as well as a proximal poly(A) site in a tandem reporter construct in vivo (45).
In this study we have asked whether the RNA binding potential of REF2 was necessary for 3′ end processing, its only known role within the cell. The characterization of a severe RNA binding mutant that was still able to fully complement a Δref2 strain suggests that it is not. To identify other proteins that are potentially involved in the 3′ end cleavage reaction, we carried out a two-hybrid screen using REF2 as bait and one positive was obtained. FIR1 is another nonessential gene whose disruption does not result in the same poly(A) site readthrough phenotype that is observed in Δref2 (Fig. 5). It may, however, code for a protein that also stimulates the 3′ end cleavage reaction as processing of a CYC1 poly(A) site within the ACT1-HIS4 reporter gene was decreased in a Δref2, Δfir1 double mutant compared to Δref2 alone (Fig. 5B). Although the RNA binding domain of REF2 is not critical for stimulating 3′ end processing in vivo, we cannot conclude that it lacks function altogether. In this regard, it is interesting to note that REF2268, which is completely inactive in RNA binding, is also nonfunctional in vivo, whereas proteins that maintain full RNA binding capability, such as REF2406 and REF2345, retain partial function (Fig. 3). Furthermore, although FgREF2ΔRBD is severely defective in RNA binding, it was still able to shift some RNA in our somewhat crude in vitro assay, making it possible that the residual RNA binding activity of this protein was sufficient for in vivo function. Therefore, although we believe that the primary role of REF2 in 3′ end processing is not the recognition of RNA signals, but rather the interaction with other proteins associated with the pre-mRNA substrate (see discussion below), being “RNA-philic” may certainly be accommodating.
The electrophoretic mobility-shift data presented in Fig. 2 suggested that the major determinants for RNA binding extended over a region of 125 amino acids from residues 250 to 275, and that these elements were redundant. This conclusion is based on the observation that partial deletions within this region do not significantly influence RNA binding. Examination of the sequence within this domain revealed four regions rich in lysine residues (see Figs. 1 and 2) extending from residues 253–263, 276–284, 342–349, and 360–369. Furthermore, clusters 1 and 2 are spaced 12 amino acids apart whereas clusters 3 and 4, found 58 amino acids downstream, have a conserved spacing of 11 amino acids. Several RNA binding proteins have been demonstrated to recognize RNA via elements rich in basic amino acids. These include retroviral Gag proteins, the nucleoprotein of lymphocytic choriomeningitis virus, the Rev and Tat proteins of human immunodeficiency virus, the E4 protein of adenovirus, and a number of ribosomal proteins, all of which contain a single arginine-rich cluster [reviewed in (23)]. More recently, a class of proteins has been discovered that carries two such basic motifs. For example, the hepatitis delta antigen, an RNA binding protein required for viral RNA replication, contains two arginine-rich elements separated by 29 amino acids. Not only are the two basic elements required for RNA binding, but the spacer region is important as well, because replacing it with four unrelated amino acids abolishes RNA binding activity (24). Also, the mammalian translation initiation factor eIF-4B carries two clusters of basic amino acids, spaced 30 amino acids apart within its carboxyl-terminal domain. Although this protein contains a canonical RNA recognition motif (RRM) [reviewed in (21)] in its amino-terminus, most of the RNA binding activity resides within the described carboxyl-terminal elements (32,38). The yeast homologue of eIF-4B, called TIF3/STM1, displays the same organization of RNA binding motifs, except that its two basic elements are separated by 19 residues (2,12). Although these elements are enriched for arginine residues, it is possible that the lysines found within the central domain of REF2 may also facilitate stable binding to RNA. As noted above, there appears to be some correlation between the number of K-clusters and REF2 RNA binding activity in vitro.
In lieu of an RNA-interactive role, it is possible that REF2 could stimulate RNA processing by providing additional protein–protein contacts. The identification of an interacting protein FIR1 that is also involved in determining the efficiency of poly(A) site use lends support to this model. It is now well established that a number of protein interactions are necessary for the cooperative assembly of cleavage and polyadenylation factors with their substrate RNA. In the mammalian polyadenylation reaction, for example, the affinity of poly(A) polymerase for substrate RNA is relatively weak, but in the presence of CPSF, forms a strong tertiary structure (4,36) that is further stabilized by the addition of PABII (4). Interestingly, U1A, which has been shown to be a stimulatory factor in 3′ end processing (27) and whose homologue in yeast is encoded by another nones-sential gene (MUD1) (25), can also stabilize an RNA–CPSF–poly(A) polymerase complex (28). Similarly, in the cleavage reaction CPSF and CstF form a stable ribonucleoprotein structure (14,15,36,50,54), which is further strengthened by the addition of CF Im (43). Because most of these factors consist of multiple proteins, it is probable that the cumulative effects of a great number of individual protein–protein contacts, many of them weak, may ultimately drive efficient 3′ end processing. Many of these interactions are now being identified and, in mammals, include the binding of U1A to the 160-kd subunit of CPSF (28), which, in turn, has been demonstrated to bind both the 77-kd subunit of CstF and poly(A) polymerase (37). The 77-kd protein also interacts with both of the other two components (50 and 64 kd) of CstF (51). In yeast, a subunit of PF I, FIP1, has been shown to interact with both RNA14 and poly(A) polymerase (41).
Based on the genetic analysis presented here, a number of important conclusions, inherent to their synergistic relationship, can be made concerning REF2 and FIR1. The observation that levels of 3′ end processing in Δref2 and Δfir1 strains are quite different (Fig. 5), the latter being virtually wild-type, indicates that REF2 is able to function in the absence of FIR1 binding. Conversely, decreased processing of the CYC1 poly(A) site in a double mutant compared to Δref2 alone implies that FIR1 must impart an influence in the absence of REF2. To extrapolate further, we interpret that the carboxyl-terminal region of REF2 from amino acids 406 to 533, which has been shown to interact with FIR1 (Fig. 6), must possess an additional function because the expression of REF2406, missing only those amino acids, still results in a null Hol+ phenotype (compared to the Δfir1 strain, which is Hol–). One explanation for this result is that the carboxyl-terminal half of REF2, which we know is capable of interacting with at least one protein identified here in a two-hybrid screen, may, in fact, associate with a number of proteins in vivo. These proteins could be related members of the same family to which FIR1 also belongs and whose redundancy in function would be consistent with the slight effects observed with disruptions in the FIR1 gene alone.
An alternative to this structural role is that REF2 contains some unknown catalytic activity that is required for optimal levels of 3′ end cleavage. In this light it is interesting to speculate that FIR1 or other interacting proteins may be involved in the modification or regulation of this activity. To help distinguish between these two possibilities, it will be valuable to assign REF2 and FIR1 to either known yeast factors such as CF Iy and CF IIy or to new, biochemically distinct ones. With the continued use of the genetic system we have described, it should be possible to identify other yeast genes that, together with REF2 and FIR1, help to maintain highly efficient poly(A) site use within the cell. It remains to be determined whether some new factor(s) or class of proteins is required for this task; however, the identification of two functionally related, nonessential genes that complement those being discovered in the basal cleavage machinery may speak to the emerging complexity of the 3′ end cleavage reaction.
ACKNOWLEDGEMENTS
We are greatly indebted to Steve Baker for the construction and gift of pSB20. We are grateful also to Mike Briggs and Matt Robinson for two-hybrid plasmids and strains, to Matt Robinson for pGAD.F library DNA and computer aid, and to Sandy Consaul for expert technical assistance. Additional thanks goes to Eric Phizicky, Matt Robinson, Scott Butler, and all the members of the Butler laboratory for valuable discussions. This work was supported in part by Public Health Service grant 5-R01-GM35658 to T.P. and a National Cancer Institute of Canada Senior Fellowship to R.R.
REFERENCES
- 1. Ahmed Y. F.; Gilmartin G. M.; Hanly S. M.; Nevins J. R.; Greene W. C. The HTLV-I Rex response element mediates a novel form of mRNA polyadenylation. Cell 64:727–737; 1991. [DOI] [PubMed] [Google Scholar]
- 1a. Amrani N.; Minet M.; Wyers F.; Dufour M.; Aggerbeck L. P.; Lacroute F. PCF11 encodes a third protein component of yeast cleavage and polyadenylation factor I. Mol. Cell. Biol. 17:1102–1109; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Altmann M.; Muller P. P.; Wittmer B.; Ruchti F.; Lanker S.; Trachsel H. A S. cerevisiae homologue of mammalian translation initiation factor 4B contributes to RNA helicase activity. EMBO J. 12:3997–4003; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bartel P. L.; Fields S. Analyzing protein-protein interactions using two-hybrid system. Methods Enzymol. 254:241–263; 1995. [DOI] [PubMed] [Google Scholar]
- 4. Bienroth S.; Keller W.; Wahle E. Assembly of a processive messenger RNA polyadenylation complex. EMBO J. 12:585–594; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Butler J. S.; Platt T. RNA processing generates the mature 3′ end of CYC1 mRNA in vitro . Science 242:1270–1274; 1988. [DOI] [PubMed] [Google Scholar]
- 6. Butler J. S.; Sadhale P. P.; Platt T. RNA processing in vitro produces mature 3′ ends of a variety of Saccharomyces cerevisiae mRNAs. Mol. Cell. Biol. 10:2599–2605; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chanfreau G.; Noble S. M.; Guthrie C. Essential yeast protein with unexpected similarity to subunits of mammalian cleavage and polyadenylation specificity factor (CPSF). Science 274:1511–1514, 1996. [DOI] [PubMed] [Google Scholar]
- 8. Chen D.-C.; Yang B.-C.; Kuo T.-T. One-step transformation of yeast in stationary phase. Curr. Genet. 21:83–84; 1992. [DOI] [PubMed] [Google Scholar]
- 9. Chen J.; Moore C. Separation of factors required for cleavage and polyadenylation of yeast pre-mRNA. Mol. Cell. Biol. 12:3470–3481; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chiang C-M.; Roeder R. G. Expression and purification of general transcription factors by FLAG epitope-tagging and peptide elution. Pept. Res. 6:62–64; 1993. [PubMed] [Google Scholar]
- 11. Chien C.-T.; Bartel P. L.; Sternglanz R.; Fields S. The two hybrid system: A method to identify and clone genes for proteins that interact with a protein of interest. Proc. Natl. Acad. Sci. USA 88:9578–9582; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Coppolecchia R.; Buser P.; Stotz A.; Linder P. A new yeast translation initiation factor suppresses a mutation in the eIF-4A RNA helicase. EMBO J. 12:4005–4011; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gietz R. D.; Sugino A. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74:527–534; 1988. [DOI] [PubMed] [Google Scholar]
- 14. Gilmartin G. M.; Nevins J. R. An ordered pathway of assembly of components required for polyadenylation site recognition and processing. Genes Dev. 2:578–587; 1989. [DOI] [PubMed] [Google Scholar]
- 15. Gilmartin G. M.; Nevins J. R. Molecular analyses of two poly(A) site-processing factors that determine the recognition and efficiency of cleavage of the pre-mRNA. Mol. Cell. Biol. 11:2432–2438; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gilmartin G. M.; Fleming E. S.; Oetjen J. Activation of HIV-1 pre-mRNA 3′ processing in vitro requires both an upstream element and TAR. EMBO J. 11:4419–4428; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guarente L. Yeast promoters and lacZ fusions designed to study expression of cloned genes in yeast. Methods Enzymol. 101:181–191; 1983. [DOI] [PubMed] [Google Scholar]
- 18. Hou W.; Russnak R.; Platt T. Poly(A) site selection in the yeast Ty retroelement requires an upstream region and sequence-specific titratable factors) in vitro . EMBO J. 13:446–452; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jenny A.; Minvielle-Sebastia L.; Preker P. J.; Keller W. Sequence similarity between the 73- kilodalton protein of mammalian CPSF and a subunit of yeast polyadenylation factor I. Science 274:1514–1517; 1996. [DOI] [PubMed] [Google Scholar]
- 20. Keller W. No end yet to messenger RNA 3′ processing! Cell 81:829–832; 1995. [DOI] [PubMed] [Google Scholar]
- 21. Kenan D.; Query C.; Keene J. RNA recognition: Towards identifying determinants of specificity. Trends Biochem. Sci. 16:214–220; 1991. [DOI] [PubMed] [Google Scholar]
- 21a. Kessler M. M.; Zhao J.; Moore C. L. Purification of the Saccharomyces cerevisiae cleavage/polyadenylation factor I. J.. Biol. Chem. 271:27167–27175; 1996. [DOI] [PubMed] [Google Scholar]
- 22. Lai M. H.; Bard M.; Kirsch D. R. Identification of a gene encoding a new Ypt/Rab-like monomeric G-protein in Saccharomyces cerevisiae . Yeast 10:399–402; 1994. [DOI] [PubMed] [Google Scholar]
- 23. Lazinski D.; Grzadzielska E.; Das A. Sequence-specific recognition of RNA hairpins by bacteriophage antiterminators requires a conserved arginine-rich motif. Cell 59:207–218; 1989. [DOI] [PubMed] [Google Scholar]
- 24. Lee C. Z.; Lin J.-H.; Chao M.; McKnight K.; Lai M. M. C. RNA-binding activity of hepatitis delta antigen involves two arginine-rich motifs and is required for hepatitis delta virus RNA replication. J. Virol. 67:2221–2227; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liao X. C.; Tang J.; Rosbash M. An enhancer screen identifies a gene that encodes the yeast U1 snRNP A protein: Implications for snRNP protein function in pre-mRNA splicing. Genes Dev. 7:419–428, 1993. [DOI] [PubMed] [Google Scholar]
- 26. Lingner J.; Keller man J.; Keller W. Cloning and expression of the essential gene for poly(A) polymerase from S. cerevisiae. Nature 354:496–498; 1991. [DOI] [PubMed] [Google Scholar]
- 27. Lutz C. S.; Alwine J. C. Direct interaction of the U1 snRNP-A protein with the upstream efficiency element of the SV40 late polyadenylation signal. Genes Dev. 8:576–586; 1994. [DOI] [PubMed] [Google Scholar]
- 28. Lutz C. S.; Murthy K. G. K.; Schek N.; O’Connor J. P.; Manley J. L.; Alwine J. C. Interaction between the U1 snRNP-A protein and the 160-kD subunit of cleavage-polyadenylation specificity factor increases polyadenylation efficiency in vitro . Genes Dev. 10:325–337; 1996. [DOI] [PubMed] [Google Scholar]
- 29. Mandart E.; Dufour M.-E.; Lacroute F. Inactivation of SSM4, a new Saccharomyces cerevisiae gene, suppresses mRNA instability due to rnal4 mutations. Mol. Gen. Genet. 245:323–333; 1994. [DOI] [PubMed] [Google Scholar]
- 30. Mandart E.; Parker R. Effects of mutations in the S. cerevisiae RNA 14, RNA 15 and PAP1 genes on polyadenylation in vivo . Mol. Cell. Biol. 15:6979–6986; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Manley J. L.; Takagaki Y. The end of the message—another link between yeast and mammals. Science 274:1481–1482; 1996. [DOI] [PubMed] [Google Scholar]
- 32. Methot N.; Pause A.; Hershey J. W. B.; Sonnenberg N. The translation initiation factor eIF-4B contains an RNA-binding region that is distinct and independent from its ribonucleoprotein consensus sequence. Mol. Cell. Biol. 14:2307–2316; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Minvielle-Sebastia L.; Winsor B.; Bonneaud N.; Lacroute F. Mutations in the yeast RNA 14 and RNA 15 genes result in an abnormal mRNA decay rate; sequence analysis reveals an RNA-binding domain in the RNA15 protein. Mol. Cell. Biol. 11:3075–3087; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Minvielle-Sebastia L.; Preker P. J.; Keller W. RNA 14 and RNA 15 proteins are components of a yeast pre-mRNA 3′-end processing factor. Science 266:1702–1705; 1994. [DOI] [PubMed] [Google Scholar]
- 35. Mitchelson A.; Simonelig M.; Williams C.; O’Hare K. Homology with Saccharomyces cerevisiae RNA 14 suggests that phenotypic suppression in Drosophila melanogaster by suppressor of forked occurs at the level of RNA stability. Genes Dev. 7:241–249; 1993. [DOI] [PubMed] [Google Scholar]
- 36. Murthy K. G. K.; Manley J. L. Characterization of the multisubunit cleavage polyadenylation specificity factor from calf thymus. J. Biol. Chem. 267:14804–14811, 1992. [PubMed] [Google Scholar]
- 37. Murthy K. G. K.; Manley J. L. The 160 kDa subunit of human cleavage-polyadenylation specificity factor coordinates pre-mRNA 3′-end formation. Genes Dev. 9:2672–2683; 1995. [DOI] [PubMed] [Google Scholar]
- 38. Naranda T.; Strong W. S.; Menaya J.; Fabbri B. J.; Hershey J. B. W. Two structural domains of initiation factor eIF-4B are involved in binding to RNA. J. Biol. Chem. 269:14465–14472; 1994. [PubMed] [Google Scholar]
- 39. Patel D.; Butler J. S. Conditional defect in mRNA 3′ end processing caused by a mutation in the gene for poly(A) polymerase. Mol. Cell. Biol. 12:3297–3304; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Philippsen P.; Stotz A.; Scherf C. DNA of Saccharomyces cerevisiae . Methods Enzymol. 194:169–182; 1991. [DOI] [PubMed] [Google Scholar]
- 41. Preker P. J.; Lingner L.; Minvielle-Sebastia L.; Keller W. The FIP1 gene encodes a component of a yeast pre-mRNA polyadenylation factor that directly interacts with poly(A) polymerase. Cell 81:379–389; 1995. [DOI] [PubMed] [Google Scholar]
- 42. Proudfoot N. J. Ending the message is not so simple. Cell 87:779–781, 1996. [DOI] [PubMed] [Google Scholar]
- 43. Ruegsegger U.; Beyer K.; Keller W. Purification and characterization of human cleavage factor Im involved in the 3′ end processing of messenger RNA precursors. J. Biol. Chem. 271:6107–6113; 1996. [DOI] [PubMed] [Google Scholar]
- 44. Ruohola H.; Baker S. M.; Parker R.; Platt T. Orientation-dependent function of a short CYC1 DNA fragment in directing mRNA 3′ end formation in yeast. Proc. Natl. Acad. Sci. USA 85:5041–5045; 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Russnak R.; Nehrke K. W.; Platt T. REF2 encodes an RNA-binding protein directly involved in yeast mRNA 3′-end formation. Mol. Cell. Biol. 15:1689–1697; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sambrook J.; Fritsch E. F.; Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 47. Sherman F.; Fink G. R.; Lawrence C. Methods in yeast genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1986. [Google Scholar]
- 48. Smith V.; Chou K. N.; Lashkari D.; Botstein D.; Brown P. O. Functional analysis of the genes of yeast chromosome V by genetic footprinting. Science 274:2069–2074; 1996. [DOI] [PubMed] [Google Scholar]
- 49. Stumpf G.; Domdey H. Dependence of yeast premRNA 3-end processing on CFT1: A sequence homolog of the mammalian AAUAAA binding factor. Science 274:1517–1520; 1996. [DOI] [PubMed] [Google Scholar]
- 50. Takagaki Y.; Manley J. L.; MacDonald C. C.; Wilusz J.; Shenk T. A multisubunit factor CstF is required for polyadenylation of mammalian premRNAs. Genes Dev. 4:2112–2120; 1990. [DOI] [PubMed] [Google Scholar]
- 51. Takagaki Y.; Manley J. L. A polyadenylation factor subunit is the human homologue of the Drosophila suppressor of forked protein. Nature 372:471–474; 1994. [DOI] [PubMed] [Google Scholar]
- 52. Wahle E. 3′-end cleavage and polyadenylation of mRNA precursors. Biochem. Biophys. Acta 1261:183–194; 1995. [DOI] [PubMed] [Google Scholar]
- 53. Weiss E. A.; Gilmartin G. M.; Nevins J. R. Poly(A) site efficiency reflects the stability of complex formation involving the downstream element. EMBO J. 10:215–219; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wilusz J.; Shenk T.; Takagaki Y.; Manley J. L. A multisubunit complex is required for the AAUAAA dependent cross-linking of a 64-kilodalton protein to polyadenlylation substrates. Mol. Cell. Biol. 10:1244–1248; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhao J.; Kessler M. M.; Moore C. L. Cleavage factor II of S. cerevisiae contains homologues to subunits of the mammalian cleavage/polyadenylation specificity factor and exhibits sequencespecific, ATP-dependent interaction with precursor RNA. J. Biol. Chem. 272:10831–10838; 1997. [DOI] [PubMed] [Google Scholar]