

Abstract
The phosphoprotein (P) of vesicular stomatitis virus (VSV) is a subunit of the RNA polymerase (L) that transcribes the negative strand genome RNA into mRNAs both in vitro and in vivo. We have recently shown that the P protein of VSV, New Jersey serotype (PNJ), expressed in E. coli, is biologically inactive unless phosphorylated at specific serine residues by cellular casein kinase II (CKII). In the present work, we are studying the role of phosphorylation in the activation of the P protein of Indiana serotype (PIND), which is highly nonhomologous in amino acid sequence yet structurally similar to its New Jersey counterpart. Despite the fact that E. coli-expressed PIND required phosphorylation by CKII for activation, the phosphorylation negative P protein mutants generated by altering the phosphate acceptors S and T to alanine, surprisingly, showed transcription activity similar to wild-type in vitro. Alteration of S and T residues to phenylalanine, similarly, supported substantial transcription activity (approx. 60% of wild-type), whereas substitution with arginine residue abrogated transcription (approx. 5% of wild-type). In contrast, the same mutants, when expressed in eucaryotic cells, exhibited greatly reduced transcription activity in vitro. This disparate display of transcription phenotype by the PIND mutants expressed in bacteria and eucaryotic cells suggests that these mutants are unique in assuming different secondary structure or conformation when synthesized in two different cellular milieu. The findings that, unless phosphorylated by CKII, the bacterially expressed unphosphorylated (P0) form of PIND, as well as the phosphorylation negative mutants expressed in eucaryotic cells, demonstrates transcription negative phenotype indicate that, like PNJ, phosphorylation of PIND is essential for its activity.
Keywords: Vesicular stomatitis virus, P protein, Phosphorylation, Casein kinase II
THE 29-kDa phosphoprotein (P) of vesicular stomatitis virus (VSV), a negative strand RNA virus, is an essential regulatory component of the virion-associated RNA polymerase complex (1,2). The P protein and the 241-kDa RNA polymerase (L) are both needed to transcribe the linear, single-stranded viral RNA genome, which is tightly wrapped with the nucleocapsid (N) protein (N-RNA template) (1,2). Genetic and biochemical studies have suggested that the L protein encodes all the basic transcriptional activities, whereas the P protein serves as an auxiliary component in both transcription and replication of the genome RNA (1,2,11,39). The N-RNA complex, the P protein, and the L protein, which form the transcribing ribonucleoprotein (RNP) core, can each be separately isolated and purified from the virions and effective mRNA synthesis occurs when these components are mixed in vitro (11–13).
Initial studies with P protein of Indiana serotype (PIND) isolated either from the virions or VSV-infected cell lysates indicated that the protein exists in a variety of phosphorylated states and that phosphorylation of P protein is important for the transcriptional activity of L protein (20,21,23, 27,28). Recently, the study of the P protein of New Jersey serotype (PNJ) expressed in E. coli in an unphosphorylated form (P0) provided the opportunity to explore, for the first time, the biological role of phosphorylation of P using an in vitro transcription-reconstitution system with L protein and N-RNA template. Barik and Banerjee (3) showed that purified P0 is transcriptionally inactive unless phosphorylated in vitro by cellular casein kinase II (CKII), which converted P0 to a transcriptionally active PI form (4). A second phosphorylation event also took place presumably near the C-terminal end, domain II (6), which was mediated by an L protein associated protein kinase activity (31) (designated LAK), which converted PI to P2 form (5). Alteration of Ser236 and Ser242 residues within domain II to alanine was shown to abrogate transcription in vitro (6). Moreover, the conversion of PI to P2 occurred sequentially for PNJ (5) (i.e., prior conversion of PI was obligatory to P2 formation). The CKII-mediated phosphorylation occurred at Ser59 and Ser61 within the N-terminal acidic domain I in PNJ (36), which activated the P protein and facilitated oligomerization with substantial increase in its α-helical structure, which may be involved in imparting its biological function (8). Interestingly, phosphorylation of P protein is found not to be required for N-P complex formation (37); the latter is implicated in the replication of the genome RNA (9,18,19,25,30,37).
It has been known for some time that PIND bears only 25% amino acid sequence similarity with respect to PNJ; nevertheless, both proteins are structurally similar with regard to having a highly acidic N-terminal domain and a conserved basic C-terminal domain (16). Because VSV Indiana serotype has been widely used in many laboratories for studying virus gene expression, it is of interest to study the role of phosphorylation, if any, in PIND function. Recently, Gao and Lenard (15), using the P0 protein of PIND, have confirmed the essential role of phosphorylation by CKII in its biological activity. However, in their studies LAK-mediated phosphorylation appeared not to be required because LAK-free L protein purified from the virion supported transcription in vitro (15). Nevertheless, alteration of Ser227 and Ser233 (the presumptive PNJ counterpart of LAK phosphorylation sites) to alanine abrogated transcription (14), similar to PNJ, suggesting that these phosphorylation sites may have a separate role in VSV life cycle. Jackson et al. (22) and Spadafora et al. (34) have also recently analyzed the phosphorylated states of PIND protein in vitro and in vivo. Their studies, in contrast, showed that phosphorylation negative PIND mutant (S60A/S64A) (the presumptive CKII sites), when expressed in BHK cells, supported transcription in vitro by about 28% compared to the wild-type when cell extracts containing expressed mutant P and L proteins were used in a transcription reconstitution reaction. This transcription activity of the P mutant was decernible only at a higher concentration; at a lower concentration, however, the mutant was much less active (< 5% of wild-type). These authors concluded that for PIND constitutive phosphorylation may not be essential for VSV RNA synthesis but may have a role in multimerization or complex formation with the L protein.
Recently, we initiated a detailed study of PIND with respect to both the phosphorylated states of the protein and the role of phosphorylation in the transcriptive process. We have mapped the CKII-mediated phosphorylation sites in PIND at Ser60, Thr62, and Ser64 using recombinant CKII and bacterially expressed P0 (7). In contrast, using BHK cell extract as the source of CKII or P protein expressed in COS cells, the phosphorylation sites are mapped at Ser60 and Ser64 only. These results are similar to the findings of Jackson and Perrault (21), but not in accordance with the P0 phosphorylation data in vitro of Gao and Lenard (14), who mapped the phosphorylation sites only at S60 and T62. We further confirmed that PI (CKII-mediated phosphorylation) is identical to the previously reported NS1 species and P2 (LAK-mediated phosphorylation) is identical to the NS2 species (7). The latter phosphorylation is mapped at Ser226 and Ser227 within domain II (7). Interestingly, in contrast to PNJ (5), the PI to P2 conversion in PIND was not sequential (i.e., P2 can be obtained without prior conversion to PI). These data strongly suggested that PNJ and PIND are structurally different.
In this report, we present evidence that PIND expressed in E. coli is indeed structurally different from similarly expressed PNJ with regard to the transcriptive properties of the phosphorylation defective mutants. We demonstrate here that, whereas bacterially expressed P0 is transcription-ally inactive unless phosphorylated by CKII, surprisingly, the alanine-substituted PIND mutants are as active as the wild-type in the transcription reaction. In direct contrast, when these mutants are expressed in eucaryotic cells they demonstrate transcriptionally inactive phenotype, indicating that E. coli-derived PIND mutants are perhaps structurally and conformationally different from eucaryotic cell-expressed mutants, and phosphorylation of PIND, like PNJ, is indeed essential for its activity.
MATERIALS AND METHODS
Cells, Cultures, and Virus
COS-1 and BHK-21 cells were grown as mono-layers in Dulbecco’s modified Eagle medium supplemented with 10% FBS, penicillin (50 U/ml), and streptomycin (50 μg/ml). The BHK-21 cells were infected with VSV (Indiana serotype) (Mudd-Summers strain), at a multiplicity of infection of 0.05, and the virus was purified as described previously (8). The recombinant baculovirus Bac-PAK6-L containing the L gene of VSV was propagated in spodoptera frugiperda cells (IPLB-sf-21). Sf21 cells were grown in a monolayer culture at 27°C in TNM-FH medium supplemented with 10% FBS, 50 U of penicillin per ml, and 50 μgof streptomycin per ml (29).
Construction of Plasmids
The site-directed mutagenesis within domain I of P gene was carried out by megaprimer PCR method (7). The plasmid pET-3a-P containing P gene was used as a template in all PCR amplifications. In the first step two primers, where one contains the mutation, were used to create a megaprimer product, which was then used along with a third primer in the second step PCR to obtain the full-length mutant P gene. The PCR-amplified products were restricted with NdeI and BamHI and ligated into a pET-3a vector DNA at the NdeI and BamHI sites. All mutations were confirmed by dideoxy sequencing.
For expression in COS cells the wild-type and mutant P genes were subcloned into mammalian expression vector pECE at SalI and SacI sites, by PCR method using primers encoding appropriate restriction sites. pSV-VSLl, which contains the L gene of the Indiana serotype of VSV under the transcriptional control of the Simian virus 40 late promoter in the transient expression vector pJC119 (35), was generously provided by Manfred Schubert, National Institutes of Health (32). The L gene was subcloned into pECE for expression in COS cells.
Purification of Recombinant P Protein From E. coli
Various recombinant P mutant proteins cloned in pET-3a were expressed in E. coli and purified from the inclusion bodies using guanidine hydro-chloride denaturation method as described previously (3). To obtain the soluble P protein from cytoplasmic extract, DE-3 cells were freshly transformed with the P plasmid at room temperature for 16 h. Colonies were scraped and inoculated into LB/amp and allowed to grow at 25°C up to 0.3 OD. Cells were then induced with 0.4 mM IPTG for 16 h at 25°C. Cells were suspended in a buffer containing 5 mM imidazole, 500 mM sodium chloride, 20 mM Tris-HCl, pH 7.0, and treated with 100 μg/ml lysozyme, 0.1% Triton-X-100, and incubated at room temperature for 15 min. MgCl2 was added up to 10 mM followed by DNase treatment at room temperature for 20 min. The lysate was then centrifuged at 39,000 × g for 20 min. The supernatant was passed through 0.45 μm filter and purified by nickel affinity column according to the manufacturer’s protocol (Novagen).
Purification of VSV Transcription Components
The N-RNA template and L protein were isolated and purified as described previously (10). Purified VSV (10 mg) was disrupted in a buffer containing 0.4 M NaCl, 10 mM Tris-HCl, pH 8.0, 5% glycerol, 2% Triton X-100, 1 mM DTT, by incubation on ice for 90 min with occasional stirring. The viral RNP was then purified by centrifugation onto a 100% glycerol cushion through 30% glycerol containing 10 mM Tris-HCl, pH 8.0, 10 mM NaCl, 2 mM MgCl2, 1 mM DTT for 2 h at 200,000 × g in SW 60 rotor at 4°C. RNP was collected from the top of the glycerol cushion and was treated with the same buffer containing 1 M NaCl and 0.5% Triton X-100 to dissociate L and P proteins from N-RNA template. The N-RNA was purified by centrifugation through 30% glycerol onto a 100% glycerol cushion in the same manner as described above. The high salt supernatant containing L and P proteins was stored at –80°C until further use. N-RNA was further purified by an additional high salt wash, centrifuged through 15% renographin onto a 76% renographin cushion followed by three serial banding in CsCl gradient. N-RNA was finally dialyzed against Tris-EDTA. The purity of N-RNA template was determined initially by silver staining of gels after SDS-PAGE and finally by reconstitution of transcription in vitro with recombinant (29) or viral L protein and bacterially expressed P protein (3).
The high salt fraction containing L and P was dialyzed against phosphocellulose (PC) buffer (20 mM Tris-HCl, pH 7.5, 10% glycerol, 1 mM DTT) and loaded onto a 2.0-ml phosphocellulose column preequilibrated with the same buffer. The column was washed with PC buffer and the bound L protein was eluted with a 0.1–1.0 M NaCl gradient in the same buffer. Fractions in which L was completely free of P protein (as identified by silver staining) were pooled and further chromatographed twice onto a phosphocellulose column to remove any contaminating CKII (as checked by phosphorylation of bacterially expressed VSV P protein as substrate).
Expression of Recombinant Baculovirus L Protein
The recombinant L protein was expressed in spodoptera frugiperda cells infected with recombinant baculovirus BacPAK6-L containing the L gene under the control of a polyhedrin promoter and cycloplasmic extracts containing L activity were prepared as described in detail earlier (29).
Expression of Recombinant L and P Proteins in COS Cells
COS cells were transfected with L or P containing plasmids at a confluency of 50–70% in 60-mm culture dishes. Supercoiled DNA (3 μg of L and 1 μg of P) was either cotransfected or transfected separately using lipofectamine (Gibco-BRL) via the proprietary protocol. Cells were incubated for 24 h in opti-MEM (Gibco-BRL) medium to allow uptake of transfected DNA and then incubated in Dulbecco’s modified Eagle media supplemented with 10% FBS for additional 24 h to allow expression of L and P proteins.
Preparation of Cytoplasmic Extracts
Cytoplasmic extracts of COS cells containing recombinant L and P proteins were prepared as described previously (33). Cells were washed three times with phosphate-buffered saline and then incubated on ice for 5 min in 500 μl of ice-cold hypotonic homogenization buffer [20 mM N-2-hydroxyethylpiperazine-N′-2-ethane sulfonic acid (HEPES, pH 7.4), 25 mM KC1, 1.5 mM MgCl2, 1 mM dithiothreitol (DTT)]. Cells were scraped and transferred to a dounce homogenizer and disrupted by 50 strokes. The lysate was centrifuged at 12,000 × g at 4°C for 3 min. The supernatant was analyzed for the presence of L or P protein by Western blot analysis and stored at –80°C.
PAGE and Western Blot Analyses
Proteins were subjected to 10% SDS-poly-acrylamide gel electrophoresis as described by Laemmli (24). The resolved proteins were transferred to nitrocellulose membrane by electroblotting. After blocking and probing with primary antibody raised against a synthetic peptide of the L protein of VSV (generously provided by Manfred Schubert, N.I.H.), or anti-P antibody, the membrane was probed with peroxidase-linked goat anti-rabbit immunoglobulin G, and the complex was detected by the procedure described by Towbin et al. (38).
Transcription Reconstitution In Vitro
Transcription reaction in vitro was carried out as described earlier (3). The reaction mixture (25 Atl) contained 50 mM Tris-HCl, pH 8.0, 0.1 M NaCl, 5 mM MgCl2, 4 mM DTT, 100 mM UTP, 1 mM (each) ATP, GTP, and CTP, 10 μCi of [α-32P]UTP (specific activity, 800 Ci/mmol, Du-Pont), 0.1 μg P protein, 0.2 μg of N-RNA, 1–10 μg of insect cell cytoplasmic extract containing L, 40 U of RNase inhibitor, and 50 ng of actinomycin D. The reaction mixture was incubated at 30°C for 2 h, 200 ng of oligo(dT) and 0.5 U of RNase H were added, and the reaction mixture was further incubated at 37°C for 15 min to remove the poly(A) tail. The transcripts were extracted with phenol-chloroform and precipitated with 2.5 volumes of ethanol at –80°C. The RNA products were visualized by autoradiography following electrophoresis in a 5% polyacrylamide gel containing 7 M urea. The transcripts were quantitated by densitometric scanning of the autoradiogram. The transcription levels for mutants were calculated based on P WT as 100%.
In Vitro Synthesis ofP Proteins
[35S]Methionine-labeled P proteins were made using TNT system according to the manufacturer’s protocol (Promega) (17). The proteins were checked by SDS-PAGE and then loaded on to 30% glycerol and centrifuged at 100,000 × g for 3 h. The protein was collected from the top of the 30% glycerol and verified by SDS-PAGE. The reticulocyte lysate made P protein was then used in an in vitro transcription reaction as described earlier (17).
RESULTS
Role of CKII in PIND Protein Activation
We have shown previously that phosphorylation of PNJ protein by CKII is essential for its ability to function as a transcriptional activator in VSV transcription reaction reconstituted with L protein and viral N-RNA template free of cellular protein kinase activity (4). We wanted to establish and confirm the role of CKII-mediated phosphorylation of PIND protein in VSV transcription. To achieve a kinase-free transcription reaction in vitro, we purified viral L free of cellular kinase as described previously (5). The N-RNA template devoid of protein kinase activity was obtained by banding three times in CsCl. Bacterially expressed wild-type P was then tested for transcriptional activity using kinase-free viral L and N-RNA template. As shown in Fig. 1, bacterially expressed P protein (P0) was virtually inactive in supporting transcription. However, transcription was restored when recombinant CKII was included in the reaction mixture, and, as expected, addition of heparin (a specific inhibitor of CKII) inhibited transcription. These results demonstrate that, like PNJ, phosphorylation of bacterially expressed PIND protein by CKII is essential for its activation and in this respect our data confirm the findings of Gao and Lenard (15).
FIG. 1.
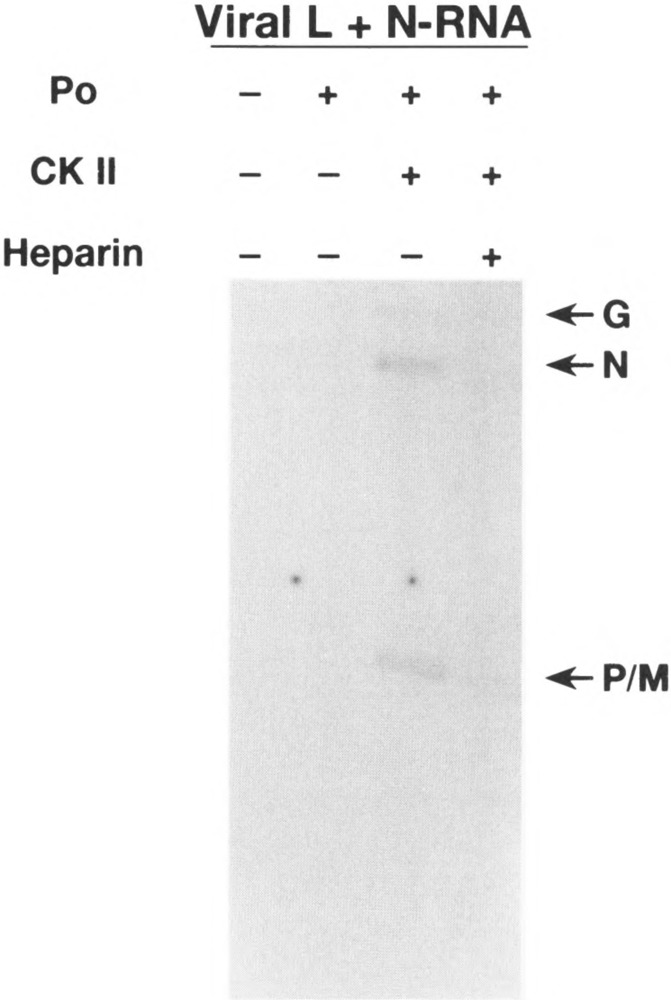
Role of CKII in VSV transcription. In vitro VSV transcription reaction contained bacterially expressed P protein (P0), host kinase-free N-RNA template and viral L protein. After incubation for 2 h at 30°C, the poly(A) tails of nascent transcripts were removed by treatment with RNase H, and the transcripts were purified by phenol-chloroform extraction and precipitation with ethanol. [32P]UTP-labeled transcripts were visualized by autoradiography of 5% polyacrylamide gel containing 7 M urea. Transcripts of different genes are indicated by G, N, and P/M. CKII (0.01/mU) and heparin (5 μg/ml) were added as indicated.
Transcriptional Ability of Bacterially Expressed P Mutants
Next, we wanted to carry out mutational analyses of PIND to directly link phosphorylation to its activation by altering CKII phosphorylation sites, Ser60, Thr62, and Ser64 (7), within the acidic domain I to different amino acids. First, we determined whether a negatively charged amino acid (e.g., glutamic acid) substituting for S and T residues can mimic the function of phosphorylated P protein in transcription reaction. For this and other subsequent experiments, we used recombinant baculovirus-expressed L protein (29) in the transcription reconstitution reactions. As shown in Fig. 2, single glutamic acid-substituted PIND mutants (P60E, P62E, P64E) demonstrated almost the same transcriptional activity as wild-type. Similarly, doubly substituted mutants (P60E/62E, P62E/64E, P60E/64E) as well as triple mutant P3E were all active as wild-type P protein; the triple mutant P3E was most active in transcription (167% wild-type). We conclude that glutamic acid-substituted PIND mutants are transcriptionally active, suggesting that the negative charge imparted by the glutamic acid substitution must have provided the similar acidic environment and structure as the phosphorylated P protein and thus favored transcription.
FIG. 2.
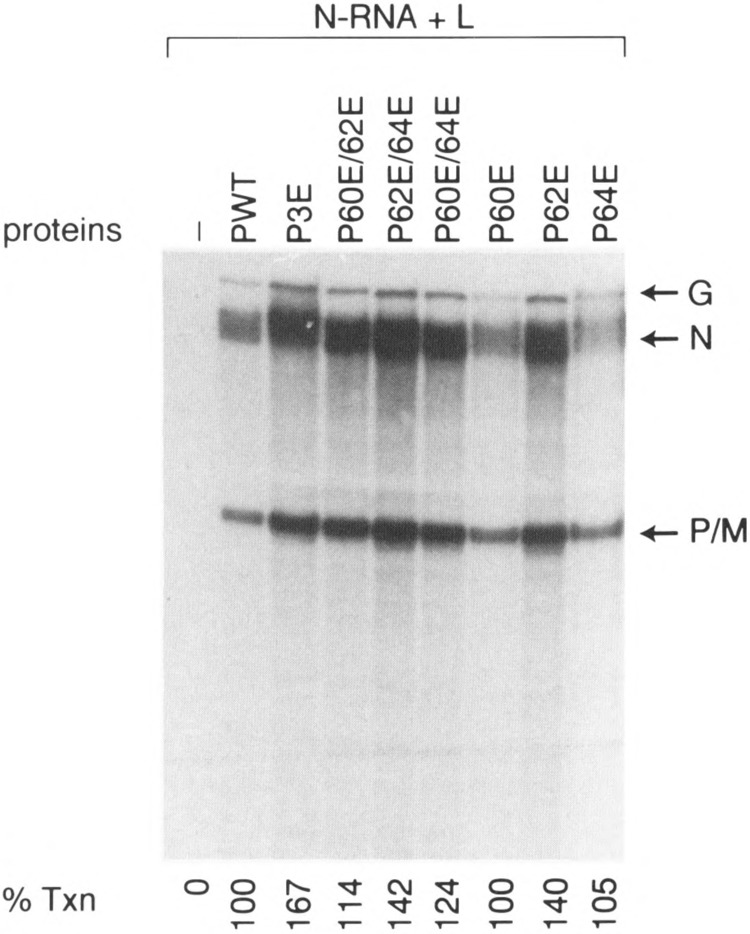
Transcriptional ability of glutamic acid-substituted P mutants. In vitro transcription reconstitution was carried out using 0.2 μg N-RNA template, 0.1 μg of recombinant P protein, and ≈1.0 μg of cytoplasmic extract from Sf21 cells expressing the L protein. mRNAs were processed as described above and separated on a 5% polyacrylamide gel containing 8 M urea. P0 stands for bacterially expressed unphosphorylated P wild-type. The various mutants are P3E (Ser60, Thr62, and Ser64 mutated to Glu), P60E/62E (Ser60, Thr62 mutated to Glu), P62E/64E (Thr62, Ser64 mutated to Glu), P60E/64E (Ser60, Ser64 mutated to Glu). P60, P62E, and P64E are single mutants in which Ser60, Thr62, or Ser64 is mutated to Glu, respectively.
Unexpected results were obtained when we determined the effect of alanine substitution in PIND protein and measured their activities in vitro. As shown in Fig. 3, bacterially expressed P mutants in which single amino acid Ser60, Thr62, or Ser64 was altered to alanine were as active as P wild-type, suggesting that the remaining unaltered residues have been fully phosphorylated (7) to yield active P protein. Surprisingly, double Ala-substituted mutants (P60A/64A, P60A/62A, P62A/ 64A) were also similarly active in transcription. More important, even the phosphorylation defective triple mutant (P3A) is fully active in transcription. These observations are in direct contrast to that obtained for PNJ where mutation of Ser59, Ser61 to Ala completely abolished transcription (36). Thus, it seems that whereas unphosphoryiated P protein (i.e., P0) is transcriptionally inactive unless phosphorylated by CKII, substitution of all three residues with Ala somehow preserves the full activity of P protein in an in vitro transcription-reconstitution assay. These unexpected results suggested to us that Ala substitution must have retained the overall structure of the P protein in a manner that behaves like phosphorylated P. Next, we addressed the possibility that P-Ala mutants may have assumed altered structure due to their extraction from the inclusion bodies in E. coli during extraction by guanidine-HCl denaturation followed by renaturation. To avoid this drastic step, we modified the induction step by which the expressed mutant proteins were obtained in a soluble form (see Materials and Methods). As shown in Fig. 3B, the P3A purified from E. coli in a soluble form is also transcriptionally active, indicating that both soluble and guanidine-HCl-solubilized P mutants are equally active in transcription. To explore further the possibility that the observed activity of P3A mutant may be due to using a high concentration of the mutant protein (34), we measured its activity over a wide range of concentration ranging from 5 ng up to 100 ng in a transcription reconstitution experiment. As shown in Fig. 4, both Pwt and P3A displayed a linear increase in transcription with increasing concentration of P, indicating that P3A indeed behaves like the Pwt at all concentrations used.
FIG. 3.
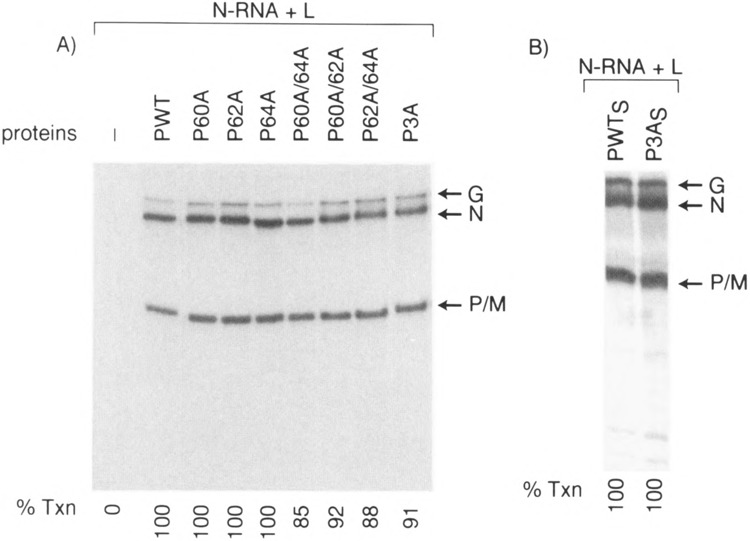
Transcriptional ability of Ala-substituted P mutants. (A) In vitro transcription reconstitution was carried out as described in Fig. 2. P60A, P62A, and P64A are single mutants where Ser60, Thr62, or Ser64 is mutated to Ala, respectively. The double mutants are P60A/64A (Ser60, Ser64 mutated to Ala), P60A/62A (Ser60, Thr62 mutated to Ala), and P62A/64A (Thr62, Ser64 mutated to Ala). P3A is the triple mutant where Ser60, Thr62, and Ser64 mutated to Ala. (B) Transcription of N-RNA using soluble PO and P3A. S stands for soluble.
FIG. 4.
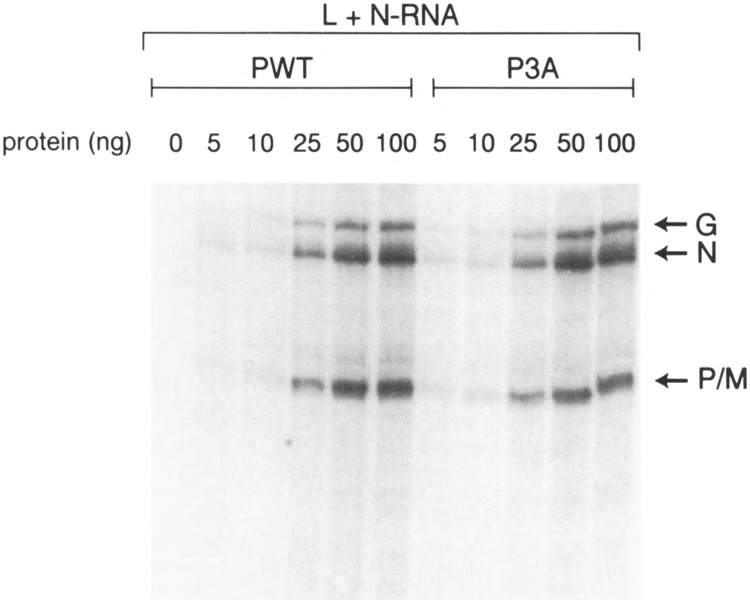
Effect of P protein concentration on transcription. In vitro transcription reconstitution was carried out using 0.2 μg of N-RNA, 1.0 μg of L extract from Sf21 cells, and bacterially expressed P protein ranging from 5 ng up to 100 ng.
We then examined whether alteration of CKII sites to some other amino acids may have any effect on their biological activities. Accordingly, we altered the three residues Ser60, Thr62, Ser64 to Thr (a phosphate acceptor), Arg (a basic amino acid), or Phe (an aromatic amino acid). As shown in Fig. 5, substitution by Thr (P3T) and Phe (P3F) yielded approximately 30% and 60% of wild-type transcriptional activity, respectively. However, substitution of all three residues by Arg (P3R) virtually abolished its transcriptional ability (approx. 5%). These results indicate that substitution by a positively charged amino acid disrupts the structure to an extent that it is inactive in transcription. However, substitution with T and F maintains the active structure, albeit, improperly. These results underscore the point that the S and T residues in PIND can accommodate other amino acids without drastically affecting its structure. However, unphosphoryiated PIND remains inactive unless phosphorylated at these crucial acceptor sites.
FIG. 5.
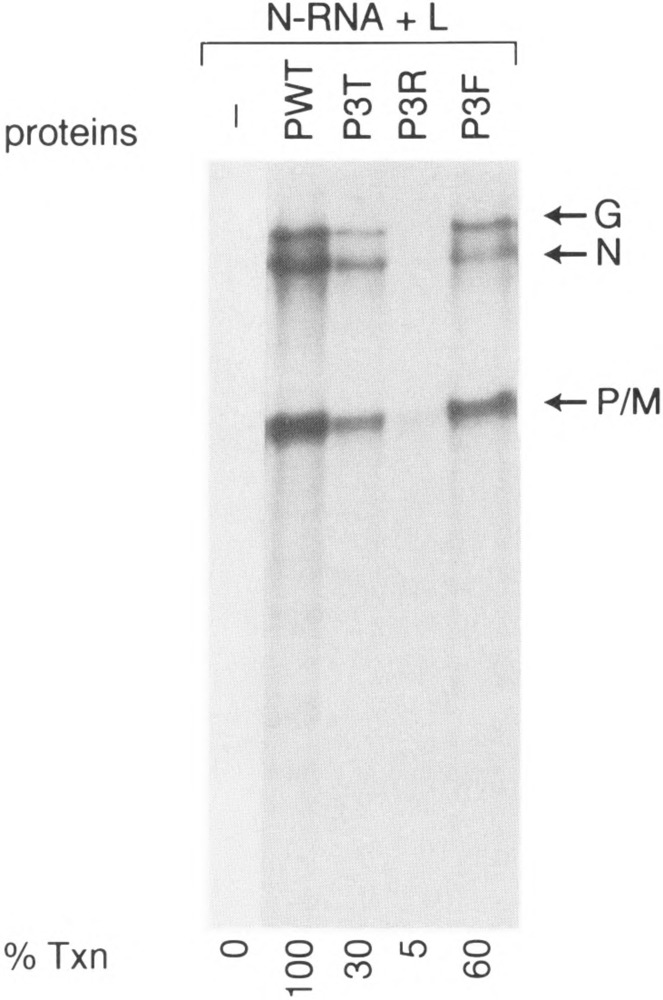
Transcriptional ability of other P mutants. In vitro transcription reconstitution was carried out as described in Fig. 2. P3T, P3R, and P3F stand for triple mutants in which Ser60, Thr62, and Ser64 are all mutated to either Thr or Arg.
Transcriptional Ability of P Mutants Expressed in COS Cells
Because the phosphate-defective bacterially expressed P3A PIND mutant is fully active in supporting transcription in vitro, we wanted to study the transcriptional ability of the same mutant expressed in mammalian cells. To address this question, we transiently coexpressed P mutants in COS cells along with L protein and the L-P-containing cell extract was then used in a transcription reconstitution experiment. In direct contrast to the E. coli-expressed P mutant, the P3A expressed in COS cells supported approx. 5% of transcriptional activity compared to the wild-type P protein (Fig. 6A). Western blot analyses showed that the amounts of expressed L and P protein were the same in both cases, indicating that the poor transcription capability of the P mutant is not due to the low expression of L and P proteins. We also carried out mixing experiments in which individually expressed L and P proteins in COS cells were mixed and the activity of the P protein assayed by transcription-reconstitution reactions. As shown in Fig. 6B, the activity of P3A was approx. 5% compared to wild-type, indicating again that, unlike the bacterially expressed P protein, the P3A, when expressed in the eucaryotic cellular milieu, perhaps has its structure altered, thereby exhibiting a true phosphorylation negative phenotype. It further indicates that phosphorylation indeed plays a vital role for the P protein to attain the appropriate structure in the cellular milieu; for example, folding and/or oligomerization. We next wanted to see whether increasing concentration of P3A protein expressed in COS cells will affect its transcriptional ability. As shown in Fig. 7, when the concentration of Pwt was increased (from 0.25 to 5 μg of COS extract) there was a dose-dependent increase of transcription. In contrast, virtually no increase in transcriptional activity was observed when P3A concentration was similarly increased to as high as 5 μg extract of P3A, indicating that phosphorylation negative mutant (P3A) remains inactive at all concentrations used.
FIG. 6.
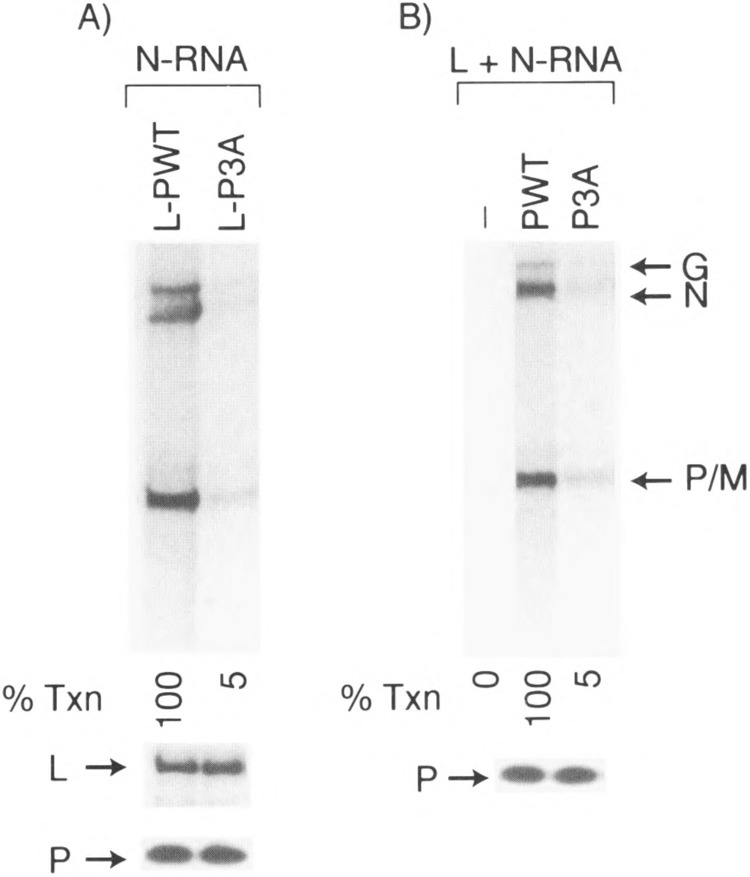
Transcriptional ability of P3A mutant expressed in COS Cells. (A) In vitro transcription reconstitution was carried out using 0.2 μg N-RNA and 6 μg of COS extract coexpressing L and P, L-Pwt (coexpressed L and Pwt), or L-P3A (coexpressed L and P3A). Cell extract (18 μg) coexpressing L and P protein was subjected to SDS-10% PAGE. Western blot of L and P proteins in each extract is shown below the transcripts. (B) L- and P-expressing COS extract (3 μg each) was mixed in a transcription reconstitution experiment. Western blot analysis of Pwt and P3A using P antibody is shown below the transcription figure.
FIG. 7.
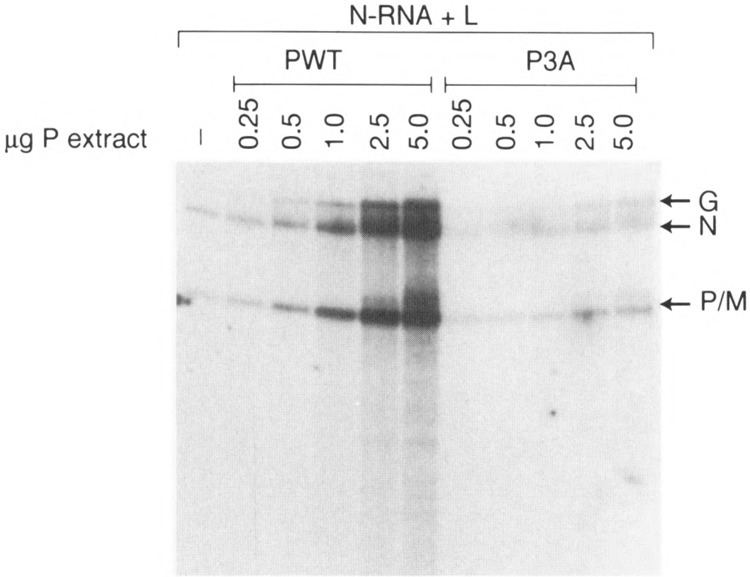
Effect of concentration of COS-expressed P protein on transcription. In vitro transcription reconstitution was carried out using 0.2 μg of N-RNA and 3 μg of COS extract expressing L protein. Various amounts of COS extract expressing P protein are indicated.
We also tested the transcriptional ability of other P mutants, including P60A/64A, P3E, or P3T, and coexpressed these mutants with L plasmid in COS cells. As shown in Fig. 8, the P mutants P60A/64A, P3E, and P3T exhibited significantly reduced activity, approx. 15%, 35%, and 20% of wild-type, respectively, compared to the corresponding E. coli-expressed P mutants. Western blot analyses showed that the expression levels of L and P mutant proteins were similar in all experiments (Fig. 8). These results indicate that even the phosphate-defective P3E mutant, which was transcriptionally more active than wild-type protein when expressed in bacteria, showed decreased transcriptional activity (35%) when expressed in COS cells, strongly implicating that phosphorylation of P protein is important in transcription. It is interesting to note that P60A/64A, double mutant, exhibited activity consistently higher than the P3A mutant (15% vs. 5%, respectively), suggesting that phosphorylation of T62 may be important in P function.
FIG. 8.
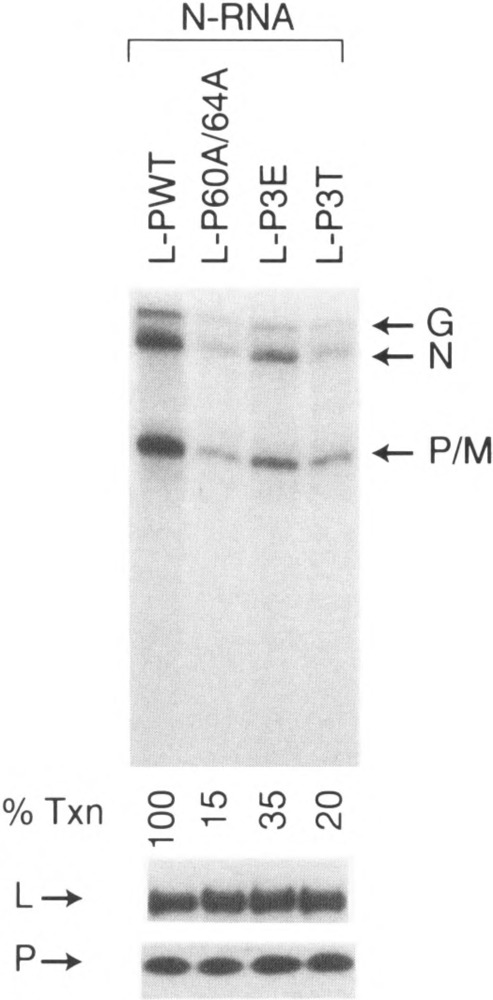
Transcriptional ability of coexpressed L and other P mutants in COS cells. In vitro transcription reconstitution was carried out using 0.2 μg N-RNA and 6 μg of COS extract coexpressing L and P mutants, as described in Materials and Methods. Western blot of L and P protein in each extract is shown below the figure.
Finally, to confirm that eucaryotic cellular milieu indeed influences the proper structural configuration of P proteins, we expressed P protein (Pwt and P3A) in rabbit reticulocyte lysate by translation in vitro of P mRNAs and the transcriptional activity of the P protein was studied. As shown in Fig. 9, the mutant P3A translated in vitro was virtually inactive in supporting transcription. This result further supports our contention that phosphorylation is important when the mutant P proteins are expressed in a eucaryotic cellular environment and the same mutant P proteins acquire different conformation when expressed in bacteria similar to the phosphorylated wild-type P protein expressed in either environment.
FIG. 9.
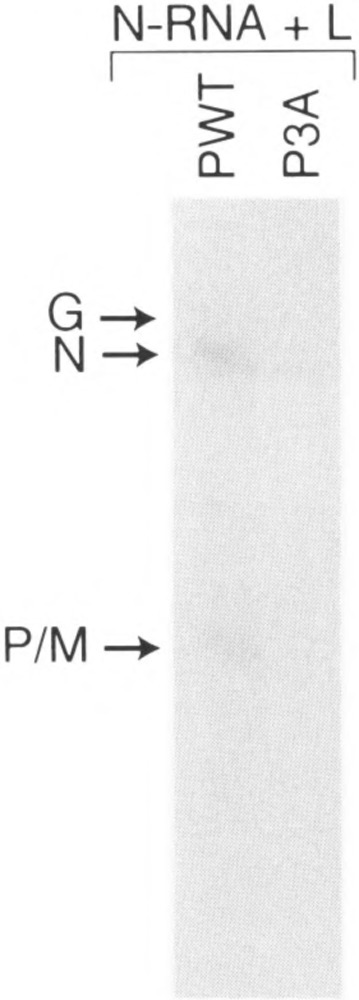
Transcriptional ability of P protein made in reticulocyte lysate. In vitro transcription reconstitution was carried out using equal amounts of in vitro rabbit reticulocyte lysate-translated Pwt and P3A (based on densitometric scanning of labeled P band) as described in Materials and Methods.
DISCUSSION
Availability of E. coli-expressed VSV PNJ in an unphosphorylated form has greatly facilitated the study of the role of phosphorylation in P protein function in transcription (3). These studies have led to the discovery of cellular CKII and elucidation of its role in phosphorylation of specific serine residues within the N-terminal acidic domain (domain I) of PNJ that are directly involved in activating the P protein (36). Alteration of the S residues (S59 and S61) to alanine completely abrogated PNJ activity in a transcription-reconstitution reaction. Moreover, these findings led to the establishment of a cascade phosphorylation for PNJ in which prior phosphorylation of serine residues in domain I was necessary to phosphorylate specific serine residues (S236 and S242) within the C-terminal domain II by an L-associated kinase (5,6,31), recently characterized as a cellular kinase (7). However, using E. coli-expressed P protein of VSV Indiana serotype to study the role of phosphorylation in P function produced perplexing results. Here, we have presented evidence that, in contrast to the unphosphorylated PIND expressed in E. coli, which required CKII for activation (Fig. 1), the alanine-substituted phosphorylation negative mutants expressed in E. coli are fully active in transcription in vitro (Fig. 3). However, the same mutants expressed in eucaryotic cells exhibited significantly reduced transcriptional activity in vitro (Figs. 6, 7, and 8).
This display of disparate transcription phenotype by PIND expressed in E. coli and eucaryotic cells underscores a profound structural difference between the same protein expressed in two different cellular milieu. The unphosphorylated PIND (P0) expressed in E. coli requires phosphorylation by CKII to manifest its biological activity presumably due to its conversion to a structurally competent form. In this respect, both PNJ and PIND demonstrate identical phenotype. Contrary to PNJ mutants (36), the alanine-substituted, phosphorylation negative PIND mutants must have assumed a structure or conformation that is similar, if not identical, to the fully active wild-type phosphorylated form. Similarly, substituting the S residues to F, the derived mutant still maintained the transcription positive phenotype, albeit at a reduced level, suggesting that they are structurally competent to act as a transcription factor. Interestingly, the PTTT mutant (where S60 and S64 were altered to T) supported transcription to approximately 30%, underscoring the importance of phosphorylation of the S residues in the P protein; the PTTT mutant is phosphorylated to approx. 80% by CKII in vitro (data not shown). Similarly, the single P mutants in which one of the serines was intact for phosphorylation (P60A or P64A) supported appreciable transcription (50–60%) (data not shown) underscoring the importance of phosphorylation in P protein function. Substitution with R, a basic amino acid, on the other hand, drastically altered the structure of the P protein to convert it into inactive form. As expected, substitution with glutamic acid (E) increases the net negative charge of the acidic domain, thus maintaining the biological activity of the P protein similar to PNJ [(15), unpublished data]. Thus, it seems that there is a fundamental structural difference between the P0 and P3A or P3F proteins of PIND. The latter proteins presumably mimic PI with regard to both structure and function, whereas corresponding alanine-substitutted mutant of PNJ is structurally different from the P1 form and functionally inactive (8,36). These results underscore the point that the P proteins of two serotypes bear inherent structural dissimilarity yet display functional similarity, at least to bind to their cognate L and N-RNA template to carry out transcription. Detailed structural and biophysical analyses of PIND wild-type and its mutants would be stimulating to gain insight into this phenomena.
Interestingly, the same P mutants behaved disparately when they were expressed in eucaryotic cells (Fig. 6, 7, and 8). The A-substituted double and triple mutants in COS cell extracts showed drastically low transcription activity, suggesting that the P mutants must have assumed a structure incompatible to function as the transactivator. This contention is further supported by the observation that the reticulocyte lysate-expressed P3A mutant was also inactive in transcription (Fig. 9).Similarly, in contrast to fully active bacterially expressed P3E, the COS cell-expressed mutant displayed significantly reduced transcriptional activity. Thus, it seems that there is a profound structural difference between the P mutants when expressed in E. coli and in mammalian cells. It is important to note that addition of the COS cell extract to the E. coli-expressed PIND mutants did not reduce its transcriptional activity (data not shown), suggesting that inactivity of the PIND mutants in the cellular milieu must have occurred de novo during their synthesis in COS cells. We recently used the phosphorylation negative P mutants for their ability to support transcription of a plasmid, p9BN, which synthesizes N mRNA upon replication using vaccinia virus-based mini genome system (Paitnaik, unpublished observation). These studies similarly showed that phosphorylation is indeed required for P protein functioning in vivo. These data clearly establish a fundamental structural difference between the P protein of two serotypes. The precise basis for this difference in structure remains to be determined. It will be interesting to map the domain within the PIND that regulates and or influences its structure by systematically introducing increasing lengths of PIND in PNJ and to study whether the latter can be converted into transcription positive phenotype.
Finally, the fact that, unlike PNJ (36), PIND can be converted directly to the P2 form without the obligate intermediate of P1 (7) also points to an important structural difference between these two proteins. For PNJ, biophysical and biochemical studies have provided strong evidence (8) that there is an alteration of secondary structure in response to phosphorylation leading to P1 form. Similar studies will be insightful to understand the structure of P1 and P2 forms of PIND. Additionally, it will be interesting to study how the differences in structure between PNJ and PIND play a role in the observed serotype specificity in transcription (10) and in the superinfection exclusion of one serotype over the other (26).
ACKNOWLEDGMENTS
This work was supported by Public Service Grant AI26585 (A.K.B.). We thank Laura Tripepi for her secretarial assistance.
REFERENCES
- 1. Banerjee A. K. Transcription and replication of rhabdoviruses. Microbiol. Rev. 51:66–87; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Banerjee A. K.; Barik S. Gene expression of vesicular stomatitis virus genome RNA. Virology 188:417–428; 1992. [DOI] [PubMed] [Google Scholar]
- 3. Barik S.; Banerjee A. K. Cloning and expression of the vesicular stomatitis virus phosphoprotein gene in Escherichia coli: Analysis of phosphorylation status versus transcriptional activity. J. Virol. 65:1719–1726; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barik S.; Banerjee A. K. Phosphorylation by cellular casein kinase II is essential for transcriptional activity of vesicular stomatitis virus phosphoprotein P. Proc. Natl. Acad. Sci. USA 89:6570–6574; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barik S.; Banerjee A. K. Sequential phosphorylation of the phosphoprotein of vesicular stomatitis virus cellular and viral protein kinases is essential for transcriptional activation. J. Virol. 66:1109–1118; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chattopadhyay D. J.; Banerjee A. K. Phosphorylation within a specific domain of the phosphoprotein of vesicular stomatitis virus regulates transcription in vitro. Cell 49:407–414; 1987. [DOI] [PubMed] [Google Scholar]
- 7. Chen J.-L.; Das T.; Banerjee A. K. Phosphorylated states of vesicular stomatitis virus P protein in vitro and in vivo. Virology 228:200–212; 1997. [DOI] [PubMed] [Google Scholar]
- 8. Das T.; Gupta A. K.; Sims P. W.; Gelfand C. A.; Jentoft J. E.; Banerjee A. K. Role of cellular casein kinase II in the function of the phosphoprotein (P) subunit of RNA polymerase of vesicular stomatitis virus. J. Biol. Chem. 270:24100–24107; 1995. [DOI] [PubMed] [Google Scholar]
- 9. Davis N. L.; Wertz G. W. Synthesis of vesicular stomatitis virus negative strand RNA in vitro: Dependence on viral protein synthesis. J. Virol. 41:821–832; 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De B. P.; Banerjee A. K. Specific interactions of L and NS proteins of vesicular stomatitis virus with heterologous genome ribonucleoprotein template lead to mRNA synthesis in vitro. J. Virol. 51:628–634; 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. De B. P.; Banerjee A. K. Requirements and functions of vesicular stomatitis virus L and NS proteins in the transcription process in vitro. Biochem. Biophys. Res. Commun. 26:40–49; 1985. [DOI] [PubMed] [Google Scholar]
- 12. Emerson S. U.; Wagner R. R. L protein requirements for in vitro RNA synthesis by vesicular stomatitis virus. J. Virol. 12:1325–1335; 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Emerson S. U.; Yu Y. H. Both NS and L proteins are required for in vitro RNA synthesis by vesicular stomatitis virus. J. Virol. 15:1348–1356; 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gao Y.; Lenard J. Cooperative binding of multimeric phosphoprotein (P) of vesicular stomatitis virus to polymerase (L) and template: Pathways of assembly. J. Virol. 69:7718–7723; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gao Y.; Lenard J. Multimerization and transcriptional activation of the phosphoprotein (P) of vesicular stomatitis virus by casein kinase II. EMBO J. 14:1240–1247; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gill D. S.; Banerjee A. K. Vesicular stomatitis virus NS protein: Structural similarity without extensive sequence homology. J. Virol. 55:60–66; 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gill D. S.; Chattopadhyay D.; Banerjee A. K. Identification of a domain within the phosphoprotein of vesicular stomatitis virus that is essential for transcription in vitro. Proc. Natl. Acad. Sci. USA 83:8873–8877, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gupta A. K.; Banerjee A. K. Expression and purification of vesicular stomatitis virus N-P complex from Escherichia coli: Role in genome RNA transcription and replication in vitro. J. Virol. 71:4264–4271; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Howard M.; Wertz G. W. Vesicular stomatitis virus RNA replication: A role for the NS protein. J. Gen. Virol. 70:2683–2694; 1989. [DOI] [PubMed] [Google Scholar]
- 20. Hsu C. H.; Kingsbury D. W. Constitutively phosphorylated residues in the NS protein of vesicular stomatitis virus. J. Biol. Chem. 260:8990–8995; 1985. [PubMed] [Google Scholar]
- 21. Hsu C. H.; Morgan E. M.; Kingsbury D. W. Site specific phosphorylation regulates the transcriptive activity of vesicular stomatitis virus NS protein. J. Virol. 43:104–112; 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jackson R. L.; Spadofora D.; Perrault J. Hierarchy constitutive phosphorylation of the vesicular stomatitis virus P protein and lack of effect on PI to P2 conversion. Virology 214:189–197; 1995. [DOI] [PubMed] [Google Scholar]
- 23. Kingsford L.; Emerson S. U. Transcriptional activities of different phosphorylated species of NS protein purified from vesicular stomatitis virions and cytoplasm of infected cells. J. Virol. 33:1097–1105; 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Laemmli U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685; 1970. [DOI] [PubMed] [Google Scholar]
- 25. LaFerla F. M.; Peluso R. W. The 1:1 N-NS protein complex of vesicular stomatitis virus is essential for efficient genome RNA replication. J. Virol. 63:3852–3857; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Legault D. D.; Takayasu D. D.; Prevec L. Heterotypic exclusion between vesicular stomatitis viruses of the New Jersey and Indiana serotypes. J. Gen. Virol. 35:53–65; 1977. [DOI] [PubMed] [Google Scholar]
- 27. Marnell L. L.; Summers D. F. Characterization of the phosphorylated small enzyme subunit, NS, of the vesicular stomatitis virus RNA polymerase. J. Biol. Chem. 259:13518–13524; 1984. [PubMed] [Google Scholar]
- 28. Masters P. S.; Banerjee A. K. Phosphoprotein NS of vesicular stomatitis virus: Phosphorylated states and transcriptional activities of intracellular and viral forms. Virology 154:259–270; 1986. [DOI] [PubMed] [Google Scholar]
- 29. Mathur M.; Das T.; Banerjee A. K. Expression of L protein of vesicular stomatitis virus Indiana serotype from recombinant baculovirus in insect cells: Requirement of a host factor(s) for its biological activity in vitro. J. Virol. 4:2252–2259; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peluso R. W.; Moyer S. A. Viral proteins required for the in vitro replication of vesicular stomatitis virus defective interfering particle genome RNA. Virology 162:369–376; 1988. [DOI] [PubMed] [Google Scholar]
- 31. Sanchez A.; De B. P.; Banerjee A. K. In vitro phosphorylation of the NS protein by the L protein of vesicular stomatitis virus. J. Gen. Virol. 66:1025–1036; 1985. [DOI] [PubMed] [Google Scholar]
- 32. Schubert M.; Harmison G. G.; Richardson C. D.; Meier E. Expression of a cDNA encoding a functional 241-kilodalton vesicular stomatitis RNA polymerase. Proc. Natl. Acad. Sci. USA 82:7984–7988; 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sleat D. E.; Chikkala N.; Gautam S.; Banerjee A. K. Restricted replication of vesicular stomatitis virus in T lymphocytes is coincident with a deficiency in a cellular protein kinase required for viral transcription. J. Gen. Virol. 73:3125–3132; 1992. [DOI] [PubMed] [Google Scholar]
- 34. Spadafora D.; Canter D. M.; Jackson R. L.; Perrault J. Constitutive phosphorylation of the vesicular stomatitis virus P protein modulates polymerase complex formation but is not essential for transcription or replication. J. Virol. 70:4538–4548; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sprague J.; Condra J. H.; Arnheiter H.; Lazzarini R. A. The expression of a recombinant DNA gene coding for the vesicular stomatitis virus nucleocapsid protein. J. Virol. 45:773–781; 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Takacs A. M.; Barik S.; Das T.; Banerjee A. K. Phosphorylation of specific serine residues within the acidic domain of the phosphoprotein of vesicular stomatitis virus regulates transcription in vitro. J. Virol. 66:5842–5848; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Takacs A. M.; Das T.; Banerjee A. K. Mapping of interacting domains between the nucleocapsid protein and the phosphoprotein of vesicular stomatitis virus by using a two-hybrid system. Proc. Natl. Acad. Sci. USA 90:10375–10379; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Towbin H.; Staehelin T.; Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 76:4350–4354; 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wertz G. W.; Davis N. L.; Patton J. The role of proteins in vesicular stomatitis virus RNA replication. In: Wagner R. R., ed. The rhabdoviruses; New York: Plenum; 1987:271–296. [Google Scholar]