

Abstract
Kidney proximal tubular cells (PTCs) are highly specialized for ultrafiltrate reabsorption and serve as paradigm of apical epithelial differentiation. Vps34/PI3-kinase type III (PI3KC3) regulates endosomal dynamics, macroautophagy and lysosomal function. However, its in vivo role in PTCs has not been evaluated. Conditional deletion of Vps34/PI3KC3 in PTCs by Pax8-Cre resulted in early (P7) PTC dysfunction, manifested by Fanconi-like syndrome, followed by kidney failure (P14) and death. By confocal microscopy, Vps34∆/∆ PTCs showed preserved apico-basal specification (brush border, NHERF-1 versus Na+/K+-ATPase, ankyrin-G) but basal redistribution of late-endosomes/lysosomes (LAMP-1) and mis-localization to lysosomes of apical recycling endocytic receptors (megalin, cubilin) and apical non-recycling solute carriers (NaPi-IIa, SGLT-2). Defective endocytosis was confirmed by Texas-red-ovalbumin tracing and reduced albumin content. Disruption of Rab-11 and perinuclear galectin-3 compartments suggested mechanistic clues for defective receptor recycling and apical biosynthetic trafficking. p62-dependent autophagy was triggered yet abortive (p62 co-localization with LC3 but not LAMP-1) and PTCs became vacuolated. Impaired lysosomal positioning and blocked autophagy are known causes of cell stress. Thus, early trafficking defects show that Vps34 is a key in vivo component of molecular machineries governing apical vesicular trafficking, thus absorptive function in PTCs. Functional defects underline the essential role of Vps34 for PTC homeostasis and kidney survival.
Introduction
Kidney proximal tubular cells (PTCs) show exquisite structural differentiation that supports their huge reabsorption activity and serve as paradigm for apical polarity1. Efficient reabsorption of water and solutes from the ultrafiltrate is ensured by abundance of channels and transporters (NaPi-IIa for phosphate, SGLT-2 for glucose) at the highly developed apical brush border membrane. These transporters are anchored by ezrin and by multi-PDZ scaffold proteins including Na+/H+-exchanger regulatory factor-1 (NHERF-1)2–5. Transepithelial transport is achieved by complementary sets of channels and transporters inserted into infolded basolateral membrane and complemented by Na+/K+-ATPase as primary transport driving force, itself stabilized by ankyrin-G scaffold. Full reabsorption of ultrafiltrated proteins by apical receptor-mediated endocytosis is no less remarkable, considering the brief contact of PTCs with rapidly passing urine. Full reabsorption is made possible by high expression of multiligand tandem receptors, megalin and cubilin6, combined with extremely fast apical endocytic recycling rate7. Megalin trafficks via sequential endosomal compartments that define the fast, Rab11-dependent apical recycling in epithelial cells8. Megalin/cubilin ligands dissociate in endosomes and are selectively transferred to lysosomes for degradation. Albumin internalization by the FcRn receptor follows a parallel pathway leading to transcytosis of intact protein9.
Establishment and maintenance of differentiated PTC polarity depend on the orchestrated action of primary polarity clues; specification of junctional, apical, lateral and basal membrane domains; recognition of protein targeting/retention motives; and further membrane domain expansion combined with regulation of sequential endosomal sorting. Biosynthetic apical sorting of membrane proteins can occur at the trans-Golgi complex via two distinct sets of vesicles, based on either inclusion of GPI-anchored proteins into rafts, or raft-independent recognition of complex N-glycans by galectin-310. Galectin-3 has recently received further recognition as luminal marker of damaged endolysosomes11. Endosomes play not only a key role in endocytic trafficking but also for the biosynthetic control of plasma membrane composition8,12–14. Mutations affecting vesicular trafficking have severe consequences in kidney function15. In PTCs, impaired apical trafficking due to defective recycling causes a generalized reabsorption deficit, known as kidney Fanconi syndrome, in Dent’s disease16,17, Lowe disease18 and possibly cystinosis19.
The vacuolar protein sorting-34 (Vps34) gene product was discovered in budding yeast as component of the machinery governing traffic to the vacuole, the equivalent of lysosomes, and is the ortholog and sole member of the class III subgroup of PI3K in multicellular organisms (PI3KC3)20. Vps34 lipid-kinase can only use phosphatidylinositol as substrate to produce phosphatidylinositol 3-phosphate (PtdIns3P, or PI3P) and is considered its main producer. However, other pathways e.g. via class II PI3K can also produce PI3P21–23. Alternatively, PI3P can be generated at endosomes by dephosphorylation of diphosphoinositides by PdIns(3,5P)2 phosphatases24.
Vps34 functions in two types of multiprotein complexes, regulating endocytic trafficking and autophagy, respectively20. A key role of Vps34 in endocytic trafficking was originally identified from in vitro fusion assays, as catalyzing recruitment of the early endosomal effectors, EEA1 and Rab525, but Vps34 acts at multiple endocytic trafficking levels, including late endosomes/lysosomes26,27. Vps34 impact on endosomal signaling28 may explain its requirement for kidney compensatory growth after unilateral nephrectomy29, thus potentially for kidney development.
Vps34 is also known as a core component of the macroautophagy initiation machinery30. Macroautophagy delivers cytoplasmic contents, including mitochondria (mitophagy), to lysosomes for degradation via double-membrane structures called autophagosomes which depend on phospholipid recruitment via LC3. Selective autophagy of aggregated ubiquitinylated cytosolic proteins is achieved by p62-dependent recruitment of LC331. Chaperone-mediated autophagy (CMA) is a distinct process whereby individual HSP-chaperoned proteins are translocated across the lysosomal membrane by a dedicated transporter, LAMP-2A32. After cell stress or injury, activation of CMA normally follows macroautophagy but CMA can compensate defective macroautophagy33. CMA is strongly upregulated in renal epithelial cells by EGF and TGFβ-1, which thereby govern the level of transcription factors such as Pax-234, thus impacting on tissue differentiation.
Since full gene Vps34 KO causes early embryonic lethality35, its role in vivo has been addressed by conditional inactivation in various lineages: myocytes36, cardiomyocytes37, hepatocytes37, T-cells38, neurons39 and kidney podocytes40–42. In podocytes, conditional Vps34 deletion caused podocyte dysfunction after ∼3 weeks and death after 3–9 weeks40,41. Interestingly, although also impacting autophagy, deleterious effects of Vps34 deletion were specifically ascribed to altered endocytosis42. The role of Vps34 in PTCs in vivo has not been studied. In the polarized OK cell lines as a PTC model43, we reported that acute pharmacological inhibition of Vps34 did not impair apical endocytosis per se (megalin internalization) but prevented megalin recycling from endosomes, without affecting lysosomes44. Remarkably, inhibitor withdrawal unleashed a burst of endosomal tubules, indicating that Vps34 indeed supports tubular endosome recycling. Here, we conditionally ablated Vps34 in PTCs by Wnt4- or Pax8-Cre-dependent excision of Vps34 exon 21 leading to in frame deletion (hereafter abbreviated as Vps34Δ/Δ). Wnt4-driven excision caused high perinatal mortality, but provided first structural clues. Pax8-driven excision did not alter mouse growth until postnatal day 14 (P14), allowing structural and functional studies. We found that Vps34Δ/Δ did not prevent specification of PTC membrane polarity, but impaired apical membrane protein trafficking, thus causing a renal Fanconi-like syndrome with polyuria and low-molecular weight proteinuria, detected at P7. Vps34Δ/Δ also impaired lysosome size/positioning from P7 onward, and blocked autophagy, thereby causing cell vacuolization at P14 and leading to kidney failure and death. We conclude that Vps34 is a crucial component of the trafficking machinery necessary for differentiated PTC function and is essential for overall PTC homeostasis.
Results
Conditional inactivation of Vps34 in PTCs
To inactivate Vps34 in kidney PTCs in vivo, we used a mouse line (Vps34fl) where exon 21, encoding a critical stretch (Ala730 to Thr754) of the Vps34 kinase domain, is flanked by loxP sites, allowing in-frame deletion by Cre-recombinase45. However, in the megakaryocyte lineage, this minimal truncation strongly decreases stability of the Vps34 protein and its associated regulatory protein Vps1545. For conditional deletion in kidney PTCs, Vps34fl mice were crossed with Wnt4-Cre mice (mouse model A, Wnt4-Cre;Vps34fl/fl, further referred to as Wnt4-Vps34Δ/Δ) or Pax8-Cre mice (mouse model B, Pax8-Cre;Vps34fl/fl, further referred to as Pax8-Vps34Δ/Δ). Genomic excision was verified by a two-fold decrease of the amplicon spanning exon 21–22 boundary, but normal abundance of 5′ and 3′ amplicons in Pax8-Vps34Δ/Δ as compared with wild-type (WT) total kidney extracts at P7 (see Supplementary Fig. S1), consistent with tissue-restricted and extensive but incomplete recombination in Pax8+ cells. Mosaicism provided informative pattern comparison: tubular sections of Vps34Δ/Δ mice with some cells showing undistinguishable features as in WT mice were denoted by # in the figures. Evaluation of Vps34 protein expression by western blotting was not possible because two commercially available antibodies did not generate convincing Vps34 band at the expected position (100 kDa).
Redistribution of apical membrane markers and perinatal mortality in Wnt4-Vps34Δ/Δ mice
We first used Wnt4-Cre recombinase which yields virtually complete excision of the megalin-floxed gene in PTCs46. Genotyping at P1 indicated high perinatal mortality within the expected 25% of Vps34Δ/Δ offspring from the Vps34fl/fl x Wnt4-Cre;Vps34fl/+ intercrosses (Fig. 1a, left panel). Vps34Δ/Δ survivors (5.8%) showed impaired growth and vigor, and were euthanized for study at P5. A representative survivor is shown in Fig. 1a, right panel. Vps34Δ/Δ kidneys of surviving mice showed normal colour and surface but were distinctly smaller and hypotrophic. Histologically normal glomeruli and tubules were recognized, yet with weak brush border (BB) staining by PAS, suggesting impaired apical differentiation (Fig. 1b). We next immunolocalized ezrin (highly concentrated at brush border), NaPi-IIa (main Na+/phosphate co-transporter) and megalin (paradigm for apical receptor-mediated endocytosis6 and fast apical recycling pathway8) (Fig. 1c). In WT P5, all PTCs showed strict apical stratification between two layers: (i) ezrin and NaPi-IIa co-localized at the brush border, and (ii) megalin signal concentrated in a subapical endosomal layer, as previously reported47–49. As shown in Supplementary Fig. S6a, NaPi-IIa was restricted to BB in inner cortex PTCs, but also occurred as longitudinal cytoplasmic stripes in PTCs from superficial cortex, indicating an immature pattern. In Vps34Δ/Δ survivors at P5, most PT sections showed coexistence of normally differentiated cells next to cells exhibiting punctate relocalization of NaPi-IIa alone, or combined with megalin throughout the cytoplasm.
Figure 1.
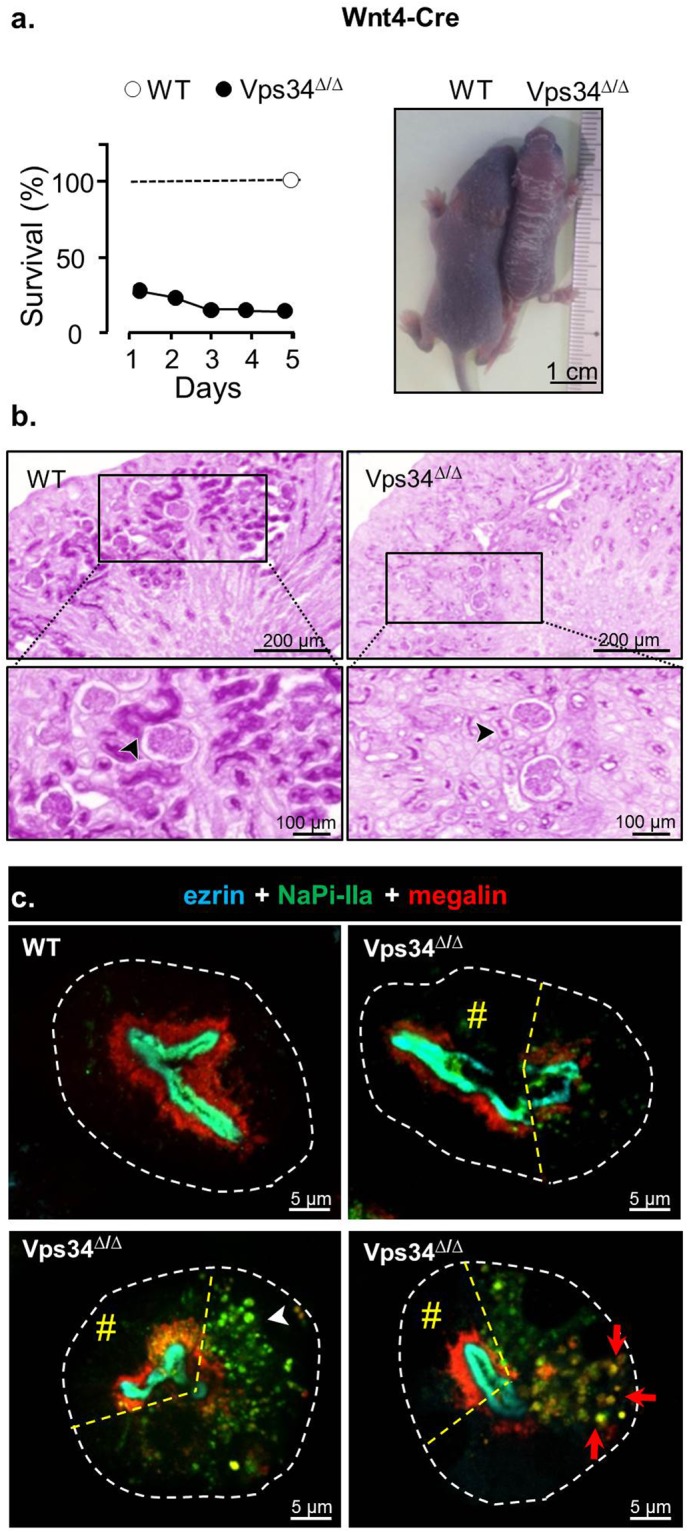
Mouse model A. Wnt4-driven excision of Vps34 exon 21 (Wnt4-Vps34Δ/Δ) causes perinatal kidney lesions and death. (a) Survival. Offspring of Vps34fl/fl x Wnt4-Cre; Vps34fl/+ was genotyped within 24 h after birth and surviving mice were sacrificed at P5. At the left, the survival of Vps34Δ/Δ mice is expressed as percentage of WT littermates (n = 25 WT pups versus 5 Vps34Δ/Δ at P1 and 3 Vps34Δ/Δ at P5, euthanized at this age). At the right, a representative surviving Vps34Δ/Δ pup is compared with a WT littermate at P5. (b) Histology after PAS staining at P5. Boxed areas are enlarged below. Arrowheads point to apical tubular PAS staining. Note that Wnt4-driven excision alters kidney growth but is compatible with normal morphogenesis of glomeruli and kidney tubules, yet with much reduced apical PAS staining. (c) Confocal immunofluorescence at P5. WT (upper left) vs Vps34Δ/Δ kidney deep (mature) cortices were immunolabeled for brush border (ezrin, cyan), NaPi-IIa (green) and megalin (red). Contours of proximal tubules recorded by bright-field microscopy are delineated by dashed white lines. Three representative views from discrete (upper right), moderate (lower left) to extensive (lower right) mis-localization of (sub)apical membrane proteins in deep cytoplasm of some PTCs contrasting with preserved localization in adjacent cells of the same tubular section shown at left of dashed dashed lines (#, mosaicism). Notice stratification in mature WT PTCs between pale green layer (full co-localization of ezrin and NaPi-IIa at brush border), and subapical red layer showing major megalin pool, with “hairy” deeper extensions. In altered Vps34Δ/Δ PTCs, preferential mislocalization of NaPi-IIa yields green dots (lower left, arrowhead); shared mis-localization of NaPi-IIa and megalin generates yellow dots (lower right, red arrows). Whole cortical views are shown in Supplementary Fig. 2.
We concluded that Wnt4-Cre driven Vps34 excision caused major perinatal lethality; and abnormalities in kidney cortex development and PTC apical differentiation in the surviving mice. To better analyze PTC apical differentiation and reabsorptive function in representative Vps34Δ/Δ pups, we next analyzed mice with Pax8-Cre-driven Vps34 excision42.
Redistribution of apical membrane markers and postnatal mortality in Pax8-Vps34Δ/Δ mice
Pax8-Cre is expressed two days later (at embryonic day (e) 16.5) than Wnt4-Cre activity50, in nephrogenic tubules and in nonvascular components of glomeruli51. As shown in Fig. 2a, Pax8-Vps34 mice were born at the expected Mendelian frequency (25/98) and grew normally up to ~2 weeks of age. Then, despite normal overall appearance and behavior, body weight levelled off and all pups died at ~3–5 weeks of age. We thus analyzed Vps34Δ/Δ pups at P7 days (normal growth) and P14 (growth arrest; compatible for 6-h urine collections).
Figure 2.

Mouse model B. Pax8-driven excision of Vps34 exon 21 (Pax8-Vps 34Δ/Δ) causes delayed postnatal kidney lesions and death. (a) Survival, growth and phenotype. In this mouse model, Vps34Δ/Δ pups are born at the expected Mendelian frequency and gain weight normally until ~P14 (weaning), then level off until death. Picture shows a typical appearance at P21. Vps34Δ/Δ pups are distinctly smaller but appear otherwise normal. They exhibit marked polyuria. (b) Western blotting of urine (n = 4 WT; 5 Vps34Δ/Δ), collected by bladder punction at sacrifice (P7) or upon 6-h placement in adapted metabolic cage (P14), for transferrin (TR), albumin (ALB) and vitamin D-binding protein (DBP). Loading was normalized for equal creatinine content. Positions of corresponding protein signals (in kDa) are indicated at the right. Low molecular-weight proteinuria is found in Vps34Δ/Δ samples. At the right, intensity of albuminuria at P14 was compared to urinary loss of IgG, both quantified by reference to serial dilutions of normal mice plasma, and IgG/albumin molar ratio was calculated as index of selective proteinuria. (c) Histology after PAS staining. Strong apical PAS stain is observed in all WT PTCs (arrowhead), contrasting with paler Vps34Δ/Δ pups PTCs at P7 and marked vacuolization at P14 (arrows).
At P14, Vps34Δ/Δ pups were markedly polyuric (~40% of body weight/day). Urinalysis (Table 1) indicated loss of glucose but preserved renal filtration at P7, then evolution to kidney failure at P14. To evaluate receptor-mediated endocytosis, urinary samples were analyzed by western blotting for the so-called low-molecular weight (LMW) proteins (transferrin, albumin, vitamin D-binding protein), characteristic of tubular proteinuria. As shown in Fig. 2b, LMW proteinuria was detected in 4/5 Vps34Δ/Δ mice at P7 and in 5/5 at P14. Selectivity of LMW proteinuria by the glomerular filter was confirmed by a low (0.15) IgG/albumin molar ratio. Taken together, polyuria, glucosuria and LMW proteinuria indicated that Pax8-driven Cre deletion of Vps34 caused an early global PTC dysfunction, i.e. renal Fanconi-like syndrome.
Table 1.
.
| Weight | Urine | Plasma | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| body (g) |
kidneys (mg) |
volume (mL/24 h) |
creatinine (mg/dL) |
creatininuria (mg/24 h) |
glucose (mg/dL) |
glucosuria (mg/24 h) |
phosphate (mg/dL) |
phosphaturia (mg/24 h) |
creatinine (mg/dL) |
BUN (mg/dL) |
||||
| P7 1 | ||||||||||||||
| WT | n = 13 | 4.39 ± 0.61 | 29 ± 5 | n = 4* | N/A | 9.5 ± 1.5 | N/A | 9.6 ± 0.8 | N/A | 301 ± 28 | N/A | n = 5 | 0.12 ± 0.05 | 31.5 ± 4.5 |
| Vps34∆/∆ | n = 13 | 4.12 ± 0.39 | 31 ± 4 | n = 4* | N/A | 8.6 ± 1.4 | N/A | 17.9 ± 4.9 | N/A | 209 ± 58 | N/A | n = 8 | 0.17 ± 0.05 | 53.2 ± 8.4 |
| P = 0.0286 | P = 0.0016 | |||||||||||||
| P14 2 | ||||||||||||||
| WT | n = 6 | 6.38 ± 1.05 | 51 ± 10 | n = 6 | 0.44 ± 0.13 | 11.9 ± 7.5 | 0.04 ± 0.03 | 9.7 ± 4.2 | 0.04 ± 0.02 | 386 ± 181 | 1.8 ± 1.1 | n = 10 | 0.11 ± 0.03 | 48.0 ± 14.0 |
| Vps34∆/∆ | n = 11 | 5.50 ± 0.89 | 63 ± 13 | n = 11 | 2.06 ± 0.82 | 5.3 ± 2.8 | 0.11 ± 0.07 | 5.6 ± 2.2 | 0.09 ± 0.04 | 216 ± 163 | 3.8 ± 2.8 | n = 10 | 0.32 ± 0.18 | 156.8 ± 86.6 |
| P = 0.0011 | P = 0.0119 | P = 0.0256 | P = 0.0019 | P < 0.0001 | ||||||||||
*n = 4 pools, each of 4 pups.
Pax8-Vps34Δ/Δ kidneys at P7 appeared normal and showed preserved histology, except for lack of PAS staining in ~half tubular sections (Fig. 2c). At P14, kidneys had become pale and bulging, indicating tissue swelling with reduced perfusion and were heavier (Table 1). RT-qPCR analysis displayed no changes in podocin mRNA up to P14, indicating preservation of glomeruli, but a significant decrease after P7 for AQP-1, megalin, cubilin, NaPi-IIa and SGLT-2 (indicating selective impairment in PTCs, originating from metanephric mesenchyme) and AQP-2 (in collecting ducts, originating from ureteric bud) (Supplementary Fig. S2). By histology, contrasting with preserved glomeruli, many tubular Pax8-Vps34Δ/Δ sections showed loss of apical PAS staining and strong vacuolization at P14. By electron microscopy, (Supplementary Fig. S3), Vps34Δ/Δ PTCs exhibited at P7 disconnected apical endosomes indicating loss of dense apical tubules. At P14, Vps34Δ/Δ PTCs further showed enlarged multivesicular late endosomes/lysosomes and gigantic lysosomes/residual bodies filled with structured (undigested) material, but always devoid of mitochondrial remnants. Mitochondrial sections were preserved except in disrupted tubules. To look at tight junction integrity, we performed tracing from the interstitium of ruthenium-red, a readily diffusing electron-dense molecule providing labelling of accessible membranes (Supplementary Fig. S4). Ruthenium-red labeled the entire basolateral membrane of Vps34Δ/Δ PTCs and was arrested at the position of tight junctions, even at strongly vacuolated cells, indicating a tight/sealed epithelium.
We next examined apical-basal PTC polarity by confocal immunofluorescence for the brush border linker, ezrin, and the apical domain marker, NHERF-1 (PDZ-scaffold for transporters), combined with the basolateral membrane marker, Na+/K+-ATPase, and its scaffold, ankyrin-G (Fig. 3a). Both WT and Vps34Δ/Δ pups showed extensive apical co-localization of NHERF-1 with ezrin and perfect segregation of these proteins from basolateral domains. In WT pups, basal infoldings bearing Na+/K+-ATPase were tortuous at P0 and P7, but orderly perpendicular (comb-like) at P14, suggesting terminal basal differentiation. Basal infoldings were less conspicuous in Vps34Δ/Δ compared to WT mice at P7 and disappeared at P14, at which time Na+/K+-ATPase was mostly at lateral membranes with a straight radial orientation, consistent with cell swelling. We concluded that Vps34 deletion in PTCs preserved apico-basal polarity, but altered terminal basolateral differentiation.
Figure 3.

Pax8-Vps34Δ/Δ mice show preserved apico-basal polarity in kidney proximal tubules but increasing mislocalization of megalin and NaPi-II from P7 to P14. (a) Confocal imaging of epithelial polarity markers: ezrin (apical brush border; red), NHERF-1 (apical PDZ scaffold; cyan), Na+/K+-ATPase subunit α1 (basolateral domain; green). Almost perfect colocalization of ezrin with NHERF-1 generates an apical white layer at P7 and P14, irrespective of conditional Vps34 excision. In WT PTCs and in most Vps34Δ/Δ PTCs up to P7, notice extensive invaginations of basal membrane labeled for Na+/K+-ATPase. These structures have vanished in Vps34Δ/Δ PTCs at P14. (b) Confocal imaging of apical transmembrane proteins: megalin (red), NaPi-IIa (green), by reference to ezrin (cyan). In WT PTCs, NaPi-IIa perfectly colocalizes with ezrin (pale green signal) while megalin is concentrated in a subapical layer. In Vps34Δ/Δ PTCs, megalin and NaPi-IIa are progressively mislocalized to separate (red, green) or common (yellow) deep cytoplasmic dots. Yellow dashed line indicates mosaicism with preserved cell indicated by #. For cubilin and SGLT-2, see Supplementary Fig. 5.
In the context of renal Fanconi-like syndrome, we next assessed by confocal multiplex immunofluorescence NaPi-IIa (non-recycling, green) and megalin (recycling, red) by reference to ezrin (brush border, cyan) (Fig. 3b). In WT pups up to P14, we confirmed the extensive co-localization of NaPi-IIa with ezrin, thus at brush border, and the distinct subapical megalin layer. In contrast, in Vps34Δ/Δ cells, NaPi-IIa and megalin appeared well apically localized in differentiated PTCs at P0 and became mislocalized into cytoplasmic compartments at P7 and more severely at P14. Again, mosaicism provided informative intercellular comparisons. Mislocalization was first detected for NaPi-IIa (mostly green punctae), then extended to megalin (mostly yellow punctae). As shown in Supplementary Fig. S5, a similar mislocalization was observed for the main PTC glucose transporter, SGLT-23, and the tandem endocytic receptor, cubilin52. Considering the marked heterogeneity of protein relocation, we performed morphometry based on NaPi-IIa, which showed the earliest mislocalization and allowed to distinguish immature (presence of lateral cytoplasmic stripes) from mature pattern (brush border only), (Supplementary Fig. S6b). As shown in Supplementary Fig. S7, tubulogenesis was not arrested in Vps34Δ/Δ kidney (comparable proportion of immature over mature patterns), and mislocalization of NaPi-IIa at P14 proved highly significant. We concluded that Pax8-Cre-mediated Vps34 deletion was compatible with tubulogenesis and preserved overall PTC polarity but caused extensive mislocalization of both recycling and non-recycling apical membrane proteins into cytoplasmic compartments.
Apical endocytosis is severely impaired in Pax8-Vps34Δ/Δ PTCs
To directly bridge LMW proteinuria with mislocalization of endocytic receptors away from the apical membrane, we looked at endocytic cargo uptake. First, we performed at P14 a 20-min pulse study of Texas-Red-labeled ovalbumin (TR-ova), validated to trace PTC endocytosis and lysosomal accessibility in PTCs; and its alteration in disease49. As shown by low-power views of sagittal kidney sections (Fig. 4a), the entire cortex of WT mice showed intense and homogenous radial TR-ova signal, with abrupt arrest at the cortico-medullary junction. In Pax8-Vps34Δ/Δ kidneys, much fewer nephrons were labeled but signal extended into the outer stripe of the outer medulla, indicating compensatory uptake by more distal cells. These sections were further immunolabeled for megalin as endocytic receptor and LAMP-1 as membrane marker for lysosomes, the final compartment of the degradative pathway where free Texas red is also sequestered, and analysed at high magnification. In WT tissue, TR-ova was concentrated in apical dots below the continuous megalin layer, largely co-localizing with LAMP-1. In Pax8-Vps34Δ/Δ PTCs showing megalin mislocalization, no TR-ova signal was detected.
Figure 4.

Vps34 deletion impairs acute endocytosis of injected ovalbumin-texasRed. WT or Vps34∆/∆ mice were injected at P14 with 180 µg TexasRed-labelled ovalbumin (TR-Ova) in the orbital plexus and euthanized at 20 min. Kidneys were fixed by immersion and whole cryostat sagittal sections were collected. (a) Low-power view of TR-Ova signal by stitched images of the two upper thirds of the sagittal sections recorded by Zeiss Mirax Midi and examined for red emission. Dashed white and yellow lines define the kidney contours and the cortico-medullary junctions, respectively. Boxes are enlarged below. Notice in WT mice the homogenous cortical signal indicating extensive recapture by all PTCs, with abrupt arrest at the cortico-medullary boundary (between cortex and outer stripe of outer medulla, OSOM). In Vps34∆/∆, much fewer PTCs are labeled and red signal extends deeper into the OSOM. (b) High magnification analysis by confocal microscopy with triple PTC imaging for TR-ovalbumin as endocytic cargo (red), megalin as endocytic receptor (white) and LAMP-1 as membrane marker of lysosomes where undigested tracer accumulates and free Texas Red is trapped (green). PTCs are circumscribed by broken white lines. Fields in the boxed rectangles are shown enlarged below with separate emission channels in white then merged channels in colors. Preserved PTC due to mosaicism in cKO is indicated by two radial broken yellow lines. In WT PTCs, megalin signal is restricted to a continuous apical layer. Intensely red dots indicating endocytosed TR-ovalbumin are mostly surrounded by LAMP-1, still in the subapical layer, indicating extensive endocytic uptake and transfer to lysosomes. In Vps34∆/∆ PTCs, LAMP-1 objects are much more numerous and become mislocalized to the deeper cytoplasm. Megalin has mostly disappeared from the apical layer and is instead redistributed into cytoplasmic dots colocalizing with LAMP-1. TR-Ova is not detected, except in the few preserved PTCs at the bottom of the section, where megalin and LAMP-1 show normal localization and intensity.
We also assessed by immunofluorescence the steady-state albumin content, as alternative reporter of PTC endocytic activity48. As shown by Supplementary Fig. S8, we found a severe reduction of albumin recapture in Vps34Δ/Δ PTCs (protected adjacent PTCs without deletion being positive controls). Thus, mislocalization of endocytic receptors (and presumably solutes co-transporters) provided a straightforward explanation for the Fanconi-like syndrome.
Disruption of apical Rab11- and perinuclear galectin-3-compartments in Pax8-Vps34Δ/Δ PTCs provides two mechanistic clues for apical membrane protein redsistribution
In OK cell lines as PTC model, acute Vps34 inhibition reversibly abrogates apical megalin recycling44. Since the fast apical megalin recycling pathway critically depends on Rab118, we thus examined by immunofluorescence whether Vps34 deletion would impact the Rab11 apical recycling compartement. As shown in Fig. 5, Rab11 occurred in WT PTCs as a dense punctate apical compartment, partially overlapping with the megalin signal and extending closer to the lumen, as expected for rapidly recycling apical endosomes/dense apical tubules. Vps34 excision totally disrupted this compartment. The sharp mosaique contrast between affected and preserved PTCs evidenced the cell-autonomous effect of Vps34 deletion. Disruption of the Rab11 apical compartment thus provided a straightforward explanation for suppressed megalin and cubilin recycling.
Figure 5.

Vps34 deletion in PTCs disrupts the Rab11-labeled, apical recycling endosomal compartment. Kidney sections from WT and Vps34∆/∆ P14 pups were double-immunolabeled for megalin (red) and Rab11 (green). Broken white lines delineate PTC contours. The pair of radial broken yellow lines defines in the upper field unusual PTCs with normal appearance in Vps34∆/∆ mice, attributed to mosaicism (notice preservation of continuous megalin layer without dotty cytoplasmic redistribution, contrasting to other PTCs of the same tubule). Boxed rectangles are enlarged below. In WT PTCs, Rab11 antibodies produce a specific finely dotty apical signal in the most apical half of the continuous megalin layer (Lu, PTC lumen), labelling the apical, rapidly recycling endosomal compartment. In Vps34∆/∆ PTCs showing megalin mislocalization into deep cytoplasmic dots (lower cells, best visible below the yellow broken line in the enlargements), the Rab11 signal is not detectable, indicating disruption of the apical recycling endosomal compartment.
However, this could not explain mislocalization of the non-recycling NaPi-IIa and SGLT-2, anchored to the NHERF-1/apical actin scaffold. We therefore looked for a primary defect of apical biosynthetic trafficking. Non-raft dependent apical sorting of N-glycan bearing glycoproteins at the trans-Golgi depends on luminal clustering by galectin-310. By immunofluorescence, galectin-3 in WT PTCs showed a remarkable perinuclear ribbon-like pattern, suggesting its concentration by retrograde trafficking into the Golgi/TGN complex. This pattern was reminiscent of galectin-3-GFP in Cos7 cells, especially upon TGN retention by a 20 °C block (see ref.10, Fig. 2b). In Vps34Δ/Δ PTCs, the perinuclear galectin-3 ribbons disappeared into dispersed dots, suggesting disruption of the biosynthetic pathway supporting apical trafficking of N-glycan bearing membrane proteins, including NaPi-IIa and SGLT-2.
In Pax8-Vps34Δ/Δ PTCs, late endosomes/lysosomes are mislocalized to basal perinuclear cytoplasm
To define the fate of apical membrane proteins once mislocalized into the cytoplasm, we first looked at lysosomal position, since Vps34 suppression or inhibition in cell culture was reported to cause late endosome/lysosome perinuclear mispositioning53. As shown in Fig. 6a, LAMP-1 immunolabelling in WT PTCs was restricted to a distinct subapical zone, below the megalin-enriched layer. Vps34 deletion strongly increased LAMP-1 signal (see also Fig. 7b), as reported in Vps34 KO podocytes40,41. In Pax8-Vps34Δ/Δ PTCs, LAMP-1 organelles became basally located and enlarged (P14, arrows). These data are consistent with multivesicular body swelling and the huge residual bodies evidenced by electron microscopy (Supplementary Fig. S3). This observation is also consistent with the reported enlargement of LAMP-1-labeled late endosomes/lysosomes in Vps34 KO MEFs37 and upon pharmacological inhibition of Vps34 by SAR405 in cultured cells54. Occasionally, LAMP-1 labelling filled the lumen of huge basal structures (boxed in Fig. 6a; see also below, small boxes in Fig. 7b,c), corresponding to large residual bodies as identified by electron microscopy and suggesting defective LAMP-1 catabolism within lysosomes. A good marker of endolysosomal damage is provided by galectin-3 redistribution into their lumen11. However, we never detected galectin-3 co-localization with LAMP-1 in Pax8-Vps34Δ/Δ PTCs, indicating endolysosome membrane integrity.
Figure 6.

Mislocalization of late endosomes/lysosomes, followed by vacuolisation, and progressive redistribution of NaPi-IIa and megalin to late endosomes/lysosomes in Pax8-Vps34Δ/Δ. (a) Confocal imaging of LAMP-1 (green) by reference to PTC apex (lotus-tetragonolobus lectin, LT-lectin, white) and basal profile (dotted white contours). Arrowheads, radial LAMP-1 redistribution; arrows, late endosomes/lysosomes vacuolization; yellow box, huge lysosome filled with LAMP-1 (see also Fig. 7b,c). (b,c) NaPi-IIa (b, red) and megalin (c, red) are compared to LAMP-1 (green). Areas boxed in white are enlarged below with split or merge channels. Areas boxed in yellow for Vps34Δ/Δ mice illustrate clusters of basally-located late endosomes/lysosomes. Yellow dotted lines in inserts distinguish two concentric apical cytoplasmic layers in PTCs. Asterisks, tubular lumen. In WT PTCs, the diffuse NaPi-IIa layer (brush border) is separated by unstained zone from deeper LAMP-1 zone made of discrete small dots. The diffuse megalin layer contacts, yet does not overlaps with, the LAMP-1 layer. In Vps34Δ/Δ, NaPi-IIa redistributes to deep cytoplasmic dots, many also labeled for LAMP-1 (arrowheads, yellow signal). Megalin may retain its normal subapical location even in cells where LAMP-1-labeled late endosomes/lysosomes extend into the basal cytoplasm, or is mislocalized to mispositioned lysosomes like NaPi-IIa.
Figure 7.

Abrogation of macro-autophagy in Pax8-Vps34Δ/Δ PTCs. (a) Mitophagy. In WT PTCs, megalin (white), late endosomes/lysosomes (LAMP-1, green) and mitochondria (TOM20, red) define distinct layers. In Vps34Δ/Δ PTCs, note the loss of stratification (central panel, right to dashed yellow line; right panel, below dashed yellow line). TOM20 is never detected in mislocalized late endosomes/lysosomes. (b) Selective p62-dependent autophagy. In WT PTCs, LAMP-1-labeled late endosomes/lysosomes (green) in subapical zone are segregated from the LT-lectin-labeled apical cytoplasmic layer (white). No p62 (red) and LC3 (blue) puncta is visible. Vps34Δ/Δ PTCs sections usually show several punctae double-labeled for p62 and LC3 in the basal cytoplasm (compare with preserved cell indicated by yellow #). For P7, see Supplementary Fig. 7. (c) Chaperone-mediated autophagy. In WT PTCs, LAMP-2A labels similar structures as LAMP-1, polarized in the subapical cytoplasm, and likewise segregated from megalin. In Vps34Δ/Δ, LAMP-2A-surrounded structures are much more abundant, extend into basal cytoplasm where they are often enlarged (arrows in right panel; area boxed in yellow) and become labeled for megalin (arrowheads in central panel). The enlarged boxed area shows lysosomal lumen filled by LAMP-2A-labeled objects.
In Pax8-Vps34Δ/Δ PTCs, apical markers become redistributed to mispositioned lysosomes
We next performed double-labelling studies of LAMP-1 with NaPi-IIa (Fig. 6b) or megalin (Fig. 6c). In WT mice, NaPi-IIa (brush border) and megalin (predominating pool below brush border) were fully resolved from the subapical LAMP-1 zone, defining a triple stratification (see inserts). In Pax8-Vps34Δ/Δ PTCs, redistributed NaPi-IIa and megalin became partially detected in LAMP-1-labeled, basally mispositioned late endosomes/lysosomes. Together, these data indicate that, as a result of defective Vps34-dependent apical targeting, transporters and endocytic receptors were mislocalized to lysosomes in Vps34Δ/Δ mice where they accumulated probably due to a defective proteolysis, as previously suggested54.
Macroautophagy is abortive in Pax8-Vps34Δ/Δ PTCs
We finally speculated that mislocation of apical transporters, presumably including amino-acid transporters, combined with suppression of macroautophagy, should cause a severe stress and trigger compensatory mechanisms. We hypothesized that without such adaptation, cellular homeostasis would be impaired, causing cell swelling and death. In view of alternative mechanisms for PI3P production21–23,28, and for entry into macroautophagy55, we first determined whether macroautophagy was altered in Vps34Δ/Δ PTCs by looking for co-localization at LAMP-1 organelles of the outer mitochondrial membrane protein, TOM20, as validated index of mitophagy56. As shown in Fig. 7a, TOM20 labeled elongated mitochondria in the basal cytoplasm of WT PTCs. In Vps34Δ/Δ PTCs at P14, TOM20-labeled structures appeared smaller and never showed colocalization with LAMP-1 organelles, and/or containing megalin (insert of central panel in Fig. 7a), compatible with full arrest of the mitophagic flux.
We also assessed p62-mediated autophagy of aggregated polyubiquitinylated cytosolic proteins31. To this aim, we performed quadruple fluorescent labeling using p62, LC3 as autophagic marker, LAMP-1 and Lotus Tetragonolobus lectin as signature of PTCs and general marker of apical membrane structures (brush border and apical endosomes). As shown in Fig. 7b, p62 and LC3 punctae were essentially absent from WT PTCs. In contrast, small punctae, all double-labeled for p62 and LC3, could be detected scattered in the cytoplasm of Vps34Δ/Δ PTCs at P7 (for time-dependent progression of alterations, see Supplementary Fig. S10). At P14, much larger punctae, mostly double-labeled for p62 and LC3, were easily found in the basal cytoplasm of Vps34Δ/Δ PTCs at P14, but again were never detected in LAMP-1-labeled structures. We concluded that LC3 could be recruited on p62 aggregates but that progression to autophagosome maturation and lysosomal fusion was arrested. This suggested that deletion of Vps34 in PTCs abrogated the autophagic flux, thus suppressing amino acid supply derived from autophagy completion.
Since chaperone-mediated autophagy (CMA) (i) can be up-regulated when macroautophagy is impaired33; (ii) relies on direct translocation of individual chaperoned proteins across the lysosomal membrane by LAMP-2A transporter; and thus (iii) does not depend of de novo membrane formation32, we assessed CMA in Vps34 mutant PTCs. As shown in Fig. 7c, LAMP-2A-labeled structures were indeed more abundant in Vps34Δ/Δ PTCs at P14 compared to WT controls and LAMP-2A extensively co-localized with LAMP-1. There was no preferential expression of the CMA-specific LAMP-2A isoform over LAMP-2B isoform as measured by RT-qPCR (data not shown). Thus, we could not find evidence that Vps34Δ/Δ PTCs could compensate defective macroautophagic flux by triggering CMA.
Discussion
Here, we tested the role of the Vps34 isoform of PI3K on the differentiation and maintenance of PTCs in vivo, by conditional deletion using Cre driven by two different promoters expressed at different time points during embryonic development. Our results show that Vps34 deletion allows proper apico-basal specification but causes primary mislocalization of apical membrane solute carriers and endocytic receptors, resulting in an early Fanconi-like syndrome. Vps34Δ/Δ PTCs also show several lysosomal alterations and abortive autophagy, with the eventual impact on PTC homeostasis leading to fatal kidney insufficiency.
Although no mosaicism was reported when inactivating Vps34fl/fl alleles in podocytes with Podocin- or Nephrin-driven Cre recombinase40,41, mosaicism was evident in tubular cells upon Pax8-Cre driven recombination. We consider unlikely this was due to the modified Vps34 locus itself, because (i) the floxed 21 exon used here was in the same region as in previously reported Vps34 KO mice40,41, and (ii) we observed similar mosaicism with our Pax8-Cre line on the Rosa26 locus (Rosa26-LSL-EYFP, data not shown). In addition, using indirect Pax8-driven deletion of mTORC components, Grahammer and collaborators57 also observed evident mosaicism in kidney tubules. We therefore favor the view that, in the rapidly expanding PTC pool, Pax8 promoter elements do not yield sufficient Cre activity in every tubular cell. Since Wnt4-Cre-mediated megalin recombination is virtually complete in adult mice46, mosaic Wnt4-Cre recombination of Vps34 locus might also be surprising, but again could be explained by the very young age of pups analysed (P5). Selectivity of tubular excision of Pax8-Cre was confirmed by preserved podocin expression up to P14, but decreased AQP-1 expression from P7 to P14. Loss of AQP-2 at P14 might indicate secondary effects.
Besides incomplete genetic recombination, phenotypic tissue heterogeneity upon Vps34 excision could be attributed to metabolic (compensatory) mechanisms of PI3P production. Although the Vps34-lipid kinase is the main producer of PI3P, other lipid kinases (e.g. class II PI3K isoforms) are capable of catalyzing this reaction23 and dephosphorylation of PI3,4P2 or PI3,5P2 also lead to PI3P production21,22,28.
Kidney PTCs are a paradigm of highly differentiated absorptive epithelium, reflected by concentric stratification of organelle layers from the apical to the basal pole, well-documented by electron microscopy1. In WT PTCs, immunofluorescence showed exclusive signal for NaPi-IIa to the brush border, mixed signal for SGLT-2 at the brush border and an upper subapical layer, and prominent signal for megalin and cubilin at a comparable upper subapical layer, as previously reported in mice and rats47–49. These distinct subcellular patterns of transporters and endocytic receptors reflect the balance between the surface pool (not readily detected by immunofluorescence for megalin and cubilin) and the prevailing recycling pool at apical endosomes in vivo. In mouse PTCs, late endosomes/lysosomes are localized in a slightly deeper zone, just below the dense megalin/cubilin endosomal layer and vacuoles. We identified by immunofluorescence a major pool of PI3P at the endosomal/lysosomal interface, where Rab11-rich endosomes have also been described58. Rab11 endosomes are the main compartment for apical membrane sorting in polarized MDCK cells8.
We found that the main early effect of Vps34 inactivation in PTC was the mislocalization of NaPi-IIa, SGLT-2, megalin and cubilin into cytoplasmic organelles, directly accounting for the renal Fanconi-like syndrome. At P14, mislocalization was further compounded by decreased expression, reflecting loss of cell homeostasis. Mislocalization of surface components undergoing multiple fast cycles such as megalin can be explained by defective recycling due to observed disruption of the Rab11 compartment. In addition, disruption of the galectin-3 compartment which governs N-glycan-dependent apical biosynthetic targeting, provides a tentative explanation for NaPi-IIa and SGLT-2 mistargetting. Furthermore, since membrane biosynthetic and endocytic pathways intersect at endosomes where polarity signals are primarily decoded14, endosomal misorientation of newly synthesized NaPi-IIa into non-recycling apical components would provide a second explanation. Apical PTC trafficking defects related to altered endosomal acidification or inositol-5-phosphatase acting on endosomes are known to cause Fanconi syndrome in Dent and Lowe diseases16–18,21, but further in vitro and in vivo studies are required for comprehensive molecular understanding of apical trafficking machineries. Surprisingly, PTC apico-basal specification appeared perfectly preserved in Vps34Δ/Δ mice. This contrasts with 3D epithelial organoids, where pharmacological inhibition of endosomal PI3K was recently reported to impair apical polarity, resulting into multilumen-cysts59.
Peripheral positioning of late endosomes/lysosomes has emerged as central feature for the regulation of cell homeostasis by mTORC160. Vps34/PI3KC3 regulates this positioning by tethering late endosomes/lysosomes to the anterograde/apical directed microtubule motor, kinesin-1, via the PI3P-binding proteins, FYCO-1 and protrudin53,61. However, to the best of our knowledge, the relation between mislocalization of late endosomes/lysosomes, mTORC1 signalling and autophagy has not been documented in polarized epithelia. We have taken advantage of mouse PTC stratification and found that Vps34 inactivation also caused basal/perinuclear lysosomal mispositioning in a polarized epithelium in vivo. However, mispositioning failed to promote autophagy completion since PI3KC3 is an essential component of autophagy initiation machinery.
In addition to vesicular trafficking defects, Vps34 deletion also caused functional impairment of late endosomes/lysosomes, as reflected by swelling and lysosomal accumulation of undigested material (electron microscopy; retention of various proteins identified by immunofluorescence). We also found that Vps34 inactivation in PTCs impaired autophagic flux (absence of TOM20 colocalization with LAMP-1 indicating lack of mitoghagy completion; colocalization of p62 with LC3 but not LAMP-1, demonstrating abortive autophagy of ubiquitylated cytosolic proteins). All these observations are consistent with in vitro evidence that Vps34 inhibition causes LAMP-1 organelle vacuolization and impairs maturation of the lysosomal enzyme, cathepsin D54. As expected, impaired macroautophagy was observed in Vps34Δ/Δ PTCs and alternative pathways to produce PI3P or to initiate macroautophagy30 were clearly insufficient to support detectable autophagy in Vps34Δ/Δ PTCs. Likewise, CMA could not compensate for abortive macroautophagy. Suppression of autophagy likely explains the loss of cellular homeostasis. PTC vacuolization and swelling resulted into increased intrarenal tension, as reflected by increased kidney size and weight, organ bulging and decreased perfusion, eventually leading to kidney insufficiency and death. These secondary effects were not detected at P7 but obvious at P14, thus calling for caution on interpretation at the late stage.
Materials and Methods
Mice
Vps34fl/fl mice and Wnt4-Cre mice have been described45,62. Pax8-Cre mice were obtained from Dr. M. Busslinger51. Vps34fl/fl mice were crossed with Wnt4-Cre;Vps34fl/+ or Pax8-Cre;Vps34fl/+ mice to generate conditional targeted excision of Vps34 exon 21 in the proximal tubules of 25% of offspring. Of note, Wnt4-Cre also causes excision of floxed alleles in the pituitary gland and Pax8-Cre in thyrocytes. Vps34fl/fl mice were used as control, denoted WT. Wnt4-Cre or Pax8-Cre driven Vps34fl/fl were denoted Vps34Δ/Δ. Mice were raised and treated according to the NIH Guide for Care and Use of Laboratory Animals, and experiments were approved by the local Ethical Committee of UCL (2016/UCL/MD/005).
Plasma, tissue collection and histology
Blood was collected at sacrifice, under anesthesia by xylazine 2% and ketamine 50 mg/ml, by heart (P7) or eye sinus puncture (P14). Kidneys were excised, decapsulated, and weighted then hemi-sagittally sectioned kidneys were fixed by immersion in neutral-buffered formaldehyde (4% F) at 4 °C under stirring overnight. Samples were paraffin-embedded or equilibrated overnight in 20% sucrose and embedded in Tissue-Tek Optimal Cutting Medium (Sakura Finetek) for 5 µm-thick cryostat sections.
Kidney function and western blotting
P14 pups were kept in metabolic cages for a duration of 6 h (10.00 a.m. to 4.00 p.m.) under adapted access to drink and urine was collected on ice with protease inhibitors cocktail (CompleteTM Protease Inhibitor Cocktail Tablets, Roche). Urine was weighted. Urinary creatinine was measured by improved colorimetric Jaffe method (QuantiChromTM BioAssay System). Glucosuria and phosphaturia were measured by automated assays by Roche Cobas Integra 6000. Plasma levels of creatinine and urea were measured on a COBAS 6000 C501 device (Roche-Hitachi®). For western blotting, the equivalent of 0.5 µg creatinine urine was resolved on precasted gels (Mini protean TGX 4–15%, 456–1083, Bio Rad). Antibodies are listed in Supplementary Table II.
Immunofluorescence
Immunofluorescence was performed on 5 µm-thick frozen sections or on 6 µm-thick paraffin sections. Antigen retrieval was promoted in citrate buffer, pH 6.0, at 98 °C for 20 min using a Lab Vision Pretreatment Module™ (Thermo Scientific). After permeabilization with PBS/0.3% Triton-X100 for 5 min, non-specific sites were blocked by 1-h incubation in PBS/0.3% Triton-X100 with 10% bovine serum albumin (BSA) and 3% milk, followed by primary antibodies in blocking buffer at 4 °C overnight. After extensive washing, sections were incubated with the indicated AlexaFluor-secondary antibodies in 10% BSA/0.3% Triton-X100 at room temperature for 1 h, extensively washed, mounted with Faramount Aqueous Mounting Medium (Dako) and imaged on a spinning disk confocal microscope using a Plan Apochromat 100×/1.4 Oil DIC objective (Cell Observer Spinning Disk; Zeiss). For whole kidney section recording, images were acquired using Zeiss Mirax Midi slide scanner, stitched and analyzed using Pannoramic Viewer software.
Electron microscopy
Kidneys were perfusion-fixed in situ as described49 using 0.1% (v/v) glutaraldehyde, then post-fixed therein overnight at 4 °C. Very small blocks were post-fixed with 1% (w/v) OsO4 in 0.1 M cacodylate buffer for 1 h, rinsed in veronal buffer (4 × 5 min) and stained overnight “en bloc” in 1% neutral uranyl acetate, all at 4 °C. Alternatively, samples were soaked in 2% glutaraldehyde containing 0.3% ruthenium-red to test for impermeability of junctions, as described63. After extensive washing in veronal buffer, blocks were dehydrated in graded ethanol and embedded in Spurr. Ultrathin (70 nm nominal) sections were obtained with a Reichert ultramicrotome (Reichert), collected on rhodanium 400 mesh grids and contrasted with 3% uranyl acetate followed by lead citrate, 10 min each. Grids were washed with water, dried, and examined in a FEI CM12 electron microscope operating at 80 kV.
RT-qPCR
Total RNA was extracted (SV total RNA isolation system; Promega). Aliquots of 300 ng RNA were reverse-transcribed by M-MLV reverse transcriptase (Invitrogen) with random hexamers, as described64. Primer sequences used are described in Supplementary Table III. Real-time qPCR was performed as described64, in presence of 250 nM of specific primers with Kappa SYBR Fast qPCR Master Mix (Kapa Biosystems) on a CFX96 touch real-time PCR Detection System (Bio-Rad). Results are presented as Delta Ct values, normalized to HPRT-1, used as internal standard. Values of 0 indicate identical target mRNA levels and higher values indicate lower levels, assuming equal amplification efficiency.
Western blotting
Kidneys were homogenized in lysis buffer containing 10 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate (CHAPS), 20 mM HEPES, pH 7.4, 150 mM NaCl, 2 mM CaCl2 with freshly added protease inhibitors (CompleteTM, Roche) as well as sodium orthovanadate, sodium pyrophosphate and sodium fluoride (2 mM each, Sigma-Aldrich). Kidneys were homogenized using a mortar-pestle and sonicated for 15 sec and lysates were centrifuged at 3000 rpm (Eppendorf centrifuge 5430 R) for 3 min at 4 °C and protein concentration in the supernatant was estimated by BCA. Proteins (20 µg protein/lane) were resolved into precasted gels (Mini protean TGX 4–15%, 456–1083, Bio Rad). For urines, sample volumes equivalent of 0.5 µg creatinine were resolved on precasted gels (Mini protean TGX 4–15%, 456–1083, Bio Rad). Antibodies are also listed in Supplementary Table II. Peroxidase-labelled secondary antibodies were revealed by enhanced chemiluminescence. Quantification was performed using imageJ software.
Statistical analysis
Statistical significance was tested using Mann-Whitney test for single comparisons, or Tuckey test and Kolmogorov-Smirnov for multiple comparisons. Differences were considered significant if p < 0.05.
Electronic supplementary material
Acknowledgements
This work was supported by grants from the Fonds pour la Recherche Scientifique (F.R.S-FNRS, Belgium), Université catholique de Louvain (Actions de Recherche concertées to CEP and PJC). FJ is a Fellow of the F.R.S.-FNRS and received support from the University of Liège (Fonds Spéciaux à la Recherche, Fonds Léon Fredericq) and the F.R.S.-FNRS (Research Credits 2013 and 2016). BV was supported by grants MRC (G0700755) and BBSRC (BB/I007806/1 and BB/M013278/1). The core facility for Imaging Cells and Tissues was also financed by National Lottery, Région bruxelloise, Région wallonne, Université catholique de Louvain and de Duve Institute (to PJC). We thank Dr. M. Busslinger (IMP, Vienna) for Pax8-Cre mice. We are very grateful to Drs P. Verroust, R. Kozyraki, J. Biber and C. Wagner and C. Yun for providing invaluable antibodies and Dr. P. Henriet for statistical analysis. GG held a fellowship from the Fonds pour la formation à la Recherche dans l′Industrie et l′Agriculture (FRIA, Belgium); HGC was Postdoctoral Researcher and CEP is Senior Research Associate at F.R.S-FNRS; VJ is supported by the Cystinosis Research Foundation. This work was presented as parts at the Belgian Society for Cell & Developmental Biology, November 2014, Antwerp, Belgium; British Society for, Cell & Developmental Biology, April 2016, Warwick, UK; and International Society for Cell Biology, July 2016, Prag, Czech Republic.
Author Contributions
B.V. is a consultant to Karus Therapeutics (Oxford, UK) and iOnctura (Geneva, Switzerland). The other authors have no competing financial or non-financial interests. C.E.P. and P.J.C. conceived the project, supervised the experiments and wrote the paper. G.G. performed experiments supervised by H.P.G.C. and assisted by V.J. and T.W. (immunofluorescence), F.N. (biochemistry) and P.V.D.S. (electron microscopy). F.J. performed urinalyses. S.V. and J.S. provided Wnt4-Cre mice. B.B. and B.V. provided Vps34-floxed mice and critically revised the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Christophe E. Pierreux and Pierre J. Courtoy jointly supervised this work.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-32389-z.
References
- 1.Christensen EI, Wagner CA, Kaissling B. Compr Physiology. 2012;2:805–861. doi: 10.1002/cphy.c100073. [DOI] [PubMed] [Google Scholar]
- 2.Murer H, Hernando N, Forster I, Biber J. Regulation of Na/Pi transporter in the proximal tubule. Annual review of physiology. 2003;65:531–542. doi: 10.1146/annurev.physiol.65.042902.092424. [DOI] [PubMed] [Google Scholar]
- 3.Vallon V, et al. SGLT2 mediates glucose reabsorption in the early proximal tubule. Journal of the American Society of Nephrology: JASN. 2011;22:104–112. doi: 10.1681/ASN.2010030246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delacour D, Salomon J, Robine S, Louvard D. Plasticity of the brush border - the yin and yang of intestinal homeostasis. Nature reviews. Gastroenterology & hepatology. 2016;13:161–174. doi: 10.1038/nrgastro.2016.5. [DOI] [PubMed] [Google Scholar]
- 5.He P, Zhao L, No YR, Karvar S, Yun CC. The NHERF1 PDZ1 domain and IRBIT interact and mediate the activation of Na+/H+ exchanger 3 by ANG II. American journal of physiology. Renal physiology. 2016;311:F343–351. doi: 10.1152/ajprenal.00247.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Christensen EI, Birn H. Megalin and cubilin: multifunctional endocytic receptors. Nature reviews. Molecular cell biology. 2002;3:256–266. doi: 10.1038/nrm778. [DOI] [PubMed] [Google Scholar]
- 7.Christensen EI, et al. Segmental distribution of the endocytosis receptor gp330 in renal proximal tubules. European journal of cell biology. 1995;66:349–364. [PubMed] [Google Scholar]
- 8.Perez Bay AE, et al. The fast-recycling receptor Megalin defines the apical recycling pathway of epithelial cells. Nature communications. 2016;7:11550. doi: 10.1038/ncomms11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tenten V, et al. Albumin is recycled from the primary urine by tubular transcytosis. Journal of the American Society of Nephrology: JASN. 2013;24:1966–1980. doi: 10.1681/ASN.2013010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delacour D, et al. Requirement for galectin-3 in apical protein sorting. Current biology: CB. 2006;16:408–414. doi: 10.1016/j.cub.2005.12.046. [DOI] [PubMed] [Google Scholar]
- 11.Chauhan S, et al. TRIMs and Galectins Globally Cooperate and TRIM16 and Galectin-3 Co-direct Autophagy in Endomembrane Damage Homeostasis. Developmental cell. 2016;39:13–27. doi: 10.1016/j.devcel.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mellman I, Nelson WJ. Coordinated protein sorting, targeting and distribution in polarized cells. Nature reviews. Molecular cell biology. 2008;9:833–845. doi: 10.1038/nrm2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodriguez-Boulan E, Macara IG. Organization and execution of the epithelial polarity programme. Nature reviews. Molecular cell biology. 2014;15:225–242. doi: 10.1038/nrm3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stoops EH, Caplan MJ. Trafficking to the apical and basolateral membranes in polarized epithelial cells. Journal of the American Society of Nephrology: JASN. 2014;25:1375–1386. doi: 10.1681/ASN.2013080883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stein M, Wandinger-Ness A, Roitbak T. Altered trafficking and epithelial cell polarity in disease. Trends in cell biology. 2002;12:374–381. doi: 10.1016/S0962-8924(02)02331-0. [DOI] [PubMed] [Google Scholar]
- 16.Christensen EI, et al. Loss of chloride channel ClC-5 impairs endocytosis by defective trafficking of megalin and cubilin in kidney proximal tubules. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8472–8477. doi: 10.1073/pnas.1432873100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gorvin CM, et al. Receptor-mediated endocytosis and endosomal acidification is impaired in proximal tubule epithelial cells of Dent disease patients. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:7014–7019. doi: 10.1073/pnas.1302063110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vicinanza M, et al. OCRL controls trafficking through early endosomes via PtdIns4,5P(2)-dependent regulation of endosomal actin. The EMBO journal. 2011;30:4970–4985. doi: 10.1038/emboj.2011.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cherqui S, Courtoy PJ. The renal Fanconi syndrome in cystinosis: pathogenic insights and therapeutic perspectives. Nature reviews. Nephrology. 2017;13:115–131. doi: 10.1038/nrneph.2016.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nature reviews. Molecular cell biology. 2010;11:329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 21.McCrea HJ, De Camilli P. Mutations in phosphoinositide metabolizing enzymes and human disease. Physiology. 2009;24:8–16. doi: 10.1152/physiol.00035.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marat AL, Haucke V. Phosphatidylinositol 3-phosphates-at the interface between cell signalling and membrane traffic. The EMBO journal. 2016;35:561–579. doi: 10.15252/embj.201593564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alliouachene S, et al. Inactivation of the Class II PI3K-C2beta Potentiates Insulin Signaling and Sensitivity. Cell reports. 2015;13:1881–1894. doi: 10.1016/j.celrep.2015.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schink KO, Raiborg C, Stenmark H. Phosphatidylinositol 3-phosphate, a lipid that regulates membrane dynamics, protein sorting and cell signalling. BioEssays: news and reviews in molecular, cellular and developmental biology. 2013;35:900–912. doi: 10.1002/bies.201300064. [DOI] [PubMed] [Google Scholar]
- 25.Christoforidis S, McBride HM, Burgoyne RD, Zerial M. The Rab5 effector EEA1 is a core component of endosome docking. Nature. 1999;397:621–625. doi: 10.1038/17618. [DOI] [PubMed] [Google Scholar]
- 26.Stein MP, Feng Y, Cooper KL, Welford AM, Wandinger-Ness A. Human VPS34 and p150 are Rab7 interacting partners. Traffic. 2003;4:754–771. doi: 10.1034/j.1600-0854.2003.00133.x. [DOI] [PubMed] [Google Scholar]
- 27.Lindmo K, Stenmark H. Regulation of membrane traffic by phosphoinositide 3-kinases. Journal of cell science. 2006;119:605–614. doi: 10.1242/jcs.02855. [DOI] [PubMed] [Google Scholar]
- 28.Cao C, Backer JM, Laporte J, Bedrick EJ, Wandinger-Ness A. Sequential actions of myotubularin lipid phosphatases regulate endosomal PI(3)P and growth factor receptor trafficking. Molecular biology of the cell. 2008;19:3334–3346. doi: 10.1091/mbc.E08-04-0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen JK, et al. Phosphatidylinositol 3-kinase signaling determines kidney size. The Journal of clinical investigation. 2015;125:2429–2444. doi: 10.1172/JCI78945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nascimbeni Anna Chiara, Codogno Patrice, Morel Etienne. Phosphatidylinositol-3-phosphate in the regulation of autophagy membrane dynamics. The FEBS Journal. 2017;284(9):1267–1278. doi: 10.1111/febs.13987. [DOI] [PubMed] [Google Scholar]
- 31.Cohen-Kaplan V, Ciechanover A, Livneh I. p62 at the crossroad of the ubiquitin-proteasome system and autophagy. Oncotarget. 2016;7:83833–83834. doi: 10.18632/oncotarget.13805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends in cell biology. 2012;22:407–417. doi: 10.1016/j.tcb.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaushik S, Massey AC, Mizushima N, Cuervo AM. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Molecular biology of the cell. 2008;19:2179–2192. doi: 10.1091/mbc.E07-11-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Franch HA, Sooparb S, Du J, Brown NS. A mechanism regulating proteolysis of specific proteins during renal tubular cell growth. The Journal of biological chemistry. 2001;276:19126–19131. doi: 10.1074/jbc.M101777200. [DOI] [PubMed] [Google Scholar]
- 35.Zhou X, Takatoh J, Wang F. The mammalian class 3 PI3K (PIK3C3) is required for early embryogenesis and cell proliferation. PloS one. 2011;6:e16358. doi: 10.1371/journal.pone.0016358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reifler A, et al. Conditional knockout of pik3c3 causes a murine muscular dystrophy. The American journal of pathology. 2014;184:1819–1830. doi: 10.1016/j.ajpath.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaber N, et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2003–2008. doi: 10.1073/pnas.1112848109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Willinger T, Flavell RA. Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naive T-cell homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:8670–8675. doi: 10.1073/pnas.1205305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang L, Budolfson K, Wang F. Pik3c3 deletion in pyramidal neurons results in loss of synapses, extensive gliosis and progressive neurodegeneration. Neuroscience. 2011;172:427–442. doi: 10.1016/j.neuroscience.2010.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen J, Chen MX, Fogo AB, Harris RC, Chen JK. mVps34 deletion in podocytes causes glomerulosclerosis by disrupting intracellular vesicle trafficking. Journal of the American Society of Nephrology: JASN. 2013;24:198–207. doi: 10.1681/ASN.2012010101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bechtel W, et al. Vps34 deficiency reveals the importance of endocytosis for podocyte homeostasis. Journal of the American Society of Nephrology: JASN. 2013;24:727–743. doi: 10.1681/ASN.2012070700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bechtel W, et al. The class III phosphatidylinositol 3-kinase PIK3C3/VPS34 regulates endocytosis and autophagosome-autolysosome formation in podocytes. Autophagy. 2013;9:1097–1099. doi: 10.4161/auto.24634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lima WR, et al. ZONAB promotes proliferation and represses differentiation of proximal tubule epithelial cells. Journal of the American Society of Nephrology: JASN. 2010;21:478–488. doi: 10.1681/ASN.2009070698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carpentier S, et al. Class III phosphoinositide 3-kinase/VPS34 and dynamin are critical for apical endocytic recycling. Traffic. 2013;14:933–948. doi: 10.1111/tra.12079. [DOI] [PubMed] [Google Scholar]
- 45.Valet Colin, Levade Marie, Chicanne Gaëtan, Bilanges Benoit, Cabou Cendrine, Viaud Julien, Gratacap Marie-Pierre, Gaits-Iacovoni Frédérique, Vanhaesebroeck Bart, Payrastre Bernard, Severin Sonia. A dual role for the class III PI3K, Vps34, in platelet production and thrombus growth. Blood. 2017;130(18):2032–2042. doi: 10.1182/blood-2017-04-781641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weyer K, et al. Mouse model of proximal tubule endocytic dysfunction. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2011;26:3446–3451. doi: 10.1093/ndt/gfr525. [DOI] [PubMed] [Google Scholar]
- 47.Christensen EI, Verroust PJ. Megalin and cubilin, role in proximal tubule function and during development. Pediatric nephrology. 2002;17:993–999. doi: 10.1007/s00467-002-0956-5. [DOI] [PubMed] [Google Scholar]
- 48.Thelle K, Christensen EI, Vorum H, Orskov H, Birn H. Characterization of proteinuria and tubular protein uptake in a new model of oral L-lysine administration in rats. Kidney international. 2006;69:1333–1340. doi: 10.1038/sj.ki.5000272. [DOI] [PubMed] [Google Scholar]
- 49.Gaide Chevronnay HP, et al. Time course of pathogenic and adaptation mechanisms in cystinotic mouse kidneys. Journal of the American Society of Nephrology: JASN. 2014;25:1256–1269. doi: 10.1681/ASN.2013060598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shan J, Jokela T, Skovorodkin I, Vainio S. Mapping of the fate of cell lineages generated from cells that express the Wnt4 gene by time-lapse during kidney development. Differentiation; research in biological diversity. 2010;79:57–64. doi: 10.1016/j.diff.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 51.Bouchard M, Souabni A, Busslinger M. Tissue-specific expression of cre recombinase from the Pax8 locus. Genesis. 2004;38:105–109. doi: 10.1002/gene.20008. [DOI] [PubMed] [Google Scholar]
- 52.Amsellem S, et al. Cubilin is essential for albumin reabsorption in the renal proximal tubule. Journal of the American Society of Nephrology: JASN. 2010;21:1859–1867. doi: 10.1681/ASN.2010050492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hong Zhi, Pedersen Nina Marie, Wang Ling, Torgersen Maria Lyngaas, Stenmark Harald, Raiborg Camilla. PtdIns3P controls mTORC1 signaling through lysosomal positioning. The Journal of Cell Biology. 2017;216(12):4217–4233. doi: 10.1083/jcb.201611073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ronan B, et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nature chemical biology. 2014;10:1013–1019. doi: 10.1038/nchembio.1681. [DOI] [PubMed] [Google Scholar]
- 55.Dupont N, Nascimbeni AC, Morel E, Codogno P. Molecular Mechanisms of Noncanonical Autophagy. International review of cell and molecular biology. 2017;328:1–23. doi: 10.1016/bs.ircmb.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 56.Soubannier V, et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Current biology: CB. 2012;22:135–141. doi: 10.1016/j.cub.2011.11.057. [DOI] [PubMed] [Google Scholar]
- 57.Grahammer F, et al. mTOR Regulates Endocytosis and Nutrient Transport in Proximal Tubular Cells. Journal of the American Society of Nephrology: JASN. 2017;28:230–241. doi: 10.1681/ASN.2015111224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mattila PE, Raghavan V, Rbaibi Y, Baty CJ, Weisz OA. Rab11a-positive compartments in proximal tubule cells sort fluid-phase and membrane cargo. American journal of physiology. Cell physiology. 2014;306:C441–449. doi: 10.1152/ajpcell.00236.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O’Farrell F, et al. Class III phosphatidylinositol-3-OH kinase controls epithelial integrity through endosomal LKB1 regulation. Nat Cell Biol. 2017;19:1412–1423. doi: 10.1038/ncb3631. [DOI] [PubMed] [Google Scholar]
- 60.Korolchuk VI, Rubinsztein DC. Regulation of autophagy by lysosomal positioning. Autophagy. 2011;7:927–928. doi: 10.4161/auto.7.8.15862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raiborg C, Wenzel EM, Stenmark H. ER-endosome contact sites: molecular compositions and functions. The EMBO journal. 2015;34:1848–1858. doi: 10.15252/embj.201591481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shan J, Jokela T, Peltoketo H, Vainio S. Generation of an allele to inactivate Wnt4 gene function conditionally in the mouse. Genesis. 2009;47:782–788. doi: 10.1002/dvg.20566. [DOI] [PubMed] [Google Scholar]
- 63.Mettlen M, et al. Src triggers circular ruffling and macropinocytosis at the apical surface of polarized MDCK cells. Traffic. 2006;7:589–603. doi: 10.1111/j.1600-0854.2006.00412.x. [DOI] [PubMed] [Google Scholar]
- 64.Hick AC, et al. Reciprocal epithelial:endothelial paracrine interactions during thyroid development govern follicular organization and C-cells differentiation. Developmental biology. 2013;381:227–240. doi: 10.1016/j.ydbio.2013.04.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.