

Abstract
In Burkitt’s lymphoma (BL) cells the proto-oncogene c-myc is transcriptionally activated by chromosomal translocation to the immunoglobulin (Ig) gene loci. This activation is characterized by preferential transcription from the c-myc promoter PI and accomplished by juxtaposed Ig enhancer elements. To identify promoter elements required for enhancer-activated PI transcription, we studied the activation of c-myc reporter gene constructs by the Ig κ intron and 3′ enhancers. Deletion analysis defined the core promoter with a TATA box and two adjacent GC/GT boxes upstream sufficient for basal and enhancer-activated transcription. Gel retardation assays revealed Sp1’s binding affinity to the GC/GT box proximal to the TATA box to be higher than to the distal one. This difference correlated well with the resulting levels of transcription mediated by Sp1 in cotransfection experiments in BL and Sp1-deficient SL2 cells. Sp3 also bound to the core promoter in vitro, but failed to transactivate in vivo. Mutation of the distal Sp1 site moderately affected basal transcription concomitant with a modest decrease in enhancer stimulation. Mutation of the proximal Sp1 site almost entirely abolished basal as well as enhanced transcription. A considerable level of basal transcription was maintained upon mutation of the TATA box, whereas enhancer-activated transcription largely was abolished. Stable transfection of the BL cell line Raji with constructs containing core promoter mutations confirmed that the proximal Sp1 site and the TATA box are essential for the activation of promoter P1 by the Ig κ enhancers.
Keywords: Burkitt’s lymphoma, Translocation genetics, c-myc, Promoter P1 transcription, Immunoglobulin κ enhancers, Sp1, Sp3, TATA box, SL2 cells
A wide range of hematological malignancies are associated with specific chromosomal translations that frequently involve the immunoglobulin (Ig) or the T-cell receptor gene loci [reviewed in (52)]. In Burkitt’s lymphoma (BL) cells the proto-oncogene c-myc on chromosome 8 is juxtaposed to one of the Ig gene loci on chromosomes 2, 14, or 22, resulting in constitutive activation of c-myc transcription. The c-myc gene encodes a nuclear phosphoprotein whose expression is closely linked to proliferation, differentiation, and survival of cells. Therefore, perturbation of c-myc gene expression is assumed to be a key step in the pathogenesis of BL and other naturally occurring tumors [reviewed in (44,60)].
Transcription of c-myc is controlled by two closely spaced promoters, PI and P2. More than 80% of steady-state c-myc RNA levels are derived from promoter P2 in normal cells. However, a preferential activation of promoter P1 is consistently observed for the translocated allele in many BL cell lines, a phenomenon referred to as promoter shift (59,60,66).
It is assumed that c-myc activation is accomplished by the physical linkage of the c-myc gene to enhancer elements within the juxtaposed Ig heavy or light chain gene loci in BL cells (8,30,40). This view is supported by several lines of evidence: i) in all BL cells c-myc is predominantly expressed from the translocated allele, whereas the unre-arranged allele is transcriptionally silent or expressed at very low levels (46); ii) c-myc alleles cloned from BL cell lines are only weakly transcribed with a normal promoter usage upon rein-troduction into BL cells (39,45,50); and iii) c-myc constructs are expressed and tumorigenic in trans-genic mice when driven by Ig enhancers (1,37). The most direct evidence for Ig enhancer-mediated c-myc activation and induction of the promoter shift derived from studies of c-myc constructs under the control of Ig enhancers stably transfected into BL cells. In this type of experiment preferential activation of PI transcription by the Ig heavy chain enhancer (39) as well as the Ig κ (32,49) and X llght chain enhancers (25) was demonstrated. Moreover, the Ig κ intron (κEi) and 3′ ehancers (κE3′) activated c-myc in a cooperative action when placed either adjacent to or separated from the c-myc promoters by as far as 30 kb, demonstrating long distance activation of c-myc by Ig κ enhancers in a position-independent manner in vivo (45).
Studies of the distribution of chromosomal breakpoints within the c-myc gene suggested that the cis-elements of c-myc involved in Ig enhancer-activated transcription reside downstream of 340 bp relative to the PI initiation start site (69). Several overlapping binding sites for Zn-finger proteins such as Spl, Sp3, MAZ, and CTCF have been described upstream and downstream of promoter PI and implicated in the activation and repression of basal transcription from the c-myc promoters in various cell lines (9,13,21,42). Currently, it is unclear whether Ig enhancer-activated transcription from promoter PI in BL cells is due to a distinct arrangement or subset of factors binding to the promoter. The TATA box element, which mediates binding of TATA box binding protein (TBP) during promoter recognition by the basal transcription initiation complex (71), is the only sequence motif described so far that is shared among all Ig κ enhancer-stimulated promoters. The TATA box has also recently been implicated in enhancer-mediated activation of transcription (43).
In an attempt to identify PI promoter elements mediating Ig enhancer-activated transcription, we analyzed the expression of c-myc promoter-reporter gene constructs under the control of the Ig κEi and κE3′ enhancers in transient and stable transfections. In addition, we identified the transfactors expressed in BL cells that bind to the enhancer-responsive promoter region in vitro and have the ability to activate basal transcription in vivo.
MATERIALS AND METHODS
Plasmids
Reporter gene constructs were based on pCG362 encompassing the HindIII/PvuII fragment of c-myc, which was inserted in front of the firefly luciferase gene (LUC) in pblueluc [described in (32)]. Deletion constructs pRF235, PCG345-1, pRF211-l, pRF278-2, and pRF278-l were made essentially by double digest of pCG362 by use of restriction enzymes HindIII (−2332), KpnI (−1058), PstI (−409), Smal (−101), XhoI ( + 66), and PvuII ( + 513), followed by a fill-in reaction of protruding ends and religation (all positions relative to the P1 initiation site). pCG21-4 was generated by PCR technology using forward primer #1067 -GCGGGGTACCCCGTATAATG CGAGGGTCTGGACGGCTGAG- (KpnI site underlined), sequence -CGAGAATCTGACGCAG GCAG- derived from LUC as reverse primer, and pRF211-l as template. The PCR fragment was then inserted as a XhoI/HindIII fragment into pblueluc. pCG90-4 (−101/+132) was generated by PCR using forward primer #1067 and reverse primer #1092 -GCCCCGAAAACCGGCAAGCT TACTCAGCGCGATCCCTCCC-, which contains a HindIII site (underlined) instead of the TATA box of promoter P2. The PCR fragment was then inserted as a XhoI/HindIII fragment into pRF278-l, digested with XhoI/HindIII. To obtain pCG79-7 and pCG84-7, sequences from −65 to −57, -AAGATCCTC-, were changed to -AGGTACCCC- in pRF278-l (−101/+66), thereby creating a KpnI site in subclone pCG37-3. The KpnI/SacI fragment of pCG21-4 covering sequences downstream of −28 was then substituted with a KpnI/SacI fragment of pCG37-3 covering sequences downstream of −57, giving raise to pCG79-7. pCG84-7 was obtained by substituting the KpnI/SacI fragment of pCG37-3 covering sequences downstream −57 with the KpnI/SacI fragment of pCG21-4 covering sequences downstream of −28. To generate enhancer-containing constructs, the Ig κEi and κE3′enhancers were inserted downstream of LUC as described (32). To obtain pCG319, a HindIII/SacI fragment of pCG362 encompassing the HindIII/PvuII fragment of c-myc, the LUC gene, and the Ig κ enhancers were cloned into the Epstein-Barr virus (EBV)-derived vector pHEBOpl (64). To avoid the production of excessive amounts of luciferase, which might interfere with cell growth upon stable transfection, a frameshift mutation was generated in the LUC gene by cutting with BstEII, followed by a Klenow fill-in reaction and religation. The c-myc sequences from −53 to −32 (proximal Spl site) and −96 to −75 (distal Spl site) were mutated by substitution in their steads, the sequence -AAGGATCAGCTTGCATGCATGA- of cloning vector pBR322 according to the Muta-Gene Phagemid mutagenesis kit (Bio-Rad, München, Germany). The TATA box was changed from -TATAAT- to -TAAGCC-. Mutations were verified by automatic sequencing (Applied Biosystems). Plasmids pCMV-hSp1, pCMV-Sp3, and pPacSp3 were kindly provided and described by G. Hagen (29). pCMV-βgal was obtained from Stratagene. pPacSp1 and pPac0 were kindly provided by K. J. Goodrich and D. Tautz, respectively.
Cell Culture, Transfection, and Reporter Gene Assay
BL Raji cells (51) were grown in RPMI-1640 medium supplemented with 10% fetal bovine serum, penicillin/streptomycin, and L-glutamine. Transfection was performed by electroporation as described (68). For transient and stable transfec-tions a total of 10 μg of DNA was used. In cotrans-fection experiments, 5 μg of reporter gene constructs and 5 μg of the appropriate expression plasmids were added to the cells. Drosophila mela-nogaster SL2 cells (56) (kindly provided by R. Rivera-Pomar) were grown to 60% confluency at 26 °C in Schneider cell medium (GIBCO) supplemented with 10% fetal bovine serum, penicillin/ streptomycin, and L-glutamine. Transfection of SL2 cells was carried out by the calcium phosphate method as described (15). Reporter gene constructs (4 μg) and variable amounts of expression plasmids pPacSp1 or pPacSp3 were transfected. Plasmid pPac0 was added to maintain a constant amount of 10 μg DNA total. Cells were harvested 48 h posttransfection, and the resulting extracts were normalized for total protein content by using a protein quantification kit (Bio-Rad), and equal amounts of protein were measured in duplicates for luciferase activity by using the Luciferase Assay System according to the manufacturers instructions (Promega Corp., Madison, WI). For generation of stably transfected cell lines, cells were grown for 48-72 h posttransfection before selection with 300 μg/ml hygromycin. At least four independent cell lines derived from single cell clones for each construct were established.
Nuclear Extract Preparation and Gel-Shift Analysis
Crude nuclear cell extracts were prepared by the method described by Dignam et al. (14) with slight modifications (73). Radiolabeled DNA (0.2−1 ng) was incubated with 5 μg protein extract at room temperature for 20 min in a buffer consisting of 12.5 mM HEPES-KOH (pH 7.5), 6.25 mM MgCl2, 5 μM ZnSO4, 50 mM KC1, 50 μg/ml BSA, 10% glycerol, and 2 itg poly(dl-dC). The reaction products were separated on 4% native polyacryl-amide gels containing 0.5 × TBE buffer at 135 V for 3 h. Gels were subsequently dried under vacuum and exposed to X-ray films or scanned with a Fuji BAS1000 phosphoimager system (Raytest, Straubenhardt, Germany). In competition and su-pershift experiments either unlabeled oligonucleo-tides or 1 μl of the appropriate antiserum were added to the reaction mixture 10 min prior to addition of the radiolabeled DNA. Rabbit poly-clonal antiserum against Spl (PEP2X) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-Sp3 and preimmune serum were kindly provided by G. Hagen. Affinity-purified human Spl prepared from HeLa cells was purchased from Promega Corp.
DNase I Footprinting
DNA (5 μg) of pRF211-1 was first digested with XhoI, labeled with polynucleotide kinase, and than digested with PstI. The appropriate fragment was gel purified and incubated under gel-shift conditions (see above) either with 25 ng purified Spl or 100 μg Raji nuclear extract, which was prepurified by phosphocellulose column fractionation as described previously (74). Kunitz units (0.1 and 50) of DNase I (Boehringer Mannheim, Germany) were added for Spl and Raji extracts, respectively. Digestion was allowed to proceed for 4 min on ice. The reaction was terminated with 100 μl of stop buffer [50 mM Tris-HCl (pH 8.0), 2% SDS, 10 mM EDTA (pH 8.0), 0.4 mg/ml proteinase K, 10 μg/ml glycogen] and ethanol precipitation. Samples were resolved on a 6% sequencing gel, which was subsequently dried and subjected to autoradiography.
RNA Analysis
Total cellular RNA samples were prepared by extraction with guanidinium thiocyanate followed by centrifugation in cesium chloride (54). RNA quantitation and nuclease SI analysis with a probe specific for the first exon of c-myc was carried out as described (17). Signals were captured and quantitated using a Fuji BAS1000 phosphoimager system (Raytest, Straubenhardt, Germany).
RESULTS
Definition of the c-myc P1 Core Promoter Sufficient for Basal and Ig κ Enhancer-Activated Transcription
To delineate the promoter elements required for Ig κ enhancer stimulation of P1 transcription, we generated reporter gene constructs containing the 2.8 kb HindIII/PvuII fragment of c-myc or progressive truncations thereof fused to the lucif-erase gene (LUC). The Ig κ intron (κEi) and 3′ enhancers (κE3′) were inserted downstream of LUC as a 1.9-kb enhancer cassette as described previously (32) (Fig. 1). Promoterless constructs with and without the enhancers were generated as negative controls. Basal transcription from pCG362 encompassing the P1 and P2 promoters was activated by κEi and κE3′ by about 10- to 15-fold (pCG363). Deletion of promoter PI did not significantly affect basal transcription (pRF235), but dropped enhancer-mediated reporter gene activity by about twofold (pCG508). This finding suggested that transcription from promoters P1 and P2 is activated almost equally by the Ig κ enhancers. Basal transcription from promoter P1 alone was activated at least 30-fold by κEi and κE3′ (pCG345-l and pCG351-2). The enhanced transcription level was comparable to that of the construct with both P1 and P2, pCG363. Thus, P1 is activated by κEi and κE3′ more effectively than promoter P2, suggesting a promoter preference of the Ig κ enhancers.
FIG. 1.
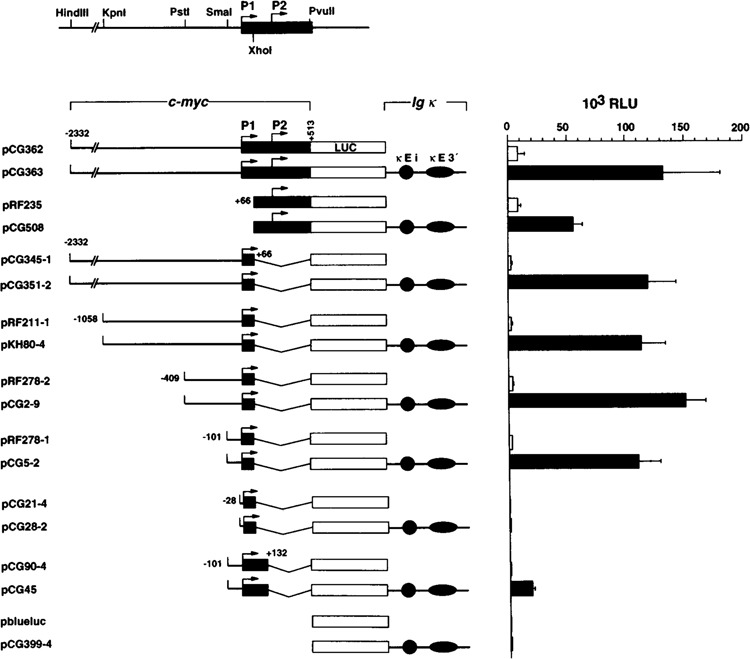
Basal and enhancer-activated transcription from c-myc promoter P1 is mediated by the core promoter from −101 to +66. The left-hand part shows a schematic representation of the c-myc constructs with and without the Ig κ intron (κEi) and 3′ enhancers (κE3′). The restriction enzyme sites used to generate the different plasmids are indicated in the map at the top. The positions of c-myc promoter fragments denote their 5′ and 3′ ends relative to the P1 transcription start site. Open boxes represent the reporter gene luciferase (LUC) and shaded boxes the first exon of c-myc. The «Ei and kE3 ’ enhancers are shown as filled circles and ellipses, respectively. Relative light units (RLU) measured after transient transfection of the c-myc constructs into Raji cells are shown to the right. Open and filled bars represent the activities measured in relative light units (RLU) of enhancer-less and enhancer-containing c-myc constructs, respectively. The median activities of eight independent experiments and standard deviations are indicated.
Deletion of sequences upstream of −101 (relative to the P1 initiation site) did not affect basal and enhancer-mediated P1 transcription (pRF278-l and pCG5-2). Notably, highest absolute reporter gene levels resulted from the -409/ + 66 promoter. Removal of sequences from −101 to −29 completely abolished basal transcription as well as enhanced PI transcription (pCG21-4 and pCG28-2). Sequences between +66 and + 132, including the region upstream of the P2 TATA box, repressed both basal and enhancer-driven P1 transcription by at least fivefold (pCG90-4 and pCG45). This observation might reflect the presence of elements between PI and P2, which negatively affect transcription from promoter P1. Taken together, these results demonstrated that basal transcription from the P1 promoter is lower than basal transcription from the P2 promoter but the effect of the enhancers on promoter P1 is greater than that on promoter P2. The core promoter sufficient for enhancer-stimulated PI transcription was defined as the region −101 to +66.
Sp1 and Sp3 Bind With Different Affinities to Two Adjacent GC/GTBoxes Within the Core Promoter and to a CT-Rich Element Upstream
Electrophoretic mobility shift assays (EMSA) were performed to identify the trans-factors expressed in BL cells that interact with the Ig κ enhancer-responsive P1 core promoter. A double-stranded oligonucleotide spanning the region from position −101 to −29 was labeled and incubated with nuclear extracts from several BL cell lines. EMSA revealed an identical mobility pattern of several retarded complexes with extracts of DG75, BL60, and Raji cells, indicating that similar or the identical protein(s) bind to the PI core promoter in different BL cells (Fig. 2B, lanes 2 to 4). Competition with a 10- or 20-fold molar excess of unla-beled probe −101/−29 impeded the formation of the prominent complexes designated I–V, whereas a 50-fold molar excess of oligonucleotides -66/ -48 did not affect any of the complexes (Fig. 2B, lanes 5, 6, and 11). This result suggested that the protein(s) present in complexes I-V bind to one or both of the overlapping consensus GC box and GT motif, which resemble binding sites for members of the Spl transcription factor family (28,34,35) (Fig. 2A). Complexes I and II were competed by oligonucleotides -101/-67 and -66/-29 covering the GC/GT box proximal (-44 to -36) and distal (-85 to -77) to the TATA box equally well (Fig. 2B, lanes 7-10). Formation of complex III was predominantly affected by oligonucleotides −66/−29, whereas oligonucleotides −101/−67 preferentially reduced the faster migrating complexes IV and V. To test whether the proximal and distal GC/GT boxes in fact bind Spl or Spl-related factors, we performed EMSA with oligonucleotides −101/−67 and −66/ −29 in the presence of specific antisera directed against Sp1 family members Sp1 and Sp3 (Fig. 2A, lanes 12-21). Preincubation of Raji extracts with an anti-Spl antiserum abolished complex III concomitant with a supershifted complex. Complexes IV and V disappeared in the presence of an antiserum raised against Sp3 (29), whereas the intensity of complex III increased. The presence of both anti-Sp1 and anti-Sp3 antisera blocked formation of all three complexes (Fig. 2A, lanes 15 and 20). The remaining faint band, which comigrated with complex III, may reflect endogenous Sp4 (29). Taken together, these data indicated that Sp1 binds with high affinity to the proximal GC/GT box, whereas the distal one is preferentially bound by Sp3.
FIG. 2.

Binding of Sp1 and Sp3 to multiple binding sites upstream of the P1 TATA box. (A) Schematic representation of the oligonucleotides used in the EMSA and their positions corresponding to the P1 transcription start site indicated by an arrow. The CT element and the two overlapping GC box and GT motifs (Sp) are marked by shaded boxes. (B) EMSAs using double-stranded oligonucleotides corresponding to the c-myc regions -101/-29 (lanes 1-11), −101/−67 (lanes 12-16), −66/−29 (lanes 17-21), and −167/−138 (lanes 22-26) as radioactively labeled probes and nuclear extracts of BL cell lines DG75, BL60, and Raji. For competition experiments unlabeled oligonucleotides were added in 10-, 20-, or 50-fold molar excess to the reaction mixture as indicated (lanes 5-11). One microliter of preimmune serum (pre), antiserum directed against Sp1 (αSp1), against Sp3 (αSp3), or a mixture of both (αSp1/3) were preincubated with Raji extracts prior to addition of the labeled probes as indicated above. (C) DNase I footprinting of nuclear factor binding sites within the P1 promoter. DNA probe (−409/+ 66) was end labeled on the coding strand and either chemically cleaved at G and A residues as a marker lane (G/A) or digested with DNase I without (−) and with ( + ) prior incubation with Raji nuclear extracts, or purified Sp1. The protected sequences are indicated next to each footprint. DNase I hypersensitive sites are marked by arrows.
Formation of complexes I and II was not detected on oligonucleotides containing only one of the Spl/Sp3 sites (−101/−67 and −66/−29), indicating that occupation of both sites is required for the formation of complexes I and II. The migration behavior of these complexes is reminiscent of dimers or multimers of Sp1 protein bound to two adjacent Sp1 sites (47). Incubation of probe −101/−29 harboring two Sp1 binding sites with purified Sp1 resulted in the formation of complexes comigrating with complexes I and II (data not shown). Therefore, it is likely that these complexes represent high-order complexes of Spl.
Because the region −409 to −101 increased basal and enhancer-activated transcription from the core promoter (see Fig. 1), we scanned this region for nuclear factor binding sites of BL cells. DNase I footprinting experiments with fragment −409 to + 66 and nuclear extracts of Raji cells revealed protection of the regions −93 to −70 and −55 to −33 encompassing the GC/GT boxes within the core promoter (Fig. 2C). In addition, protection of the region −160 to −138 was revealed, which resembles the most upstream of five repeated CT elements. The CT region was described previously to increase c-myc promoter activity in transfection assays (13,65). Protection experiments with purified Sp1 resulted in footprints almost indistinguishable from that observed with Raji nuclear extract. As shown by EMSA with a probe corresponding to the protected CT element, Sp3 has the ability to bind to this element as well (Fig. 2B, lanes 22-26). Thus, the region upstream of the PI TATA box can be envisaged as an arrangement of multiple sites with binding affinity for Sp1 and Sp3.
Activation of Basal P1 Transcription by Sp1
To determine whether the binding of Sp1 and Sp3 to promoter P1 in vitro had functional relevance in vivo, we first transfected Raji cells with c-myc constructs shown in Fig. 3A along with CMV-promoter driven expression vectors for Sp1 and Sp3. Transcription from the −1058 to +66 promoter (pRF211-l) was transactivated by Sp1 similar to the −409/+ 66 promoter (pRF278-2), indicating that no further functional Sp1 sites are located upstream of −409. Removal of the CT element reduced Sp1-mediated transcription levels by about 30% (pRF278-l). Deletion of the distal low-affinity Sp1 site (−93 to −70) dropped Sp1-activated transcription levels by about 20% (compare pRF278-l and pCG79-7). Sp1-promoted transcription derived from construct pCG79-7 solely reflected binding of Sp1 to the proximal GC/GT box, as construct pCG21-4 lacking any Sp1 binding sites was unaffected by exogenous Sp1. To compare the abilities of the distal and proximal core promoter sites to mediate transacti-vation by Sp1 more directly, we fused the distal site from −101 to −64 to the TATA box. A 5-bp spacer inserted directly upstream of the TATA box ascertained that the distal GC/GT motif was in the same phase topologically to the TATA box as the proximal motif. A modest activation of transcription by Sp1 acting from the distal GC/ GT box in pCG84-7 was observed. The activation level from the core promoter with two Sp1 sites (pRF278-l) was greater than the sum of the two sites acting independently (pCG79-7 and pCG84-7), suggesting that activation of transcription from the core promoter is mediated by a syner-gistic interaction between the two Spl sites. In contrast to Sp1, coexpression of Sp3 had no significant effect on P1 transcription in BL cells, similar to what has been observed in other cell types (42).
FIG. 3.

Activation of basal P1 transcription by Sp1, but not Sp3. (A) Raji cells were transiently transfec-ted with 5 μg of c-myc constructs shown in the left-hand part along with 5 μg of CMV promoter-driven expression plasmids for Sp1, Sp3, or β-galactosidase (−). (B) Four micrograms of c-myc constructs shown above and increasing amounts (0, 0.5, 2, and 5 μg) of pPacSp1 and pPacSp3 were cotransfected in Drosophila melanogaster SL2 cells. A constant amount of 10 μg total was maintained by transfection of pPac0. Results are presented as median activities of four independent experiments. Standard deviations were less than 10%.
We repeated the cotransfection experiments in Drosophila melanogaster SL2 cells to avoid any interfering background activities from endogenous Sp1 and Sp3 proteins. These insect cells serve as an ideal cell system for this purpose due to the lack of Sp1 and Sp1-like proteins (12,29). Consistent with the results obtained in Raji cells, Sp1 activated transcription at a modest level from the promoter with the distal GC/GT box only, pCG84-7. The promoter with the proximal GC/ GT box, pCG79-7, was activated by Sp1 in a dose-dependent manner at a level about half of the promoter with both Sp1 sites, pRF278-l. Again, this suggests a synergistic action of Sp1 acting from two adjacent binding sites. Sp1-mediated transcription levels further increased in the presence of the CT element in pRF278-2 and pRF211-l. Sp3 was unable to activate PI transcription significantly, similar to what is observed in Raji cells. We therefore now refer to the distal and proximal GC/GT box as Sp1 sites.
The Integrity of the Sp1 Site Proximal to the TATA Box Is Crucial for Basal and Ig κ Enhancer-Activated P1 Transcription
Our next aim was to determine the role of the Sp1 sites within the core promoter for enhancer-mediated activation of transcription. Substitution mutation of either one of the Sp1 sites within the −101/+ 66 promoter dramatically impaired basal and enhancer-activated transcription (Fig. 4). In the context of the −1058/+ 66 promoter, however, mutation of the distal Sp1 site (−93 to −70) moderately affected basal promoter activity (pCG66). Moreover, the enhancement was only slightly reduced (pCG35), suggesting that sequences upstream of −101 including the CT element substituted for the distal Sp1 site. None of the mutations impaired Sp1 binding to the unmutated sites nor to the CT element in vitro (data not shown). Mutation of the proximal Sp1 site abolished basal and enhancer-mediated transcription almost entirely. Thus, binding of Sp1 in the proximity of the TATA box but not the spacing between the proximal site and a second Sp1 site upstream appears to be crucial for basal and enhancer-mediated P1transcription.
FIG. 4.

Effects of mutations within the core promoter Sp sites and TATA box on basal and Ig κ enhancer-activated P1 transcription. Mutation of the Sp sites and the TATA box, indicated by a large X, was carried out by substitutional and site-directed mutagenesis, respectively. Activities of enhancer-less constructs measured after transient transfection of Raji cells in relative light units (RLU) are indicated by open bars. Filled bars represent LUC activities of corresponding Ig κ enhancer-containing constructs. Standard deviations were less than 10%.
The TATA Box Is an Essential Element for Ig κ Enhancer-Activated But Not Basal Transcription
The arrangement of multiple Sp1 binding sites close to the initiation site is characteristic for many promoters of viral and cellular genes (i.e., housekeeping genes), some of which lack a consensus TATA box motif [(6) and references therein]. To determine the role of the TATA box in basal and enhanced transcription we changed the TATAAT motif at position −28 to −23 to TAAGCC in the context of two or three Sp1 sites upstream (Fig. 4). Though basal transcription from a TATA-less PI promoter was maintained at considerable levels (pCG85-l, pCG29), a significant stimulation of promoter activity by the Ig κ enhancers was not observed (pCG47, pCG13). These results suggest that the TATA box is required for P1 transcription when stimulated by the Ig κ enhancers, but appears to be dispensable for basal transcription.
The TATA Box and the Proximal Sp1 Site Are Required for Enhancer-Mediated P1 Activation on Stably Replicating Constructs
As noted above, the TATA box and the proximal Sp1 site were absolutely required for activation of P1 transcription by Ig κ enhancers in transient transfection experiments. However, this type of experiment does not allow the assesment of the role of the core promoter elements in enhancer-activated transcription associated with chromatin, an integral part of the transcription process in vivo. Therefore, we generated constructs based on the Epstein-Barr virus (EBV)-derived vector pHEBO encompassing the HindIII/PvuII fragment of c-myc with and without core promoter mutations, the LUC gene, and the Ig κ enhancers (Fig. 5A). Vector pHEBO was chosen because it replicates stably as an episome and permits the study of gene regulation without interfering position effects (64). Moreover, pHEBO-derived constructs carrying the c-myc gene under the control of the Ig k enhancers have been shown to adopt a chromatin structure within the promoter region characteristic for an actively transcribed c-myc gene (32,58,62). Constructs were transfected into the BL cell line Raji and several independent stable transfectants of each construct were obtained after selection with hygromycin B. Transcripts derived from the endogenous translocated allele and transcripts from the transfected constructs were distinguished by a large deletion at the 3′ end of the first exon of the translocated c-myc allele in Raji cells by nuclease SI protection analysis using an exon 1-specific probe (Fig. 5C) (53). Because the 3′ part of this probe contains vector sequences that hybridize only with transcripts from the transfected c-myc constructs, transcripts from the nontranslocated allele can be distinguished from those of the transfected constructs. The result of two representative cell lines of each construct is shown in Fig. 5B. Transfectants of pCG319 revealed a strong induction of P1-derived transcripts with a P1/P2 ratio of about 1, similar to what has been described for a construct encompassing the complete c-myc gene under the control of the Ig κ enhancers (32) (Fig. 5D, and data not shown). Mutation of the distal Sp1 binding site (pCG336) modestly decreased the amount of transcripts from P1, whereas P2-derived transcripts slightly increased. Mutation of the TATA box (pCG324) and the proximal Sp1 binding site (pCG338) abolished hybridization signals of PI transcripts with the expected length and, in addition, created a new initiation site about 30 bp further upstream. Quantification of these signals as PI-derived transcripts revealed signal intensities of about 10% compared to the wild-type promoter, pCG319. Interestingly, a reduced PI transcription rate was concomitant with a threefold increase in P2-derived transcripts. In accordance with the results obtained by transient transfection experiments, the proximal Sp site and the TATA box are absolutely required for the activation of promoter PI by Ig κ enhancers.
FIG. 5.

Enhancer-activated transcription of c-myc promoter P1 requires the TATA box and the proximal Sp site on stably replicating constructs. (A) Schematic representation of constructs used for stable transfection of the BL cell line Raji based on the episomal replicating vector pHEBO (64). Mutations within the Sp sites and the TATA box in the context of the HindIII/PvuII fragment of c-myc (−2332/+ 513) are indicated with a large X. The luciferase gene contains an internal frameshift mutation as indicated (see the Materials and Methods section). (B) Nuclease SI analysis of total cellular RNA of the respective transfectants and untransfected Raji cells. The result of two out of four independent cell lines for each construct is shown. Transcripts were analyzed using a uniformly labeled, single-stranded DNA probe spanning the c-myc promoter region shown in (C). The deletion at the end of exon 1 of the translocated allele of Raji and the vector sequences at the 3’ end of the probe (hatched box) allow to differentiate between transcripts derived from the translocated alleles (P1t, P2t) and nontranslocated allele (P2) and those derived from the constructs (Plconstr; P2constr). The new PI transcription start site on pCG324 and pCG338 is indicated with an asterisk. The size of the fragments protected by c-myc transcripts initiated at the different promoters is given below. As internal control a GAPDH probe was added to the hybridization mixture as described (50). (D) Quantification of the signals captured by a phosphoimager and plotted as a histogram.
DISCUSSION
The preferential activation of c-myc transcription from promoter P1 in BL cells has been established to be a consequence of the juxtaposition of c-myc to Ig enhancer elements by chromosomal translocations. By reconstruction of a (2;8) trans-location we have previously identified the Ig κEi and κE3′ enhancers as sufficient for activation of P1 transcription in BL cells (49). Our first aim was to identify the minimal Ig κ enhancer-responsive region within promoter P1 by transfection of various c-myc promoter-reporter gene constructs with and without κEi and κE3′. By this approach we were able to define the P1 core promoter −101/ + 66 as sufficient for basal and enhancer-activated transcription. Moreover, we found that P1 is activated by the combination of κEi and κE3′ more effectively than promoter P2. Recent studies have shown that κEi and κE3′ act synergistically in transcriptional activation of c-myc P1 and murine Vκ promoters, but not the murine κ germline promoter (22,32). The differential activation of the c-myc promoters by Ig k enhancers raised the intriguing possibility that promoter P2 does not support this enhancer synergism. Direct comparison of PI and P2 transcription rates driven by each individual κ enhancer and their combination demonstrated that the synergism is not confined to one of the c-myc promoters; however, *κEi and κE3′ alone stimulated promoter P1 twice as effectively as P2 [(32), and data not shown). Thus, it appears that the κ enhancers display a c-myc promoter preference rather than specificity, similar to what has been described for other Ig κ enhancer-activated promoters (23,24).
The closely related transcription factors Sp1 and Sp3 were identified as the t rans -factors expressed in BL cells, which bind in the core promoter −101/+ 66 at two sites with overlapping GC/GT motifs. In addition, these factors bind to one of the CT elements in the region upstream of -101. A similar distribution of binding sites for Sp1 and Sp1-related factors upstream of the PI TATA box has been described for the murine P1 promoter (3).
Four independent sets of experiments indicated that binding of Sp1 in the proximity of the TATA box plays an important role in basal as well as enhancer-activated P1 transcription: i) competition experiments indicated that Sp1 binds to the proximal GC/GT box with higher affinity than to the distal box; ii) exogenous Sp1 acting from the proximal site is able to strongly activate basal transcription in BL and insect cells; iii) mutation of the proximal site, which blocks binding of Sp1 in vitro, abolishes basal and enhancer-activated P1 transcription in transient transfection experiments; and iv) mutation of this site eliminates enhancer-activated transcription in the context of an ordered chromatin structure on stably replicating episomes. Comparison of various Sp1-dependent promoters has revealed a correlation between the ability of a given Sp1 site to activate transcription, its affinity for Sp1, and proximity to the TATA box (2,33,57). In this regard, it is important that Sp1-mediated transcription is assumed to be accomplished by a direct interaction of the glutamine-rich activation domains of Sp1 with at least two components of the TFIID complex, the TATA box binding protein (TBP) and TBP-associated factor TAFII110 (19,26,31).
The role of the distal GC/GT box, separated from the TATA box by about 50-60 bp, in basal and enhanced PI transcription is quite different from the proximal one. Spl appears to bind with lower affinity to the distal site than to the proximal site in vitro. This finding explains well the modest activation of transcription by Sp1 in vivo acting from the distal site, even when placed at the position of the proximal site. These observations further support the notion that the functional difference between the two adjacent Sp1 sites is due to different affinities for Spl rather than to the position relative to the TATA box. Basal and enhancer-driven promoter activity is dramatically affected upon mutation of the distal site in the context of the core promoter −101/+ 66; however, the presence of the third Sp1 site within the CT region upstream substitutes for the distal site in a transient assay. On stably replicating episomes, enhancer-driven PI transcription was also only moderately reduced. Taken together, these results suggest that activation of promoter PI by Ig κ enhancers requires at least two binding sites for Sp1, the one in proximity of the TATA box and one located further upstream.
In contrast to Sp1, we failed to detect any significant activation effect of Sp3, in line with previous reports (29). Recent data indicated that over-expression of Sp3 in HeLa and SL2 cells is able to repress Spl-mediated transcription from c-myc promoter P2 (42). Overexpression of Sp3 has been reported to repress Spl transactivation, most likely by competing with Spl for a given Spl/ Sp3 binding site (29,41). Though Sp3 has binding affinity to promoter PI in vitro, especially to the distal site, the overexpression of Sp3 did not have a significant negative effect on basal and κ enhancer-activated P1 transcription (Figs. 2A and 3, and data not shown). A recent report clearly demonstrates that Spl transactivation of different promoters is selectively repressed by Sp3 dependent on the context and/or number of functional Sp1 binding sites (5). Data presented here indicate that differences in the relative affinities of Sp1 versus Sp3 for specific Sp binding sites may also contribute to promoter context-dependent effects of Sp3 on Spl transactivation. However, the precise role of endogenous Sp3 on activation and/ or repression of transcription still remains to be elucidated.
A considerable level of basal transcription was maintained upon mutation of the TATA box, suggesting that Spl sites are sufficient for activity of a TATA-less P1 promoter. However, we clearly observed a TATA box requirement for Ig κ enhancer-activated P1 transcription. It is assumed that binding of the TFIID complex to the promoter and thereby the alignment of the transcription initiation complex is facilitated by a TATA box [for review, see (71)]. Promoters of several genes apparently lack a TATA box, but often contain multiple Sp1 sites, which have been implicated in efficient and accurate initiation of transcription of these promoters [(6), and references therein]. TATA-dependent activation of transcription by enhancer elements has been reported for the hsp70 (27), the herpes simplex virus (HSV) thymidine kinase (43), and Drosophila actin 5C gene distal promoters (11). Thus, it appears that a TATA box is required for transcription when stimulated by enhancer elements, which might reflect binding of different forms of the TFIID complex to different TATA elements (67).
The nature of the stimulatory signal provided by enhancer elements is still a matter of speculation. Enhancers may either facilitate the assembly of the transcription initiation complex by increasing the density of transcription factors on the promoter or prevent repression of promoters by altering the chromatin structure, or both (20). Recent data have invoked another possible function of enhancers, which is the stimulation of the processivity of RNA polymerase II (pol II) complexes during transcription elongation (70). This appears to be a very attractive mechanism by which the Ig κ enhancers might accomplish activation of c-myc transcription as well as formation of the shift in promoter usage from P2 to PI in BL cells. The transcriptional activity of c-myc is regulated at the level of transcript elongation by the rate of release of pol II complexes from the c-myc promoter P2 (4,16,61). Elongation of polymerases initiated at promoter P1 appears to be impeded by a paused transcription complex at the P2 promoter (62). Recently, it has been demonstrated that the transactivation domains of Sp1 activate the initiation of transcription complexes with low processivity, which tend to stall and/or fall off shortly after initiation (7,36,72). The ability of Sp1 to activate transcript initiation rather than elongation has been correlated with its inability to interact with TFIIH (7). TFIIH, which is directly contacted by the activation domains of different activators such as VP16 and p53, has not only been implicated in transcription initiation, but also in the elongation process [(71), and references therein]. Because PI can be envisaged as an Sp1-dependent promoter, these findings may explain why P1-derived transcripts, successfully reading through promoter P2, are hardly detectable on unrearranged c-myc genes. In BL cells, however, the situation is quite different. Retention of pol II at promoter P2 is abolished in BL cells (10,18,62). Moreover, the Ig κ enhancers are able to relieve pol II complexes pausing at promoter P2 in addition to inducing the BL-specific promoter shift (32). The large amounts of P1-derived transcripts appear not to correlate with an increased density of pol II at the PI promoter, but rather, result from an increased rate of elongation competent polymerases successfully reading into the c-myc gene (62). Thus, it is most likely that the Ig k enhancers provide a stimulatory signal that not only mobilizes pausing pol II complexes at the P2 promoter, but also increases the processivity of initiation complexes at promoter P1.
The prevailing opinion seems to be that the putative stimulatory signal for processive transcription provided by enhancers is brought to a promoter by a looping mechanism (55). This implies direct protein-protein interaction of enhancer binding proteins with either factors of the transcription initiation complex, activator proteins, or both. Interestingly, Sp1 has the ability to interact with itself bound to a proximal and remote site, thereby looping out intervening DNA (63). Moreover, Sp1 can directly interact with a number of other transcription factors including NFκB (48), an indispensable factor for κEi function (38). Thus, Spl appears to be a prime candidate for a mediator of c-myc promoter-Ig enhancer interactions.
ACKNOWLEDGEMENTS
We are grateful to Dirk Eick, David Levens, and Alison Deckhut for helpful discussion and critical revision of the manuscript. We would like to thank Ulla Zimber-Strobl, Lothar Strobl, and Charlotte Meitinger for help with DNase I foot-printing, and Dietmar Tautz, Karen J. Goodrich, Gustaf Hagen, and Regina Feederle for providing plasmids. This work was supported by “Die Deut-sche Forschungsgemeinschaft” (P0325/1-3) and “Fonds der Chemischen Industrie.” C.G. is a fellow of “Die Studienstiftung des deutschen Volkes.”
REFERENCES
- 1. Adams J. M.; Harris A. W.; Pinkert C. A.; Corcoran L. M.; Alexander W. S.; Cory S.; Palmiter R. D.; Brinster R. L. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318:533–538; 1985. [DOI] [PubMed] [Google Scholar]
- 2. Anderson G. M.; Freytag S. O. Synergistic activation of a human promoter in vivo by transcription factor Sp1. Mol. Cell. Biol. 11:1935–1943; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Asselin C; Nepveu A.; Marcu K. B. Molecular requirements for transcriptional initiation of the marine c-myc gene. Oncogene 4:549–558; 1989. [PubMed] [Google Scholar]
- 4. Bentley D. L.; Groudine M. A block to elongation is largely responsible for decreased transcription of c-myc in differentiated HL60 cells. Nature 321:702–706; 1986. [DOI] [PubMed] [Google Scholar]
- 5. Birnbaum M. J.; van Wijnen A. J.; Odgren P. R.; Last T. J.; Suske G.; Stein G. S.; Stein J. L. Sp1 trans-activation of cell cycle regulated promoters is selectively repressed by Sp3. Biochemistry 34:16503–16508; 1995. [DOI] [PubMed] [Google Scholar]
- 6. Blake M. C.; Jambou R. C.; Swick A. G.; Hahn J. W.; Azizkhan J. C. Transcriptional initiation is controlled by upstream GC-box interactions in a TATAA-less promoter. Mol. Cell. Biol. 10:6632–6641; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blau J.; Xiao H.; McCracken S.; O’Hare P.; Greenblatt J.; Bentley D. Three functional classes of transcriptional activation domains. Mol. Cell. Biol. 16:2044–2055; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bornkamm G. W.; Polack A.; Eick D. c-myc deregulation by chromosomal translocation in Burkitt’s lymphoma. In: Klein G., ed. Cellular oncogene activation. New York: Marcel Dekker; 1988:223–273. [Google Scholar]
- 9. Bossone S. A.; Asselin C.; Patel A. J.; Marcu K. B. MAZ, a zinc finger protein, binds to c-myc and C2 gene sequences regulating transcriptional initiation and termination. Proc. Natl. Acad. Sci. USA 89:7452–7456; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cesarman E.; Dalla-Favera R.; Bentley D.; Groudine M. Mutations in the first exon are associated with altered transcription of c-myc in Burkitt lymphoma. Science 238:1272–1275; 1987. [DOI] [PubMed] [Google Scholar]
- 11. Chung Y. T.; Keller E. B. The TATA-dependent and TATA-independent promoters of the Drosophila melanogaster actin 5C-encoding gene. Gene 106:237–241; 1991. [DOI] [PubMed] [Google Scholar]
- 12. Courey A. J.; Tjian R. Analysis of Spl in vivo reveals multiple transcriptional domains, including a novel glutamine-rich activation motif. Cell 55:887–898; 1988. [DOI] [PubMed] [Google Scholar]
- 13. DesJardins E.; Hay N. Repeated CT elements bound by zinc finger proteins control the absolute and relative activities of the two principal human c-myc promoters. Mol. Cell. Biol. 13:5710–5724; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dignam J. D.; Lebovitz R. M.; Roeder R. G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids. Res. 11:1475–1489; 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. DiNocera P. P.; Dawid I. B. Transient expression of genes introduced into cultured cells of Drosophila . Proc. Natl. Acad. Sci. USA 80:7095–7098; 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eick D.; Bornkamm G. W. Transcriptional arrest within the first exon is a fast control mechanism in c-myc expression. Nucleic Acids Res. 14:8331–8346; 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eick D.; Bornkamm G. W. Expression of normal and translocated c-myc alleles in Burkitt’s lymphoma cells: Evidence for different regulation. EMBO J. 8:1965–1972; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eick D.; Polack A.; Kofler E.; Bornkamm G. W. The block of elongation in c-myc exon 1 is abolished in Burkitt’s lymphoma cell lines with variant translocation. Oncogene 3:397–403; 1988. [PubMed] [Google Scholar]
- 19. Emili A.; Greenblatt J.; Ingles C. J. Species-specific interaction of the glutamine-rich activation domains of Sp1 with the TATA box-binding protein. Mol. Cell. Biol. 14:1582–1593; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Felsenfeld G. Chromatin assembly as an esential part of the transcriptional mechanism. Nature 355:219–224; 1992. [DOI] [PubMed] [Google Scholar]
- 21. Filippova G. N.; Fagerlie S.; Klenova E. M.; Myers C.; Dehner Y.; Goodwin G.; Neiman P. E.; Collins S. J.; Lobanenkov V. V. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc finger to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol. Cell. Biol. 16:2802–2813; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fulton R.; Van Ness B. Kappa immunoglobulin promoters and enhancers display developmentally controlled interactions. Nucleic Acids Res. 21:4941–4947; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fulton R.; Van Ness B. Selective synergy of immunoglobulin enhancer elements in B-cell development. A characteristic of kappa light chain enhancers, but not heavy chain enhancers. Nucleic Acids Res. 22:4216–4223; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Garcia J. V.; Bich-Thuy L. T.; Stafford J.; Queen C. Synergism between immunoglobulin enhancers and promoters. Nature 322:383–385; 1986. [DOI] [PubMed] [Google Scholar]
- 25. Gerbitz A.; Mautner J.; Geltinger C.; Hörtnagel K.; Polack A. unpublished data.
- 26. Gill G.; Pascal E.; Tseng Z. H.; Tjian R. Aglutamine-rich hydrophobic patch in transcription factor Sp1 contacts the dTAFIIl 10 component of the Drosophila TFIID complex and mediates transcriptional activation. Proc. Natl. Acad. Sci. USA 91:192–196; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Green J. M.; Kingston R. E. TATA-dependent and TATA-independent function of the basal and heat shock elements of a human hsp70 promoter. Mol. Cell. Biol. 10:1319–1328; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hagen G.; Müller S.; Beato M.; Suske G. Cloning by recognition site screening of two novel GT box binding proteins: A family of Sp1 related genes. Nucleic Acids Res. 20:5519–5525; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hagen G.; Müller S.; Beato M.; Suske G. Sp1-mediated transcriptional activation is repressed by Sp3. EMBO J. 13:3843–3851; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hayday A. C.; Gillies S. D.; Saito H.; Wood C.; Wiman K.; Hayward W. S.; Tonegawa S. Activation of a translocated human c-myc gene by an enhancer in the immunoglobulin heavy-chain locus. Nature 307:334–340; 1984. [DOI] [PubMed] [Google Scholar]
- 31. Hoey T.; Weinzierl R. O. J.; Gill G.; Chen J.-L.; Dynlacht B. D.; Tjian R. Molecular cloning and functional analysis of Drosophila TAF110 reveal properties expected of coactivators. Cell 72:247–260; 1993. [DOI] [PubMed] [Google Scholar]
- 32. Hörtnagel K.; Mautner J.; Strobl L. J.; Wolf D. A.; Christoph B.; Geltinger C.; Polack A. The role of immunoglobulin κ elements in c-myc activation. Oncogene 10:1393–1401; 1995. [PubMed] [Google Scholar]
- 33. Kadonaga J.; Jones L.; Tjian R. Promoter-specific activation of RNA polymerase II transcription by Sp1. Trends Biochem. Sci. 11:20–23; 1986. [Google Scholar]
- 34. Kadonaga J. T.; Carner K. R.; Masiarz F. R.; Tjian R. Isolation of cDNA encoding transcription factor Sp1 and functional analysis of the DNA binding domain. Cell 51:1079–1090; 1987. [DOI] [PubMed] [Google Scholar]
- 35. Kingsley C.; Winoto A. Cloning of GT box-binding proteins: A novel Sp1 multigene family regulating T-cell receptor gene expression. Mol. Cell. Biol. 12:4251–4261; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krumm A.; Hickey L.; Groudine M. Promoter-proximal pausing of RNA polymerase II defines a general rate-limiting step after transcription initiation. Genes Dev. 9:559–572; 1995. [DOI] [PubMed] [Google Scholar]
- 37. Lavenu A.; Pournin S.; Babinet C.; Morello D. The cis-acting elements known to regulate c-myc expression ex vivo are not sufficient for correct transcription in vivo . Oncogene 9:527–536; 1994. [PubMed] [Google Scholar]
- 38. Lenardo M.; Pierce J W.; Baltimore D. Protein-binding sites in Ig gene enhancers determine transcriptional activity and inducibility. Science 236:1573–1577; 1987. [DOI] [PubMed] [Google Scholar]
- 39. Madison L. and Groudine M. Identification of a locus control region in the immunoglobulin heavy-chain locus that deregulates c-myc expression in plasmacytoma and Burkitt’s lymphoma cells. Genes Dev. 8:2212–2226; 1994. [DOI] [PubMed] [Google Scholar]
- 40. Magrath I. The pathogenesis of Burkitt’s lymphoma. Adv. Cancer Res. 55:134–270; 1990. [DOI] [PubMed] [Google Scholar]
- 41. Majello B.; De Luca P.; Hagen G.; Suske G.; Lania L. Different members of the Spl multigene family exert opposite transcriptional regulation of the long terminal repeat of HIV-1. Nucleic Acids Res. 22:4914–4921; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Majello B.; De Luca P.; Suske G.; Lania L. Differential transcriptional regulation of c-myc promoter through the same DNA binding sites targeted by Spl-like proteins. Oncogene 10:1841–1848; 1995. [PubMed] [Google Scholar]
- 43. Majumder S.; DePamphilis M. L. TATA-dependent enhancer stimulation of promoter activity in mice is developmentally acquired. Mol. Cell. Biol. 14:4258–4268; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Marcu K. B.; Bossone S. A.; Patel A. J. myc function and regulation. Annu. Rev. Biochem. 61:809–860; 1992. [DOI] [PubMed] [Google Scholar]
- 45. Mautner J.; Behrends U.; Hörtnagel K.; Brielmeier M.; Hammerschmidt W.; Strobl L.; Bornkamm G. W.; Polack A. c-myc expression is activated by the immunoglobulin κ-enhancers from a distance of at least 30 kb but not by elements located within 50 kb of the unaltered c-myc locus in vivo . Oncogene 12:1299–1307; 1996. [PubMed] [Google Scholar]
- 46. Nishikura K.; ar Rushdi A.; Erikson J.; Watt R.; Rovera G.; Croce C. Differential expression of the normal and of the translocated human c-myc oncogenes in B cells. Proc. Natl. Acad. Sci. USA 80:4822–4826; 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pascal E.; Tjian R. Different activation domains of Sp1 govern formation of multimers and mediate transcriptional synergism. Genes Dev. 5:1646–1656; 1991. [DOI] [PubMed] [Google Scholar]
- 48. Perkins N. D.; Edwards N. L.; Duckett C. S.; Agranoff A. B.; Schmid R. M.; Nabel G. J. A cooperative interaction between NF-kappa B and Sp1 is required for HIV-1 enhancer activation. EMBO J. 12:3551–3558; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Polack A.; Feederle R.; Klobeck G.; Hortnagel K. Regulatory elements in the immunoglobulin kappa locus induce c-myc activation and the promoter shift in Burkitt’s lymphoma. EMBO J. 12:3913–3920; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Polack A.; Strobl L.; Feederle R.; Schweizer M.; Koch E.; Eick D.; Wiegand H.; Bornkamm G W. The intron enhancer of the immunoglobulin kappa gene activates c-myc but does not induce the Burkitt specific promoter shift. Oncogene 6:2033–2040; 1991. [PubMed] [Google Scholar]
- 51. Pulvertaft R. I. V. A study of malignant tumors in Nigeria by short term tissue culture. J. Clin. Pathol. 18:261–273; 1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rabbitts T. H. Chromosomal translocations in human cancer. Nature 372:143–149; 1994. [DOI] [PubMed] [Google Scholar]
- 53. Rabbitts T. H.; Hamlyn P. H.; Baer R. Altered nucleotide sequences of a translocated c-myc gene in Burkitt’s lymphoma. Nature 306:760–765; 1983. [DOI] [PubMed] [Google Scholar]
- 54. Sambrook J.; Fritsch E. F.; Maniatis T. Molecular cloning: A laboratory manual. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 55. Schleif R. DNA looping. Annu. Rev. Biochem. 61:199–223; 1992. [DOI] [PubMed] [Google Scholar]
- 56. Schneider I. Cell lines derived from late embryonic stages of Drosophila melanogaster . J. Embryol. Exp. Morphol. 27:353–365; 1972. [PubMed] [Google Scholar]
- 57. Segal R.; Berk A. J. Promoter activity and distance constraints of one versus two Sp1 binding sites. J. Biol. Chem. 266:20406–20411; 1991. [PubMed] [Google Scholar]
- 58. Siebenlist U.; Hennighausen L.; Battey J.; Leder P. Chromatin structure and protein binding in the putative regulatory region of the c-myc gene in Burkitt lymphoma. Cell 37:381–391; 1984. [DOI] [PubMed] [Google Scholar]
- 59. Spencer C. A.; Groudine M. Transcription elongation and eucaryotic gene regulation. Oncogene 5:777–785; 1990. [PubMed] [Google Scholar]
- 60. Spencer C. A.; Groudine M. Control of c-myc regulation in normal and neoplastic cells. Adv. Cancer Res. 56:1–48; 1991. [DOI] [PubMed] [Google Scholar]
- 61. Strobl L. J.; Eick D. Hold back of RNA polymerase II at the transcription start site mediates down-regulation of c-myc in vivo. EMBO J. 11:3307–3314; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Strobl L. J.; Kohlhuber F.; Mautner J.; Polack A.; Eick D. Absence of a paused transcription complex from the c-myc P2 promoter of the translation chromosome in Burkitt’s lymphoma cells: Implication for the c-myc P1/P2 promoter shift. Oncogene 8:1437–1447; 1993. [PubMed] [Google Scholar]
- 63. Su W.; Jackson S. P.; Tjian R.; Echols H. DNA looping between sites for transcriptional activation: Self-association of DNA-bound Sp1. Genes Dev. 5:820–826; 1991. [DOI] [PubMed] [Google Scholar]
- 64. Sugden B.; Marsh K.; Yates J. A vector that replicates as a plasmid and can be efficiently selected in B-lymphoblasts transformed by Epstein-Barr virus. Mol. Cell. Biol. 5:410–413; 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Takimoto M.; Quinn J. P.; Farina A. R.; Staudt L. M.; Levens D. fos/jun and octamer-binding protein interact with a common site in a negative ele ment of the human c-myc gene. J. Biol. Chem. 264:8992–8999; 1989. [PubMed] [Google Scholar]
- 66. Taub R.; Moulding C.; Battey J.; Murphy W.; Vasicek T.; Lenoir G. M.; Leder P. Activation and somatic mutation of the translocated c-myc gene in Burkitt lymphoma cells. Cell 36:339–348; 1984. [DOI] [PubMed] [Google Scholar]
- 67. Timmers H. T.; Sharp P. A. The mammalian TFIID protein is present in two functional distinct complexes. Genes Dev. 5:1946–1956; 1991. [DOI] [PubMed] [Google Scholar]
- 68. Toneguzzo F.; Hayday A. C.; Keating A. Electric field-mediated DNA transfer: Transient and stable gene expression in human and mouse lymphoid cells. Mol. Cell. Biol. 6:703–706; 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wiman K. G.; Clarkson B.; Hayday A. C.; Saito H.; Tonegawa S.; Hayward W. S. Activation of a translocated c-myc gene: Role of structural alterations in the upstream region. Proc. Natl. Acad. Sci. USA 81:6798–6802; 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yankulov K.; Blau J.; Purton T.; Roberts S.; Bentley D. L. Transcriptional elongation by RNA polymerase II is stimulated by transactivators. Cell 77:749–759; 1994. [DOI] [PubMed] [Google Scholar]
- 71. Zawel L.; Reinberg D. Common themes in assembly and function of eukaryotic transcription complexes. Annu. Rev. Biochem. 64:533–561; 1995. [DOI] [PubMed] [Google Scholar]
- 72. Zhou Q. A.; Sharp P. A. Novel mechanism and factor for regulation by HIV-1 tat. EMBO J. 14:321–328; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zimber-Strobl U.; Kremmer E.; Grasser F.; Mar-schall G.; Laux G.; Bornkamm G. W. The Epstein-Barr virus nuclear antigen 2 interacts with an EBNA2 responsive cis-element of the terminal protein 1 gene promoter. EMBO J. 12:167–175; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zimber-Strobl U.; Strobl L. J.; Meitinger C.; Hinrichs R.; Sakai T.; Furukawa T.; Honjo T.; Bornkamm G. W. Epstein-Barr virus nuclear antigen 2 exerts its transactivating function through interaction with recombination signal binding protein RBP-Jk, the homologue of Drosophila suppressor of hairless. EMBO J. 13:4973–4982; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]