

Abstract
Alu interspersed repetitive elements possess internal RNA polymerase III promoters that are transcribed in vitro and in transfected mouse cells but are nearly silent in human HeLa cells. Transcriptional repression of these elements is to some extent reversible, as pol III-dependent Alu expression can be induced with herpes simplex or adenovirus. To assess whether sequence-specific DNA binding proteins might contribute to Alu transcriptional silencing, we examined the internucleosomal spacer region surrounding the B box of the Alu pol III promoter in HeLa cell nuclei for evidence of proteins bound at specific sites in vivo. We identified a DNase I-hypersensitive site 5′ to the B box and a DNase I-resistant region 3′ to the B box in nuclei. An Alu-specific repressor binds to a 5-bp inverted repeat motif overlapping the 5′ end of the TFIIIC binding site and may inhibit pol III transcription through competitive displacement. The level of Alu-specific pol III repressor activity is significantly reduced in adenovirus-infected HeLa cells, suggesting that the repressor may contribute to Alu transcriptional silencing in vivo. The 3′ DNase I-resistant region coincided with a binding site for the pol II transcription factor YY1 in vitro. YY1 is one of the major proteins in HeLa cells having binding specificity for Alu elements. YY1 bound to tandem arrays of genomic Alu elements may play a role in chromatin organization and silencing.
Keywords: Adenovirus, Alu repetitive sequence, Chromatin, DNA binding proteins, Pol III
FAMILIES of short, interspersed, repetitive elements (SINES) are a prominent feature of mammalian genomes. The major SINE family of primates, the Alu sequence family, contains ∼5 × 105 elements, ∼300 bp in length, having 87% sequence identity and representing ∼5% of the human genome (3,51). Alu elements appear to be derived from the gene coding for 7SL RNA (the RNA component of the signal recognition particle) and to have been amplified and dispersed in genome through retroposition (37,42). The Alu element has a dimeric structure, consisting of two internally deleted 7SL RNA genes, termed the left (5′) and right (3′) monomers, joined head-to-tail through an A-rich linker and followed by a 3′ A-rich tail. The Alu left monomer contains a type II (i.e., “split”) internal pol III promoter consisting of A and B blocks, similar to tRNA and adenovirus VA genes (3,9,15,34,36,42,51,53).
Alu pol III transcripts are relatively scarce in human cells and tissues (e.g., ∼10–103 Alu transcripts vs. 106 7SL RNA transcripts in HeLa cells) (26,27). The reason for this scarcity is not obvious, considering the abundance of Alu elements in the genome. It has been proposed that many Alu elements are transcriptionally silent either due to genetic drift (28,42) or the lack of pol III activator elements in 5′-flanking sequences (25,42,48). However, individual cloned Alu elements are efficiently transcribed by pol III in vitro (14,34,36,39) and are expressed in transfected mouse 3T3 cells, but not in transfected HeLa cells (Humphrey et al, in preparation). The level of Alu pol III transcripts in HeLa cells is increased > 50-fold, to ∼5 × 104 transcripts per cell, by infection with either herpes simplex virus (18,32) or adenovirus (33,39), suggesting that some Alu elements are subject to reversible modes of transcriptional repression.
The induction of Alu expression by adenovirus infection is accompanied by an increase in the accessibility of Alu elements in isolated nuclei to exogenous pol III transcription factors (14,39), suggesting that induction is either due to a change in chromatin structure or to depletion of Alu-specific transcriptional repressors. Alu elements are capable of positioning nuclesomes in chromatin reconstituted in vitro (7) and confer rotational positioning on nucleosomes in vivo (6). Such positioning inhibits pol III transcription from cloned Alu elements in vitro and very likely contributes to transcriptional silencing of Alu elements in vivo. However it seems unlikely that nucleosome positioning alone can account for the profound transcriptional silencing of Alu elements in vivo.
In this study, we have identified protein binding sites located within the internucleosomal spacer region in nuclei surrounding the B box in the Alu pol III promoter. One site, located 5′ to the B box, is located within the in vitro binding site for a pol III transcriptional repressor acting selectively on Alu templates. A second site, located 3′ to the B box, coincides with an in vitro binding site for pol II transcription factor YY1, one of the major proteins in HeLa cell nucleus having binding specificity for Alu elements. The level of pol III repressor activity is significantly reduced in adenovirus-infected HeLa cells, suggesting that the repressor may contribute to Alu transcriptional silencing in vivo.
MATERIALS AND METHODS
Cell and Virus Culture
HeLa S3 cells, obtained from American Type Culture Collection (Rockville, MD), were grown as 2-1 suspension cultures in DMEM (Gibco/BRL) containing 10% calf serum (Gibco/BRL), penicillin (50 U/ml), and streptomycin (50 U/ml). Large-scale HeLa S3 suspension cultures, 20 1, were grown at the National Cell Culture Center (Minneapolis, MN). Adenovirus strain 2 was kindly provided by Dr. R. Padmanabhan (Univ. of Kansas Medical Center). HeLa S3 cells were infected at 50 pfu/cell as described by Panning and Smiley (33).
Pol III-Dependent In Vitro Transcription Assay
As a source of pol III transcription factors, nuclear extract was prepared from HeLa S3 cells, harvested at a density of 106 cells/ml, as described by Dignam et al. (4). The transcription extract was concentrated by precipitation with 33% (w/v) (NH4)2S04, dialyzed against storage buffer [20 mM HEPES (pH 7.9), 100 mM KCl, 1.5 mM MgCl2, 2 mM DTT, 17% glycerol], and stored as aliquots at –70°C (protein concentration: ∼10 μg/μl). Pol III templates were subcloned in plasmid vector mini-s, 1.85 kb, which was constructed by ligating together the “super-ori” region from pHC624, the AmpR gene from pBR322, and the pUC20/21 polylinker (Humphrey et al., in preparation). Plasmid mini-s-AluK was constructed by ligating the HindIII/EcoRI fragment from plasmid pUC19.AluK [which contains a synthetic Alu consensus sequence (22,40) between the BamHI/ EcoRI sites in the polylinker] into the HindIII/ EcoRI site of mini-s (Humphrey et al., in preparation). Plasmid mini-s-AFP was constructed by insertion of a HindIII/EcoRI fragment (658 bp) representing a segment from the fourth intron of the human α-fetoprotein gene (10) containing an Alu element previously subcloned in pUC20 (7). Plasmid mini-s-VAI was constructed by ligating the adenovirus 2 genomic Xbal/BaII fragment (232 bp) containing the VAI RNA gene (20) into the XbaI/SmaI site of mini-s. In vitro transcription reactions were performed in a 25-μl volume containing 6 mM HEPES (pH 7.9), 5 mM MgCl2, 75 mM KCl, 0.6 mM DTT, 5% glycerol, 10 ng mini-s-AluK or mini-s-AFP template, and 1 ng mini-s-VAI template. The concentration of nucleotides was 250 μM for UTP, CTP, and ATP, and 25 μM for GTP (Pharmacia); 5 mCi of [α-32P]GTP (3000 Ci/mmol, Dupont-NEN) was used per reaction. An aliquot of nuclear extract protein was added to the reaction mixture on ice (see the Results section); all dialyzable buffer components were held constant. The reaction was initiated by incubating the samples at 30°C, 60 min. The reaction was stopped by addition of 200 pi of stop buffer [50 mM Tris (pH 7.4), 5 mM EDTA, 250 mM NaCl, 1% SDS, and 10 mg/ml yeast RNA]. Radiolabeled RNA was purified by phenolchloroform extraction and ethanol precipitation and analyzed by electrophoresis in polyacrylamide-urea gels as described previously (7,39). Radiolabeled transcripts were visualized in dried gels by autoradiography. Incorporation of 32P cpm into Alu and VAI transcripts was measured by scanning the dried gels on a Molecular Dynamics Phosphorlmager and analysis with ImageQuant software.
Solid-Phase Adsorbents for Depletion of DNA Binding Proteins
Complementary oligonucleotides for preparation of dsDNA probes were synthesized on an Applied Biosystems DNA synthesizer. All oligonucleotides were 63 bases long except (5′-ORI)2 and (3′-ORI)2, 53 bases. For attachment to streptavidin-agarose, two biotinylated residues were added to the 5′ end of the (+) strand with Biotin-ON phosphoramidite (Clontech). Complementary oligonucleotides were annealed in 10 mM Tris (pH 7.4), 50 mM NaCl, 10 mM MgCl2 by heating at 90°C, 1 min, followed by gradual cooling from 75 °C to 23 °C over 1–2 h. Double-stranded product (ds-oligo) was isolated by electrophoresis in nondenaturing 5% polyacrylamide-TBE gels, located by UV shadowing, recovered by electrocution, and quantitated by optical density. All ds-oligos had an unpaired T at the 5′ ends to facilitate attachment to streptavidin-agarose and radiolabeling for EMSA (see below). Three ds-oligos representing overlapping segments of the AluK left monomer (22,40), K1–57, K40–97, and K63–121, contained the sequence segment specified by position numbers plus flanking “TAT” motifs on either end. RAND is a 63-bp randomized control sequence that contains no matches of greater than 3 bp with either strand of the AluK sequence; in ds-oligos designated RAND+ (#–#) a region in the middle of RAND was modified to match the sequence of the designated segment from the AluK (see Fig. 3); (5′-ORI)2 contains two tandem copies of sequence segment 40–57 in AluK; (3′-ORI)2 contains two tandem copies of sequence segment 174–196 in AluK. Solid-phase affinity adsorbents were prepared by washing an aliquot (100 μl packed volume) of streptavidin-agarose (Pierce) with PBS, resuspending in a 100 μl volume of PBS containing 0.5 nmol ds-oligo (50% slurry), and incubation for 30 min at 23°C on a rotator. The ds-oligo-agarose was washed 3 × with PBS and 1 × with extract storage buffer (see above) containing 0.005% Tween-20 (Bio-Rad). An aliquot (50 μl) of transcription extract (see above) with addition of 0.005% Tween-20 (to prevent nonspecific protein depletion) was mixed with a 10-μl packed volume of ds-oligo-agarose and incubated on a rotator for 45 min at 4°C (2 ×). The adsorbent was removed by centrifugation at 15,000 × g and the protein concentration of the depleted extract determined using the Coomassie G250 dye binding (Bradford) assay (Bio-Rad). Solid-phase immunoaffinity adsorbents were prepared using affinity-purified rabbit IgG raised to Sp1 (sc-59), YY1 (sc-281), and egr-1 (sell 0) peptides obtained from Santa Cruz Biotechnology. A 200-μl aliquot of IgG bound to a 5 μl packed volume of protein A/G-agarose (Pierce) was used to deplete a 50-μl aliquot of transcription extract.
FIG. 3.
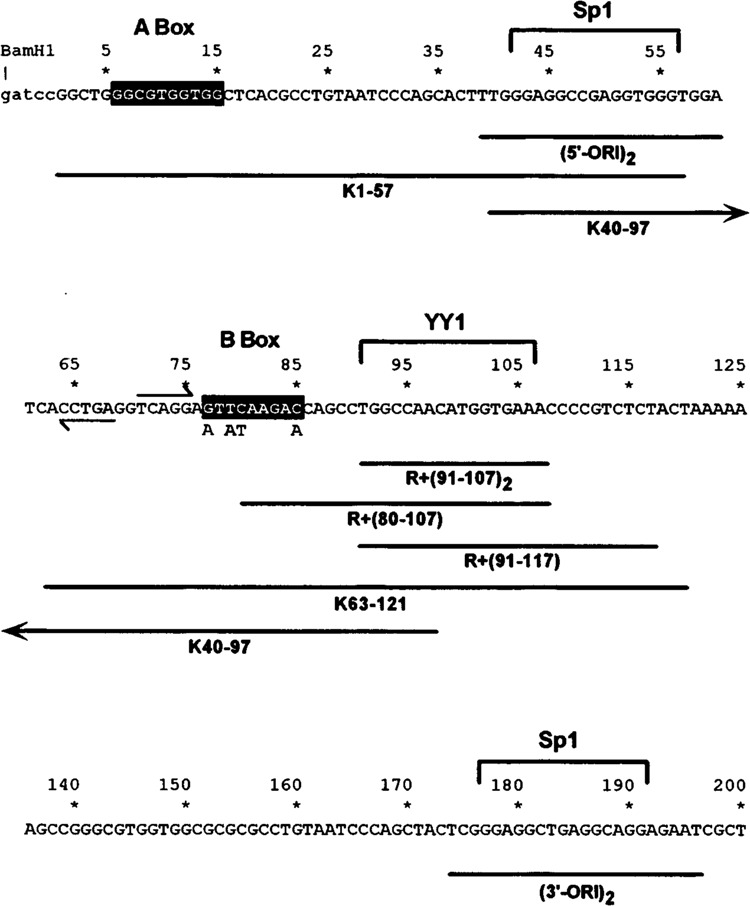
Sequence of AluK pol III promoter and ds-oligo probes used as solid-phase affinity adsorbents. Nucleotide sequence of AluK template from the 5′ BamHI linker (lower case) through the 3′ end of left monomer. Position 1 is the pol III transcriptional start site. Regions of homology to A and B box elements of tRNA and VAI genes are highlighted. Left and right arrows indicate 5-bp inverted repeat. Brackets enclose binding sites for pol II transcription factors Sp1 and YY1. Below: horizontal lines indicate sequence segments represented by ds-oligo probes used as solid-phase affinity adsorbents and as probes for EMSA. The four indicated base substitutions were made in the B box sequence of ds-oligos K40–97, K63–121, and R +(63–97) to prevent depletion of TFIIIC.
Electrophoretic Mobility Shift Analysis
For mobility shift assays (8), nonbiotinylated ds-oligo probes (see above) were radiolabeled using Klenow enzyme and [α-32P]dATP. Binding assays were performed by mixing 10–20 fmol of 32P-radiolabeled probe with 100 ng transcription extract protein in a 10-μl volume of binding buffer [20 mM HEPES (pH 7.9), 50 mM KCl, 1.5 mM MgCl2, 2 mM DTT, 0.05% Tween-20 (v/v), 10% glycerol (v/v), and 3 μg BSA]. For competition assays, the unlabeled ds-oligo competitor (see the Results section) was mixed with the radiolabeled probe, prior to addition of extract protein. After incubation for 20 min on ice, the binding reaction was loaded on a 4% polyacrylamide gel containing 0.5 × TBE and 5% glycerol and was electrophoresed in 0.5 × TBE at 2.5 W constant power for 2–3 h, until bromphenol blue tracking dye loaded in a separate marker lane reached the bottom of the gel. After drying, mobility shift complexes were visualized by autoradiography using Kodak X-Omat film. The amount of radiolabeled probe in the retarded complex was measured by scanning the dried gels on a Molecular Dynamics PhosphorImager and analysis with ImageQuant software.
Purification of Sequence-Specific DNA Binding Proteins
For affinity chromatography, the transcription extract was clarified by centrifugation at 105 × g and Tween-20 was added to 0.005% (v/v). An aliquot, 0.5 ml (5.0 mg protein), of transcription extract was mixed with 4.0 mg yeast RNA (Sigma) and 5.0 nmol ds-oligo competitor, incubated on ice for 10 min, and applied to the RAND+(91–107)2-agarose affinity adsorbent equilibrated with binding buffer [20 mM HEPES (pH 7.9), 100 mM NaCl, 1 mM MgCl2, 2 mM DTT, 0.005% Tween-20 (v/v), 10% glycerol (v/v)] at 4°C. Pairs of columns were run in parallel using either specific [RAND+(91–107)2] or nonspecific (RAND) competitor. The column was washed with 5.0 ml binding buffer and then eluted with four 250-μl aliquots of elution buffer (binding buffer with 500 mM NaCl). The eluate was concentrated and adjusted to 100 mM NaCl using a Centricon-10 (Amicon) filtration device. To prepare protein for amino acid sequencing, the procedure was scaled-up by a factor of five: a 2.5-ml (25 mg protein) aliquot of transcription extract applied to a 1.0 ml column; the eolumn was eluted with 10 mM Tris (pH 7.4), 1 mM EDTA, 0.5% SDS at 23°C. Column fractions were analyzed by electrophoresis in 10% SDS-polyacrylamide gels. Proteins were visualized either by staining with silver (29) or with Coomassie R250 in 10% methanol/10% acetic acid.
Amino Acid Sequence Determination
The 60-kDa polypeptide was prepared for amino acid sequencing by electrophoresing the eluate from the RAND+(91–107)2-agarose column (see above) in a 1.5-mm-thick 8% SDS-polyacrylamide gel using procedures recommended by Hunkapiller et al. (16) to minimize decomposition. The gel was polymerized overnight, the sample was heated in SDS sample buffer at 55 °C for 15 min, and 0.1 mM sodium thioglycolate was added to the upper (cathode) buffer reservoir. Proteins were transferred to nitrocellulose by electroblotting for 16 h at 20 V (47); blotted proteins were visualized by staining with 0.1% Ponceau S dye in 1% acetic acid. A 0.2-cm2 piece of nitrocellulose containing ∼100 pmol of 60 kDa polypeptide was excised, washed with 1.0 ml HPLC-grade water, and stored at –20°C. Amino acid sequencing was performed by William S. Lane of the Harvard Microchemistry Facility as described by Aebersold et al. (1). Briefly, the blotted protein was digested in situ with trypsin, the resulting peptides were separated by reverse-phase HPLC, and two peptides (NT55 and NT57) were selected for analysis by Edman degradation in a gas-phase sequenator (see Table 1).
TABLE 1.
AMINO ACID SEQUENCE ANALYSIS OF TRYPTIC PEPTIDES NT55 AND NT57 ISOLATED FROM 60-kDa BP-2 POLYPEPTIDE
| Cycle | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NT55 | T | L | E | G | E | F | S | V | T | M | – | S | S | D | E | K | |
| YY1, 213–229 | K | T | L | E | G | E | F | S | V | T | M | W | S | S | D | E | K |
| NT57 | F | S | L | D | F | N | L | R | |||||||||
| YY 1,363–371 | R | F | S | L | D | F | N | L | R |
The peptide sequence is aligned with predicted tryptic peptides from cDNA sequence of human YY1 protein (12,35). Tryptic sites are C-terminal to boldface K and R residues. – indicates no residue was identified in peptide NT55 at cycle 11, corresponding to W in the predicted sequence, which is difficult to detect due to chemical instability (16).
Linear PCR-Mediated DNase I Footprint Analysis
Nuclei were isolated from HeLa cells by modification of established methods (6). Briefly, cells were incubated for 30 min on ice in a hypotonic buffer (10 mM, HEPES, pH 7.9, 1 mM PMSF, 1 mM DTT) and Dounce homogenized (20 strokes) in the presence of 3 mM MgCl2 and 0.1% NP-40. Nuclei were recovered by centrifugation through a 0.25-M and subsequently 1-M sucrose layer in a swinging bucket rotor and resuspended in a storage buffer (0.25 M sucrose, 10 mM HEPES, pH 7.9, 3 mM MgCl2, 0.4 mM PMSF, and 1 mM DTT, 50% glycerol). Genomic DNA was purified following lysis of HeLa cells at 37°C for 3 h in the presence of 0.5% SDS and 0.2 mg/ml proteinase K. DNA was extracted several times with phenol/ chloroform and precipitated by spooling from 70% ethanol. The primer used for linear PCR-mediated DNase I foot printing analysis was designed using the major Alu family consensus sequence, which is expected to represent ∼65% of Alu repeats (52). It spans residues 8 to 24 (5′-GCGGTGGCTCACGCCT-3′) within the left monomer and harbors a 3-bp mismatch with the corresponding region in the right monomer. Template DNA was purified following DNase I (Worthington) digestion in nuclei; a typical digest was at 25°C with 0.04 U/μg DNA in nuclei or with 0.001 U/μg naked DNA. To verify the absence of endogenous nuclease activity in a given preparation, samples were incubated without DNase I for the corresponding periods of time and examined by primer extension. Linear amplification reactions were carried out with an end-labeled primer (6): template DNA (50–100 ng) was amplified by 22 cycles of Vent polymerase (New England Biolabs) mediated linear PCR. Cycles were as follows: 1 min at 90°C, 1 min at 58°C, and 30 s at 72°C. Products of amplification were resolved in 6% polyacrylamide 7 M urea gel.
RESULTS
Intranuclear DNase I Footprinting on Genomic Alu Elements
Our previous studies have examined nucleosome positioning on Alu elements in vitro and in vivo (see the Introduction). When cloned Alu elements are reconstituted with histone octamers or tetramers, nucleosomes are positioned over the A box at the 5′ end of the left monomer and over the middle of the right monomer, leaving an internucleosomal spacer region extending from the B box through the A-rich linker (7). Analysis of genomic Alu elements in HeLa cell nuclei by DNase I footprinting and micrococcal nuclease digestion suggested that nucleosome positioning on a readily detectable fraction of Alu elements in vivo is similar to that observed in vitro (6).
To assess whether proteins are bound at specific sites within the internucleosomal spacer region in vivo, we performed a linear PCR-mediated DNase I footprinting analysis in HeLa cell nuclei. The nuclei were isolated without using chelators to avoid possible depletion of Zn+2 finger DNA binding proteins. We used a primer that is specific for the major Alu subfamily to extend in the sense orientation on the lower strand of the left monomer (see the Materials and Methods section). Similar patterns of DNase I cleavage were observed in this region in either nuclei or naked DNA, except that in nuclei we observed: 1) a hypersensitive site at position ∼60, located ∼15 bp 5′ to the B box, 2) a region of diminished DNase cleavage, located 10–20 bp 3′ to the B box, and 3) greatly diminished DNase cleavage in the A-rich spacer, as in our previous study (Fig. 1). These results suggest that proteins are bound at specific sites flanking the B box in a readily detectable fraction of Alu elements in vivo. The absence of footprinting within the B box is consistent with the absence of bound TFIIIC and the transcriptional inactivity of Alu elements in vivo.
FIG. 1.
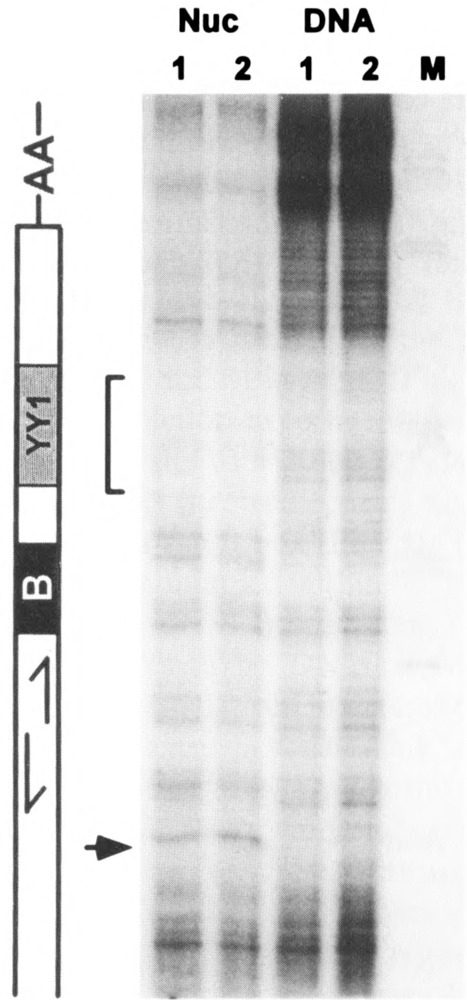
Intranuclear footprinting of protein binding sites in human genomic Alu elements. Linear PCR-mediated DNase I footprinting analysis using either nuclei or genomic DNA prepared from human HeLa S3 cells. The end-labeled primer spanned residues 8 to 24 of the Alu Major subfamily consensus sequence. Right: autoradiogram shows extension products corresponding to the lower strand of the left monomer from positions 50 to 135. M, size markers from Msp I digest of pBR322. Left: schematic representation of the relevant portion of the Alu consensus sequence, as in Fig. 3; inverted repeat motif, positions 64–75; B box, positions 77–85; YY1 binding site, positions 91–104; A-rich linker, positions 121–135. Arrow indicates DNase I-hypersensitive site at position 60. Bracket indicates DNase I-insensitive region in nuclei corresponding to YY1 binding site.
In Vitro Transcription Assay for Alu-Specific Pol III Repressor
To identify proteins that may bind within the internucleosomal spacer region and repress transcription from Alu elements, we employed a standard pol III-dependent in vitro transcription assay. As a source of pol III factors, a transcription extract was prepared from HeLa cell nuclei by extraction with 0.4 M NaCl and concentrated by (NH4)2SO4 precipitation (see the Materials and Methods section). The template for pol III transcription was a synthetic Alu element, AluK (40), based on the consensus of Kariya et al. (22) and similar to the major subfamily consensus, sub-cloned in plasmid vector mini-s (see the Materials and Methods section). This construct, mini-s-AluK, contains a pol III terminator at the 3′ end of the AluK sequence and so codes for a 283-nt pol III transcript. To detect repressors acting selectively on the Alu template, we included the well-characterized adenovirus VAI RNA gene (20), also subcloned in mini-s, as a reference template. Pol III transcription complexes are assembled on Alu and VAI promoters through similar TFIIIC-dependent pathways (see the Discussion section).
To detect specific repressor activity, we used the lowest amount of template supporting detectable transcription: 10 ng mini-s-AluK, 1 ng mini-s-VAI/assay. The Alu and VAI templates were both transcribed efficiently when a low ratio of transcription extract protein to template DNA was used (i.e., 10 μg protein) (Fig. 2A). As the ratio of extract protein to template DNA was increased to 150 μg protein/assay, while holding other buffer components constant, transcription from the Alu template was completely inhibited whereas transcription from the VAI template varied less than twofold (Fig. 2A). Transcription from other pol III templates [i.e., 5S (55) and 7SL RNA (49) genes] either increased or remained constant, similar to VAI (data not shown). This indicates that a nondialyzable component of the transcription extract (e.g., protein) specifically inhibits pol III transcription from the Alu template. In transcription assays performed at high protein to template ratios, we sometimes observed large (> 1 kb) transcripts near the origin of the polyacrylamide gel (Fig. 2). These transcripts were observed using different plasmid vectors as templates, with or without a pol III gene insert, and thus appear to be unrelated to transcription initiating from the Alu promoter.
FIG. 2.
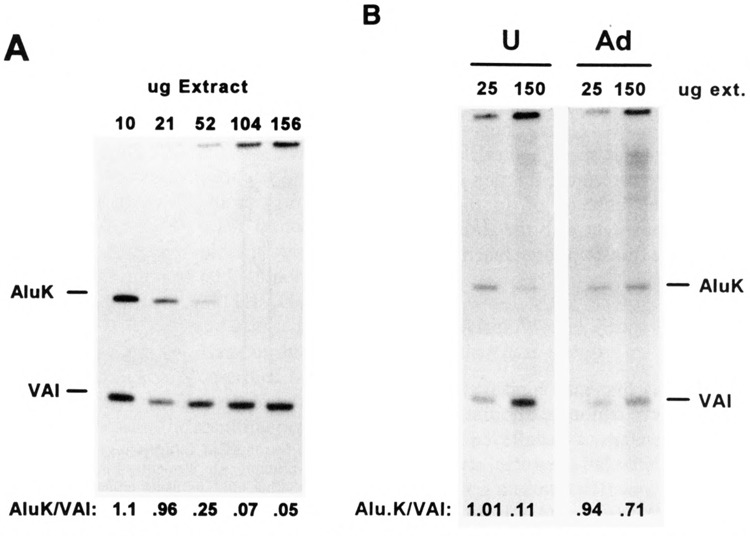
Effect of extract protein/template DNA ratio on Alu, VAI transcription in vitro. Increasing amounts (10–156 μg protein) of transcription extract prepared from HeLa S3 cells were added to pol III-dependent in vitro transcription assays containing 10 ng mini-s-AluK and 1 ng mini-s-VAI template DNA. (A) Radiolabeled AluK and VAI transcripts were resolved by electrophoresis in a 6% polyacrylamide-urea gel and visualized by autoradiography. Incorporation of [α-32P]GTP into Alu and VAI transcripts was quantified by phosphorimaging and is expressed as the ratio of radiolabeled AluK/VAI transcripts in each lane. (B) Pol Ill-dependent in vitro transcription assays, as in (A), performed at either low (25 μg/assay) or high (150 μg/assay) extract protein/template ratios, using either uninfected HeLa cell extract (U) or extract prepared from HeLa cells 24 h after infection with adenovirus (Ad); autoradiography: 45 h.
Depletion of Alu Transcriptional Repressor in Adenovirus-Infected Cells
The level of Alu pol III transcripts in HeLa cells is increased approximately 50-fold 24 h after infection with adenovirus (33). The induction of Alu transcription in vivo is accompanied by an increase in the accessibility of Alu promoters in nuclei to exogenous pol III factors in vitro (39). To assess whether depletion of Alu-specific repressor activity in vitro correlated with the induction of Alu transcription in vivo, we prepared transcription extract from adenovirus-infected HeLa cells. In contrast to extracts prepared from uninfected cells, the infected cell extract supported efficient transcription from the Alu template at either low or high protein/template ratios. A sixfold increase in protein concentration caused only a small decrease in the Alu/VAI transcription ratio (Fig. 2B). The absence of Alu transcriptional repressor activity from the infected cell extract suggests that adenovirus infection leads to either depletion or modification of the repressor protein(s) in HeLa cells, supporting the idea that the repressor may contribute to transcriptional silencing of Alu elements in uninfected cells.
The Alu Transcriptional Repressor Is a Sequence-Specific DNA Binding Protein
The specificity of the pol III transcriptional repressor for the Alu template could be due to site-specific binding within the Alu promoter. To test this idea we prepared three double-stranded 63-bp oligonucleotides representing overlapping segments of the AluK left monomer: Kl-57, positions 1–57; K40–97, positions 40–97; K63–121, positions 63–121 (Fig. 3). The plus strand contained two biotinylated residues at the 5′ end, permitting attachment to streptavidin-agarose to prepare a solid-phase affinity adsorbent. Four base substitutions were made within the B box in K40–97 and K63–121 to prevent depletion of TFIIIC (Fig. 3). A 63-bp ds-oligonucleotide, RAND, served as a specificity control (Fig. 5).
FIG. 5.
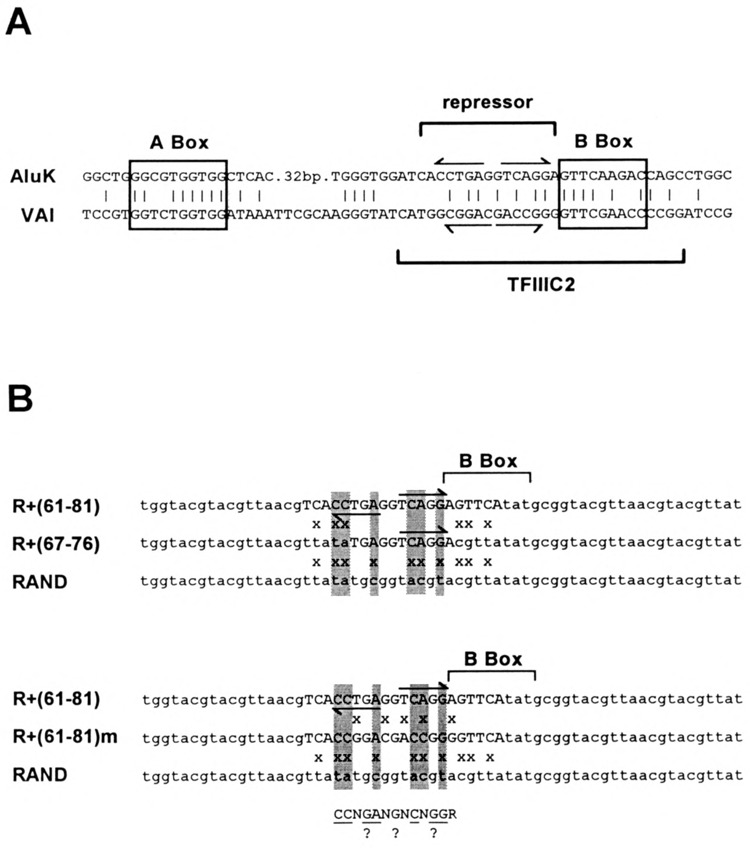
Sequence homology between Aluk and VAI pol III transcriptional promoters. (A) Sequence alignment between VAI gene pol III promoter and homologous regions in the Aluk left monomer. A and B box elements are enclosed by rectangles. Binding site for TFIIIC2, as defined by depletion experiments, are indicated by brackets. (B) Sequence alignment between RAND+(61–81) ds-oligo, containing the minimal Alu repressor binding site as defined by depletion experiments, and the RAND control ds-oligo. Base substitutions that define the minimal repressor binding site are highlighted. Arrows indicate a 5-bp inverted repeat motif at the repressor binding site. Upper alignment includes RAND+(67–76) ds-oligo defining a core repressor binding site; lower alignment includes RAND+(61–81)m ds-oligo with five base substitutions, making the region between positions 65 and 80 match the homologous region of VAI. Below: consensus binding site for Alu pol II transcriptional repressor.
Localization of the Alu Transcriptional Repressor Binding Site
The above extract depletion experiments suggested that the binding site(s) for the Alu transcriptional repressor was located in the overlapping region between K40–97 and K63–121 (i.e., positions 63–97). Based on this inference, we prepared a ds-oligo, RAND+(63–97), in which a central 34-bp region of the 63-bp RAND ds-oligo was modified to match the overlapping sequence in ds-oligos K40–97 and K63–121 (Fig. 3). The partial depletion of repressor activity by ds-oligo K1–57 suggested that the repressor binding site might overlap the 5′ end of the 63–97 region. A perfect 5-bp inverted repeat in this region (positions 64–75, Fig. 5), near the DNase I-hypersensitive site in nuclei further suggested a protein binding site. To encompass this region, we prepared a ds-oligo, RAND+(61–81), in which a 20-bp region of RAND was modified to match this region of AluK between positions 61 and 81 (Figs. 3 and 5B). Because RAND+(61–81) does not include the entire B box, no base substitutions were made in the B box as in ds-oligos K40–97, K63–121, and RAND+(63–97). The RAND+(63–97)-agarose and RAND+(61–81)-agarose adsorbents both depleted Alu transcriptional repressor activity equally well, similarly to the K63–121-agarose positive control (Table 2), indicating that the repressor binding site is located in the region between positions 61 and 81 in AluK (Fig. 5).
TABLE 2.
Alu TRANSCRIPTIONAL REPRESSOR ACTIVITY
| Blank | RAND | R61–81 | R63–97 | K63–121 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 10 μg | 110 μg | 10 μg | 110 μg | 10 μg | 110 μg | 10 μg | 110 μg | 10 μg | 110 μg | |
| AluK/VAI | 1.1 | 0.06 | 1.3 | 0.09 | 2.2 | 0.67 | 2.4 | 0.75 | 2.6 | 0.89 |
| Ads./blank | – | – | 1.1 | 1.6 | 2.0 | 1.2 | 2.1 | 13 | 2.3 | 16 |
| Blank | RAND | R61–81 | R67–76 | R61–81M | ||||||
| 10 μg | 110 μg | 10 μg | 110 μg | 10 μg | 110 μg | 10 μg | 110 μg | 10 μg | 110 μg | |
| AluK/VAI | 1.0 | 0.21 | 1.2 | 0.31 | 1.8 | 1.9 | 1.7 | 1.6 | 1.8 | 1.3 |
| Ads./blank | – | – | 1.2 | 1.5 | 1.8 | 9.2 | 1.7 | 7.8 | 1.8 | 6.1 |
Quantitation of 32P-radiolabeled Alu in vitro transcripts relative to internal VAI controls, as in Fig. 4, using low vs. high concentrations of transcription extract depleted with ds-oligo adsorbents as indicated. AluK/VAI is the ratio of radiolabeled Alu transcripts to VAI transcripts; Ads./blank is the ratio of AluK/ VAI for extracts depleted with a specific adsorbent compared to AluK/VAI for a control depletion using blank streptavidin-agarose.
Depletion of transcription extract with either blank streptavidin-agarose (no attached oligo) or RAND-agarose had no effect on the Alu/VAI transcription ratio (Fig. 4). In contrast, depletion with segments of the Alu left monomer increased Alu transcription at the high protein/template ratio, compared to the controls. Depletion with the K40–97 and K63–121 adsorbents had the greatest effect, increasing the Alu/VAI transcription ratio sixfold compared to depletion with blank agarose. Extract depletion had no effect on VAI transcription, indicating that nonspecific inhibitors of pol III transcription, present in transcription extracts (54,55), are not depleted by these solid-phase adsorbents.
FIG. 4.
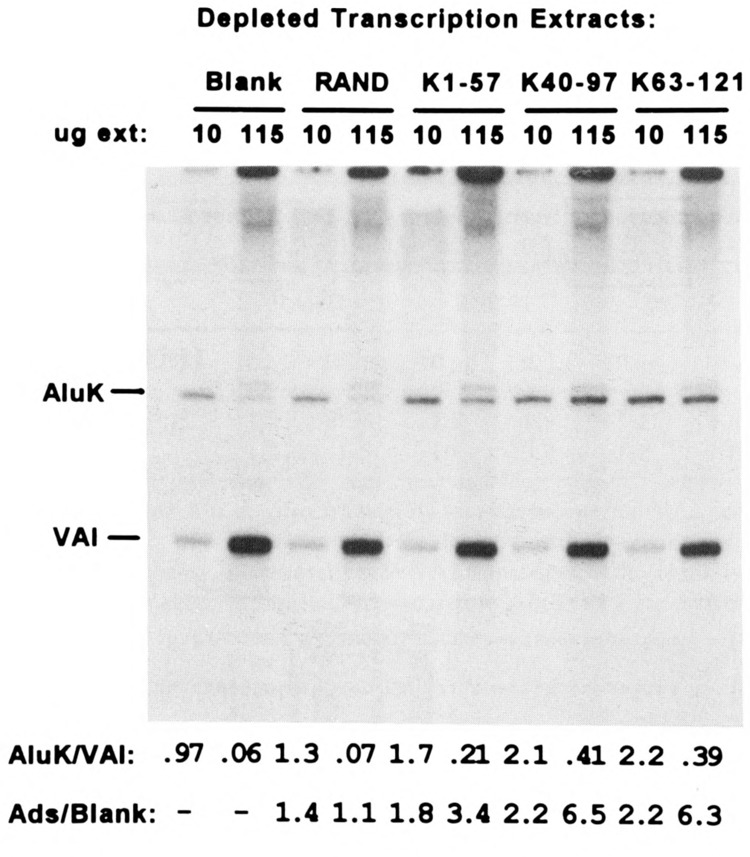
Effect of extract depletion with ds-oligo adsorbents on pol III-dependent transcription from AluK and VAI templates. Pol III-dependent in vitro transcription assays, as in Fig. 2, performed at either low (10 μg/assay) or high (115 μg/assay) extract protein/template ratios using HeLa extracts depleted with either blank streptavidin-agarose or streptavidin-agarose with biotinylated ds-oligo ligand: RAND, K1–57, K40–97, or K63–121; autoradiography: 15 h. Below: ratio of radiolabeled AluK/VAI transcripts determined as in Fig. 2; the enhancement of Alu transcription, relative to VAI, is described by the parameter “Ads/Blank,” which is the Alu/VA transcription ratio in a reaction using extract depleted with ds-oligo-agarose divided by the Alu/VA transcription ratio in the reaction using extract depleted with blank agarose.
The extract depletion experiments localized the repressor binding site to a 20-bp region of AluK, centered on a 5-bp inverted repeat, and overlapping the 5′ side of the B box (Fig. 5). The repressor binding site is contained within the binding site for TFIIIC2 (i.e., the B Box binding component of TFIIIC) as defined by DNase I footprinting (23,43,56) and methylation interference (2). Sequence homology exists between AluK and VAI in the B box region (Fig. 5A). To determine whether the repressor can bind to the homologous region in VAI, we made five base substitutions in ds-oligo RAND+(61–81), to generate ds-oligo RAND+(61–81)m, in which the region between positions 65 and 80 matches the homologous VAI sequence (Fig. 5B). To better define the repressor binding site, we also “truncated” the AluK sequence in ds-oligo RAND+(61–81) to an 11-bp region, making ds-oligo RAND+(67–76).
Extract depletion with either RAND+(61–81)m-agarose or RAND+(67–76)-agarose resulted in partial depletion of Alu transcriptional repressor activity [i.e., less than the RAND+(61–81)-positive control but significantly greater than the negative controls] (Table 2). Partial depletion by RAND+(61–81)m-agarose indicates that the repressor can bind to the homologous region in the VAI promoter, but more weakly than to the Alu promoter (see the Discussion section). Partial depletion by RAND+(67–76)-agarose indicates that this ds-oligo contains the core repressor binding site, but that either flanking region (61–66 or 77–81) enhances binding affinity. It is more likely that the 5′-flanking region (61–66) is required for full binding affinity, because base substitutions within the B box (76–84) had no effect on repressor binding (see above). Comparison of the ds-oligos that deplete repressor activity with the inactive RAND control sequence suggests that the repressor binds preferentially to a 4–5-bp inverted repeat motif of the type: “(CCNGN)NN(NCN GG).”
The conserved Precise and Precise Variant Alu subfamilies have a 2-bp deletion in the 5′ half of the inverted repeat, changing CCTGA to CATGA (28,52). To examine whether this deletion affects transcriptional repression, we used an Alu element (with this deletion) from the fourth intron of the human α-foetoprotein gene (10), Alu-AFP, as a template for in vitro transcription. Pol III transcription from the Alu-AFP promoter was specifically inhibited at a high extract protein/template ratio, similar to transcription from the AluK promoter and transcriptional repression was relieved by extract depletion with RAND+(61–81)-agarose (data not shown). This result indicates that the Alu transcriptional repressor may act on both the major and conserved Alu subfamilies.
Localization of Protein Binding Sites Within the Alu Pol III Promoter
As another means to identify proteins that may contribute to Alu transcriptional silencing in vivo, we employed EMSA to identify proteins in the HeLa nuclear extract binding at specific sites in the AluK left monomer. Using K1–57 as the radio-labeled probe, we identified a protein present in the HeLa transcription extract that formed a discrete, relatively slow-migrating complex (Fig. 6A). The same complex was observed using ds-oligo K40–97 (Fig. 6C), but was not observed using ds-oligo K63–121. The K63–121 probe formed a more rapid-migrating complex, not seen using either the K1–57 or K40–97 probes (Fig. 6B). In addition, a cluster of three minor complexes was only observed using ds-oligo K40–97 (Fig. 6C).
FIG. 6.
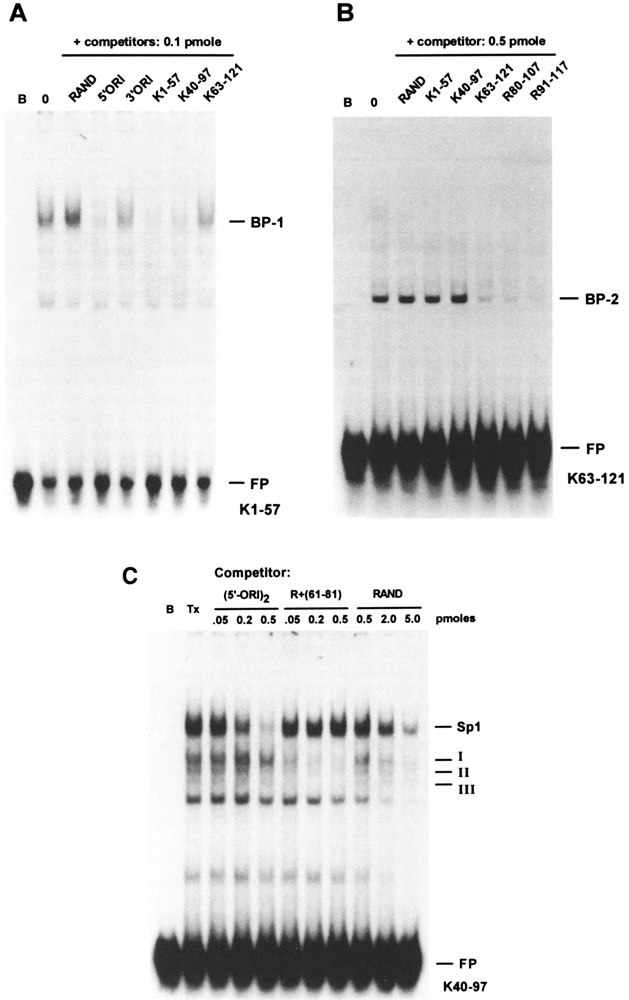
Identification of protein binding sites in AluK left monomer. For electrophoretic mobility shift analysis (EMSA), 0.1 μg extract protein was mixed with 10 fmol 32P-radiolabeled ds-oligo probe in binding buffer and electrophoresed in a 4% polyacrylamide gel; for competition assays, 100 fmol unlabeled ds-oligo was mixed with the radiolabeled probe prior to addition of extract protein. Mobility shift complexes were visualized by autoradiography, 2 h; B, blank binding reaction (no extract protein); 0, no competitor ds-oligo; FP, free probe. (A) Radiolabeled probe: K1–57 ds-oligo containing binding site for specific complex BP-1; (B) radiolabeled probe: K63–121 ds-oligo containing binding site for specific complex BP-2. (C) Probe is 32P-radiolabeled K40–97 ds-oligo; FP, free probe; B, blank binding reaction (no extract protein); Tx, 0.1 μg HeLa extract protein; competition assays contained 0.05–0.5 pmol unlabeled (5′-ORI)2, 0.05–0.5 pmol unlabeled RAND +(61–81) ds-oligo, or 0.5–5.0 pmol unlabeled RAND ds-oligo; positions of mobility shift complexes containing Sp1 and proteins binding to Alu repressor site are indicated.
These results indicated that two distinct and relatively abundant proteins were present in the HeLa cell nuclear extract binding to different sites within the AluK left monomer: one protein, termed BP-1, binding within the overlapping region of K1–57 and K40–97 (i.e., positions 40–57), and the other, termed BP-2, binding between positions 97 and 121 (Fig. 3). To assess whether BP-1 and BP-2 were binding to the probes in a sequence-specific manner, we performed competition assays. Binding of BP-1 to the K1–57 probe was competed by ds-oligos K1–57 and K40–97, but not by K63–121 or the RAND ds-oligo. Similarly, binding of BP-2 to the K63–121 probe was competed by ds-oligo K63–121, but not by K1–57, K40–97, or RAND (Fig. 6A, B).
A data base search indicated that the binding site for BP-1 in the AluK left monomer was a potential binding site for transcription factor Sp1 (21). The region between positions 40 and 60 in AluK, represented by ds-oligo (5′-ORI)2, is similar to an Sp1 binding site in the human snRNA gene enhancer (19), particularly in the location of G-C base pairs identified as contact points by methylation-interference footprinting (Fig. 7A). The ds-oligo (5′-ORI)2 contains the minimal binding site for BP-1 (Fig. 6A, C) and is a stronger competitor than the homologous region in the right monomer (positions 174–196) represented by ds-oligo (3′-ORI)2. The identification of BP-1 as Sp1 was confirmed by EMSA supershift (Fig. 8A) and specific depletion with anti-Sp1 IgG immunoadsorbent (Fig. 9A). The three minor complexes detected by ds-oligo K40–97 were specifically competed by ds-oligo RAND+(61–81), corresponding to the repressor binding site (Fig. 6C).
FIG. 7.

Identification of minimal binding sites for BP-1 and BP-2 in AluK. (A) Lower: sequence alignment of ori-like regions in AluK left (5′-ORI) and right (3′-ORI) monomers; the X indicates mismatches. Upper: Sp1 binding site 2 in human U2 snRNA gene (19); vertical lines indicate identities; underlining indicates G residues on either strand identified as contact points for Sp1 by methylation-interference footprinting. (B) Lower: minimal binding site for BP-2 in AluK, positions 91–107, aligned with modified regions in ds-oligos R+(91–117)PR, R+(91–117)PV, and unmodified RAND ds-oligo; the X indicates mismatches. Upper: YY1 binding site 2 in mouse rpL 30 gene (12); the vertical lines indicate identities; underlining indicates G residues on either strand identified as contact points for YY1 by methylation-interference footprinting.
FIG. 8.
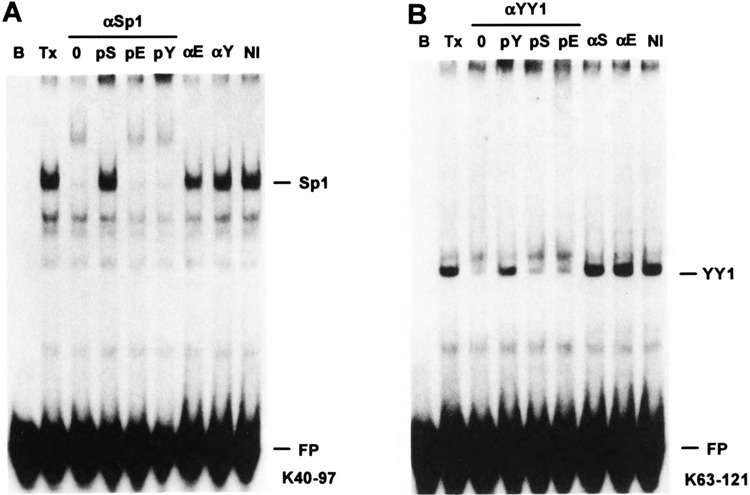
Immunochemical identification of Sp1 and YY1. For EMSA supershift assays, 0.2 μg affinity-purified rabbit IgG raised to either Sp1 (αS), YY1 (αY), or egr-1 (αE), or nonimmune IgG (Nl) was added to a mobility shift binding reaction as in Fig. 6 using either (A) ds-oligo K40–97 as the radiolabeled probe to assay for Sp1 complex or (B) ds-oligo RAND+(91–107)2 as the radiolabeled probe to assay for YY1 complex. As a specificity control, the rabbit αSp1 or αYY1 IgG was preincubated with 2 μg of either Sp1 (pS), YY1 (pY), or egr-1 (pE) peptide antigen or a PBS blank (0) prior to addition to the binding reaction.
FIG. 9.
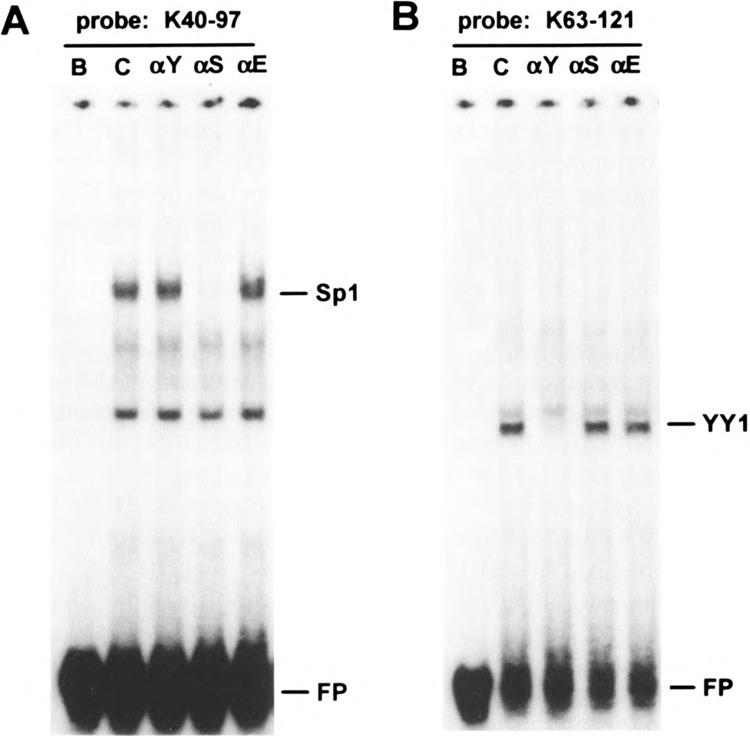
Immunodepletion of Sp1 and YY1. HeLa transcription extracts, depleted with immunoaffinity adsorbents containing affinity-purified rabbit IgG raised to either Spl (αS), YY1 (αY), or egr-1 (αE) or nonimmune IgG (C) were added to mobility shift binding reactions as in Fig. 6 using either (A) ds-oligo K40–97 as the radiolabeled probe to assay for Sp1 complex or (B) ds-oligo RAND+(91–107)2 as the radiolabeled probe to assay for YY1 complex.
Identification of BP-2 as Transcription Factor YY1
To further define the binding site for BP-2, we prepared two ds-oligos in which the central region of the RAND ds-oligo was modified to match the sequence between positions 80 and 107, R+(80–107), or 91 and 117, R+(91–117) (Fig. 3). Both ds-oligos R+(80–107) and R+(91–117) competed binding of BP-2 to the K63–121 probe (see Fig. 6B). Competition by ds-oligo R+(91–107)2 (data not shown) localized the binding site for BP-2 to the region between positions 91 and 107 in AluK (Fig. 3), which corresponds to a protein binding site in vivo (Fig. 1). The data base search (see above) did not identify any known transcription factor binding sites in this region of the AluK left monomer. Therefore, we isolated BP-2 for identification by direct amino acid sequencing. We prepared an affinity adsorbent by attaching biotinylated R+(91–107)2 ds-oligo to streptavidin-agarose (see the Materials and Methods section). HeLa nuclear extract was applied to R+(91–107)2-agarose affinity columns in the presence of either specific R+(91–107)2 or nonspecific RAND competitor ds-oligo competitor. BP-2 binding activity was eluted from the column loaded in the presence of nonspecific competitor but not from the column loaded in the presence of specific competitor and was correlated with a ∼60-kDa polypeptide, present only in the eluate from the column loaded with nonspecific competitor (data not shown). Two tryptic peptides (NT55 and NT57) were analyzed by Edman degradation (see the Materials and Methods section, Table 1). These two peptides match the sequence of two predicted tryptic peptides from human transcription factor YY1 (12,35,45).
Immunochemical Identification of Spl and YY1
We employed an EMSA supershift assay to confirm the identity of mobility shift complexes BP-1 and BP-2 as Sp1 and YY1, respectively. Affinity-purified rabbit IgG raised to peptides from Sp1, YY1, and another Zn+2 finger protein, egr-1 (46), or nonimmune rabbit IgG were added to EMSA binding reactions. Anti-Sp1 IgG specifically supershifted the BP-1 complex and this supershift was specifically blocked by preincubation with the Sp1 peptide antigen (Fig. 8A). Anti-YY1 IgG specifically inhibited formation of the BP-2 complex (apparently, the IgG–YY1 complex does not bind to the DNA probe) and this inhibition was prevented by preincubation with the YY1 peptide antigen (Fig. 8B).
Mobility shift complexes BP-1 and BP-2 were also specifically depleted from the HeLa extract by adsorption with anti-Sp1 or anti-YY1 IgG, respectively, bound to protein A/G-agarose (Fig. 9). Blank and anti-egr-1 (46) protein A/G-agarose served as specificity controls. Depletion of the transcription extract by the immunoaffinity adsorbents to Sp1, YY1, and egr-1 had no significant effects on the Alu/VAI transcription ratio, demonstrating that these transcription factors do not interfere directly with TFIIIC binding to the Alu promoter. Similarly, extract depletion with ds-oligos representing the Sp1 or YY1 binding sites had little or no effect on the Alu/VAI transcription ratio (data not shown).
YY1 Binds Selectively to “Old” Alu Elements
Base substitutions within the 90–110 region of the Alu element, termed the DB region, distinguish different Alu subfamilies (26,52). The AluK sequence is characteristic of the Major subfamily, which includes ∼65% of human Alu elements and includes the older, more divergent elements. To assess whether YY1 can bind to the more recently inserted Alu elements, we made two base substitutions in R+(91–117): C95→T and T100→C to match the sequence of the Precise subfamily in this region, creating ds-oligo R+(91–117)PR, and two additional substitutions: T91→C and C98→A to match the sequence of the Precise Variant subfamily, creating ds-oligo R+(91–117)PV (Fig. 7B). The Precise subfamily includes 10% of human Alu elements and represents more conserved elements inserted during the period of anthropoid radiation. The Precise Variant subfamily represents a small number of Alu elements (<1%), which are currently undergoing amplification and dispersal in the human genome (28,42).
The R+(91–117) ds-oligo is ∼200-fold more effective than the RAND ds-oligo as a competitor of YY1 binding in EMSA (Fig. 10). The two base substitutions in ds-oligo R+(91–117)PR reduce the binding stability of YY1 ∼10-fold. The two additional base substitutions in R+(91–117)PV reduce the binding stability ∼200-fold, to the same level as the RAND competitor (Fig. 10). Thus, YY1 binds preferentially to the Major Alu subfamily and appears not to bind at all to the minor PV subfamily. Of the well-characterized YY1 binding sites, the site in the +3 to +41 region of the mouse rpL 30 gene (12) is the most similar to the Alu DB region, with 10 identities in 14 residues, including contact points identified by methylation interference footprinting (see Fig. 7B). Base substitutions in the Precise and Precise Variant DB regions eliminate contact points at C95→T and C98→A, thus consistent with the reduced binding affinity of YY1 for these Alu subfamilies (Fig. 7B).
FIG. 10.
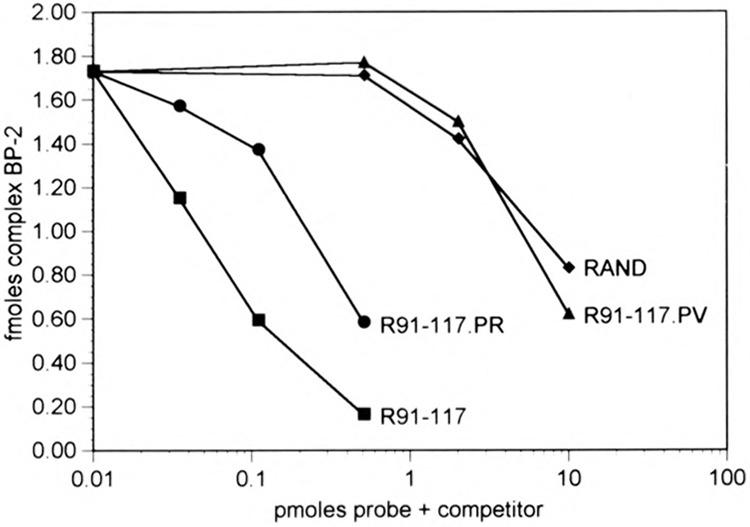
Binding affinity of YY1 for different Alu subfamilies. EMSA, as in Fig. 6, using 10 fmol radiolabeled R+(91–107)2 ds-oligo probe plus increasing amounts of unlabeled ds-oligo competitors: R+(91–117), R+(91–117)PR, R+(91–117)PV, and RAND as in Fig. 7B. The amount of radiolabeled probe present as BP-2 complex in each lane was quantified by phosphorimaging and is plotted as femtomoles complex vs. picomoles ds-oligo probe plus competitor in each binding reaction.
DISCUSSION
Previous reports have established that the vast majority of transcriptionally competent Alu elements in nuclei are masked from the pol III transcriptional machinery, and that induction of Alu transcription by adenovirus infection can be accounted for by an increased accessibility of these elements to general pol III factors (14,18,32,33,39). Alu elements contain intrinsic nucleosome positioning information, and such positioning results in efficient repression of template activity on cloned Alu elements reconstituted with histone octamers in vitro (7). Nucleosomes are positioned on a detectable fraction of Alu elements in vivo, implying that nucleosome positioning is largely responsible for the transcriptional silencing of Alu elements in vivo (6). However, sequence-specific DNA binding proteins may also contribute to silencing, either by direct interference with transcription complex assembly or indirectly, by stabilizing nucleosome positioning. In this study, we have identified specific binding sites for two proteins within the internucleosomal spacer region, which may mediate reversible modes of transcriptional repression in vivo.
We have evidence for one (or more) transcriptional repressor proteins present in HeLa nuclear extracts binding at an inverted repeat motif (TCAGG) located just 5′ to the B box, near the DNase I-hypersensitive site observed in nuclei. The repressor exhibits a relatively loose binding specificity and can repress transcription from both the less highly conserved Alu Major subfamily (representing 65% of genomic Alu elements) and the more highly conserved Alu Precise subfamily (representing 10% of genomic Alu elements). The Alu repressor activity is much reduced in transcription extracts prepared from adenovirus-infected HeLa cells, implying that the repressor protein may contribute to Alu transcriptional silencing in vivo.
The Alu repressor binding site overlaps the 5′ end of the TFIIIC2 binding site as defined by DNase I footprinting (2,23,43,56), thus the repressor could block transcription by competitive displacement of TFIIIC from the B box. The Alu repressor can also bind to the homologous region 5′ to the VAI B box, but does not repress transcription from the VAI template. As TFIIIC binds to the VAI B box with greater stability than to the Alu B box (36), the repressor may be able to displace TFIIIC from the Alu B box but not from the VAI B box. The specificity of the repressor for Alu templates could also be due to conformational differences in the pol III transcription complex; the A and B boxes are further apart in the Alu promoter than in the VAI promoter.
In particular Alu elements, the region just 5′ to the Alu B box can be a binding site for either retinoic acid receptor (50) or estrogen receptor (31) if direct repeats of the hormone response element (HRE), GGTCA, are present. The Alu transcriptional repressor is unlikely to be an HRE binding protein, as ds-oligos containing HRE motifs were less effective in depleting repressor activity than the RAND+(61–81) ds-oligo (Humphrey and Howard, unpublished). It is possible that retinoic acid receptor and/or estrogen receptor bound at this site may contribute to formation of the DNase I-hypersensitive site 5′ to the Alu B box.
We have identified binding sites for the Zn+2 finger transcription factors Spl and YY1 in the human Alu Major subfamily consensus sequence, representing ∼65% of genomic Alu elements (52). Sp1 and YY1 appear to be the most abundant proteins in the HeLa nuclear extract having binding specificity for the Alu left monomer. Of these two, YY1 is of greater potential significance, as its binding site is located within the internucleosomal spacer region and corresponds to a DNase I-insensitive region in nuclei. It is possible that the Sp1 binding site may be accessible in particular Alu elements having alterations in chromatin structure.
YY1 can either activate or repress pol II transcription depending on the context of its binding site within a promoter or enhancer (11,12,35,41,45). Proposed mechanisms for transcriptional activation and repression by YY1 include specific binding to other transcription factors (11,24,44), DNA bending (30), and nucleosome positioning (45). In vivo, pol III transcripts from Major Alu subfamily members are relatively scarce compared to transcripts from PR and PV subfamily members (26–28,42). The inverse correlation between YY1 binding and pol III expression could imply that YY1 contributes to transcriptional silencing of Alu elements in vivo, but we were unable to demonstrate any direct effect of YY1 on pol III transcription in vitro. YY1 binding within the internucleosomal spacer region might have an indirect effect on Alu transcription by contributing to stable positioning of a nucleosome over the 5′ region of the left monomer, as has been suggested for the yeast α2 repressor (38).
In addition to local cis-acting effects, YY1 binding to Alu elements may have implications for general chromatin structure. Some Alu elements exist in long arrays (17). Insertions of P elements in the Drosophila genome as tandem arrays can induce formation of heterochromatin, possibly due to pairing of adjacent repeats mediated by sequence-specific DNA binding proteins (5,13). The DNase I footprint at the YY1 binding site in nuclei suggests that YY1 may be bound to a significant fraction of the ∼3 × 105 genomic Alu elements in vivo. Based on our recovery of YY1 protein using affinity chromatography, we estimate that a HeLa cell nucleus contains at least 5 × 104 YY1 molecules with Alu-specific binding activity; thus, a significant fraction of YY1 molecules may be sequestered by Alu elements. It is interesting to speculate that YY1 and other proteins bound to adjacent Alu repeats in tandem arrays might mediate pairing of Alu elements leading to heterochromatin formation, analogous to the mechanism proposed for Drosophila.
ACKNOWLEDGEMENTS
The authors wish to thank Dr. Richard Maraia for helpful discussions and critical reading of the manuscript; R. Padmanabhan for adenovirus strain 2; T. Howard for assistance with cell culture; and V. Russanova for providing adenovirus-infected cells.
REFERENCES
- 1. Aebersold R. H.; Leavitt J.; Saavedra R. A.; Hood L. E.; Kent S. B. H. Internal amino acid sequence analysis of proteins separated by one- or two-dimensional gel electrophoresis after in situ protease digestion on nitrocellulose. Proc. Natl. Acad. Sci. USA 84:6970–6974; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cromlish J. A.; Roeder R. G. Human transcription factor HIC (TFIIIC). J. Biol. Chem. 264:18100–18109; 1989. [PubMed] [Google Scholar]
- 3. Deininger P. SINEs: Short interspersed repeated DNA elements in higher eucaryotes. In: Berg D. E.; Howe M. M., eds. Mobile DNA. Washington, DC: American Society for Microbiology; 1989:619–636. [Google Scholar]
- 4. Dignam J. D.; Lebovitz R. M.; Roeder R. G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11:1475–1489; 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dorer D. R.; Henikoff S. Expansions of transgene repeats cause heterochromatin formation and gene silencing in Drosophila . Cell 77:993–1002; 1993. [DOI] [PubMed] [Google Scholar]
- 6. Englander E. W.; Howard B. H. Nucleosome positioning by human Alu elements in chromatin J. Biol. Chem. 270:10091–10096; 1995. [DOI] [PubMed] [Google Scholar]
- 7. Englander E. W.; Wolffe A. P.; Howard B. H. Nucleosome interactions with a human Alu element: Transcriptional repression and effects of template methylation. J. Biol. Chem. 268:19565–19573; 1993. [PubMed] [Google Scholar]
- 8. Fried M.; Crother D. M. Equilibrium and kinetics of lac repressor-operator interactions by polyacrylamide gel electrophoresis. J. Mol. Biol. 172:263–282; 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Geiduschek E. P.; Tocchini-Valentini G. P. Transcription by RNA polymerase III. Annu. Rev. Biochem. 57:873–914; 1988. [DOI] [PubMed] [Google Scholar]
- 10. Gibbs P. E. M.; Zielinski R.; Boyd C.; Dugaic-zyk A. Structure, polymorphism, and novel repeated DNA elements revealed by a complete sequence of the human α-fetoprotein gene. Biochemistry 26:1332–1343; 1987. [DOI] [PubMed] [Google Scholar]
- 11. Hahn S. The yin and yang of mammalian transcription. Curr. Biol. 2:152–154; 1992. [DOI] [PubMed] [Google Scholar]
- 12. Hariharan N.; Kelley D. E.; Perry R. P. δ, a transcription factor that binds to downstream elements in several polymerase II promoters, is a functionally versatile zinc finger protein. Proc. Natl. Acad. Sci. USA 88:9799–9803; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Henikoff S. A reconsideration of the mechanism of position effect. Genetics 138:1–5; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Howard B. H.; Russanova V. R.; Englander E. W. Alu silencing mechanisms: Implications for the modulation of local chromatin structure. In: Maraia R. J., ed. The impact of short interspersed elements (SINES) on the host genome. Austin, TX: R. G. Landes Co.; 1995:133–141. [Google Scholar]
- 15. Humphrey G. W. SINEs and trans-acting factors. In: Maraia R. J., ed. The impact of short interspersed elements (SINES) on the host genome. Austin, TX: R. G. Landes Co.; 1995:197–222. [Google Scholar]
- 16. Hunkapiller M. W.; Lujan E.; Ostrander F.; Hood L. E. Isolation of microgram quantities of proteins from polyacrylamide gels for amino acid sequence analysis. Methods Enzymol. 91:227–236; 1983. [DOI] [PubMed] [Google Scholar]
- 17. Iris F. J. M.; Bougueleret L.; Prieur S.; Caterina D.; Primas G.; Perrot V.; Jurka J.; Rodriguez-Tome P.; Claverie J. M.; Dausset J.; Cohen D. Dense Alu clustering and a potential new member of the NFκB family within a 90 kilobase HLA class III segment. Nat. Genet. 3:137–145; 1993. [DOI] [PubMed] [Google Scholar]
- 18. Jang K. L.; Latchman D. S. HSV infection induces increased transcription of Alu repeated sequences by RNA polymerase III. FEBS Lett. 258:255–258; 1989. [DOI] [PubMed] [Google Scholar]
- 19. Janson L.; Bark C.; Pettersson U. Identification of proteins interacting with the enhancer of human U2 small nuclear RNA genes. Nucleic Acids Res. 15:4997–5016; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jutterman R.; Hosokawa K.; Kochanek S.; Doer-fler W. Adenovirus type 2 VAI RNA transcription by polymerase III is blocked by sequence-specific methylation. J. Virol. 65:1735–1742; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kadonaga J. T.; Carner K. R.; Masiarz F. R.; Tjian R. Distinct regions of Spl modulate DNA binding and transcriptional activation. Cell 51:1079–1090; 1987. [DOI] [PubMed] [Google Scholar]
- 22. Kariya Y.; Kato K.; Hayashizaki Y.; Himeno S.; Tarui S.; Matsubara K. Revision of consensus sequence of human Alu repeats-a review. Gene 53:1–10; 1987. [DOI] [PubMed] [Google Scholar]
- 23. Kochanek S.; Renz D.; Doerfler W. Probing DNA-protein interactions in vitro with the CpG DNA methyltransferase. Nucleic Acids Res. 21:2339–2342; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee J.-S.; Galvin K. M.; Shi Y. Evidence for physical interaction between the zinc-finger transcription factors YY1 and Spl. Proc. Natl. Acad. Sci. USA 90:6145–6149; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Leeflang E. P.; Liu W.-M.; Chesnokov I. N.; Schmid C. W. Mobility of short interspersed repeats within the chimpanzee lineage. J. Mol. Evol. 37:559–565; 1993. [DOI] [PubMed] [Google Scholar]
- 26. Liu W.-M.; Maraia R. J.; Rubin C. M.; Schmid C. W. Alu transcripts: Cytoplasmic localisation and regulation by DNA methylation. Nucleic Acids Res. 22:1087–1095; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maraia R. J.; Driscoll C. T.; Bilyeu T.; Hsu K.; Darlington G. J. Multiple dispersed loci produce small cytoplasmic Alu RNA. Mol. Cell. Biol. 13:4233–4241; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matera A. G.; Hellmann U.; Schmid C. W. A transpositionally and transcriptionally competent Alu subfamily. Mol. Cell. Biol. 10:5424–5432; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morrissey J. H. Silver stain for proteins in polyacrylamide gels: A modified procedure with enhanced uniform sensitivity. Anal. Biochem. 117:307–310; 1981. [DOI] [PubMed] [Google Scholar]
- 30. Natesan S.; Gilman M. Z. DNA bending and orientation-dependent function of YY1 in the c-fos promoter. Genes Dev. 7:2497–2509; 1993. [DOI] [PubMed] [Google Scholar]
- 31. Norris J.; Fan D.; Aleman C.; Marks J. R.; Futreal P. A.; Wiseman R. W.; Iglehart J. D.; Deininger P. L.; McDonnell D. P. Identification of a new subclass of Alu DNA repeats which can function as estrogen receptor-dependent transcription enhancers. J. Biol. Chem. 270:22777–22782; 1995. [DOI] [PubMed] [Google Scholar]
- 32. Panning B.; Smiley J. R. Activation of RNA polymerase III transcription of human Alu elements by herpes simplex virus. Virology 202:408–417; 1994. [DOI] [PubMed] [Google Scholar]
- 33. Panning B.; Smiley J. R. Activation of RNA polymerase III transcription of Human Alu repetitive elements by adenovirus type 5: Requirement for the Elb 58-kilodalton protein and the products of E4 open reading frames 3 and 6. Mol. Cell. Biol. 13:3231–3244; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paolella G.; Lucero M. A.; Murphy M. H.; Baralle F. E. The Alu family repeat promoter has a tRNA-like bipartite structure. EMBO J. 2:691–696; 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Park K.; Atchison M. L. Isolation of a candidate repressor/activator, NF-El (YY-1, δ), that binds to the immunoglobulin κ3′ enhancer and the immuno-globulin heavy-chain μE1 site. Proc. Natl. Acad. Sci. USA 88:9804–9808; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Perez-Stable C.; Shen C.-K. J. Competitive and cooperative functioning of the anterior and posterior elements of an Alu family repeat. Mol. Cell. Biol. 6:2041–2052; 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Quentin Y. Origin of the Alu family: A family of Alu-like monomers gave birth to the left and the right arms of the Alu elements. Nucleic Acids Res. 20:3397–3401; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roth S. Y.; Dean A.; Simpson R. T. Yeast α2 repressor positions nucleosomes in TRP1/ARS1 chromatin. Mol. Cell. Biol. 10:2247–2260; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Russanova V. R.; Driscoll C. T.; Howard B. H. Adenovirus type 2 preferentially stimulates polymerase III transcription of Alu elements by relieving repression: A potential role for chromatin. Mol. Cell. Biol. 15:4282–4290; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sakamoto K.; Fordis C. M.; Corsico C. D.; Howard T. H.; Howard B. H. Modulation of HeLa cell growth by transfected 7SL RNA and Alu gene sequences. J. Biol. Chem. 266:3031–3038; 1991. [PubMed] [Google Scholar]
- 41. Satyamoorthy K.; Park K.; Atchison M. L.; Howe C. C. The intracisternal A-particle upstream element interacts with transcription factor YY1 to activate transcription: Pleitropic effects of YY1 on distinct DNA promoter elements. Mol. Cell. Biol. 13:6621–6628; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schmid C.; Maraia R. Transcriptional regulation and transpositional selection of active SINE sequences. Curr. Opin. Genet. Dev. 2:874–882; 1992. [DOI] [PubMed] [Google Scholar]
- 43. Schneider H. R.; Waldschmidt R.; Jahn D.; Seifart K. H. Purification of human transcription factor HIC and its binding to the gene for ribosomal 5S RNA. Nucleic Acids Res. 17:5003–5015; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Seto E.; Lewis B.; Shenk T. Interaction between transcription factors Spl and YY1. Nature 365:462–464; 1993. [DOI] [PubMed] [Google Scholar]
- 45. Shi Y.; Seto E.; Chang L.-S.; Shenk T. Transcriptional repression by YY1, a human GLI-Kruppel-related protein, and relief of repression by adenovirus ElA protein. Cell 67:377–388; 1991. [DOI] [PubMed] [Google Scholar]
- 46. Sukhatme V. P.; Cao X.; Chang L. C.; Tsai-Morris C.-H.; Stamenkovich D.; Ferreira P. C. P.; Cohen D. R.; Edwards S. A.; Shows T. B.; Curran T.; LeBeau M. M.; Adamson E. D. A zinc finger-encoding gene coregulated with c-fos during growth and differentiation, and after cellular depolarization. Cell 53:37–43; 1988. [DOI] [PubMed] [Google Scholar]
- 47. Towbin H.; Staehelin T.; Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 76:4350–4354; 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ullu E.; Tschudi C. Alu sequences are processed 7SL RNA genes. Nature 312:171–172; 1984. [DOI] [PubMed] [Google Scholar]
- 49. Ullu E.; Weiner A. M. Upstream sequences modulate the internal promoter of the human 7SL RNA gene. Nature 318:371–374; 1985. [DOI] [PubMed] [Google Scholar]
- 50. Vansant G.; Reynolds W. F. The consensus sequence of a major Alu subfamily contains a functional retinoic acid response element. Proc. Natl. Acad. Sci. USA 92:8229–8233; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weiner A. M.; Deininger P. L.; Efstratiadis A. Nonviral retroposons: Genes, pseudogenes, and transposable elements generated by the reverse flow of genetic information. Annu. Rev. Biochem. 55:631–661; 1986. [DOI] [PubMed] [Google Scholar]
- 52. Willard C.; Nguyen H. T.; Schmid C. W. Existence of at least three distinct Alu subfamilies. J. Mol. Evol. 26:180–186; 1987. [DOI] [PubMed] [Google Scholar]
- 53. Willis I. M. RNA polymerase III: Genes, factors and transcriptional specificity. Eur. J. Biochem. 212:1–11; 1993. [DOI] [PubMed] [Google Scholar]
- 54. Wilson E. T.; Larson D.; Young L. S.; Sprague K. U. A large region controls tRNA gene transcription. J. Mol. Biol. 183:153–163; 1985. [DOI] [PubMed] [Google Scholar]
- 55. Wolffe A. P.; Jordan E.; Brown D. D. A bacteriophage RNA polymerase transcribes through a Xenopus 5S RNA gene transcription complex without disrupting it. Cell 44:381–389; 1986. [DOI] [PubMed] [Google Scholar]
- 56. Yoshinaga S. K.; L’Etoile N. D.; Berk A. J. Purification and characterization of transcription factor IIIC2. J. Biol. Chem. 264:10726–10731; 1989. [PubMed] [Google Scholar]