

Abstract
Modulation of the epithelial Na+ channel (ENaC) activity in the collecting duct (CD) is an important mechanism for normal Na+ homeostasis. ENaC activity is inversely related to dietary Na+ intake, in part due to inhibitory paracrine purinergic regulation. Evidence suggests that H+,K+-ATPase activity in the CD also influences Na+ excretion. We hypothesized that renal H+,K+-ATPases affect Na+ reabsorption by the CD by modulating ENaC activity. ENaC activity in HKα1 H+,K+-ATPase knockout (HKα1−/−) mice was uncoupled from Na+ intake. ENaC activity on a high-Na+ diet was greater in the HKα1−/− mice than in WT mice. Moreover, dietary Na+ content did not modulate ENaC activity in the HKα1−/− mice as it did in WT mice. Purinergic regulation of ENaC was abnormal in HKα1−/− mice. In contrast to WT mice, where urinary [ATP] was proportional to dietary Na+ intake, urinary [ATP] did not increase in response to a high-Na+ diet in the HKα1−/− mice and was significantly lower than in the WT mice. HKα1−/− mice fed a high-Na+ diet had greater Na+ retention than WT mice and had an impaired dipsogenic response. These results suggest an important role for the HKα1 subunit in the regulation of purinergic signaling in the CD. They are also consistent with HKα1-containing H+,K+-ATPases as important components for the proper regulation of Na+ balance and the dipsogenic response to a high-salt diet. Such observations suggest a previously unrecognized element in Na+ regulation in the CD.
Keywords: purinergic signaling, regulated epithelial Na channel, ENaC, renal
the renal collecting duct (CD) represents the final site of Na+ reabsorption and is important for normal salt balance. Within the CD, the epithelial Na+ channel (ENaC) is the principal mechanism for Na+ reabsorption, and modulation of ENaC activity is an important mechanism for the regulation of renal Na+ homeostasis. ENaC activity is inversely related to dietary Na+ intake, in part due to inhibitory paracrine purinergic regulation in the CD. Loss of this regulation contributes to salt-sensitivity, and purinergic signaling has been implicated in mineralocorticoid escape (26). Paracrine purinergic signaling has emerged as an important regulator of Na+ excretion in the distal nephron (27, 28). ATP is secreted from intercalated cells in response to increases in urine flow and is facilitated by a flow-sensitive ATP-K+ efflux-coupled mechanism involving ATP release through the connexin hemichannels and K+ efflux through the calcium-activated big K+ channel (BKCa) (1, 8). ATP acts to decrease ENaC activity by decreasing channel open probability, Po (21, 22, 26). Urinary ATP increases proportionally with dietary Na+ intake facilitating Na+ excretion through the downregulation of ENaC (22, 26). Purinergic receptor P2Y2 knockout mice have normal ATP levels but have an impaired capacity to excrete Na+ (26). These mice failed to respond appropriately to increases in Na+ intake and exhibited salt-sensitive hypertension (22). Treatment with mineralocorticoid revealed that when similar Na+ intake levels are compared, P2Y2 knockout mice had a greater increase in blood pressure (BP) in response to desoxycorticosterone acetate (DOCA) than controls.
The renal H+,K+-ATPases localize to the apical membrane of intercalated cells of the CD and couple proton secretion and K+ absorption to the hydrolysis of ATP (6). Two renal H, K-ATPase isoforms have been identified in kidney, HKα1 and HKα2. The HKα1 isoform is predominately expressed in the stomach, where it is responsible for gastric acid secretion. The HKα2 isoform is abundantly expressed in the colon as well as in other tissues. In the kidney, both enzymes acidify the tubular fluid and reabsorb K+ from the filtrate and have been demonstrated to be upregulated by conditions of K+ restriction or depletion. The knockout of either HKα1 or HKα2 or both reduced pharmacologically defined H+,K+-ATPase activity in both A-type and B-type intercalated cells in mice on a normal diet (12). Pharmacological inhibitors have been historically used to differentiate between the function of each transporter. The HKα1 isoform is sensitive to Sch-28080, whereas the pharmacological profile of the HKα2 isoform is not completely understood and is complicated by poor primary structure conservation between mammalian species. In a study of mouse gastric and colonic mucosa, the pharmacological profile of each isoform was studied and confirmed that HKα1 is inhibited by Sch-28080; whereas HKα2 was Sch-28080 and ouabain insensitive (23). Previous studies have suggested that H+,K+-ATPases function in the regulation of Na+ balance (4, 19, 29). Our laboratory has shown that the long-acting mineralocorticoid desoxycorticosterone pivilate (DOCP) induced an increase in H+,K+-ATPase-mediated proton secretion in A-type intercalated cells of the cortical CD from WT mice (4). Eight days after DOCP treatment, HKα2 mRNA was increased, and H+,K+-ATPase-null mice exhibited an abnormal response to mineralocorticoids. The DOCP-induced changes in H, K-ATPase mRNA expression observed in our laboratory were time dependent. HKa1 mRNA expression in the medulla was also increased by DOCP but at an earlier time point (day 6) (3). Moreover, mice with knockout of the HKα1 isoform of the H+,K+-ATPase (HKα1−/−) had a greater body weight gain and urinary Na+ retention than control mice during DOCP treatment. These data strongly support a role for the HKα1 H+,K+-ATPase to regulate mineralocorticoid-induced Na+ reabsorption in the CD.
Here, we investigated the importance of the HKα1 H+,K+-ATPase in purinergic regulation of ENaC and found that purinergic regulation of ENaC by a local signaling system is abnormal in HKα1−/− mice, contributing to inappropriate ENaC responses to changes in dietary Na+. HKα1−/− mice have lower urinary ATP than WT on a high-Na+ diet and show no effect of dietary Na+ on urinary ATP, in contrast to WT mice, in which ATP is increased with a high-Na+ diet. The defect in the HKα1−/− mice is likely upstream of the inhibitory purinergic receptor, since ENaC in HKα1−/− mice responds normally to exogenous ATP. Furthermore, a high-Na+ diet resulted in greater Na+ retention in the HKα1−/− mice compared with WT controls. Thus, the current results further implicate the HKα1 H+,K+-ATPase in the regulation of Na+ homeostasis, demonstrating that this pump plays an important role in paracrine purinergic regulation of ENaC.
METHODS
Animals.
All animal use was in compliance with the American Physiological Society’s Guiding Principles in the Care and Use of Laboratory Animals, and animal use protocols were approved by the Institutional Animal Care and Use Committee of the North Florida/South Georgia Veterans Administration and the University of Texas Health Science Center at San Antonio. The generation and characterization of HKα1−/− mice were previously reported (11, 25). The HKα1−/− mice used in this report were back-crossed onto the C57BL/6J strain for more than 10 generations to generate HKα1−/− mice on a C57BL/6J genetic background.
Electrophysiology.
For patch clamp experiments, age-matched male C57Bl/6J control and HKα1−/− mice were maintained for at least 1 wk with ad libitum water and standard chow (Harlan Teklad TD.7912) containing 0.32% [Na+] or with a high-Na+ diet containing 2% [Na+] (TD.92034). A method published previously by us for isolating and opening aldosterone-sensitive distal nephrons suitable for patch-clamp electrophysiology was followed (16, 21). The activity of ENaC in murine aldosterone-sensitive distal nephrons was quantified in cell-attached patches of the apical membrane of principal cells made under voltage-clamp conditions (negative pipette potential: −60 mV) using standard procedures (16, 21). For the present study, bath and pipette solutions were (in mM) 150 NaCl, 5 mM KCl, 1 CaCl2, 2 MgCl2, 5 glucose, and 10 HEPES (pH 7.4) and 140 LiCl, 2 MgCl2, and 10 HEPES (pH 7.4), respectively. Unitary ENaC current (i) was determined, as normal, from all-point amplitude histograms fitted with single- or multi-Gaussian curves using the standard 50% threshold criterion to differentiate between events. Channel activity, defined as NPo, was calculated using the following equation: NPo = (t1 + 2t2 + . . . + ntn), where N and Po are the number of ENaC in a patch and the mean open probability of these channels, respectively, and tn is the fractional open time spent at each of the observed current levels. Po was calculated by dividing NPo by the number of active channels within a patch as defined by all-point amplitude histograms. For each experimental condition, aldosterone-sensitive distal nephrons from at least five different mice were assayed.
Urinary ATP measurement.
Urine was collected in metabolic cages under mineral oil over 24 h from age-matched, male C57Bl/6J WT and HKα1−/− mice maintained, as above, with standard chow or a high-Na+ diet. Urinary [ATP] was quantified using a luciferase bioluminescence assay (ATP Determination Kit A22066; Molecular Probes, Eugene, OR) following the manufacturer’s instructions.
Metabolic cage experiment.
Age-matched male C57Bl/6J WT and HKα1−/− mice were fed TD.99131 (0.2% base with Na+ added as NaCl to 0.32% Na+) for 7 days followed by a 2.0% Na+ diet (TD.99131 with Na+ added as NaCl) fed for 7 days. A gelled diet (45% chow, 54% water, and 1% agar) was fed with free access to a water bottle. Each day, body weight, food intake, and water intake were measured, and urine and feces were collected. Fecal samples were processed as previously described (13). Additional mice were treated identically and euthanized on day 1 of the 2.0% Na+ diet. Blood was collected via the abdominal aorta under isoflurane anesthesia into heparinized syringes and measured immediately with a blood gas analyzer (Stat Profile pHOx Plus C, Nova Biomedical, Waltham, MA). Immediately following, plasmas were separated and flash-frozen in liquid nitrogen and stored at −80°C. [Na+] and [K+] were analyzed using a digital flame analyzer (Cole-Parmer, model 2655-00, Chicago, IL). Urine and plasma osmolalities were measured using a vapor pressure osmometer (Wescor Vapro 5520). Plasma [AVP] was quantified following standard procedures (16, 17), with a competitive enzyme-linked immunoassay (Arg8-vasopressin EIA kit; Enzo Life Sciences, Farmingdale, NY) using the manufacturer’s recommended protocol.
Plasma aldosterone measurement.
Plasma from mice fed either a 0.32% Na+ diet or a 0.32% Na diet followed by a 2.0% Na+ diet for 1 day was analyzed using an aldosterone ELISA kit (catalog no. ADI-900-173, Enzo Life Sciences) according to the manufacturer’s instructions. The diet was prepared as described for the metabolic cage experiment.
Statistical analysis.
The results are expressed as means ± SE. Two-way repeated-measures ANOVA was performed with post hoc analysis by Tukey's test using Sigma Plot for Windows version 12. Effect of treatment with a single genotype was confirmed by one-way repeated-measures ANOVA when there was a significant interaction between treatment and genotype. Differences between groups were considered statistically significant at P < 0.05.
RESULTS
ENaC activity in HKα1−/− mice is uncoupled from Na+ intake.
We previously demonstrated that ENaC activity in WT mice is controlled by Na+ feeding (22). To evaluate ENaC activity in the HKα1−/− mice, patch-clamp experiments were performed under normal (0.32%) and high (2.0%) dietary Na+ conditions. Representative single-channel current traces for ENaC during high-Na+ dietary conditions for WT and HKα1−/− mice are shown in Fig. 1, A and B. In WT and HKα1−/− mice under normal and high-Na+ dietary conditions, we compared ENaC channel numbers, N, open probability, Po, and activity, NPo, as shown in Fig. 1, C and D, respectively. As expected, a high-Na+ diet significantly decreased N, Po, and NPo in WT mice compared with WT mice fed a normal Na+ diet. This coupling of ENaC activity and Na+ intake was not apparent in the HKα1−/− mice. There were no differences in N, Po, or NPo in the HKα1−/− mice fed normal or high-Na+ diets. When fed the normal Na+ diet, NPo in the HKα1−/− mice was significantly less than in the WT mice (Fig. 1E, P < 0.05). Furthermore, N, Po, and NPo were not significantly suppressed in HKα1−/− mice by a high-Na+ diet when compared with this same genotype on a normal Na+ diet. This lack of inhibition in the HKα1−/− mice was also apparent when the two genotypes were compared on the high-Na+ diet. Under these conditions N and NPo were significantly higher in the HKα1−/− mice compared with WT (Fig. 1E, P < 0.05).
Fig. 1.

Epithelial Na+ channel (ENaC) activity in mice with knockout of the HKα1 isoform of H+,K+-ATPase (HKα1−/−) is uncoupled from Na+ intake. Representative single-channel current traces for ENaC in wild-type (WT; A) and HKα1−/− mice (B) during high-Na+ dietary (2.0%) conditions. ENaC channel numbers, N, open probability, Po, and activity, NPo, are shown for WT and HKα1−/− mice (C–E). Data are shown as means ± SE. Numbers of patches are shown in columns. *P < 0.05 vs. 0.32% Na+; †P < 0.05 vs. WT, same dietary Na+ condition.
HKα1−/− mice have abnormal urinary ATP secretion but respond normally to exogenous ATP.
In the normal animal, ATP is secreted in response to dietary increases in Na+ and increased urine flow. When urinary ATP levels were measured in the HKα1−/− mice (Fig. 2A), ATP did not increase in HKα1−/− fed a high-Na+ diet. This is in contrast to WT mice, where ATP is proportionally related to dietary Na+ intake (18). Thus, urinary ATP in the HKα1−/− mice was significantly less than in WT mice under high-Na+ dietary conditions. To determine whether ENaC in the HKα1−/− mice responds normally to exogenous ATP, patch-clamp experiments were performed before and after the acute application of ATP (Fig. 2B). Po in the HKα1−/− mice responded normally to exogenous ATP, indicating that the defect in the HKα1−/− mice was likely upstream of the inhibitory purinergic receptor.
Fig. 2.

Urinary ATP does not increase in HKα1−/− mice fed a high-Na+ diet in contrast to controls where ATP is proportionally related to dietary Na+ intake. A: effect of dietary Na+ on urinary ATP in wild-type (WT) and HKα1−/− mice. WT, n = 8–20; HKα1−/−, n = 24 *P < 0.05 vs. 0.32% [Na+] same genotype. †P < 0.05 vs. WT, same condition. B: ENaC in HKα1−/− mice responds normally to exogenous ATP. Po in HKα1−/− mice is shown before and after addition of ATP (100 μM) as individual experiments and means ± SE; n = 16. *P < 0.05 vs. before ATP.
HKα1−/− mice retain more Na+ and have an impaired dipsogenic response vs. WT mice when fed a high-Na+ diet.
Since HKα1−/− mice have inappropriately high ENaC activity when fed a high-Na+ diet, we predicted that HKα1−/− mice might retain a significantly greater amount of Na+ and water than control mice. We performed metabolic cage experiments to measure Na+ and water intake and output in control and HKα1−/− mice on a normal and high-Na+ diet. Figure 3 shows fluid intake, urine osmolality, urinary Na excretion, calculated Na+ retention, and change in body weight in HKα1−/− and WT mice on a normal Na+ diet (days 5–7) followed by a high-Na+ diet fed for 7 days. There was a profound difference in the dipsogenic response to a high-Na+ diet between the genotypes. The statistical analysis confirmed a highly significant interaction term between genotype and high-Na+ treatment for water intake, urine osmolality, urinary Na excretion, and Na+ retention. Subsequently, we evaluated the difference between genotypes for these parameters on each day of the high-Na+ phase and also the effect of the high-Na+ feeding within each genotype. On a normal Na+ diet, water intake, urine osmolality, urinary Na excretion, and Na+ retention (Fig. 3, A–D) were similar between genotypes. By the first day of high-Na+ feeding, however, the WT mice more than tripled their water intake, and water intake remained significantly greater throughout the high-Na+ phase compared with all days of the normal Na+ feeding. The stimulation of thirst by the high-Na+ diet was substantially less pronounced in the HKα1−/− mice. On the first day of the high-Na+ feeding, the HKα1−/− mice increased their water intake but by less than half the increase observed in the WT (Fig. 3A, P < 0.05). After day 1, the HKα1−/− fluid intake remained relatively constant and significantly less than the intake of the WT mice until after the 4th day on the high-Na+ diet when it further increased to the level of WT mice. Consistent with fluid intake, urine osmolality (Fig. 3B) in the HKα1−/− mice was significantly greater than in the WT mice when fed the high-Na+ diet. The WT mice exhibited normal response to the high-Na+ diet by increasing water intake and lowering urine osmolality.
Fig. 3.

HKα1−/− mice have impaired dipsogenic response to a high-Na+ (HS) diet. A: water bottle intake in WT and HKα1−/− mice shown normalized for body weight. B: urine osmolality. C: urinary Na excretion. D: Na+ retention calculated as the difference between Na+ intake and urinary output. E: change in body weight. *P < 0.05, WT vs. HKα1−/−, Tukey's test; **P < 0.001 NS vs. HS within same genotype, one-way repeated-measures ANOVA. †P < 0.001 normal-Na+ diet (NS) vs. HS within WT only, one-way repeated-measures ANOVA. All data are shown as means ± SE for days 5–7 of NS and days 1–7 of HS. WT (n = 6–7); K HKα1−/− (n = 6). Solid lines, WT; dotted lines, HKα1−/−.
Urinary Na excretion in the HKα1−/− mice was significantly less than in the WT mice on days 1, 3, 4, and 7 of the high-Na+ feeding and is shown in Fig. 3D. Na+ retention was calculated as the difference between Na+ intake and urinary output and is shown in Fig. 3C. The HKα1−/− mice retained a significantly greater amount of Na+ than WT mice on days 1, 3, and 4 of the high-Na+ feeding. Na+ retention increased modestly in the WT mice with the high-Na+ feeding and was significantly greater than during normal Na+ feeding on several days [post hoc high-Na+ diet (HS) days 2, 6, and 7 vs. normal Na+ diet (NS) days 6 and 7)]. The moderate Na+ retention in the WT mice occurred simultaneously with a significant increase in body weight (Fig. 3E, post hoc HS days 5, 6, and 7) and was significantly greater than the HKα1−/− by the last 3 days of the high-Na+ diet. The HKα1−/− mice exhibited much greater Na+ retention on day 1 of the high-Na+ diet compared with WT mice, which was due to less renal Na excretion, since Na+ intake on that day was not different (Table 1). Na+ retention (Fig. 3C) was significantly increased in the HKα1−/− mice on nearly all days of the HS diet compared with normal Na+ (double asterisk, Fig. 3C; post hoc, HS days 1, 2, 3, 4 and 6 compared with NS days 5, 6, and 7). However, because fluid intake was substantially less, the HKα1−/− mice did not have a significant gain in body weight (Fig. 3E). Fecal Na+ excretion increased similarly due to the high-Na+ diet and was not different between the genotypes (data not shown).
Table 1.
Metabolic balance data of wild-type and HKα1−/− mice on a normal (0.32%) and high- (2.0%) Na diet
| Normal Na |
High Na, Day 1 |
High Na, Day 7 |
||||
|---|---|---|---|---|---|---|
| Wild Type | HKα1−/− | Wild Type | HKα1−/− | Wild Type | HKα1−/− | |
| Food intake/body weight, g/g | 0.34 ± 0.01 | 0.34 ± 0.01 | 0.35 ± 0.01 | 0.34 ± 0.01 | 0.28 ± 0.01 | 0.32 ± 0.01* |
| Fluid balance, ml/g | 0.16 ± 0.02 | 0.16 ± 0.01 | 0.23 ± 0.02† | 0.17 ± 0.01* | 0.21 ± 0.01 | 0.21 ± 0.01 |
| Na intake, mg·day−1·g−1 | 0.50 ± 0.02 | 0.49 ± 0.01 | 3.1 ± 0.1† | 3.1 ± 0.1† | 2.5 ± 0.1† | 2.9 ± 0.1* |
| Urine volume, ml/day | 3.4 ± 0.5 | 3.4 ± 0.3 | 8.0 ± 1.0† | 4.7 ± 0.3*† | 5.3 ± 0.3 | 5.9 ± 0.5 |
Data are shown as means ± SE; wild type, n = 7; knockout of the HKα1 isoform of H+,K+-ATPase (HKα1−/−), n = 6.
P < 0.05 vs. wild type, same day;
P < 0.05 vs. normal Na, same genotype.
Table 1 shows gelled-diet intake, fluid balance, Na intake, and urine volume during normal Na+ feeding and days 1 and 7 of the high-Na+ feeding. Because water intake was markedly reduced in the HKα1−/− mice compared with WT, greater positive fluid balance was not observed. On day 1, fluid balance and urine volume were significantly lower in the HKα1−/− mice than in the WT mice, and urine osmolality was elevated. A statistically significant increase in fluid balance on day 1 of the high-Na+ diet compared with normal Na+ was measured in the WT mice due to appropriately increased fluid intake in these mice. A similar increase in the HKα1−/− mice was not observed.
HKα1−/− mice are hypernatremic, with elevated plasma vasopressin on day 1 of a high-Na+ diet.
Since HKα1−/− mice had three times greater Na+ retention than WT on day 1 of the high-Na+ feeding, we euthanized mice on this day to measure electrolytes and hormones. HKα1−/− mice euthanized on day 1 of the high-Na+ feeding had significantly greater hematocrit, hemoglobin, plasma Na+, and osmolality than WT mice (Fig. 4, A–D). Plasma K+ was not different between genotypes (Fig. 4E). Plasma vasopressin levels were significantly greater in the HKα1−/− mice compared with the WT (Fig. 5A). Hematocrit, hemoglobin, plasma Na+, K+, and osmolality were not different between the genotypes after 7 days of high-Na+ feeding. Plasma aldosterone levels were not different between genotypes with normal Na+ or with high-Na+ feeding and were normally downregulated in both genotypes due to the high-Na+ diet (Fig. 5B).
Fig. 4.

HKα1−/− mice are hemoconcentrated and hypernatremic on day 1 of HS diet. Hematocrit, hemoglobin, plasma Na+, osmolality, and K+ on day 1 and day 7 of a HS diet are shown in A–E, respectively. *P < 0.05, WT (n = 5) vs. HKα1−/− (n = 5–6); †P < 0.05, day 7 HS vs. day 1 HS, same genotype. All data are shown as means ± SE; open columns, WT; hatched columns, HKα1−/−.
Fig. 5.

HKα1−/− mice have elevated plasma vasopressin on day 1 of a HS diet. Plasma aldosterone is normally regulated in HKα1−/− mice with HS feeding. A: Plasma [AVP] is shown on day 1 of HS. *P < 0.05, WT (N = 10) vs. HKα1−/− (n = 11). B: plasma aldosterone is shown during NS feeding and on day 1 of HS diet. †P < 0.05, day 1 HS vs. NS, same genotype. WT, n = 5; HKα1−/−, n = 4–5. All data are shown as means ± SE; open columns, WT; hatched columns, HKα1−/−.
DISCUSSION
The present studies collectively implicate the HKα1 subunit of the HKα1-containing H+,K+-ATPase, and presumably the holoenzyme, in the regulation of ENaC activity and, thus, Na+ balance. This statement is supported by patch-clamp analysis and metabolic balance studies. Together, they provide strong evidence for a new element in the purinergic regulation of ENaC: the renal HKα1 H+,K+-ATPase.
Purinergic regulation of ENaC involves the secretion of ATP that has been linked to potassium secretion via BKCa channels (8). Luminal ATP released through the connexin hemichannel Cx30 is triggered by increased flow rate or osmotic pressure (15, 24). Cx30 is exclusively localized to the nonjunctional apical membrane of intercalated cells in mice. Specific connexin hemichannel inhibition increased ENaC activity (1). Cx30 knockout mice have a blunted renal perfusion pressure-induced natriuresis and diuresis and exhibited ENaC-dependent, salt-sensitive increases in blood pressure. Deletion of the BK-β4 regulatory subunit of BKCa resulted in diminished urinary ATP production that is normally stimulated by increased dietary Na+ intake (1). Furthermore, ENaC activity in these mice was not normally regulated by dietary Na+ intake and was elevated compared with control mice on a high-Na+ diet. When challenged with a high-Na+ diet, these mice exhibited impaired urinary Na+ excretion. The phenotypes of the BK-β4-null and the HKα1−/− mice are strikingly similar. Regulation of ENaC-facilitated Na+ transport by purinergic signaling depends on BKCa channel K+ efflux, and the H+,K+-ATPase is an important mechanism for K+ reabsorption. During K+-replete conditions, the activity of a Sch-28080-senstive H+,K+-ATPase has been demonstrated to be dependent on an apical barium-sensitive pathway, presumably a K+ channel, and important in apical recycling of K+ (30).
Given the role for the HKα1-H+,K+-ATPase in mediating K+ recycling at the apical membrane (30) of the CD, the excessive Na+ retention in the HKα1−/− mice during mineralocorticoid treatment (4) and the marked reduction in urinary ATP of HKα1−/− mice, we speculate that the HKα1-H+,K+-ATPase is an important component in BKCa channel-mediated K+ efflux that is coupled to ATP secretion. We propose a model for K+ recycling mediated by the H+,K+-ATPase, thus facilitating downregulation of ENaC by the purinergic signaling system during high dietary Na+ intake (Fig. 6). The molecular mechanism(s) for reduced urinary ATP in HKα1−/− mice may also involve disruption of the electrochemical ATP-K+ efflux-coupled mechanism, but these events require further investigation. The diet fed in the present study was replete in K+ (0.6%) a condition when the HKα2 isoform is downregulated (6), and a K+ imbalance was not observed in this study. Plasma K+ was not different between the HKα1−/− and WT mice on day 1 of the high-Na+ diet or by the end of the study after 7 days of the high-Na+ feeding. However, our observations may be due either to the absence of the HKα1 protein or possibly the compensatory increases in HKα2 expression. These data do not preclude the effect of the loss of the HKα1 H+,K+-ATPase on local K+ recycling, since changes in K+ absorption can be compensated for by changes in K+ secretion. The basolateral Na+,K+-ATPase activity would be expected to be rate limiting for K+ secretion, especially during high-Na+ feeding. Although speculative at this time, the function of the HKα1 H+,K+-ATPase would ensure that there is enough intracellular K+ available for BK channels to allow maximal ATP release in the presence of low pump capacity. This negative feedback system localized to the distal nephron uses K+ as the feedback signal to couple ATP release from intercalated cells to inhibit Na+ reabsorption in the principal cells.
Fig. 6.
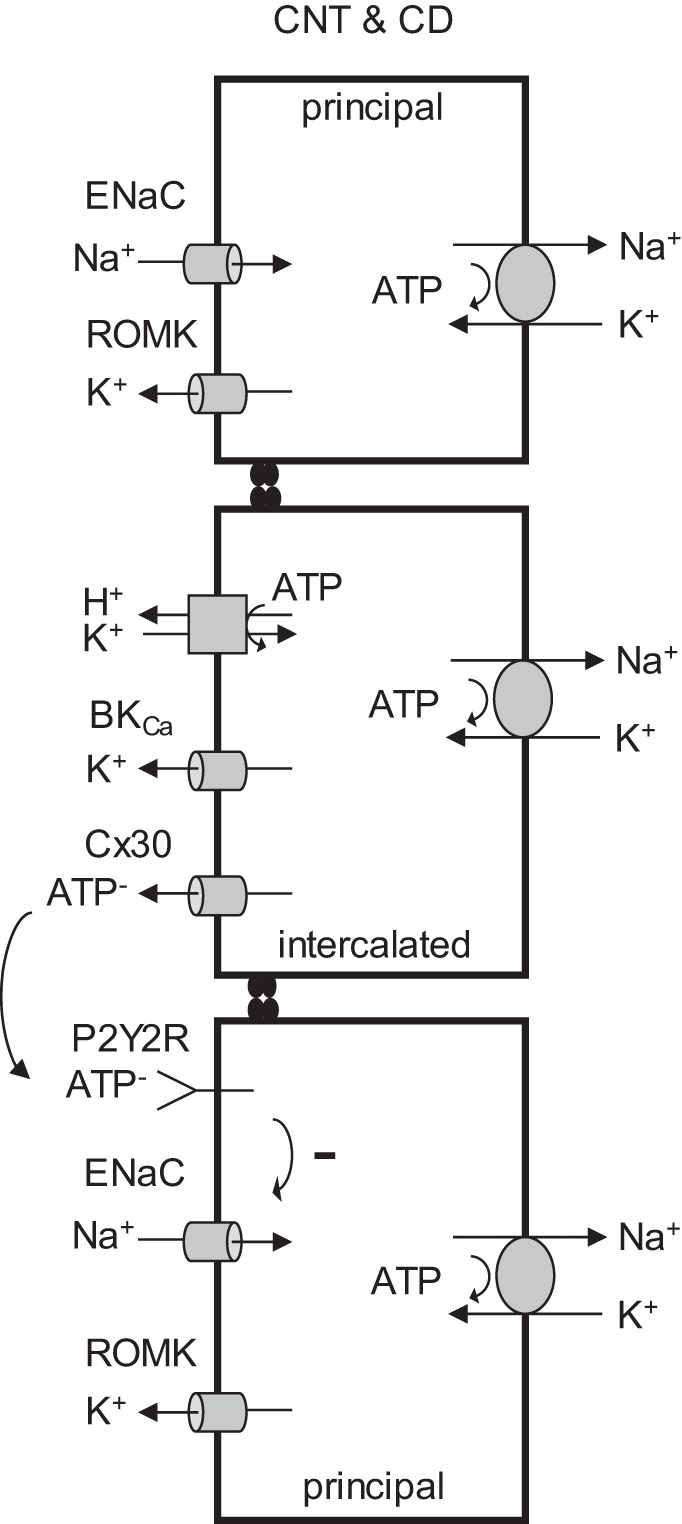
Model for K+ recycling via H+,K+-ATPase in purinergic regulation of ENaC. Apical K+ recycling by H+,K+-ATPase facilitates normal ATP secretion and downregulation of ENaC by the purinergic signaling system during HS intake. HKα1−/− mice have reduced urinary ATP, possibly due to disruption of the electrochemical ATP-K+ efflux-coupled mechanism.
ENaC activity in the HKα1−/− mice was not normally regulated by dietary Na+ intake and was greater than in WT mice on a high-Na+ diet. Similarities in the phenotype of mice with the deletion of the BK-β4 regulatory subunit of BKCa (BK-β4−/−) and in mice with the deletion of the HKα1 H+,K+-ATPase support a model of cooperative function of these two mechanisms in the regulation of ATP secretion. Furthermore, both purinergic signaling and the H+,K+-ATPases (4) have been implicated in mineralocorticoid regulation of Na+ reabsorption in the CD, and the proper control of ENaC activity by purinergic signaling may be necessary for aldosterone escape (26). Hyperaldosteronism is associated with the development of hypokalemia, which has been widely accepted to be the result of chronic kaliuresis. However, chronic balance studies on the effect of mineralocorticoids in several species (dog, rabbit, pig, and mouse) have failed to find a significant effect of either desoxycorticosterone or aldosterone to produce a significant negative K+ balance (2, 5, 9, 10, 20). Furthermore, several recent studies in mice have demonstrated mineralocorticoid-induced hypokalemia despite a positive K+ balance (4, 13). These studies support the concept that aldosterone causes the intracellular redistribution of K+ (7) with the renal H+,K+-ATPases mediating K+ conservation.
The coupling of an apical H+,K+-ATPase and an apical Ba2+-sensitive mechanism (presumably a K+ channel) has been supported by the observation that, in the presence of luminal Ba2+, stimulation by 10% CO2 does not increase either apical H+ secretion or K+ efflux from the lumen to the bath (30). Impaired Na+ clearance in HKα1−/− mice fits with our model of coupling of K+ recycling with K+/ATP secretion; however, the absence of a normal dipsogenic response in these mice was unexpected. Indeed, on the basis of the evidence suggesting that the HKα1−/− mice exhibit hypovolemia and dehydration (hyperosmolality, hypernatremia, increased hematocrit, and blood hemoglobin), one would predict a marked polydipsic response in the HKα1−/− mice. Thus, the impaired drinking response is even more striking. Furthermore, despite excessive urinary Na retention in the HKα1−/− mice during mineralocorticoid treatment, the HKα1−/− mice had significantly lower urinary output than the WT mice (4). The global nature of this knockout raises the possibility that the HKα1 subunit is also expressed in discrete cell types not previously recognized. Although after 7 days on the high-Na+ diet compensatory mechanisms likely restored normal daily Na+ balance, the acute response to the high-Na+ diet was impaired and marked by the absence of normal stimulation of thirst despite elevated plasma AVP and positive fluid balance. Although the high AVP and reduced inhibition of ENaC by ATP in the HKα1−/− mice would facilitate urine concentrating ability and in part compensate for the dispogenic response, this compensation was insufficient to restore plasma osmolality and [Na+] to normal. Such observations suggest that an additional and important input to the thirst response is present, and its elucidation may prove to be quite informative.
In summary, the present study provides strong evidence that the HKα1 H+,K+-ATPase is an important contributor to Na+ handling by the renal CD, as supported by both patch-clamp and metabolic balance studies in the same mouse model. This approach has provided consistent data demonstrating that HKα1−/− mice exhibit a clear defect in the response to a high-Na+ diet.
GRANTS
This work was supported by Merit Review Award # I01 BX001472-01A1 to C. S. Wingo from the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory Research and Development Program and by funds from the University of Florida, Department of Medicine. The contents herein do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors. Some data herein were submitted in abstract form: Stockand JD et al. (26). TH-PO433, 2015.
AUTHOR CONTRIBUTIONS
E.M., I.J.L., and J.M.B. performed experiments; E.M., I.J.L., J.M.B., and J.D.S. analyzed data; E.M., I.J.L., J.M.B., J.D.S., and C.S.W. interpreted results of experiments; E.M., I.J.L., and J.D.S. prepared figures; E.M., I.J.L., J.M.B., M.L.G., J.D.S., and C.S.W. approved final version of manuscript; I.J.L. drafted manuscript; I.J.L., M.L.G., J.D.S., and C.S.W. edited and revised manuscript.
REFERENCES
- 1.Bugaj V, Sansom SC, Wen D, Hatcher LI, Stockand JD, Mironova E. Flow-sensitive K+-coupled ATP secretion modulates activity of the epithelial Na+ channel in the distal nephron. J Biol Chem : 38552–38558, 2012. doi: 10.1074/jbc.M112.408476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dawborn JK, Ross EJ. The effect of prolonged administration of aldosterone on sodium and potassium turnover in the rabbit. Clin Sci : 559–570, 1967. [PubMed] [Google Scholar]
- 3.Greenlee MM. Physiological Function of Renal H+,K+-ATPases in Electrolyte and Acid-Base Homeostasis (PhD dissertation) Gainesville, FL: University of Florida, 2011. [Google Scholar]
- 4.Greenlee MM, Lynch IJ, Gumz ML, Cain BD, Wingo CS. Mineralocorticoids stimulate the activity and expression of renal H+,K+-ATPases. J Am Soc Nephrol : 49–58, 2011. doi: 10.1681/ASN.2010030311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grekin RJ, Terris JM, Bohr DF. Electrolyte and hormonal effects of deoxycorticosterone acetate in young pigs. Hypertension : 326–332, 1980. doi: 10.1161/01.HYP.2.3.326. [DOI] [PubMed] [Google Scholar]
- 6.Gumz ML, Lynch IJ, Greenlee MM, Cain BD, Wingo CS. The renal H+-K+-ATPases: physiology, regulation, and structure. Am J Physiol Renal Physiol : F12–F21, 2010. doi: 10.1152/ajprenal.90723.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gumz ML, Rabinowitz L, Wingo CS. An integrated view of potassium homeostasis. N Engl J Med : 1787–1788, 2015. doi: 10.1056/NEJMc1509656. [DOI] [PubMed] [Google Scholar]
- 8.Holtzclaw JD, Cornelius RJ, Hatcher LI, Sansom SC. Coupled ATP and potassium efflux from intercalated cells. Am J Physiol Renal Physiol : F1319–F1326, 2011. doi: 10.1152/ajprenal.00112.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hulter HN, Licht JH, Sebastian A. K+ deprivation potentiates the renal acid excretory effect of mineralocorticoid: obliteration by amiloride. Am J Physiol Renal Physiol : F48–F57, 1979. [DOI] [PubMed] [Google Scholar]
- 10.Hulter HN, Sigala JF, Sebastian A. K+ deprivation potentiates the renal alkalosis-producing effect of mineralocorticoid. Am J Physiol Renal Physiol : F298–F309, 1978. [DOI] [PubMed] [Google Scholar]
- 11.Judd LM, Andringa A, Rubio CA, Spicer Z, Shull GE, Miller ML. Gastric achlorhydria in H/K-ATPase-deficient (Atp4a(−/−)) mice causes severe hyperplasia, mucocystic metaplasia and upregulation of growth factors. J Gastroenterol Hepatol : 1266–1278, 2005. doi: 10.1111/j.1440-1746.2005.03867.x. [DOI] [PubMed] [Google Scholar]
- 12.Lynch IJ, Rudin A, Xia SL, Stow LR, Shull GE, Weiner ID, Cain BD, Wingo CS. Impaired acid secretion in cortical collecting duct intercalated cells from H-K-ATPase-deficient mice: role of HK isoforms. Am J Physiol Renal Physiol : F621–F627, 2008. doi: 10.1152/ajprenal.00412.2007. [DOI] [PubMed] [Google Scholar]
- 13.Lynch IJ, Welch AK, Gumz ML, Kohan DE, Cain BD, Wingo CS. Effect of mineralocorticoid treatment in mice with collecting duct-specific knockout of endothelin-1. Am J Physiol Renal Physiol : F1026–F1034, 2015. doi: 10.1152/ajprenal.00220.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lynch IJ, Welch AK, Kohan DE, Cain BD, Wingo CS. Endothelin-1 inhibits sodium reabsorption by ETA and ETB receptors in the mouse cortical collecting duct. Am J Physiol Renal Physiol : F568–F573, 2013. doi: 10.1152/ajprenal.00613.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCulloch F, Chambrey R, Eladari D, Peti-Peterdi J. Localization of connexin 30 in the luminal membrane of cells in the distal nephron. Am J Physiol Renal Physiol : F1304–F1312, 2005. doi: 10.1152/ajprenal.00203.2005. [DOI] [PubMed] [Google Scholar]
- 16.Mironova E, Bugaj V, Roos KP, Kohan DE, Stockand JD. Aldosterone-independent regulation of the epithelial Na+ channel (ENaC) by vasopressin in adrenalectomized mice. Proc Natl Acad Sci USA : 10095–10100, 2012. doi: 10.1073/pnas.1201978109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mironova E, Chen Y, Pao AC, Roos KP, Kohan DE, Bugaj V, Stockand JD. Activation of ENaC by AVP contributes to the urinary concentrating mechanism and dilution of plasma. Am J Physiol Renal Physiol : F237–F243, 2015. doi: 10.1152/ajprenal.00246.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mironova E, Peti-Peterdi J, Bugaj V, Stockand JD. Diminished paracrine regulation of the epithelial Na+ channel by purinergic signaling in mice lacking connexin 30. J Biol Chem : 1054–1060, 2011. doi: 10.1074/jbc.M110.176552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morla L, Doucet A, Lamouroux C, Crambert G, Edwards A. The renal cortical collecting duct: a secreting epithelium? J Physiol : 5991–6008, 2016. doi: 10.1113/JP272877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pan YJ, Young DB. Experimental aldosterone hypertension in the dog. Hypertension : 279–287, 1982. doi: 10.1161/01.HYP.4.2.279. [DOI] [PubMed] [Google Scholar]
- 21.Pochynyuk O, Bugaj V, Rieg T, Insel PA, Mironova E, Vallon V, Stockand JD. Paracrine regulation of the epithelial Na+ channel in the mammalian collecting duct by purinergic P2Y2 receptor tone. J Biol Chem : 36599–36607, 2008. doi: 10.1074/jbc.M807129200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pochynyuk O, Rieg T, Bugaj V, Schroth J, Fridman A, Boss GR, Insel PA, Stockand JD, Vallon V. Dietary Na+ inhibits the open probability of the epithelial sodium channel in the kidney by enhancing apical P2Y2-receptor tone. FASEB J : 2056–2065, 2010. doi: 10.1096/fj.09-151506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shao J, Gumz ML, Cain BD, Xia SL, Shull GE, van Driel IR, Wingo CS. Pharmacological profiles of the murine gastric and colonic H,K-ATPases. Biochim Biophys Acta : 906–911, 2010. doi: 10.1016/j.bbagen.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sipos A, Vargas SL, Toma I, Hanner F, Willecke K, Peti-Peterdi J. Connexin 30 deficiency impairs renal tubular ATP release and pressure natriuresis. J Am Soc Nephrol : 1724–1732, 2009. doi: 10.1681/ASN.2008101099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spicer Z, Miller ML, Andringa A, Riddle TM, Duffy JJ, Doetschman T, Shull GE. Stomachs of mice lacking the gastric H,K-ATPase alpha -subunit have achlorhydria, abnormal parietal cells, and ciliated metaplasia. J Biol Chem : 21555–21565, 2000. doi: 10.1074/jbc.M001558200. [DOI] [PubMed] [Google Scholar]
- 26.Stockand JD, Mironova E, Bugaj V, Rieg T, Insel PA, Vallon V, Peti-Peterdi J, Pochynyuk O. Purinergic inhibition of ENaC produces aldosterone escape. J Am Soc Nephrol : 1903–1911, 2010. doi: 10.1681/ASN.2010040377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toney GM, Vallon V, Stockand JD. Intrinsic control of sodium excretion in the distal nephron by inhibitory purinergic regulation of the epithelial Na(+) channel. Curr Opin Nephrol Hypertens : 52–60, 2012. doi: 10.1097/MNH.0b013e32834db4a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vallon V, Rieg T. Regulation of renal NaCl and water transport by the ATP/UTP/P2Y2 receptor system. Am J Physiol Renal Physiol : F463–F475, 2011. doi: 10.1152/ajprenal.00236.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walter C, Tanfous MB, Igoudjil K, Salhi A, Escher G, Crambert G. H,K-ATPase type 2 contributes to salt-sensitive hypertension induced by K(+) restriction. Pflügers Arch : 1673–1683, 2016. doi: 10.1007/s00424-016-1872-z. [DOI] [PubMed] [Google Scholar]
- 30.Zhou X, Wingo CS. Stimulation of total CO2 flux by 10% CO2 in rabbit CCD: role of an apical Sch-28080- and Ba-sensitive mechanism. Am J Physiol Renal Physiol : F114–F120, 1994. [DOI] [PubMed] [Google Scholar]