

Abstract
Acute kidney injury (AKI) induced by clamping of renal vein or pedicle is more severe than clamping of artery, but the mechanism has not been clarified. In the present study, we tested our hypothesis that increased proximal tubular pressure (Pt) during the ischemic phase exacerbates kidney injury and promotes the development of AKI. We induced AKI by bilateral clamping of renal arteries, pedicles, or veins for 18 min at 37°C, respectively. Pt during the ischemic phase was measured with micropuncture. We found that higher Pt was associated with more severe AKI. To determine the role of Pt during the ischemic phase on the development of AKI, we adjusted the Pt by altering renal artery pressure. We induced AKI by bilateral clamping of renal veins, and the Pt was changed by adjusting the renal artery pressure during the ischemic phase by constriction of aorta and mesenteric artery. When we decreased renal artery pressure from 85 ± 5 to 65 ± 8 mmHg, Pt decreased from 53.3 ± 2.7 to 44.7 ± 2.0 mmHg. Plasma creatinine decreased from 2.48 ± 0.23 to 1.91 ± 0.21 mg/dl at 24 h after renal ischemia. When we raised renal artery pressure to 103 ± 7 mmHg, Pt increased to 67.2 ± 5.1 mmHg. Plasma creatinine elevated to 3.17 ± 0.14 mg·dl·24 h after renal ischemia. Changes in KIM-1, NGAL, and histology were in the similar pattern as plasma creatinine. In summary, we found that higher Pt during the ischemic phase promoted the development of AKI, while lower Pt protected from kidney injury. Pt may be a potential target for treatment of AKI.
Keywords: tubular pressure, acute kidney injury, micropuncture
acute kidney injury (AKI) is a syndrome characterized by an abrupt reduction in kidney function (1, 42), resulting in failure to maintain fluid, electrolyte and acid-base homeostasis, and retention of nitrogenous waste products (6, 30). AKI occurs in ~5% of all hospital admissions and is responsible for approximately two million deaths annually worldwide (33, 45, 46). AKI increases the risk of development of chronic kidney disease (CKD) (10, 11), exacerbates preexisting CKD, and can evolve into end-stage renal disease (ESRD) (19, 25, 48). Patients who survive an episode of AKI have poorer long-term outcomes with increased mortality and extensive morbidity (3, 23). Even though AKI is extensively studied, unfortunately, there is no approved therapy to prevent or treat AKI (20). Therefore, further understanding of the pathophysiological mechanism is crucial to develop therapeutic approaches for AKI.
Renal ischemia reperfusion is a common cause of AKI (7, 24, 31). After ischemia reperfusion, tubular epithelial cells undergo serious damage, such as apical brush-border disruption, swelling, detachment from the basement membrane, and even death with acute tubular necrosis, resulting in rapid loss of kidney function (7, 13, 31). Several factors, such as hypoxia-induced ATP depletion (4, 13, 21, 28), the imbalance between superoxide (29) and nitric oxide (9, 44, 49), and the inflammatory response have been demonstrated to play important roles in renal ischemia-reperfusion injury (36, 43, 44). However, the pathophysiological mechanisms of AKI are complicated and are not well elucidated. This is especially true regarding the role of hemodynamic alterations and changes in mechanical force in the development of AKI.
The commonly used AKI models induced by occlusions of renal blood flow are typically accomplished by clamping of the renal artery, pedicle (artery and vein), or vein. Previous studies have reported that renal vein or pedicle clamping produced more severe AKI than renal artery clamping alone, but the mechanism remains to be determined (22, 24, 35). In the present study, we tested our hypothesis that increased intratubular pressure of the proximal tubule (Pt) during the ischemic phase exacerbates kidney injury and promotes the development of AKI. We first measured Pt during the ischemic phase while clamping the arteries, pedicles, or veins, respectively, and found a positive correlation between the Pt and the severity of the AKI. To determine whether the Pt during the ischemic phase is a causal factor for the kidney injury, we adjusted the Pt by altering the renal artery pressure during clamping of the renal veins. We found that increasing the Pt during the ischemic phase worsens AKI while lowering the Pt protects renal function.
METHODS
Animals and Experimental Protocols
C57BL/6 mice (male, 13–15 wk old) were purchased from Jackson Laboratory and housed in the University of South Florida Animal Facility. All protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of South Florida, College of Medicine.
Protocol I.
AKI was induced by the occlusion of renal blood flow for 18 min in the following three groups: 1) bilateral clamping of the renal arteries (RA group); 2) bilateral clamping of the renal pedicles (artery and vein; RP group); and 3) bilateral clamping of the renal veins (RV group). The sham-operated group underwent the same surgical procedures and time courses as the experimental groups except without occlusion of the renal blood flow.
The mice were anesthetized with pentobarbital sodium (50 mg/kg ip) and placed on a temperature-controlled operating table (03–02; Vestavia Scientific). The body temperature was monitored and controlled at 36.8–37.2°C during surgery with a temperature control unit. Both kidneys were exposed through a single abdominal incision. The Pt was measured as described below. The bilateral renal arteries, pedicles, or veins were carefully dissected and clamped with silver clips (FE690K; AESCULAP) for 18 min.
In separate survival experiments, the Pt was not measured. After 18 min of clamping, the clips were removed, the wounds were sutured, and the animals were allowed to recover for 24 h to evaluate the renal injury.
Protocol II.
Before the renal veins were clamped, the renal artery pressure was adjusted as described below. Then AKI was induced with bilateral clamping of the renal veins for 18 min as described in protocol I. The animals were divided into the following three groups: 1) normal renal artery pressure renal artery pressure (NP group); 2) low renal artery pressure (LP group); and 3) high renal artery pressure renal artery pressure (HP group).
np group.
The renal artery pressure was not adjusted in this group of mice. The mean arterial pressure (MAP) was measured with a femoral artery catheter.
lp group.
The aorta above the renal arteries was partially ligated to lower the renal artery pressure during the ischemic phase. The abdominal aorta above the renal arteries was then carefully separated from vena cava, and a 30-gauge needle was placed along the side of the isolated aorta segment. After a 6-0 suture was tied around the aorta and the needle, the needle was carefully removed from the ligature. The MAP was measured at the femoral artery.
hp group.
Both the superior mesenteric artery and the aorta below the renal arteries were partially ligated by using a 30-gauge needle before the renal veins were clamped to raise the renal artery pressure during the ischemic phase. This technique was similar to the steps described in the LP group. The MAP was measured at the carotid artery.
In the nonsurvival experiments, the Pt was measured as described below after the renal artery pressure was adjusted in the NP, LP, and HP groups. In the survival experiments, the Pt was not measured. The clips and ligature were removed after 18 min of ischemia, and the wound was closed. The mice were allowed to recover for 24 h to evaluate the renal injury.
Measurement of Pt During Ischemic Phase
The Pt was measured using micropuncture as we previously described (17, 27). The mice were anesthetized with pentobarbital sodium (50 mg/kg ip). A tracheostomy was performed to facilitate breathing. The femoral artery or carotid artery was catheterized for blood pressure measurements. The femoral vein was catheterized for infusion of saline with 1% bovine serum albumin (1 ml·h−1·100 g body wt−1). After an abdominal incision was made, the left kidney was exposed and immobilized in a kidney holder cup (03–12; Vestavia Scientific). The kidney orientation was positioned so that the superficial tubules could be clearly visualized under the microscope (SZX16; Olympus, Tokyo, Japan). A long superficial proximal tubule was punctured by a micropipette, which connected with a micropressure system (model 900A; World Precision Instruments). The Pt was measured and recorded during the ischemic phase while the renal veins were clamped.
Measurements of Kidney Injury Markers
The mice were anesthetized again with isoflurane 24 h after renal ischemia. The blood samples (100 µl) were collected through the retro-orbital venous sinus and centrifuged at 8,000 rpm for 5 min at 4°C. Plasma samples (50 µl/each) were obtained for creatinine, kidney injury molecule-1 (KIM-1), and neutrophil gelatinase-associated lipocalin (NGAL) measurements. The creatinine concentration was measured by HPLC at the O’Brien Center Core of the University of Alabama at Birmingham. The KIM-1 was measured with a Mouse KIM-1 Immunoassay Quantikine ELISA KIT and the NGAL was measured with a Mouse NGAL Immunoassay Quantikine ELISA KIT (R&D System) by following the manufacturer’s instructions.
Histological Study
The kidneys were harvested and fixed in 4% paraformaldehyde solution 24 h after AKI. Fixed kidney tissues were embedded in paraffin, and 4-µm kidney tissue slices were cut and stained with Periodic Acid Schiff (PAS). Ten randomly chosen fields were captured under ×200 magnification from the cortex, corticomedullary region (CMR), outer medulla (OM), and inner medullar (IM), respectively. The percentage of necrotic tubules and obstructed tubules by casts in each image was quantified as reported (32, 37). All morphometric analyses were performed in a blinded manner.
Statistical Analysis
The effects of interest were tested using a Student’s paired t-tests, or two-factor ANOVA with repeated measures when appropriate. Data were presented as means ± SE. The changes were considered to be significant if the P value was <0.05.
RESULTS
Protocol I
The Pt was measured for 2 min before occlusion of the renal blood flow and during 18 min of ischemia in the RA, RP, and RV groups. The Pt at baseline was ~10 mmHg, increased to 38.4 ± 3.6 mmHg in the RP group (P < 0.01 vs. RA group), to 53.3 ± 2.7 mmHg in the RV group (P < 0.01 vs. RP group), and decreased to 3.2 ± 2.1 mmHg in the RA group at the end of 18 min of ischemia. The Pt in the sham group was constant at ~10 mmHg throughout the experiment (Fig. 1A; P < 0.05 vs. the RA, RP, and RV groups). During the ischemic phase, the MAP was not significantly different between groups (Fig. 1B).
Fig. 1.
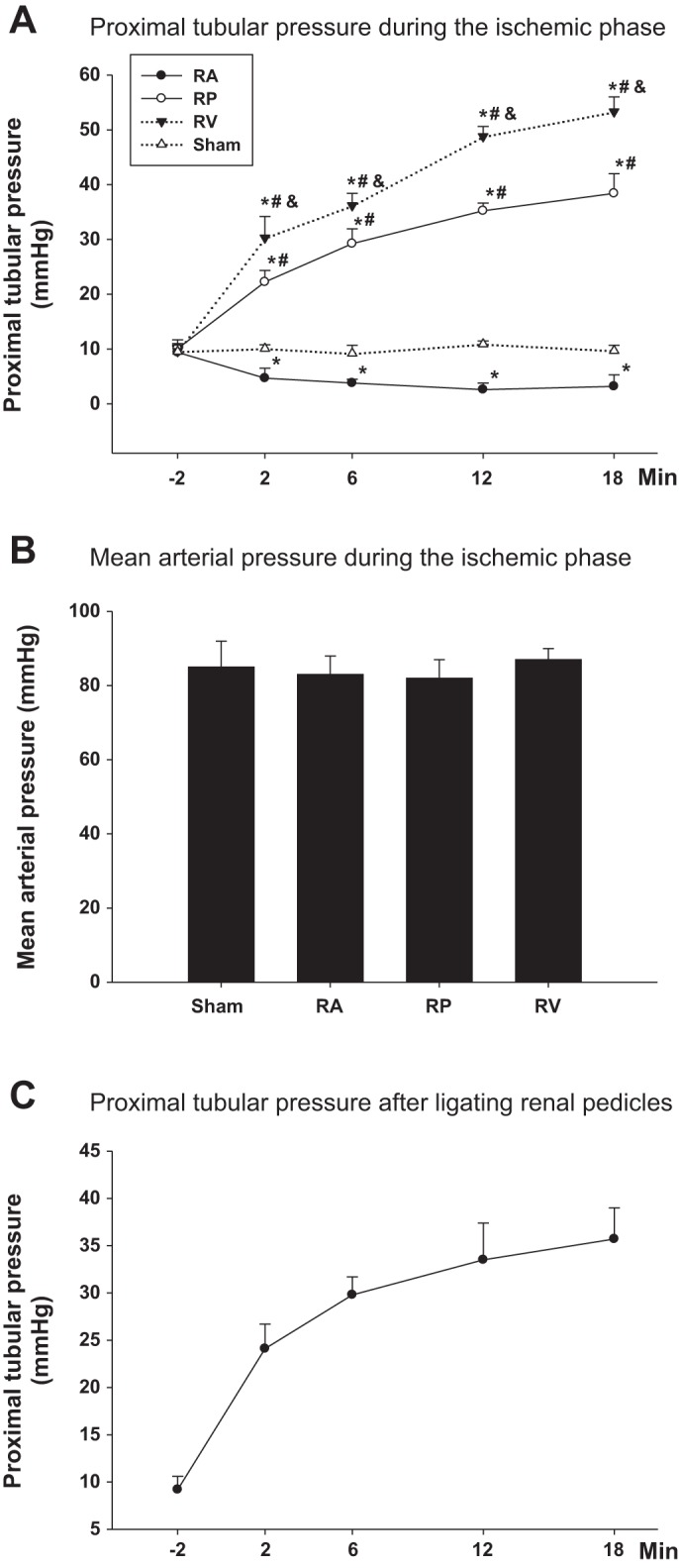
Proximal tubular pressure. Tubular pressure in proximal tubule was measured using micropuncture. A: difference of proximal tubular pressure during 18-min ischemic phase while clamping renal arteries (RA), renal pedicles (RP), and renal veins (RV). B: mean arterial pressure during the ischemic phase in all groups of protocol I (n = 10). *P < 0.01 vs. sham group; #P < 0.01 vs. RA group; &P < 0.01 vs. RP group. C: proximal tubular pressure during 18-min ischemic phase while ligating renal pedicles (n = 3).
To exclude the possibility of incomplete occlusion of the renal blood flow by clamping, we tightly ligated the renal pedicles with 6-0 suture and then measured the Pt during the ischemic phase. The Pt increased from 9.2 ± 1.4 to 35.7 ± 3.3 mmHg over 18 min (Fig. 1C).
The plasma creatinine, KIM-1, and NGAL were measured 24 h after renal ischemia. The plasma creatinine was substantially increased in all three AKI groups compared with the sham group 24 h after renal ischemia (P < 0.01 vs. sham group). The RV group was the highest (2.48 ± 0.23 mg/dl; P < 0.01 vs. RP group), followed by the RP group (1.43 ± 0.19 mg/dl; P < 0.01 vs. RA group) and the RA group (0.45 ± 0.07 mg/dl; Fig. 2A). The plasma KIM-1 and NGAL had similar patterns as the creatinine (Fig. 2, B and C).
Fig. 2.
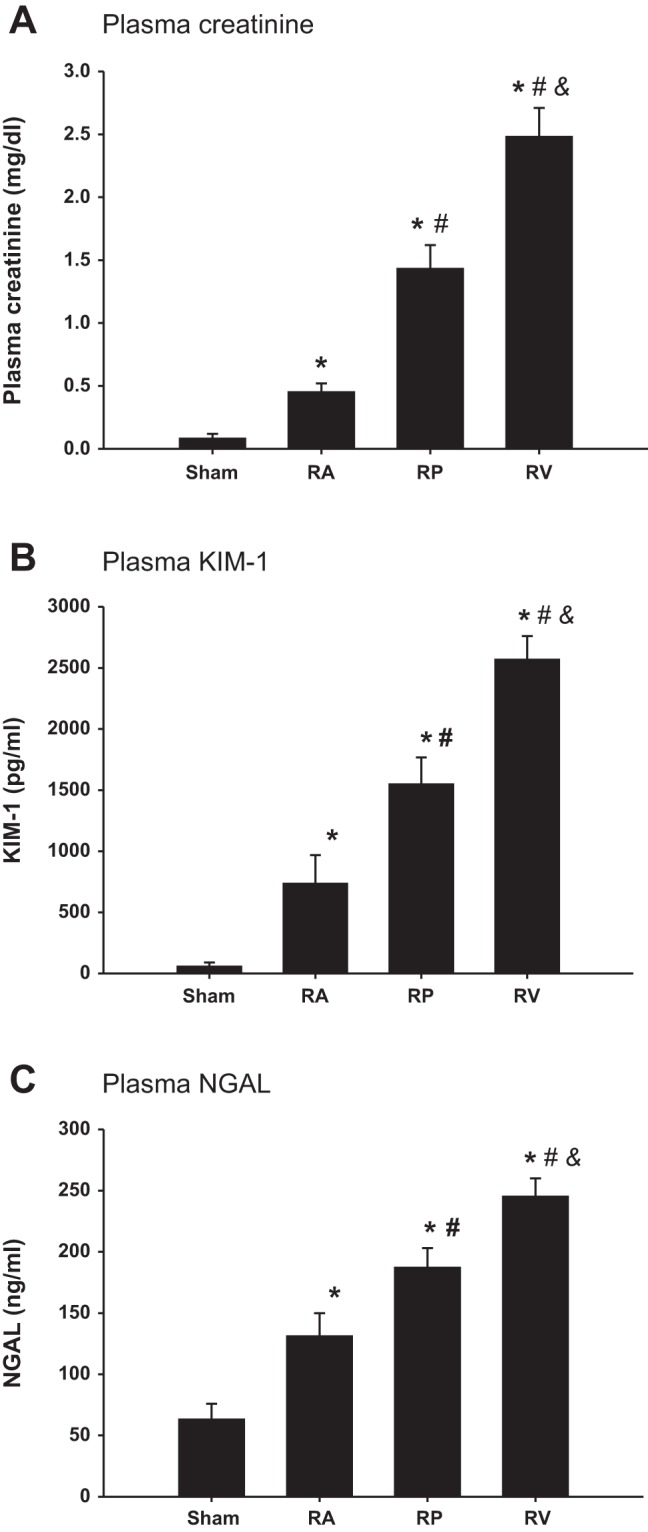
Kidney injury markers after renal ischemia. Plasma creatinine (A), plasma KIM-1 (B), and plasma NGAL (C) were measured and compared 24 h after renal ischemia while RA, RP, and RV were clamped (n = 5). *P < 0.01 vs. sham group; #P < 0.05 vs. RA group; &P < 0.05 vs. RP group.
The histology of the kidney slices stained by PAS at 24 h after ischemia reperfusion was examined among the different groups of animals. The RV mice showed the most severe tubular injury with the largest incidence of tubular obstruction by casts and tubular necrosis (P < 0.01 vs. RP and RA groups; Fig. 3A), followed by the RP mice (P < 0.05 vs. RA group) and the RA mice (Fig. 3, B and C).
Fig. 3.

Histology. Histology by PAS staining showed slices of cortex, corticomedullary region (CMR), outer medulla (OM), and inner medulla (IM) (A). Kidney injury was quantitatively measured by percentage of tubular necrosis (B) and obstructed tubules by casts (C) in the cortex, CMR, OM, and IM in acute kidney injury (AKI) groups by clamping RA, RP, and RV (n = 5). #P < 0.05 vs. RA group; &P < 0.05 vs. RP group. Quantitative analysis was performed on 10 randomly chosen fields in each kidney tissue slice.
Protocol II
The MAP was 85 ± 5 mmHg in the NP group. It decreased to 65 ± 8 mmHg after the aorta was partially ligated above the renal arteries in the LP group and increased to 103 ± 7 mmHg after both the superior mesenteric artery and the aorta were partially ligated below the renal arteries in HP group. To determine whether the Pt was altered by adjusting the renal artery pressure during the ischemic phase, we measured the Pt during 18 min of ischemia in the NP, LP, and HP groups. Clamping of the renal veins increased the Pt to 53.3 ± 2.7 mmHg in the NP group at the end of 18 min of ischemia (Fig. 4A). When the renal artery pressure decreased in the LP group, the Pt decreased to 44.7 ± 2.0 mmHg (P < 0.01 vs. NP group). When the renal artery pressure increased in the HP group, the Pt increased to 67.2 ± 5.1 mmHg at the end of 18 min of ischemia (Fig. 4B; P < 0.01 vs. NP group).
Fig. 4.
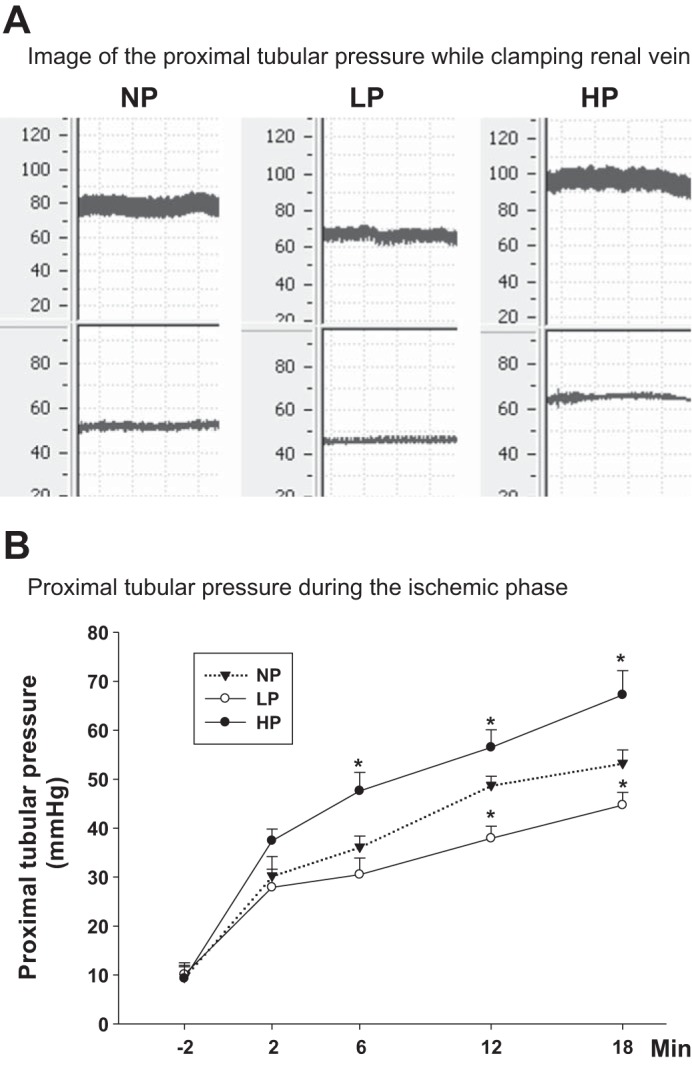
Proximal tubular pressure. Tubular pressure in proximal tubule was measured using micropuncture. A: image of the proximal tubular pressure while clamping renal vein with normal renal artery pressure (NP), low renal artery pressure (LP), and high renal artery pressure (HP) in protocol II. B: difference of tubular pressure in proximal tubules during 18-min ischemic phase in NP group, LP group, and HP group (n = 7). *P < 0.01 vs. NP group.
To determine whether the Pt during the ischemic phase had any effect on renal injury, we measured kidney injury markers 24 h after renal ischemia. The plasma creatinine was 2.48 ± 0.23 mg/dl in the NP group; it decreased to 1.91 ± 0.21 mg/dl in the LP group (P < 0.05 vs. NP); and increased to 3.17 ± 0.14 mg/dl in the HP group (Fig. 5A; P < 0.05 vs. NP). The plasma KIM-1 and NGAL were similar in pattern as the plasma creatinine (Fig. 5C).
Fig. 5.
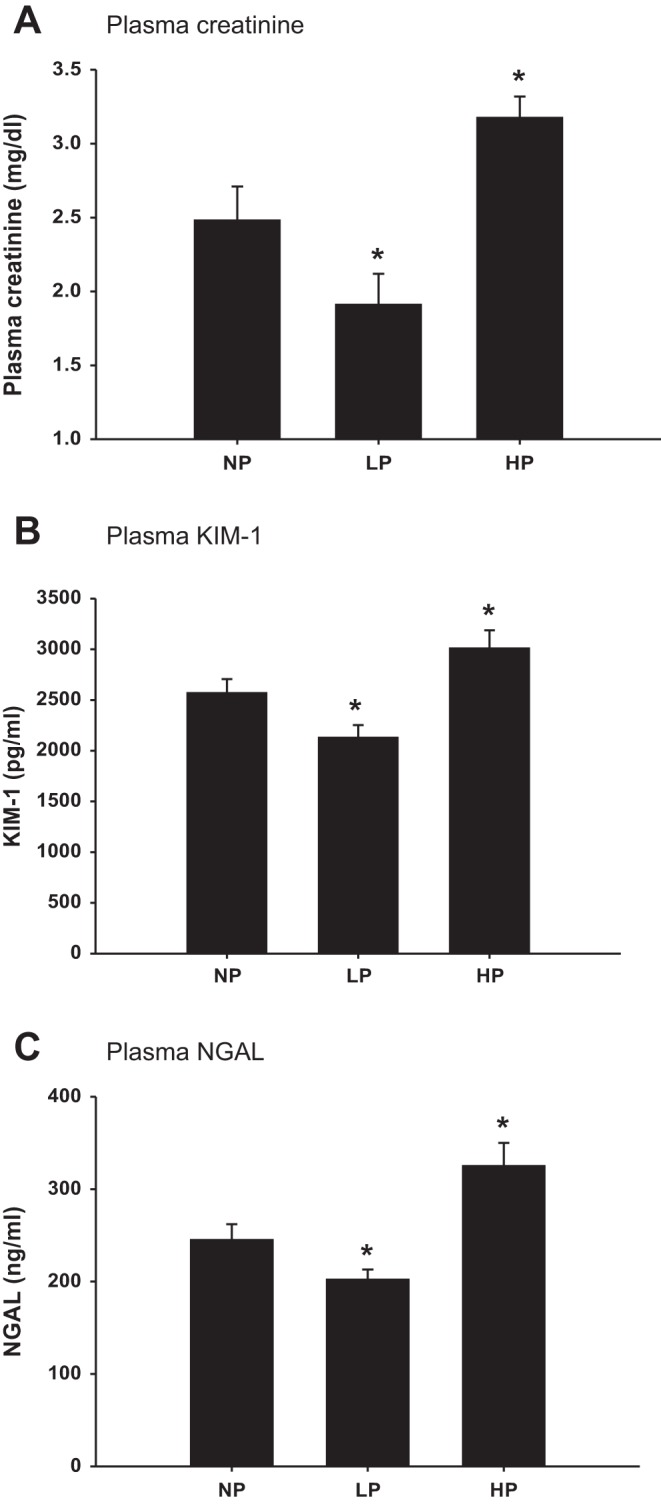
Kidney injury markers after renal ischemia. Plasma creatinine (A), plasma KIM-1 (B), and plasma NGAL (C) were measured and compared 24 h after renal ischemia while renal veins were clamped with normal renal artery pressure (NP), low renal artery pressure (LP), and high renal artery pressure (HP) (n = 6). *P < 0.05 vs. NP group.
Figure 6A shows the representative histology images from different groups of mice. The HP group exhibited the most severe tubular injury with the larger incidence of tubular obstruction by casts and tubular necrosis (P < 0.01 vs. NP group), while the LP group showed the least tubular injury among the three groups of mice (Fig. 6, B and C; P < 0.05 vs. NP group).
Fig. 6.

Histology. A: histology by PAS staining showed slices of cortex, CMR, OM, and IM. Kidney injury was quantitatively measured by percentage of tubular necrosis (B) and obstructed by tubules casts (C) in the cortex, CMR, OM, and IM in AKI groups by clamping renal veins with low renal artery pressure (LP), normal renal artery pressure (NP), and high renal artery pressure (HP) (n = 6). *P < 0.05 vs. LP group and HP grout. Quantitative analysis was performed on 10 randomly chosen fields in each kidney tissue slice.
DISCUSSION
In the present study, we found a positive association between the Pt during the ischemic phase and the severity of AKI in three widely used models by clamping of the renal arteries, pedicles, or veins. To determine the role of Pt during the ischemic phase of the development of AKI, we adjusted Pt by altering the renal artery pressure while clamping the renal veins. We found that increasing the Pt during the ischemic phase exacerbates AKI while lowering the Pt protects against kidney injury.
Tubular biomechanical forces are involved in the regulation of renal tubular function and associated with epithelial cell damage. In obstructive uropathy, increased intratubular pressure and tubular stretch upregulate transforming growth factor-β1 (TGF-β1), contributing to tubulointerstitial inflammation and fibrosis (38, 39). Also, cyclical stretch on renal epithelial cells induces apoptosis by activating JNK/SAPK and p38 SAPK-2 pathways (34). However, little is known about the role of tubular biomechanical forces in the development of AKI. Renal vein or pedicle clamping induced more severe AKI than renal artery clamping, but the mechanisms are elusive (22, 24, 35). We examined whether tubular pressure during the ischemic phase plays any significant role in the development of kidney injury. AKI was induced with 18 min occlusion of the renal blood flow at 37°C in C57BL/6 mice. First, we measured the Pt during the ischemic phase in three AKI models. We found that clamping of the renal arteries significantly lowered the Pt below the preischemia basal level. Occlusion of the renal arteries created cessation of glomerular filtration, resulting in a decrease of the Pt. On the contrary, the Pt was significantly elevated to ~50 mmHg by clamping the renal veins. The renal venous occlusion led to kidney congestion, which increased the glomerular hydrostatic pressure, consequently resulting in an increase of the Pt. Surprisingly, clamping of the renal pedicles also significantly increased the Pt to ~35 mmHg. We confirmed this finding by tightly ligating the renal pedicles with a 6-0 suture to exclude the possibility of incomplete occlusion of the renal blood flow. Suture ligation of the renal pedicles raised the Pt to a similar level as clamping of the renal pedicles. Thus, this phenotype was not due to the incomplete occlusion of the renal artery. However, we do not know the exact mechanism for this phenomenon, which might result from decreased tubular reabsorption, tubular obstruction, and continued glomerular filtration induced by residual pressure after clamping of the pedicles. To our knowledge, the present study is the first to examine the Pt during the ischemic phase. Several previous studies measured the Pt after reperfusion. Arendshorst et al. (5) examined the Pt after occlusion of the renal artery in rats for 60 min in an elegant study. They found that at 1–3 h after reperfusion, the proximal tubules were filled with fluid and dilated; the Pt elevated to ~30 mmHg with great heterogeneity (5). The Pt measured in the other studies was also found to be higher than baseline 1–3 days after IRI- or uranyl nitrate-induced AKI (12, 14, 16, 41). The heterogeneity in the Pt observed in these studies may be due to tubular obstructions (5, 12, 14, 16, 41).
Although we used the method of Isotope Dilution LC-MSMS to measure creatinine in mouse plasma to reduce the background and increase specificity (40), we realized the limitation of creatinine as an injury maker in mice, since 50% of the creatinine is eliminated via secretion in the mouse kidney tubules (15, 47). Therefore, we evaluated kidney injury combined with changes in KIM-1, NGAL, and histology in these three AKI models. We found that at 24 h after renal ischemia, clamping of the renal veins induced the most severe AKI while clamping of the renal arteries induced the least renal injury. These results were consistent with the results from previous studies (22, 24, 35). Therefore, the Pt during the ischemic phase in these three models was positively associated with the severity of AKI.
Acute reduction in renal blood flow in various clinical situations, such as in hypovolemic shock, some cardiac and abdominal surgeries, cardiac arrest, acute left heart failure, and kidney transplantation, etc. is often caused by renal arterial hypoperfusion. In such case, the renal artery clamping model should better mimic these clinical conditions. Clamping of the renal vein is more consistent with clinical situations of vein thrombosis, chronic right heart failure, or severe liver cirrhosis. Even though occlusion of both the renal artery and vein is not a typical clinical scenario, clamping of the renal pedicle is the most common choice for the rodent AKI models, probably due to the technical challenge of isolating murine renal arteries. In consideration of the significant difference in renal injury in different models, comparison and interpretation of these results should be conducted cautiously.
To determine the significance of Pt during the ischemic phase of the development of AKI, we modulated the Pt by adjusting the renal artery pressure while clamping the renal veins. To adjust the renal artery pressure, we constricted the aorta and mesenteric artery, rather than clamp them to avoid ischemic injury of tissues supplied by the aorta and mesenteric artery. Constriction of the aorta above the renal arteries lowered the renal artery pressure, which led to a decrease in the Pt compared with the normal renal artery pressure group. On the contrary, constriction of the aorta and the superior mesenteric artery raised the renal artery pressure, which resulted in an increase in the Pt. The kidney injury markers were compared at 24 h after renal ischemia. Mice with a lower Pt in the ischemic phase exhibited less renal injury than mice with a normal Pt, while mice with an elevated Pt showed more severe renal injury. Therefore, these results suggested that Pt during the ischemic phase contributes to the development of AKI. A lower Pt in the ischemic phase protects against the development of AKI, while a higher Pt aggravates the progression of AKI. Under physiological conditions, renal autoregulation maintains the renal blood flow and glomerular filtration rate relatively constant, despite fluctuations in the arterial blood pressure over a range of ~80–160 mmHg. This buffers the transmission of alterations in the arterial blood pressure into the renal tubules (8, 26). However, ischemia may impair this autoregulatory capacity, resulting in the loss of independent stability in tubular dynamics, thereby permitting the arterial pressure transmitted to the intraglomerulus and tubules (2, 18).
In summary, we found that the severity of AKI from clamping of the renal arteries, veins, or pedicles is positively associated with the Pt during the ischemic phase. Furthermore, we demonstrated that elevations of the Pt during the ischemic phase by increasing the renal artery pressure promote the development of AKI, while decreasing the Pt protects renal function. These findings not only provide novel insight into the mechanisms of AKI but also suggest that modulation of the Pt might be a potential approach for the treatment of AKI.
GRANTS
This study was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant 100000062 DK099276.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
J.W. and R.L. conceived and designed research; J.W., S.J., G.Z., J.Z., and S.W. performed experiments; J.W., J.S., D.W., and R.L. analyzed data; J.W. and S.J. interpreted results of experiments; J.W. prepared figures; J.W., S.J., and J.Z. drafted manuscript; J.W., J.S., S.J., G.Z., D.W., J.Z., E.Y.L., L.W., J.B., and R.L. edited and revised manuscript; R.L. approved final version of manuscript.
REFERENCES
- 1.Abuelo JG. Normotensive ischemic acute renal failure. N Engl J Med : 797–805, 2007. doi: 10.1056/NEJMra064398. [DOI] [PubMed] [Google Scholar]
- 2.Adams PL, Adams FF, Bell PD, Navar LG. Impaired renal blood flow autoregulation in ischemic acute renal failure. Kidney Int : 68–76, 1980. doi: 10.1038/ki.1980.111. [DOI] [PubMed] [Google Scholar]
- 3.Ali T, Khan I, Simpson W, Prescott G, Townend J, Smith W, Macleod A. Incidence and outcomes in acute kidney injury: a comprehensive population-based study. J Am Soc Nephrol : 1292–1298, 2007. doi: 10.1681/ASN.2006070756. [DOI] [PubMed] [Google Scholar]
- 4.Andreucci M, Michael A, Kramers C, Park KM, Chen A, Matthaeus T, Alessandrini A, Haq S, Force T, Bonventre JV. Renal ischemia/reperfusion and ATP depletion/repletion in LLC-PK(1) cells result in phosphorylation of FKHR and FKHRL1. Kidney Int : 1189–1198, 2003. doi: 10.1046/j.1523-1755.2003.00204.x. [DOI] [PubMed] [Google Scholar]
- 5.Arendshorst WJ, Finn WF, Gottschalk CW. Pathogenesis of acute renal failure following temporary renal ischemia in the rat. Circ Res : 558–568, 1975. doi: 10.1161/01.RES.37.5.558. [DOI] [PubMed] [Google Scholar]
- 6.Bauerle JD, Grenz A, Kim JH, Lee HT, Eltzschig HK. Adenosine generation and signaling during acute kidney injury. J Am Soc Nephrol : 14–20, 2011. doi: 10.1681/ASN.2009121217. [DOI] [PubMed] [Google Scholar]
- 7.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest : 4210–4221, 2011. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlström M, Wilcox CS, Arendshorst WJ. Renal autoregulation in health and disease. Physiol Rev : 405–511, 2015. doi: 10.1152/physrev.00042.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chatterjee PK, Patel NS, Kvale EO, Cuzzocrea S, Brown PA, Stewart KN, Mota-Filipe H, Thiemermann C. Inhibition of inducible nitric oxide synthase reduces renal ischemia/reperfusion injury. Kidney Int : 862–871, 2002. doi: 10.1046/j.1523-1755.2002.00234.x. [DOI] [PubMed] [Google Scholar]
- 10.Chawla LS, Amdur RL, Amodeo S, Kimmel PL, Palant CE. The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int : 1361–1369, 2011. doi: 10.1038/ki.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chawla LS, Kimmel PL. Acute kidney injury and chronic kidney disease: an integrated clinical syndrome. Kidney Int : 516–524, 2012. doi: 10.1038/ki.2012.208. [DOI] [PubMed] [Google Scholar]
- 12.Conger JD, Robinette JB, Kelleher SP. Nephron heterogeneity in ischemic acute renal failure. Kidney Int : 422–429, 1984. doi: 10.1038/ki.1984.191. [DOI] [PubMed] [Google Scholar]
- 13.Devarajan P. Update on mechanisms of ischemic acute kidney injury. J Am Soc Nephrol : 1503–1520, 2006. doi: 10.1681/ASN.2006010017. [DOI] [PubMed] [Google Scholar]
- 14.Eisenbach GM, Steinhausen M. Micropuncture studies after temporary ischemia of rat kidneys. Pflügers Arch : 11–25, 1973. doi: 10.1007/BF00586571. [DOI] [PubMed] [Google Scholar]
- 15.Eisner C, Faulhaber-Walter R, Wang Y, Leelahavanichkul A, Yuen PS, Mizel D, Star RA, Briggs JP, Levine M, Schnermann J. Major contribution of tubular secretion to creatinine clearance in mice. Kidney Int : 519–526, 2010. doi: 10.1038/ki.2009.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flamenbaum W, Huddleston ML, McNeil JS, Hamburger RJ. Uranyl nitrate-induced acute renal failure in the rat: micropuncture and renal hemodynamic studies. Kidney Int : 408–418, 1974. doi: 10.1038/ki.1974.126. [DOI] [PubMed] [Google Scholar]
- 17.Fu Y, Hall JE, Lu D, Lin L, Manning RD Jr, Cheng L, Gomez-Sanchez CE, Juncos LA, Liu R. Aldosterone blunts tubuloglomerular feedback by activating macula densa mineralocorticoid receptors. Hypertension : 599–606, 2012. doi: 10.1161/HYPERTENSIONAHA.111.173195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guan Z, Gobé G, Willgoss D, Endre ZH. Renal endothelial dysfunction and impaired autoregulation after ischemia-reperfusion injury result from excess nitric oxide. Am J Physiol Renal Physiol : F619–F628, 2006. doi: 10.1152/ajprenal.00302.2005. [DOI] [PubMed] [Google Scholar]
- 19.Ishani A, Xue JL, Himmelfarb J, Eggers PW, Kimmel PL, Molitoris BA, Collins AJ. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol : 223–228, 2009. doi: 10.1681/ASN.2007080837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jo SK, Rosner MH, Okusa MD. Pharmacologic treatment of acute kidney injury: why drugs haven’t worked and what is on the horizon. Clin J Am Soc Nephrol : 356–365, 2007. doi: 10.2215/CJN.03280906. [DOI] [PubMed] [Google Scholar]
- 21.Leemans JC, Stokman G, Claessen N, Rouschop KM, Teske GJ, Kirschning CJ, Akira S, van der Poll T, Weening JJ, Florquin S. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest : 2894–2903, 2005. doi: 10.1172/JCI22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li X, Liu M, Bedja D, Thoburn C, Gabrielson K, Racusen L, Rabb H. Acute renal venous obstruction is more detrimental to the kidney than arterial occlusion: implication for murine models of acute kidney injury. Am J Physiol Renal Physiol : F519–F525, 2012. doi: 10.1152/ajprenal.00011.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liangos O, Wald R, O’Bell JW, Price L, Pereira BJ, Jaber BL. Epidemiology and outcomes of acute renal failure in hospitalized patients: a national survey. Clin J Am Soc Nephrol : 43–51, 2006. doi: 10.2215/CJN.00220605. [DOI] [PubMed] [Google Scholar]
- 24.Liaño F, Pascual J;The Madrid Acute Renal Failure Study Group . Epidemiology of acute renal failure: a prospective, multicenter, community-based study. Kidney Int : 811–818, 1996. doi: 10.1038/ki.1996.380. [DOI] [PubMed] [Google Scholar]
- 25.Lo LJ, Go AS, Chertow GM, McCulloch CE, Fan D, Ordoñez JD, Hsu CY. Dialysis-requiring acute renal failure increases the risk of progressive chronic kidney disease. Kidney Int : 893–899, 2009. doi: 10.1038/ki.2009.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loutzenhiser R, Griffin K, Williamson G, Bidani A. Renal autoregulation: new perspectives regarding the protective and regulatory roles of the underlying mechanisms. Am J Physiol Regul Integr Comp Physiol : R1153–R1167, 2006. doi: 10.1152/ajpregu.00402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu Y, Wei J, Stec DE, Roman RJ, Ge Y, Cheng L, Liu EY, Zhang J, Hansen PB, Fan F, Juncos LA, Wang L, Pollock J, Huang PL, Fu Y, Wang S, Liu R. Macula densa nitric oxide synthase 1beta protects against salt-sensitive hypertension. J Am Soc Nephrol : 2346–2356, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma Z, Wei Q, Dong G, Huo Y, Dong Z. DNA damage response in renal ischemia-reperfusion and ATP-depletion injury of renal tubular cells. Biochim Biophys Acta : 1088–1096, 2014. doi: 10.1016/j.bbadis.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masztalerz M, Włodarczyk Z, Czuczejko J, Słupski M, Kedziora J. Superoxide anion as a marker of ischemia-reperfusion injury of the transplanted kidney. Transplant Proc : 46–48, 2006. doi: 10.1016/j.transproceed.2005.12.084. [DOI] [PubMed] [Google Scholar]
- 30.Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, Levin A; Acute Kidney Injury Network . Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care : R31, 2007. doi: 10.1186/cc5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Munshi R, Hsu C, Himmelfarb J. Advances in understanding ischemic acute kidney injury. BMC Med : 11, 2011. doi: 10.1186/1741-7015-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muroya Y, Fan F, Regner KR, Falck JR, Garrett MR, Juncos LA, Roman RJ. Deficiency in the formation of 20-hydroxyeicosatetraenoic acid enhances renal ischemia-reperfusion injury. J Am Soc Nephrol : 2460–2469, 2015. doi: 10.1681/ASN.2014090868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murugan R, Kellum JA. Acute kidney injury: what’s the prognosis? Nat Rev Nephrol : 209–217, 2011. doi: 10.1038/nrneph.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen HT, Hsieh MH, Gaborro A, Tinloy B, Phillips C, Adam RM. JNK/SAPK and p38 SAPK-2 mediate mechanical stretch-induced apoptosis via caspase-3 and -9 in NRK-52E renal epithelial cells. Nephron Exp Nephrol : e49–e61, 2006. doi: 10.1159/000088401. [DOI] [PubMed] [Google Scholar]
- 35.Orvieto MA, Zorn KC, Mendiola F, Lyon MB, Mikhail AA, Gofrit ON, Shalhav AL. Recovery of renal function after complete renal hilar versus artery alone clamping during open and laparoscopic surgery. J Urol : 2371–2374, 2007. doi: 10.1016/j.juro.2007.01.115. [DOI] [PubMed] [Google Scholar]
- 36.Patschan D, Patschan S, Müller GA. Inflammation and microvasculopathy in renal ischemia reperfusion injury. J Transplant : 764154, 2012. doi: 10.1155/2012/764154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pegues MA, McCrory MA, Zarjou A, Szalai AJ. C-reactive protein exacerbates renal ischemia-reperfusion injury. Am J Physiol Renal Physiol : F1358–F1365, 2013. doi: 10.1152/ajprenal.00476.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quinlan MR, Docherty NG, Watson RW, Fitzpatrick JM. Exploring mechanisms involved in renal tubular sensing of mechanical stretch following ureteric obstruction. Am J Physiol Renal Physiol : F1–F11, 2008. doi: 10.1152/ajprenal.00576.2007. [DOI] [PubMed] [Google Scholar]
- 39.Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest : 1486–1494, 2003. doi: 10.1172/JCI200319270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takahashi N, Boysen G, Li F, Li Y, Swenberg JA. Tandem mass spectrometry measurements of creatinine in mouse plasma and urine for determining glomerular filtration rate. Kidney Int : 266–271, 2007. doi: 10.1038/sj.ki.5002033. [DOI] [PubMed] [Google Scholar]
- 41.Tanner GA, Sophasan S. Kidney pressures after temporary renal artery occlusion in the rat. Am J Physiol : 1173–1181, 1976. [DOI] [PubMed] [Google Scholar]
- 42.Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med : 1448–1460, 1996. doi: 10.1056/NEJM199605303342207. [DOI] [PubMed] [Google Scholar]
- 43.Thurman JM. Triggers of inflammation after renal ischemia/reperfusion. Clin Immunol : 7–13, 2007. doi: 10.1016/j.clim.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tripatara P, Patel NS, Webb A, Rathod K, Lecomte FM, Mazzon E, Cuzzocrea S, Yaqoob MM, Ahluwalia A, Thiemermann C. Nitrite-derived nitric oxide protects the rat kidney against ischemia/reperfusion injury in vivo: role for xanthine oxidoreductase. J Am Soc Nephrol : 570–580, 2007. doi: 10.1681/ASN.2006050450. [DOI] [PubMed] [Google Scholar]
- 45.Uchino S, Bellomo R, Goldsmith D, Bates S, Ronco C. An assessment of the RIFLE criteria for acute renal failure in hospitalized patients. Crit Care Med : 1913–1917, 2006. doi: 10.1097/01.CCM.0000224227.70642.4F. [DOI] [PubMed] [Google Scholar]
- 46.Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, Schetz M, Tan I, Bouman C, Macedo E, Gibney N, Tolwani A, Ronco C; Beginning and Ending Supportive Therapy for the Kidney (BEST Kidney) Investigators . Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA : 813–818, 2005. doi: 10.1001/jama.294.7.813. [DOI] [PubMed] [Google Scholar]
- 47.Vallon V, Eraly SA, Rao SR, Gerasimova M, Rose M, Nagle M, Anzai N, Smith T, Sharma K, Nigam SK, Rieg T. A role for the organic anion transporter OAT3 in renal creatinine secretion in mice. Am J Physiol Renal Physiol : F1293–F1299, 2012. doi: 10.1152/ajprenal.00013.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wald R, Quinn RR, Luo J, Li P, Scales DC, Mamdani MM, Ray JG; University of Toronto Acute Kidney Injury Research Group . Chronic dialysis and death among survivors of acute kidney injury requiring dialysis. JAMA : 1179–1185, 2009. doi: 10.1001/jama.2009.1322. [DOI] [PubMed] [Google Scholar]
- 49.Walker LM, Walker PD, Imam SZ, Ali SF, Mayeux PR. Evidence for peroxynitrite formation in renal ischemia-reperfusion injury: studies with the inducible nitric oxide synthase inhibitor L-N(6)-(1-Iminoethyl)lysine. J Pharmacol Exp Ther : 417–422, 2000. [PubMed] [Google Scholar]