

Abstract
Aims
Mitral valve (MV) abnormalities are recognized features of hypertrophic cardiomyopathy (HCM), and there is preliminary evidence suggesting they are intrinsic phenotypic manifestations of sarcomere mutations, present in mutation carriers without left ventricular (LV) hypertrophy (subclinical HCM). However, further study is required to characterize the nature of these changes and their functional impact. Thus, we performed comprehensive echocardiographic analysis of MV structure and function on a genotyped population.
Methods and results
MV and papillary muscle echocardiographic parameters were measured in 192 genotyped individuals, including 50 overt HCM, 79 subclinical HCM, and 63 mutation-negative, healthy relatives as normal controls. Compared to controls, subclinical HCM subjects had elongated anterior MV leaflets relative to LV end-diastolic volume index (0.57 ± 0.02 vs. 0.51 ± 0.02 mm/mL/m2, P = 0.013) and anteriorly displaced papillary muscles [decreased papillary-septal separation (31.1 ± 0.7 vs. 34.2 ± 0.9 mm, P = 0.004) and relative antero-posterior position ratio of the papillary muscles (0.67 ± 0.01 vs. 0.71 ± 0.01, P = 0.011]. Similar findings were identified comparing overt HCM to controls. These MV changes were associated with an increased prevalence of systolic anterior motion (SAM) of the MV amongst subclinical HCM subjects.
Conclusions
Sarcomere mutations are associated with primary abnormalities of the MV apparatus, specifically excess anterior leaflet length relative to LV cavity size and anterior displacement of the papillary muscles; both features predisposing to SAM. These abnormalities appear to be early phenotypic consequences of sarcomere mutations, observed in mutation carriers with normal LV wall thickness.
Keywords: sarcomere mutation carriers, hypertrophic cardiomyopathy, subclinical hypertrophic cardiomyopathy, mitral valve, mitral valve abnormalities, systolic anterior motion, papillary muscles
Introduction
The mitral valve (MV) has been a focus of interest and investigation in hypertrophic cardiomyopathy (HCM) since the initial description of this heterogeneous disease over 50 years ago.1,2 Structural abnormalities of the mitral apparatus are well-recognized features of HCM, including increased leaflet length and area,3–6 leaflet thickening,4 distorted MV leaflet coaptation, papillary muscle anomalies including anterior displacement,7–10 and systolic anterior motion (SAM) of the MV11,12 producing obstruction to left ventricular (LV) outflow. Indeed, the high prevalence of MV anomalies in HCM patients has led to speculation that they may be an intrinsic manifestation of the underlying sarcomere mutation. However, MV morphology is not well characterized in individuals who have inherited disease-causing sarcomere mutations but who have not yet manifest left ventricular hypertrophy (LVH), denoted subclinical HCM. Increased anterior leaflet length (ALL) in subclinical HCM compared to control subjects has been identified in prior small studies that included a median of only 15 subclinical HCM subjects and focused predominantly on mitral leaflet length.3,13–16 However, results are inconsistent and leaflet coaptation, SAM, and papillary muscle morphology have not been systematically assessed. Therefore, more comprehensive characterization of the MV apparatus in subclinical HCM is needed to better understand how sarcomere mutations may impact the MV.
In this study, we used echocardiographic imaging of genotyped subjects to test the hypothesis that sarcomere mutation carriers manifest subtle abnormalities in MV morphology when LV wall thickness is normal. Moreover, we hypothesize that these structural abnormalities predispose individuals to future development of the stereotypical functional abnormalities of HCM, SAM and dynamic LV outflow tract obstruction, once clinical disease develops.
Methods
Study population
Genotyped HCM patients and relatives identified via research protocols or clinical evaluation at two centres in Boston, USA (n = 120), and one centre in Copenhagen, Denmark (n = 72) were studied. Genetic status was determined by sequencing sarcomere genes. Subjects were assigned to three cohorts designated overt HCM, subclinical HCM, and normal control based on genotype status and LV wall thickness. The overt HCM group consisted of sarcomere mutation carriers with a maximal LV wall thickness of ≥12 mm (Z score ≥2 in children). The subclinical group consisted of mutation carriers without LVH (maximal LV wall thickness <12 mm or Z score <2 in children). A lower criterion of 12 mm was used than the 13 mm employed by some diagnostic criteria17 to reduce the likelihood of including subjects with overt HCM into the subclinical cohort. Controls were healthy relatives who do not carry their family’s pathogenic sarcomere mutation. Individuals were excluded if they had prior septal myectomy or alcohol septal ablation, electronic ventricular pacing, or atrial fibrillation. Informed consent was obtained from all participants in protocols approved by the Institutional Review Boards of Brigham and Women’s Hospital and Boston Children’s Hospital, and the Local Science Ethics Committee in Copenhagen, Denmark.
Echocardiographic protocol
Vivid-7 ultrasound systems (GE Medical Systems, Milwaukee, WI, USA) were used to obtain standard 2D echocardiographic images. Recordings were stored digitally and analysed offline by two independent observers (J.D.G. and P.Z.G.), blinded to genotype status. Standard measurements were made on the average of 3 cardiac cycles according to established criteria of the American Society of Echocardiography.18 MV measurements reflect the average of 2 cardiac cycles.
Parasternal long-axis view of the left ventricle
ALL, posterior leaflet length (PLL), and thickness of each leaflet were measured at maximal diastolic extension19 (Figure 1A). The distance from posterior mitral hinge point to leaflet coaptation (F1) and MV annular diameter (F2) were measured in the first systolic frame demonstrating mitral coaptation (Figure 1B). MV annular diameter was measured in end diastole (peak of the QRS complex on simultaneous electrocardiographic recording). Left ventricular outflow tract (LVOT) diameter was measured at the base of the aortic valve leaflets during mid-systole. Mitral regurgitation (MR) was semi-quantitatively graded as: 0—none, 1—trace MR, 2—mild MR, 3—moderate MR, and 4—severe MR.
Figure 1.

Echocardiographic measurements in the parasternal long-axis view (A and B), and parasternal short-axis views of the left ventricle at the level of the mitral valve (C) and papillary muscles (D). A, mitral annular diameter; Ao, aortic root; B, anterior leaflet length; C, anterior leaflet thickness; D, posterior leaflet thickness; E, posterior leaflet length; F1, distance from posterior mitral hinge point to leaflet coaptation; F2, mitral annular diameter at end-systole; G1, distance from posterior wall of LV to posterior MV leaflet; G2, LV internal diameter at this level; H1, medio-lateral papillary muscle separation; H2, papillary-septal separation; H3, LV internal diameter at this level; LA, left atrium; LV, left ventricle; RV, right ventricle.
Short-axis view of the left ventricle at the level of the mitral valve
The distance between the LV posterior wall to posterior MV leaflet (G1) and the antero-posterior internal diameter of the LV at this level (G2) were measured in the first systolic frame demonstrating mitral coaptation (Figure 1C). The relative antero-posterior position of the MV was calculated as the ratio G1:G2.
Short-axis view of the left ventricle at the level of the papillary muscles
The medio-lateral papillary muscle separation (H1) was measured at end diastole (Figure 1D). The papillary-septal separation (H2) was measured as the distance between the anterior papillary muscle border and the left septal surface. Measurements were taken near the papillary muscle tips where H1 and H2 were minimal. The antero-posterior internal diameter of the LV at this level (H3) was also measured. The relative antero-posterior position of the papillary muscles was then calculated as the ratio H2:H3.
Systolic anterior motion
SAM was evaluated by visual assessment on both the parasternal long-axis and apical long-axis 2D echocardiography. SAM was defined as systolic anterior motion of the body of the anterior leaflet of the MV and/or of the MV chordae tendineae into the LVOT, and was classified in a dichotomous fashion only as either present or absent.
Statistical analysis
All statistical analyses were performed with SAS version 9.4 (SAS Institute Inc., Cary, NC, USA). Categorical variables are presented as percentages, and compared using Fisher’s exact test. Age and body surface area (BSA) are presented as unadjusted mean ± simple standard deviation, and compared using the Student’s t-test. Echocardiographic parameters are expressed as adjusted mean ± standard error derived from a generalized estimating equation approach using the GenMod procedure in SAS to account for an exchangeable correlation structure within families. When appropriate, the model was additionally adjusted for age, sex, ±BSA. A global test P-value of <0.05 was the threshold necessary to proceed to pairwise comparisons. After the age and family-correlation adjusted global analyses, pairwise comparisons between the 3 status groups were carried out using a Bonferroni-adjusted P-value of 0.017 for significance. Multivariable logistic regression was used to examine the relationship between SAM and candidate variables, controlling for aggregate confounding by age, sex, family relations, ±BSA. Unadjusted and adjusted odds ratios (OR) with 95% confidence intervals (CI) are presented.
Results
Baseline characteristics
A total of 192 individuals from 82 families were analysed, including 50 subjects with overt HCM, 79 with subclinical HCM, and 63 mutation-negative, healthy relatives as normal controls. Baseline clinical and echocardiographic characteristics are summarized in Table 1. Mutations in four sarcomere genes were represented: cardiac β-myosin heavy chain (MYH7), myosin binding protein C (MYBPC3), troponin T (TNNT2), and troponin I (TNNI3). Subclinical HCM subjects were substantially younger than both control and overt HCM subjects, and correspondingly had a smaller BSA. Standard cardiac dimensions were within normal limits for control and subclinical subjects, while overt HCM patients had significantly smaller LV cavity size and larger left atrial diameter. Left ventricular ejection fraction (LVEF) was significantly higher in overt HCM compared to control subjects (69.3 ± 1.3 vs. 65.1 ± 0.7%, P = 0.003), but there was no difference between overt and subclinical HCM subjects (69.3 ± 1.3 vs. 67.7 ± 0.8%, P = 0.311).
Table 1.
Baseline characteristics of control, subclinical HCM, and overt HCM cohorts
| Controls (n = 63) | Subclinical HCM (n = 79) | Overt HCM (n = 50) | P-value for global test | P-valuea, control vs. subclinical | P-valuea, control vs. overt | P-valuea, subclinical vs. overt | |
|---|---|---|---|---|---|---|---|
| Age (years)b | 32.9 ± 13.1 | 24.9 ± 13.1 | 44.1 ± 13.4 | 0.0004 | <0.0001 | <0.0001 | |
| Female, n (%)c | 31 (49.2%) | 47 (59.5%) | 18 (36.0%) | 0.239 | 0.184 | 0.012 | |
| BSA (m2)b | 1.89 ± 0.29 | 1.70 ± 0.33 | 1.96 ± 0.24 | 0.0004 | 0.163 | <0.0001 | |
| Septal thickness (mm) | 9.3 ± 0.2 | 9.4 ± 0.3 | 16.2 ± 0.7 | <0.0001 | 0.707 | <0.0001 | <0.0001 |
| Posterior wall thickness (mm) | 8.9±0.2 | 8.7 ± 0.2 | 10.6 ± 0.3 | <0.0001 | 0.437 | <0.0001 | <0.0001 |
| LV end-diastolic diameter (mm) | 45.8 ± 0.7 | 45.5 ± 0.5 | 40.2 ± 0.8 | <0.0001 | 0.709 | <0.0001 | <0.0001 |
| LVEDVid(mL/m2) | 59.2 ± 1.6 | 54.7 ± 1.6 | 43.8 ± 2.1 | <0.0001 | 0.023 | <0.0001 | <0.0001 |
| LVESVid (mL/m2) | 38.6 ± 1.6 | 33.4 ± 1.5 | 25.0 ± 2.3 | <0.0001 | 0.007 | <0.0001 | 0.005 |
| LVEF (%) | 65.1 ± 0.7 | 67.7 ± 0.8 | 69.3 ± 1.3 | 0.003 | 0.011 | 0.003 | 0.311 |
| LA diameter (cm) | 3.4 ± 0.1 | 3.4 ± 0.1 | 3.8 ± 0.1 | 0.013 | 0.969 | 0.002 | 0.001 |
| LVOT diameter (mm) | 21.8 ± 0.3 | 21.3 ± 0.3 | 21.8 ± 0.3 | 0.20 | |||
| Causal gene, n (%)e | |||||||
| MYH7 | Not applicable | 35 (44.3%) | 18 (36.0%) | ||||
| MYBPC3 | 35 (44.3%) | 29 (58.0%) | |||||
| TNNT2 | 5 (6.3%) | 3 (6.0%) | |||||
| TNNI3 | 4 (5.1%) | 3 (6.0%) |
BSA, body surface area; LA, left atrium; LV, left ventricle; LVEDVi, LV end-diastolic volume indexed for BSA; LVEF, left ventricular ejection fraction; LVESVi, LV end-systolic volume indexed for BSA; LVOT, left ventricular outflow tract.
P-values reflect adjustment for age, sex, BSA, and family relations unless otherwise stated. A P-value <0.017 was considered statistically significant for pairwise comparisons between status groups.
Values expressed as unadjusted mean ± simple standard deviation, and groups compared using two-tailed Student’s t-test.
Group comparisons using Fishers Exact test.
BSA was not included in multivariable models given that BSA was already incorporated in the dependent variable.
Cumulative percentage exceeds 100% in overt HCM cohort because 1 patient had a mutation in MYH7 and MYBPC3 genes, 1 patient had a mutation in MYBPC3 and TNNT2 genes, and 1 patient had a mutation in MYBPC3 and TNNI3 genes.
Compared to controls, subclinical HCM subjects had smaller left ventricular end-systolic volume index (LVESVi, 33.4 ± 1.5 vs. 38.6 ± 1.6 mL/m2, P = 0.007), and higher LVEF (67.7 ± 0.8 vs. 65.1 ± 0.7%, P = 0.011).
Mitral leaflet length
MV characteristics are summarized for each cohort in Table 2. There was no difference in ALL between control and subclinical HCM subjects. However, ALL/LVEDVi (0.57 ± 0.02 vs. 0.51 ± 0.02 mm/mL/m2, P = 0.013; Figure 2A) and ALL/LVOT diameter (1.42 ± 0.03 vs. 1.35 ± 0.02, P = 0.015) were on average 11.8% and 5.2% higher in subclinical subjects compared to controls, respectively. Absolute ALL and ALL indexed to BSA, LVEDVi, and LVOT diameter were significantly higher in overt HCM patients compared to controls. Control and subclinical HCM subjects had similar anterior leaflet thickness, but overt HCM patients had significantly thicker anterior leaflets.
Table 2.
Mitral valve characteristics of control, subclinical HCM, and overt HCM cohorts
| Controls (n = 63) | Subclinical HCM (n = 79) | Overt HCM (n = 50) | P-value for global test | P-valuea, control vs. subclinical | P-valuea, control vs. overt | P-valuea, subclinical vs. overt | |
|---|---|---|---|---|---|---|---|
| MV annular diameter at end diastole (mm) | 34.6 ± 0.4 | 33.9 ± 0.4 | 37.5 ± 0.7 | 0.002 | 0.29 | 0.0008 | <0.0001 |
| Anterior MV leaflet characteristics | |||||||
| ALL (mm) | 29.1 ± 0.5 | 29.9 ± 0.5 | 31.7 ± 0.5 | 0.0008 | 0.21 | 0.0002 | 0.014 |
| Anterior leaflet thickness (mm) | 1.34 ± 0.06 | 1.41 ± 0.08 | 1.76 ± 0.09 | 0.030 | 0.45 | 0.0003 | 0.009 |
| ALL/BSAb (mm/m2) | 16.0 ± 0.4 | 17.1 ± 0.4 | 17.9 ± 0.4 | 0.006 | 0.018 | 0.002 | 0.11 |
| ALL/LVEDVib (mm/ mL/m2) | 0.51 ± 0.02 | 0.57 ± 0.02 | 0.78 ± 0.04 | <0.0001 | 0.013 | <0.0001 | <0.0001 |
| ALL/LVOT diameter | 1.35 ± 0.02 | 1.42 ± 0.03 | 1.47 ± 0.03 | 0.006 | 0.015 | 0.001 | 0.21 |
| Posterior MV leaflet characteristics | |||||||
| PLL (mm) | 15.3 ± 0.4 | 15.6 ± 0.4 | 18.2 ± 0.6 | 0.001 | 0.45 | <0.0001 | 0.0002 |
| Posterior leaflet thickness (mm) | 1.62 ± 0.07 | 1.79 ± 0.08 | 2.12 ± 0.09 | 0.013 | 0.059 | <0.0001 | 0.012 |
| PLL/BSAb (mm/m2) | 8.3 ± 0.3 | 8.8 ± 0.3 | 10.2 ± 0.4 | <0.0001 | 0.11 | <0.0001 | 0.001 |
| PLL/LVEDVib (mm/ mL/m2) | 0.28 ± 0.01 | 0.30 ± 0.01 | 0.45 ± 0.02 | 0.0003 | 0.079 | <0.0001 | <0.0001 |
| PLL/LVOT diameter | 0.71 ± 0.02 | 0.74 ± 0.02 | 0.84 ± 0.03 | 0.001 | 0.18 | <0.0001 | 0.001 |
| Papillary muscle (PM) characteristics | |||||||
| H2: Papillary-septal separation (mm) | 34.2 ± 0.9 | 31.1 ± 0.7 | 29.4 ± 0.8 | 0.007 | 0.004 | <0.0001 | 0.033 |
| H1: Medio-lateral PM separation (mm) | 17.6 ± 0.5 | 18.4 ± 0.6 | 20.0 ± 0.7 | 0.016 | 0.27 | 0.001 | 0.08 |
| Papillary-septal separation /LV internal diameter | 0.71 ± 0.01 | 0.67 ± 0.01 | 0.64 ± 0.01 | 0.010 | 0.011 | <0.0001 | 0.065 |
| Mitral regurgitation (MR)c | |||||||
| None | 36 (57.1%) | 40 (50.6%) | 11 (22.0%) | 0.048 | 0.17 | 0.08 | 0.09 |
| Trace | 25 (39.7%) | 37 (46.8%) | 30 (60.0%) | ||||
| Mild | 2 (3.2%) | 1 (1.3%) | 7 (14.0%) | ||||
| Moderate | 2 (4.0%) | ||||||
| Severe | |||||||
Values expressed as mean adjusted for age, sex, family relations, and BSA ± standard error, unless otherwise stated.
ALL, anterior leaflet length; BSA, body surface area; LVEDVi, LV end-diastolic volume indexed for BSA; LVOT, left ventricular outflow tract; MV, mitral valve; PLL, posterior leaflet length.
P-values reflect adjustment for age, sex, BSA, and family relations unless otherwise stated. A P-value <0.017 was considered statistically significant.
BSA was not included in multivariable models given that BSA was already incorporated in the dependent variable.
MR could not be evaluated in 1 (1.3%) patient in the subclinical HCM cohort.
Figure 2.
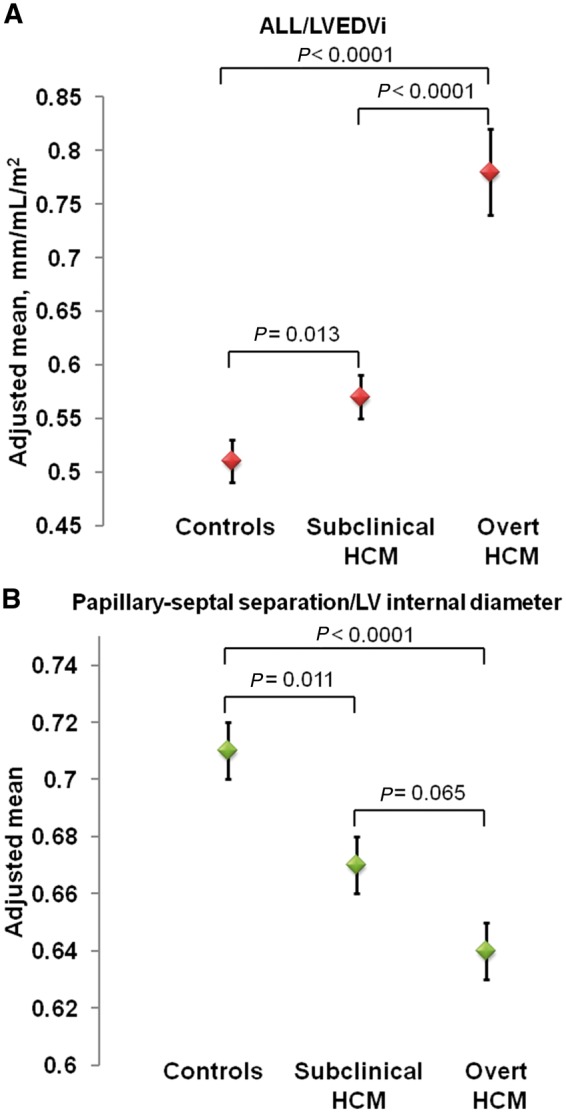
Adjusted means (±standard errors) of ALL/LVEDVi (A) and papillary-septal separation/LV internal diameter (B) across control, subclinical HCM, and overt HCM cohorts. Higher ALL/LVEDVi indicates greater elongation of ALL relative to LV volume, whereas lower papillary-septal separation/LV internal diameter indicates greater anterior displacement of papillary muscles in the LV cavity.
No significant differences were found in absolute or indexed posterior leaflet thickness or length between control and subclinical HCM subjects. Conversely, overt HCM patients demonstrated longer and thicker posterior leaflets (absolute and indexed measures) compared to the subclinical HCM and control cohorts.
Mitral leaflet coaptation
The distance from the posterior mitral hinge point to leaflet coaptation (F1) was not significantly different in subclinical HCM subjects compared to controls (8.5 ± 0.3 vs. 9.2 ± 0.2 mm, P = 0.027). In addition, neither measure of the relative antero-posterior position of leaflet coaptation (i.e. F1:F2 and G1:G2) differed significantly between these two cohorts (Supplementary data online, Table S1). F1 was significantly higher in overt HCM compared to both controls (10.8 ± 0.5 vs. 9.2 ± 0.2, P = 0.001) and subclinical HCM (10.8 ± 0.5 vs. 8.5 mm, P < 0.0001). The absence of any significant difference in F1:F2 or G1:G2 suggests that MV coaptation was not anteriorly displaced in subclinical or overt HCM compared with controls (Supplementary data online, Table S1).
Papillary muscle position
Papillary-septal separation (H2) was on average 9.1% less in subclinical HCM compared to controls (31.1 ± 0.7 vs. 34.2 ± 0.9 mm, P = 0.004). Similarly, the relative antero-posterior position ratio of the papillary muscles (H2:H3) was on average 5.6% less in subclinical HCM subjects compared to controls (0.67 ± 0.01 vs. 0.71 ± 0.01, P = 0.011) (Figure 2B). These findings suggest that the papillary muscles are anteriorly displaced in subclinical HCM. There was no difference in medio-lateral papillary muscle separation (H1) between these cohorts (Table 2).
Similar findings suggestive of anterior displacement of the papillary muscles were observed when overt HCM patients were compared to controls (Table 2). These indices were not significantly different between overt HCM and subclinical HCM subjects.
Systolic anterior motion
SAM was observed in 1 (1.6%) control subject, 12 (15.2%) subclinical HCM subjects, and 16 (32.0%) overt HCM patients (Figure 3). Of note, 5 of these 16 overt HCM patients had only chordal SAM. This frequency of SAM was significantly higher among subclinical HCM compared to controls in unadjusted analysis (P = 0.006), and there remains a strong trend after adjusting for age, sex, BSA, and family relations (P = 0.018; P-value for global test = 0.002). The frequency of SAM was not significantly different between subclinical and overt HCM in adjusted analyses (P = 0.16).
Figure 3.
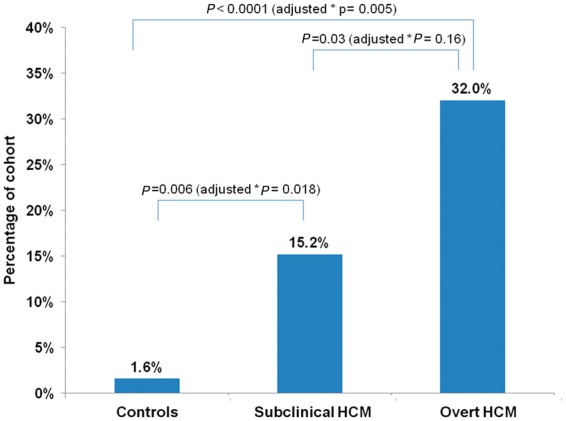
Frequency of systolic anterior motion (SAM) across control, subclinical HCM, and overt HCM cohorts. P-values for unadjusted and adjusted group comparisons are presented. (*Adjusted for age, sex, BSA, and family relations; P-value for global test = 0.002).
In an exploratory analysis of the full cohort, septal thickness, LVEDD, LVEDVi, LVESVi, LVEF, and ALL/LVEDVi were identified as univariate predictors of SAM (Table 3). After adjusting for age, sex, family relations, and BSA (where appropriate), these variables all remained predictive of SAM (Table 3). Greater LV wall thickness and smaller LV cavity size showed the strongest relationship, but MV parameters were also significant. For example, every 1 SD increase in ALL/LVEDVi was associated with an almost two-fold increased likelihood of SAM [adjusted OR 1.95 (95% CI 1.34, 2.85), P = 0.0005]. Similar findings were seen for ALL/BSA [adjusted OR 1.90 (95% CI 1.16, 3.11), P = 0.01]. Papillary-septal separation (H2) and relative antero-posterior position ratio of the papillary muscles (H2:H3) were not predictive of SAM.
Table 3.
Predictors of systolic anterior motion of the mitral valve
| Unadjusted OR (95% CI) | P-value | Adjusted ORa (95% CI) | P-value | |
|---|---|---|---|---|
| ALL, per 1 SD increase | 1.27 (0.86, 1.89) | 0.24 | 1.41 (0.94, 2.11) | 0.10 |
| Septal thickness, per 1 SD increase | 2.22 (1.55, 3.18) | <0.0001 | 3.10 (1.90, 5.04) | <0.0001 |
| LV end-diastolic diameter, per 1 SD decrease | 2.17 (1.34, 3.51) | 0.002 | 2.44 (1.37, 4.37) | 0.003 |
| LVEDVi, per 1 SD decreaseb | 2.28 (1.42, 3.67) | 0.0007 | 2.30 (1.34, 3.96) | 0.003 |
| LVESVi, per 1 SD decreaseb | 2.93 (1.63, 5.25) | 0.0003 | 2.85 (1.62, 5.01) | 0.0003 |
| LVEF, per 1 SD increase | 1.79 (1.17, 2.75) | 0.008 | 1.65 (1.05, 2.59) | 0.03 |
| ALL/LVEDVi, per 1 SD increaseb | 2.05 (1.42, 2.96) | 0.0001 | 1.95 (1.34, 2.85) | 0.0005 |
| ALL/BSA, per 1 SD increaseb | 1.39 (0.96, 2.01) | 0.08 | 1.90 (1.16, 3.11) | 0.01 |
| Papillary-septal separation, per 1 SD decrease | 1.12 (0.74, 1.68) | 0.59 | 1.05 (0.70, 1.57) | 0.82 |
| Papillary-septal separation/LV internal diameter, per 1 SD decrease | 1.20 (0.80, 1.82) | 0.38 | 1.08 (0.73, 1.59) | 0.72 |
Adjusted for age, sex, family relations, and BSA unless otherwise stated.
Adjusted for age, sex, and family relations only given that BSA was incorporated into dependent variable.
Reproducibility
Reproducibility for anterior/posterior leaflet projections, leaflet coaptation height, and leaflet thickness have previously been reported as very good with correlations exceeding 0.94.20,21 Inter-observer variability was low for leaflet coaptation height and papillary muscle characteristics (Supplementary data online, Table S2). Furthermore, intra-observer correlation coefficients for key echocardiography variables were consistent with excellent correlation (Supplementary data online, Table S3).
Discussion
To better understand whether structural abnormalities of the MV are an early phenotypic manifestation of HCM sarcomere mutations, comprehensive echocardiographic MV parameters were examined in subclinical mutation carriers, patients with overt HCM, and normal controls. Our study identifies three key findings: (i) HCM sarcomere mutation carriers have anterior MV leaflets that are disproportionately elongated relative to their LV cavity size; (ii) the papillary muscles are anteriorly displaced in sarcomere mutation carriers; and (iii) SAM was more frequently observed amongst mutation carriers compared to controls. Notably, these abnormalities are present not just in patients with clinically overt HCM, but also in sarcomere mutation carriers with normal LV wall thickness. As such, this study provides further support that structural alterations of the MV may precede LVH as part of the early phenotypic expression of HCM sarcomere mutations. Moreover, these early changes may underlie the increased susceptibility to developing dynamic outflow tract obstruction so prevalent in clinically overt HCM.
This study provides additional insight and increases the knowledge base provided by smaller prior studies investigating the MV in subclinical HCM. These earlier studies included a median of 15 subclinical subjects (only one included >20 subclinical HCM subjects)3,13–16 and most simply focused on the absolute length of the anterior and posterior mitral leaflets without objectively analysing SAM, leaflet coaptation, or papillary muscle placement. Hagege et al. did not demonstrate any difference in absolute ALL in echocardiographic comparisons of 20 subclinical HCM and 61 control subjects, in contrast to similar sized studies by Maron et al. and Peyrou et al. that included 15 and 14 subclinical HCM subjects, respectively. Similarly, Tarkiainen et al.16 did not find any difference in absolute MV leaflet lengths measured by cardiovascular magnetic resonance, or indexed leaflet lengths, in 15 phenotype negative carriers of a Finnish founder mutation (MYBPC3-Q1061X) compared to controls. Although our study did not demonstrate any significant difference in absolute ALL between the subclinical HCM and control cohorts across a variety of genotypes, we found that the anterior mitral leaflet is elongated relative to LV cavity size. Collectively, these findings highlight the need to look beyond absolute leaflet length.
Indeed, our results demonstrate that abnormalities of the MV complex in subclinical HCM subjects extend beyond the leaflets. Although anterior displacement of papillary muscles is a well-recognized abnormality in overt HCM, our study provides novel information indicating that anterior displacement of the papillary muscles also appears to be an early phenotypic consequence of sarcomere mutations. These findings indicate a more complex relationship whereby sarcomere mutations predispose to a disproportionate MV with leaflets that are excessively long for the size of the heart as well as anterior displacement of the papillary muscle. These changes occur early in disease development, independently of the development of LVH or other pathophysiologic changes associated with clinically overt HCM.
Owing to these anatomical variations of the MV apparatus in mutation carriers, a higher frequency of SAM was observed in sarcomere mutation carriers compared to control subjects. Elongation of ALL relative to LVEDVi (ALL/LVEDVi) and relative to BSA (ALL/BSA) were each strong predictors of SAM, conferring an almost two-fold increased adjusted likelihood per standard deviation increase in either ratio. In addition, smaller LV cavity size and higher LVEF, both present in this subclinical cohort, further predispose to SAM.
The pathogenesis underlying MV abnormalities in HCM is uncertain. Finding disproportionate elongation of the anterior leaflet and anterior displacement of the papillary muscles in sarcomere mutation carriers without LVH implies that these changes are not just a consequence of changes in ventricular geometry or due to flow acceleration with increased shear stress seen in clinically overt disease, but rather a primary trait driven by the underlying sarcomere mutation. We speculate that pathogenic HCM sarcomere mutations influence the developmental program of the mitral apparatus. This is supported by the demonstration that abnormal MV formation is part of the pre-hypertrophic HCM phenotype in an MYBPC3-targeted knock-out mouse model of HCM.22 It has been proposed that MV area increases with mechanical stretch created by papillary muscle displacement.23 Alternatively or additionally, relative elongation of MV leaflets and papillary muscle abnormalities in sarcomere mutation carriers may also result from abnormal paracrine effects arising from adjacent mutated myocardium.24 For example, elevated levels of periostin adjacent to the MV could promote leaflet elongation through increased collagen production.25,26 Ultimately, the pathogenesis of MV abnormalities in subclinical HCM is multifactorial, resulting from a complex interplay of above and other mechanisms. Moreover, HCM likely develops in a continuous manner.27 Early changes triggered by sarcomere mutations are subtle but may set the stage for more prominent functional abnormalities as disease progresses.
Limitations
The lack of longitudinal imaging to follow MV morphology over time in subclinical HCM sarcomere mutation carriers precludes comment on the temporal sequence of phenotypic evolution or on the clinical implications of observed MV alterations. In addition, limited power due to the relatively small number of subjects in each cohort means that failure to detect significant differences between groups in this study does not prove that no differences are present. SAM was assessed using 2D echocardiography only rather than M-mode which would have offered higher temporal resolution. Although papillary-septal separation (H2) and relative antero-posterior position ratio of the papillary muscles (H2:H3) were not predictive of SAM in our analyses, this study may not be sufficiently powered to exclude an association. Because subjects with prior septal myectomy, alcohol septal ablation, electronic ventricular pacing, and/or atrial fibrillation were excluded from this study, our findings may not be generalizable to overt HCM patients with those features and potentially more severe disease. One subject included in the subclinical HCM cohort had a c.115G>A (Val39Met) variant in the MYH7 gene, which has since been reclassified as a likely benign variant with current-day variant interpretation rules. Associations remained unchanged when analyses were repeated excluding this subject and their related control. We also acknowledge that penetrance of sarcomere mutations is not 100% and that some subclinical mutation carriers may not ultimately develop HCM. Nonetheless, our findings beget a rationale for future larger and longitudinal studies to better evaluate gene-specific influences on MV morphology, how these changes evolve over time, and how they relate to other important disease manifestations.
Conclusions
Pathological changes caused by sarcomere gene mutations in HCM are not confined to the myocardium. Primary abnormalities of the MV apparatus are identifiable, specifically excess anterior leaflet tissue relative to LV cavity size and anterior displacement of the papillary muscles; features that individually and collectively predispose to the development of SAM. These abnormalities appear to be intrinsic, early phenotypic consequences of sarcomere mutations as they are observed in mutation carriers when LV wall thickness is normal.
Funding
This work was supported by the National Heart, Lung, and Blood Institute at the National Institutes of Health [grant numbers K23 HL078901 and 1P20HL101408 to C.Y.H.]. NIH had no role in the conduct of the study or this manuscript.
Conflict of interest: None declared.
Supplementary Material
References
- 1. Ross J Jr, Braunwald E, Gault JH, Mason DT, Morrow AG.. The mechanism of the intraventricular pressure gradient in idiopathic hypertrophic subaortic stenosis. Circulation 1966;34:558–78. [DOI] [PubMed] [Google Scholar]
- 2. Simon AL, Ross J Jr, Gault JH.. Angiographic anatomy of the left ventricle and mitral valve in idiopathic hypertrophic subaortic stenosis. Circulation 1967;36:852–67. [DOI] [PubMed] [Google Scholar]
- 3. Maron MS, Olivotto I, Harrigan C, Appelbaum E, Gibson CM, Lesser JR. et al. Mitral valve abnormalities identified by cardiovascular magnetic resonance represent a primary phenotypic expression of hypertrophic cardiomyopathy. Circulation 2011;124:40–7. [DOI] [PubMed] [Google Scholar]
- 4. Klues HG, Maron BJ, Dollar AL, Roberts WC.. Diversity of structural mitral valve alterations in hypertrophic cardiomyopathy. Circulation 1992;85:1651–60. [DOI] [PubMed] [Google Scholar]
- 5. Kaple RK, Murphy RT, DiPaola LM, Houghtaling PL, Lever HM, Lytle BW. et al. Mitral valve abnormalities in hypertrophic cardiomyopathy: echocardiographic features and surgical outcomes. Ann Thorac Surg 2008;85:1527–35. [DOI] [PubMed] [Google Scholar]
- 6. Kim DH, Handschumacher MD, Levine RA, Choi YS, Kim YJ, Yun SC. et al. In vivo measurement of mitral leaflet surface area and subvalvular geometry in patients with asymmetrical septal hypertrophy: insights into the mechanism of outflow tract obstruction. Circulation 2010;122:1298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Levine RA, Vlahakes GJ, Lefebvre X, Guerrero JL, Cape EG, Yoganathan AP. et al. Papillary muscle displacement causes systolic anterior motion of the mitral valve. Experimental validation and insights into the mechanism of subaortic obstruction. Circulation 1995;91:1189–95. [DOI] [PubMed] [Google Scholar]
- 8. Kwon DH, Setser RM, Thamilarasan M, Popovic ZV, Smedira NG, Schoenhagen P. et al. Abnormal papillary muscle morphology is independently associated with increased left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. Heart 2008;94:1295–301. [DOI] [PubMed] [Google Scholar]
- 9. Klues HG, Roberts WC, Maron BJ.. Anomalous insertion of papillary muscle directly into anterior mitral leaflet in hypertrophic cardiomyopathy. Significance in producing left ventricular outflow obstruction. Circulation 1991;84:1188–97. [DOI] [PubMed] [Google Scholar]
- 10. Rowin EJ, Maron BJ, Lesser JR, Rastegar H, Maron MS.. Papillary muscle insertion directly into the anterior mitral leaflet in hypertrophic cardiomyopathy, its identification and cause of outflow obstruction by cardiac magnetic resonance imaging, and its surgical management. Am J Cardiol 2013;111:1677–9. [DOI] [PubMed] [Google Scholar]
- 11. Sherrid MV, Gunsburg DZ, Moldenhauer S, Pearle G.. Systolic anterior motion begins at low left ventricular outflow tract velocity in obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2000;36:1344–54. [DOI] [PubMed] [Google Scholar]
- 12. Lin CS, Chen KS, Lin MC, Fu MC, Tang SM.. The relationship between systolic anterior motion of the mitral valve and the left ventricular outflow tract Doppler in hypertrophic cardiomyopathy. Am Heart J 1991;122:1671–82. [DOI] [PubMed] [Google Scholar]
- 13. Peyrou J, Reant P, Reynaud A, Cornolle C, Dijos M, Rooryck-Thambo C. et al. Morphological and functional abnormalities pattern in hypertrophy-free HCM mutation carriers detected with echocardiography. Int J Cardiovasc Imaging 2016;32:1379–89. [DOI] [PubMed] [Google Scholar]
- 14. Captur G, Lopes LR, Mohun TJ, Patel V, Li C, Bassett P. et al. Prediction of sarcomere mutations in subclinical hypertrophic cardiomyopathy. Circ Cardiovasc Imaging 2014;7:863–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hagege AA, Dubourg O, Desnos M, Mirochnik R, Isnard G, Bonne G. et al. Familial hypertrophic cardiomyopathy. Cardiac ultrasonic abnormalities in genetically affected subjects without echocardiographic evidence of left ventricular hypertrophy. Eur Heart J 1998;19:490–9. [DOI] [PubMed] [Google Scholar]
- 16. Tarkiainen M, Sipola P, Jalanko M, Helio T, Laine M, Jarvinen V. et al. Cardiovascular magnetic resonance of mitral valve length in hypertrophic cardiomyopathy. J Cardiovasc Magn Reson 2016;18:33.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Michels M, Soliman OI, Phefferkorn J, Hoedemaekers YM, Kofflard MJ, Dooijes D. et al. Disease penetrance and risk stratification for sudden cardiac death in asymptomatic hypertrophic cardiomyopathy mutation carriers. Eur Heart J 2009;30:2593–8. [DOI] [PubMed] [Google Scholar]
- 18. Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA. et al. Recommendations for chamber quantification: a report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr 2005;18:1440–63. [DOI] [PubMed] [Google Scholar]
- 19. Jiang L, Levine RA, King ME, Weyman AE.. An integrated mechanism for systolic anterior motion of the mitral valve in hypertrophic cardiomyopathy based on echocardiographic observations. Am Heart J 1987;113:633–44. [DOI] [PubMed] [Google Scholar]
- 20. Freed LA, Levy D, Levine RA, Larson MG, Evans JC, Fuller DL. et al. Prevalence and clinical outcome of mitral-valve prolapse. N Engl J Med 1999;341:1–7. [DOI] [PubMed] [Google Scholar]
- 21. Delling FN, Gona P, Larson MG, Lehman B, Manning WJ, Levine RA. et al. Mild expression of mitral valve prolapse in the Framingham offspring: expanding the phenotypic spectrum. J Am Soc Echocardiogr 2014;27:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Captur G, Ho CY, Schlossarek S, Kerwin J, Mirabel M, Wilson R. et al. The embryological basis of subclinical hypertrophic cardiomyopathy. Sci Rep 2016;6:27714.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dal-Bianco JP, Aikawa E, Bischoff J, Guerrero JL, Handschumacher MD, Sullivan S. et al. Active adaptation of the tethered mitral valve: insights into a compensatory mechanism for functional mitral regurgitation. Circulation 2009;120:334–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hagege AA, Bruneval P, Levine RA, Desnos M, Neamatalla H, Judge DP.. The mitral valve in hypertrophic cardiomyopathy: old versus new concepts. J Cardiovasc Transl Res 2011;4:757–66. [DOI] [PubMed] [Google Scholar]
- 25. Norris RA, Moreno-Rodriguez R, Hoffman S, Markwald RR.. The many facets of the matricelluar protein periostin during cardiac development, remodeling, and pathophysiology. J Cell Commun Signal 2009;3:275–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Norris RA, Moreno-Rodriguez RA, Sugi Y, Hoffman S, Amos J, Hart MM. et al. Periostin regulates atrioventricular valve maturation. Dev Biol 2008;316:200–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ho CY, Day SM, Colan SD, Russell MW, Towbin JA, Sherrid MV. et al. The burden of early phenotypes and the influence of wall thickness in hypertrophic cardiomyopathy mutation carriers: findings from the HCMNet Study. JAMA Cardiol 2017;2:419–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.