

Abstract
Light chain (AL) amyloidosis is a devastating, complex, and incurable protein misfolding disease. It is characterized by an abnormal proliferation of plasma cells (fully differentiated B cells) producing an excess of monoclonal immunoglobulin light chains that are secreted into circulation, where the light chains misfold, aggregate as amyloid fibrils in target organs, and cause organ dysfunction, organ failure, and death. In this article, we will review the factors that contribute to AL amyloidosis complexity, the findings by our laboratory from the last 16 years and the work from other laboratories on understanding the structural, kinetics, and thermodynamic contributions that drive immunoglobulin light chain-associated amyloidosis. We will discuss the role of cofactors and the mechanism of cellular damage. Last, we will review our recent findings on the high resolution structure of AL amyloid fibrils. AL amyloidosis is the best example of protein sequence diversity in misfolding diseases, as each patient has a unique combination of germline donor sequences and multiple amino acid mutations in the protein that forms the amyloid fibril.
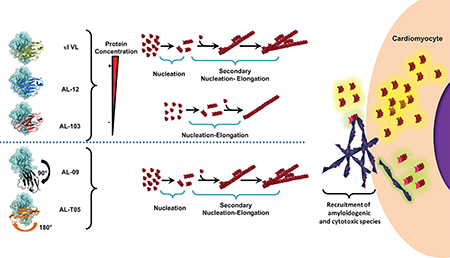
Grahical abstract
Introduction
Light chain (AL) amyloidosis was one of the first amyloid diseases characterized via the seminal work of Henry Bence Jones, Robert Kyle [1], George Glenner [2], Alan Edmundson [3], Alan Cohen [4], Merril Benson [5] and Alan Solomon [6], among others. In this article, we describe the current state of knowledge of AL amyloidosis biochemical and biophysical research in the context of the advances in clinical research. Our contributions to the field are discussed in the context of work performed by several other research groups worldwide.
We consider that a wider understanding of the role of stability needs to be acknowledged by the field. We are just starting to acknowledge and understand the role and the interactions between the two domains of immunoglobulin light chains. The toxic species identified for AL amyloidosis suggest that amyloid fibrils may play a role in cytotoxicity by affecting the cells in ways that differ from the oligomeric species, something that appears to be a unique feature among amyloidosis. Amyloid fibrils heterologous recruitment (known as cross seeding) could have profound implications for individuals with AL amyloidosis because of the potential of recruitment of normal repertoire immunoglobulins. Finally, the fibrillary structure of light chain amyloid appears to be joining the increasing number of amyloid structures from full sequences showing complex topology.
We believe that a sophisticated understanding of the biophysics, biochemistry, and cell biology of AL amyloidosis is required to fill the knowledge gap necessary to ameliorate the cellular and tissue damage associated with AL amyloidosis. In the sections below, we will describe our contributions to the main research questions of this complex disease.
Analysis of immunoglobulin light chains sequences associated with AL amyloidosis
Gene rearrangement
The functional light chain gene consists of three different segments: the Variable (V) gene, the Junction (J) gene, and the Constant (C) gene. Humans have 73 light chain V genes (40 kappa (κ) V genes and 33 lambda (λ) V genes), 5 Jκ, 4 Jλ genes, one Cκ gene and 5 Cλ genes. The functional gene arises from a recombination of the different VJC genes. Structurally, the variable domain consist of (VL) nine β-strands (abcc’c”defg) and the constant domain (CL) comprise seven β-strands (abcdefg) arranged such that four strands form one β-sheet while 5 β-strands (5 in the case of the constant domain (CL) comprise the other β-sheet (Figure 1). The sheets pack together and are joined by a disulphide bond. The topology is a form of a Greek key barrel [7]. Several groups, including ours, have shown that there are 5 V gene products that are overrepresented in AL amyloidosis: Vκ1, Vλ1, Vλ2, Vλ3, and Vλ6 [8–11]. One unresolved question in the field is whether or not these 5 V genes are inherently-prone to misfolding or whether somatic mutation contributes more to amyloid risk. A recent report studying the stability and amyloidogenic potential of several germline gene-encoded proteins showed that there is no correlation that might explain why these germline gene products are overrepresented in AL amyloidosis [12]; thus the particular properties that may explain this germline gene overrepresentation in AL amyloidosis remain unanswered.
Figure 1.

2D topology diagram of immunoglobulin light chain (LC) fold. The nine β-strands that form theframework regions. These strands are connected by unstructured loops in a Greek key pattern. The loops (blue lines) that connect strands B and C, C’, C”, and F and G are determine the specificity of the antigen-antibody interactions, and are known as the complementarity-determining regions (CDRs).
Somatic hypermutation
As mentioned above, the amino acid sequence diversity that characterizes immunoglobulin molecules arises from somatic hypermutation. Our laboratory and collaborators initially explored the possibility that there may be regions in the light chain variable domain (composed of the V and J germline gene products) that may accumulate more non-conservative mutations (defined as mutations that change the chemistry of the amino acid side chain) as compared to other light chain V domains from other plasma cell dyscrasias (such as multiple myeloma, where the prevalence of amyloidosis is uncommon) or from the normal immunoglobulin repertoire. Our results showed that in specific secondary structure elements, there are significant differences in the number of non-conservative mutations between normal and AL variable domain sequences. In addition, AL sequences from patients with different levels of secreted light chain have distinct differences in the location of non-conservative mutations, suggesting that for patients with very low levels of light chains and advanced amyloid deposition, the location of non-conservative mutations, rather than the amount of free light chain in circulation, may determine the amyloidogenic propensity of light chains [9].
Recently, we have embarked on determining the mutational hotspots in AL sequences at the amino acid level. Our results show that lambda variable domain sequences have a larger number of mutational hotspots compared to kappa sequences, resulting in large differences in the amino acid positions that are heavily mutated between the kappa and lambda light chains throughout the variable domain sequence (Ramirez-Alvarado et al., in preparation). A mutational spot region to highlight for its differences between kappa and lambda variable domains is the N-terminus β-strand A (amino acids 1–13). Our sequence analysis shows that positions 1, 3–9, and 13 in AL lambda proteins contain more non-conservative mutations as compared to lambda variable domains in the normal immunoglobulin repertoire. Studies of amino acid substitutions at position 2, 7, and 8 in lambda 6a germline variable domain show a decreased thermodynamic stability and increased rate of amyloid formation [13]. Destabilizing mutations are generally associated with increased amyloidogenicity in AL proteins, but the magnitude of the effect is context-dependent. For example, substitutions that introduce a Pro residue at the N-terminus β-strand A, F2P and H8P, decreased the amount of fibrils formed at equilibrium. In contrast, substituting Pro7 to Ser significantly increased fibrillar content [13].
Thermodynamic folding stability studies on the polymorphism R25G in lambda 6a (reported in 25% of lambda 6a sequences [14]; found to be present in 27% of lambda 6a sequences in our database) by del Pozo-Yauner et al. have shown a decreased stability associated with this polymorphism, which may enhance its propensity to form amyloid fibrils.
Mutations in residues appearing on the surface of the light chain protein that may affect electrostatic interactions can reduce thermodynamic stability and make the proteins more prone to aggregation as it has been shown for several AL proteins [15, 16].
Role of plasma cells in AL amyloidosis
Plasma cells are fully differentiated B cells responsible for the synthesis and secretion of immunoglobulins. In the context of AL amyloidosis, the abnormal proliferation of a plasma cell clone in the bone marrow that secrete an excess of monoclonal (meaning one unique protein sequence is synthesized) light chains is the initial pathological event in AL amyloidosis (Figure 2). AL amyloidosis is not considered a cancer due to the fact that the plasma cell burden (a measure of the extent of the malignant plasma cells throughout the entire bone marrow) is less than 10% of the bone marrow. It is thought that AL plasma cells are not as proliferative as those found in multiple myeloma, which explains the reason behind the lack of cellular models for AL plasma cells. In 2008, Jelinek led a team of researchers, including our laboratory, in the establishment and characterization of plasma cell lines called ALMC-1 and ALMC-2. The ALMC-1 cell line was established from the initial diagnostic bone marrow aspirate of a 50-year-old female patient with cardiac symptoms and suspicion of amyloid. The patient received a peripheral blood stem cell transplant, but relapsed 100 days after transplant. The second cell line, ALMC-2, was established at the time of relapse. Cytogenetic analysis of both cell lines compared to the primary patient cells pre- and post-transplant revealed c-myc amplification (from 30–100% cells) and p53 deletion (from 40–100% cells) among other cytogenetic abnormalities. Both cell lines express and secrete identical a monoclonal λ6a light chain with 9 mutations, 5 of those non-conservative mutations; 6 out of the 9 mutations are within mutational hotspots in lambda protein AL sequences. The secreted protein adopts the typical β-sheet structure characteristic of immunoglobulin light chains and is able to form amyloid fibrils under conditions where partially folded states are populated (incubation at the melting temperature of unfolding in the presence and absence of sodium sulphate) [17]. Other laboratories have conducted numerous studies using hybridoma technology (a hybrid cell produced by the fusion of an antibody-producing plasma cell with a tumor cell used to continuously express a specific monoclonal antibody) to shed light into the plasma cell biology in the context of multiple myeloma (for a recent review, please see [18]). While these studies are important in the field of plasma cell dyscrasias, their relevance to the role of the plasma cell biology in AL amyloidosis remains enigmatic.
Figure 2.

Light chain amyloidosis pathology. Clonal expansion of plasma cells secreting light chain (LC) dimers that deposit in vital organs as amyloid fibrils.
Structural studies of immunoglobulin light chains
Historically, structural information from immunoglobulin molecules was obtained from proteolytic fragments denoted Fc (“Fragment, crystallizes” corresponding to the heavy chain constant domains 2 and 3) and Fab (“Fragment, antigen binding” corresponding to the dimeric structure including the light and heavy chain variable domains, the light chain constant domain, and the first heavy chain constant domain). Allen Edmundson and colleagues obtained the first high resolution structures of immunoglobulin molecules using Bence Jones proteins (light chain proteins voided in the urine of patients with plasma cell dyscrasias and named for the British physician who first isolated them) [19]. The information gleaned from Edmundson’s efforts together with the x-ray structures of immunoglobulin fragments showed that immunoglobulin variable and constant domains for both heavy and light chains presented similar structural domains, which are now known as immunoglobulin folds. As we mentioned before, this structure consists of nine (seven in the case of the constant domain) β-strands arranged in a way that four strands form one β-sheet while 3 (5) strands form the other β-sheet. The sheets pack together and are joined by a disulphide bond. There is a conserved tryptophan at position 35. The dimer interface interactions involve a network of aromatic residues that are highly conserved as well.
The topology is a form of a Greek key barrel [7]. The residues that form the Greek key barrel are part of the framework region. There are three loops connecting strands B-C, strands C’-C” and strands F-G that are called the complementarity determining regions (CDRs), responsible for antigen binding and immunologic specificity (Figure 3). The CDRs are the regions in the protein that accumulate the largest number of somatic hypermutations in order to achieve the extensive diversity of reactivity required by the mammalian immunoglobulin system and high-affinity antigen binding.
Figure 3.

Primary structure of an immunoglobulin LC consisting of a variable domain (VL) and a constant domain (CL). A. Crystal structure of an immunoglobulin LC. B. Antibody basic structure, showing the interactions between the LCs and heavy chains (HC) forming a heterotetramer.
A feature from the first Bence Jones protein crystal structure solved by Edmundson that appears in all light chain structures in the protein data bank (pdb) [3, 16, 20–48] is the dimerization between the two immunoglobulin light chains. The dimer interface surface is the same surface used by the light chain to dimerize with the heavy chain, with very minor alterations in some proteins. Another feature observed in the crystal structure of Bence Jones proteins from AL amyloidosis patients was the asymmetry of the two monomers. This asymmetry occurs when the angle between the VL and the CL is different between the two monomers [49]. In the recent article by Oberti et al., they observe this asymmetry with some of their structures [50]. One of the first contributions from our laboratory to the field of AL amyloidosis came from solving the crystal structure of the recombinant bacterially produced AL-09 variable domain (AL-09 variable domain comes from a cardiac AL amyloidosis patient). The AL-09 crystal structure showed an altered dimer interface with a 90° rotation from the dimer interface geometry found in most immunoglobulin, Bence Jones proteins, and recombinant variable domain crystal structures [51]. This altered dimer interface has not been observed in any light chain crystal structure solved by any other groups or since, by our group [52] to date. Other reports with an unusual dimer interface (protein RHE and Loc) attributed the dimer alterations to crystallization conditions [43, 45, 49, 53]. Through mutational analysis, we identified the amino acid responsible for the altered dimer interface and learned that light chains have the ability to populate, at least 3 different dimer interfaces in a dynamic equilibrium that depends on the specific amino acid substitutions in the dimer interface residues. Given that this interface is a model of the light and heavy chain interface this region, is something that could be designed or modulated by genetic mutation for improved stability and better antigen binding properties of biomedically relevant antibodies [51, 54–56].
The Immunoglobulin fold is a versatile structural domain that provides stability, the ability to build highly diverse binding loops or edge β-strands without disrupting its core structure, It is in fact one of the most widely used protein topologies in nature. However, significant differences in the N-terminal region (namely, β-strand A) are observed between kappa and lambda light chains. The β-strand A in both kappa and lambda light chains adopts a characteristic conformation known as a β-sheet switch (the inclusion of a β-bulge), a known negative design element that prevents the two edge β-strands from non-native intermolecular interactions that lead to aggregation [57]. The size of the β-bulge in kappa light chains and the β-strand A hydrogen bond network is greater than in lambda light chains. Mutations that disrupt the interactions and the negative design of this particular region of the molecule can have significant effects in the thermodynamic stability and amyloidogenic properties of the proteins.
Protein stability studies
Work by our group and others have established that decreased thermodynamic stability in the VL domain is associated with an increased propensity to form amyloid fibrils [51, 54, 58–60]. The rationale behind this paradigm is that decreased stability will allow the protein to sample partially folded structured states necessary to initiate the aggregation reaction, as has been generally proposed for all amyloid formation initiation reactions [61, 62]. Surface mutations can affect thermodynamic stability. It is worth mentioning that there are data in the literature from irreversible folding transitions reported as thermodynamic parameters. This data does not conform with thermodynamic formalisms. Therefore, any reported ‘thermodynamic parameters’ shown in those publications should be considered as apparent [15]. Auton et al., have developed and applied scan rate dependency of protein folding for proteins with irreversible transitions that we have applied for AL proteins [63, 64].
The effect of solutes (e.g salts from the Hofmeister series) and sulphated glycosaminoglycans affect the thermodynamic equilibrium of AL proteins, and alter the experimental conditions required for fibril formation for AL-09 and AL-12 VL [65, 66]. Interestingly, we have found that unstable (AL-12 H32Y: the substitution of histidine for tyrosine at position 32) (or the more stable AL-12 Q96Y) AL-12 restorative mutants form fibrils, suggesting that fully unfolded AL proteins are unable to promote aggregation. This prompted us to suggest a thermodynamic range for optimal aggregation in physiological solution conditions. This was also observed with AL-T03 VL, associated with renal amyloidosis [67, 68].
In addition to thermodynamic stability, we have explored the role of kinetic stability in the process of amyloid formation using two protein systems: the AL protein AL-103 VL [69] and the full length proteins (FL) AL-09 FL, κI FL [63], as well as Bence Jones proteins [70]. AL-103 VL exhibits kinetic control in protein unfolding evident as scan rate-dependence in thermal denaturation experiments. The refolding process is reversible with hysteresis of different extents for several AL-103 VL restorative mutations. Interestingly, AL-103 VL and AL-103 del95aIns present the largest scan rate dependency [69]. AL-103 VL has a diproline motif as a result of the insertion of a Proline in position 95a which results in a trans/cis proline conformation while the del95Ins mutant has cis proline conformation. This is important because trans/cis proline isomerization directly affects the kinetic protein stability with little or no effect on the thermodynamic stability, slowing down the rate of protein folding, allowing the VL and full length light chains to sample amyloidogenic misfolded conformations. The main structural deviations found in the AL-103 structure center around the CDR3 region, where the Pro95aIns mutation is located [52], suggesting that the AL-103 CDR3 region appears to be a dynamic region based on the poor electron density data. Similar weak electron density in the CDR3 region of AL-103 delP95Ins crystal structure suggested to us that this mutant exhibits the same dynamic behaviour around CDR3 as its parent protein AL-103.
Full length AL proteins in amyloid deposits
Early reports on the composition of tissue amyloid deposits from patient-derived material indicated that the amyloid fibrils comprised fragments consisting principally of the entire variable domain and a portion of the constant domainn [71, 72]. Because the variable domain is the region of the light chain that accumulates somatic hypermutation, it was assumed that the light chain variable domain was the main component of the amyloid deposits. In 2008 and 2009, mass spectrometry analysis of patient fat aspirates and biopsy tissues reported the presence of the entire light chain (variable + constant domains) in patients’ samples [73, 74]. These mass spectrometry findings highlighted an added level of complexity associated with AL amyloid deposition, suggesting that mixed fragments of variable, variable + portions of the constant domain, and full length light chains may form part of the amyloid fibrils in AL amyloidosis. Mass spectrometry is not a quantitative approach, so the proportion of each fragment cannot be assessed and requires different approaches. However, given that highly sensitive mass spectrometry studies were required to identify the full length components, it suggests that full length proteins may be minor component of the amyloid deposit relative to the VL domain fragments.
The participation of the CL in the pathophysiology of AL amyloidosis is not well understood, although previous studies reported that the κI CL domain can form amyloid deposits in AL patients [6, 75]. In addition, work performed with a mouse immunoglobulin light chain has shown the importance of the the initial residues of the CL domain (considered a structural linker between the structural VL and CL) for protein stability and amyloid formation potential [76]
The role of the CL in the thermal stability of lambda-6a light chain AL-01–095 was assessed and compared with its corresponding variable and constant domain proteins [77]. One common characteristic of full length (FL) AL proteins is their irreversible refolding nature [50, 78–80]. In this regard, Oberti et. al [50] highlight the complexity to achieve generalizable observations about the role of the constant domain. Their results indicate that AL LCs have lower stability than the non-amyloidogenic LC, however low thermodynamic values do not correlate perfectly with LCs amyloidogenicity. Another interesting observation from the Oberti report is that structural rearrangements and the nature of the LC dimeric interface (as observed on the crystallographic structures) do not correlate with amyloid propensity. One possible explanation to the discrepancy can be the effect of solvent of crystallization, its ionic strength, and pH influence the interactions within the dimeric interface of LCs, as shown by the work of Shiffer et. al. [49] Our thermodynamic studies on AL-09 and κI FL present similar thermodynamic stability parameters compared to their VL counterparts [63].
In the case of κI FL, the thermodynamic parameters calculated in our 2015 study suggest that the VL domain is slightly more stable than the FL protein, suggesting that for AL-09 and κI FL, the presence of the CL domain did not have any stabilizing effect. Our results differ from those published by Klimtchuk and colleagues using a λ6 (IGLV 6–57) full length protein [77]. Their study used the VL, CL, and the FL versions of protein AL-01–095 (comparing their results to the corresponding urine-derived Bence Jones protein). They showed that the CL domain (LC3*04 or IGLC3) is more stable than the VL and FL AL-01–095 suggesting that the CL domain in this λ6 protein is destabilized by the VL domain, further indicating that some potential interactions between these domains exist. Heretofore, it has widely been acknowledged that these domains are completely autonomous and independent. Sequence alignment of the kappa constant domain (IGKC) and the constant domain from AL-01–095 (IGLC3) indicated that the proteins are 41% identical. The level of sequence identity among the 5 lambda constant domains (IGLC) is quite high (93–99%), varying among them by a maximum of 7 amino acids.
Our sequence comparisons between IGKC and IGLC indicate that the differences in stability behaviour observed between Klimtchuk et al. and our study could be easily explained by the differences in the amino acid composition of the constant domains. In addition, the high sequence identity among IGLC sequences suggest that their thermodynamic properties are probably very similar, implying that there may be more interactions between VL and CL domains in lambda light chains that may modulate their stability and amyloidogenic potential. This is again another example of the ways in which kappa and lambda proteins differ in their structural and stability properties and how that may impact differentially their amyloidogenic potential.
With regards to the role of FL proteins in the process of amyloid formation, our data indicate that the presence of the constant domain in κI FL modulates fibril formation, facilitating the recruitment of κI VL [56] (see section below for more detailed description of these results).
Role of co-factors
The amyloid deposits in the tissues and organs affected by AL amyloidosis consist of a number of protein co-factors. These “accessory molecules” are often ubiquitous in the amyloid deposits from patients affected by any of the 36 human amyloid diseases [73]. Tissue amyloid is located in the extracellular space and perhaps not surprisingly many of the accessory proteins are components of the extracellular matrix. Proteoglycans are glycoproteins comprising a core protein associated with glycosaminoglycans (GAGs). GAGs are a group of negatively charged, unbranched, long heteropolysaccharides composed of repeating disaccharide units consisting of uronic acid and either N-acetylglucosamine or N-acetylgalactosamine. GAGs play multiple roles related to cell signaling and adhesion and are a major component of all plasma membranes and extracellular matrices. The structures of the repeating disaccharides determine the classes of GAG, which include heparan sulfate, chondroitin sulfate A, dermatan sulfate, hyaluronic acid, and keratan sulfate. Amyloid fibrils have been found associated with GAGs from proteoglycans in AL amyloidosis and many other forms of amyloid-associated disorders (for a review on this topic, see [81]). Our laboratory has studied the effect of glycosaminoglycans and anions from the Hofmeister series in amyloid formation in vitro in AL amyloidosis [65, 66, 82, 83]. Our data indicate that charge (in the case of GAGs, this means the level of sulphation) and size (meaning number of disaccharide repeats) are important factors that contribute to the acceleration and possibly the stability of the amyloid formation reaction. We have also reported that the different somatic mutations in AL proteins may be responsible for differential enhancing effects of GAGs in amyloid formation reactions (Figure 4). Our results may explain the differential deposition between different systemic amyloidosis. For example, it is possible that cardiac AL amyloidosis is stabilized through the interaction of AL proteins with Heparan sulfate. In contrast, it is possible that joint deposition of β2-microglobulin in haemodialysis-associated amyloidosis is stabilized by the strong interaction with Chondroitin sulfate [82]. Research from Boston University has also demonstrated the potential role of sulphation for in vitro and cell culture studies in AL amyloidosis [84, 85].
Figure 4.

A–C. suggested model of GAG modeof action on AL amyloid fibril formation. The morphological designations/definitions were based on empirical/visual comparison and contrasted with the description of amyloid aggregates and intermediates in the fibril formation pathway, reported for AL proteins as well as other proteins. Adapted from Blancas-Mejia et al., 2015.
The ability of amyloid deposits to bind to GAGs has been explored by the Wall laboratory to develop molecular imaging agents. Their basic heparin-binding peptides recognize murine and human amyloid deposits in both in vivo and ex vivo tissues and therefore, have potential as radiotracers for the non-invasive molecular imaging of amyloid deposits in situ [86]. More recently, the Wall laboratory has shown potential therapeutic applications for fusion peptides based on the GAG binding peptide sequences [87].
Light chain induced toxicity in cells and model organisms
Animal models of systemic AL amyloidosis have been challenging to generate. Those that have been published do not recapitulate the complex pathophysiology of AL disease [88–93]. Therefore, cellular studies that shed light into the pathophysiological mechanisms, notably cytotoxicity, are a very important source of knowledge as it relates to AL amyloidosis. Early studies from several groups, including our laboratory [84, 94, 95,96–98] utilized rat and mouse-derived cardiac fibroblasts and cardiomyocytes to study the internalization and effects of LC soluble species on cellular homeostasis. Numerous cellular effects have been observed including, increased oxidative stress, reduced contractility and relaxation, increased apoptosis and cell death, as well as increased sulphation of extracellular glycosaminoglycans. In primary human renal mesangial cells, amyloidogenic light chains internalize, promoting what appears to be macrophage-type phenotypic transformation [99–101].
More recent studies from our laboratory in collaboration with the Wall laboratory have utilized a human-derived cardiomyocyte cell line to characterize the in vitro effect of exposure to amyloid fibrils composed of recombinant AL-09 (κI) and Wil (λ6) AL proteins. AL-09 and Wil fibrils become internalized by the cells and are also seen bound to the human cardiomyocyte plasma membrane. Membrane bound fibrils act as nucleus for fibril elongation in cell culture with enhanced kinetics compared to test tube elongation reactions. Both types of fibrils cause cell growth arrest when cultured with 12.5% fetal bovine serum, while Wil can also cause mitochondrial dysfunction in the cardiomyocyte cell line when serum starved; as found by other laboratories as well [102–105].
Non mammalian model organisms (Caenorhabditis elegans and Danio rerio) have been utilized by other groups to study AL amyloidosis and to screen molecular libraries for possible therapeutic strategies [106–111].
Recruitment of soluble proteins using preformed nucleus-seeding
The ability to self-propagate is a fundamental property of all amyloid fibrils, and self-propagation proceeds as a continuous autocatalytic process via the recruitment of additional protein molecules from the surrounding solution (reviewed in [112]). Autocatalytic reactions proceed slowly in the beginning because there is little catalyst present. As the reaction continues, the rate of the reaction accelerates with an increased amount of catalyst then slowing as the reactant is consumed. We have reported the ability of different cardiac amyloid-associated AL proteins to be recruited by homologous (also known as self-seeding: fibrils made with the same recombinant AL protein) and heterologous (also known as cross-seeding: fibrils made with different recombinant AL protein) preformed seeds [112, 113]. Our data have indicated that fibril formation is kinetically controlled by the conformation of the amyloidogenic precursor and modulated by the differential ability of each protein to either nucleate or elongate fibrils. For the most amyloidogenic light chains, the presence of preformed fibrils did not have a significant effect accelerating amyloid formation, indicating that these proteins do not need a catalyst or the seed provided cannot serve as a catalyst for these proteins.
As discussed previously (Full length protein section), the appearance of mixed populations of full length, variable domain and other fragments present in tissue amyloid deposits has been documented extensively in the literature. In 2016, we demonstrated that for the least fibrillogenic of the proteins we have studied so far, κI O18/O8, both the VL and FL fibrillar forms of the protein are able to recruit soluble κI O18/O8 (κI) VL and FL proteins with optimal efficiency compared to de novo fibril reaction at the conditions where we have observed optimal de novo fibril formation (pH 2.0 for VL, pH 7.4 for FL) [112]. We studied the heterologous recruitment of κI VL and the κI FL at different ratios within one reaction and with both preformed fibrils of VL and FL.
In 2017, we demonstrated that FL κI O18/O8 fibrils served as excellent nucleators of fibril formation in mixed reactions, while the VL κI O18/O8 acted as an excellent elongator of the reaction, particularly when VL:FL ratio is 2:1 [56].
These results have profound implications on the mechanism of disease initiation and propagation and sheds light into the complex behaviour of different forms of the same AL protein.
The ability of amyloidogenic light chains to be recruited during the amyloid fibril elongation reaction may serve as a useful tool for diagnosis. The Wall laboratory led a collaborative effort with our laboratory to develop a sensitive binding assay to quantify the recruitment of full length, patient-derived LC proteins by synthetic amyloid fibrils composed of the recombinant Vλ6 Wil protein. Eight urine derived light chains from AL and non-AL patients (multiple myeloma (MM) controls) were used and evaluated for their ability to bind to preformed amyloid fibrils. This study indicated that AL-associated light chains bound significantly more efficiently to the recombinant amyloid fibrils compared to the MM controls. Notably, the light chain from a MM patient who subsequently developed AL amyloidosis behaved as an AL associated protein in the assay, indicating the possibility for identifying MM patients at risk for developing AL amyloidosis based on the light chain recruitment efficacy [114].
Cell to cell propagation-the role of extracellular vesicles
The kidneys are the most common organs affected by AL amyloidosis. Urinary extracellular vesicles (UEX) contain kidney-derived membrane and cytosolic proteins that can be used to probe the proteomics of the entire urinary system from the glomerulus to the bladder for clinical and mechanistic questions related to organ damage [115]. In 2012, our laboratory analysed UEX to determine the possible differences between those from patients with light chain amyloidosis, multiple myeloma, monoclonal gammopathy of undetermined significance, and non-paraproteinemia related kidney disease controls. In patients with light chain amyloidosis, multiple myeloma and monoclonal gammopathy of undetermined significance, immunoreactive proteins corresponding to monomeric light chains were found in UEX by western blot.
In all of the samples from AL amyloidosis patients with active disease, high molecular weight immunoreactive species corresponding to a decamer were observed. These oligomeric species were not found in UEX from the other diseases or in amyloidosis UEX from patients in remission. These initial results showed that UEX may have tremendous potential in furthering our understanding of the pathophysiology and diagnosis of plasma cell dyscrasia related kidney diseases [116].
In 2017, we embarked on a follow-up study to further explore the role of the light chain oligomers in progression of AL amyloidosis. For this study, we isolated UEX from an AL amyloidosis patient with progressive renal disease despite achieving a complete haematological response. Light chain oligomers were identified in the UEX. MS analysis of the UEX and serum identified two monoclonal lambda light chains. Proteomics of the trypsin digested amyloid fragments in the kidney by laser microdissection and MS analysis identified a λ6 light chain. The cDNA from plasma cell clone was also from a λ6 family and matched the amino acid sequences of the amyloid peptides. The predicted mass of the peptide product of the cDNA correlated with the mass of one of the two LCs identified in the UEX and serum. UEX combined with MS were able to identify 2 monoclonal lambda LCs that current clinical methods could not. It also identified the amyloidogenic light chain which holds potential for response assessment in the future [117].
In addition to the potential for clinical applications described above, UEX have been implicated in the cell-to-cell propagation of amyloid disease and aggregated species in neurodegeneration [69, 118–120]. It is unclear to us what may be the potential role of UEX in the pathophysiology of renal amyloidosis; however, it is possible that glomerular AL amyloid could spread throughout the nephron and propagate tubular disease in this manner.
Fibril structure
Our laboratory, in collaboration with Chad Rienstra et al. have performed solid state NMR studies of isotopically labelled AL-09 VL fibrils to glean insight into the high resolution structure of these fibrils [121, 122]. Our results show that the N- and C-terminus of the variable domain are involved in the rigid portion of the fibril structure. Moreover, work in preparation has allowed us to assign >90% of the residues involved in the fibril structure (Piehl et al., in preparation). Initial analysis of the structural properties observed in these fibrils indicate that these light chain fibrils may adopt complex fibril structures, similar to what has been found for α-synuclein and the protein Fused in Sarcoma (FUS) amyloid fibrils [123, 124].
Conclusions
AL amyloidosis is a complex, heterogeneous, and devastating disease. While there has been tremendous progress in the field, there are many questions that remain unanswered in all areas of research associated with this disease. It is our belief that biochemical and biophysical studies of AL amyloidosis will shed light on features and characteristics of immunoglobulin molecule dynamics, positive and negative design considerations, and sequence selection for stability and solubility. These new insights can have enormous impact in the design and delivery of many antibodies now used as therapeutic agents.
Outlook-where do we go from here?
Structural studies to understand the dynamic interactions between the variable and constant domain in the context of AL amyloidosis.
Role of accessory proteins in the elongation of amyloid fibrils and the cytotoxicity of the amyloid deposits
Mechanistic studies of the role of extracellular vesicles in the propagation of AL amyloidosis between organs.
Molecular basis for heterologous recruitment on systemic amyloidosis. In depth investigation of possible heterologous recruitment of different amyloidogenic proteins
The role of macrophages in amyloid clearance.
Acknowledgements
We would like to thank Jon Wall for his critical reading of this review and his excellent suggestions. Elizabeth (EB) Allen helped us with excellent editing support. The work described in this article has been possible thanks to 16 years of hard work from the 70 former and current Ramirez-Alvarado team members: 1 research volunteer, 17 summer students, 1 undergraduate research employee, 4 post baccalaureate students, 2 graduate research employee students, 10 research technologists, 3 research technologist laboratory managers, 14 PhD rotation students, 6 graduate students, 12 postdocs, and 1 visiting professor. We are very thankful for all of our collaborators both within Mayo and in other institutions. In particular, we want to thank the Mayo Clinic Amyloid Interest group for the many years of synergistic collaboration and learning we have enjoyed within our multidisciplinary team.
The Ramirez-Alvarado team has been supported through the years by NIH R01 grants GM128253, GM071514, and CA111345, AHA grant 0630077N, Prothena Corporation, the Mayo Foundation, the Mayo Regenerative Medicine Program, Morie Gertz and the Seidler Professorship, the Sanford Burnham/Mayo Consortium, the Arizona State University/Mayo Consortium, the Mayo Alzheimer’s Disease Research Center, the Mayo Hematologic Malignancies Research Program, and the generous support from amyloidosis patients and their families.
Footnotes
Conflicts of interest
There are no conflicts to declare.
KRR is a graduate student in the Molecular Pharmacology and Experimental Therapeutics track; TLJ is a student in the Immunology track.
Notes and references
- [1].Kyle RA. Monoclonal gammopathy of undetermined significance. Natural history in 241 cases. Am J Med. 1978;64:814–26. [DOI] [PubMed] [Google Scholar]
- [2].Glenner GG, Terry W, Harada M, Isersky C, Page D. Amyloid Fibril Proteins: Proof of Homology with Immunoglobulin Light Chains by Sequence Analyses. Science. 1971;172:1150–1. [DOI] [PubMed] [Google Scholar]
- [3].Ely KR, Girling RL, Schiffer M, Cunningham DE, Edmundson AB. Preparation and properties of crystals of a Bence-Jones dimer with mercury inserted into the interchain disulfide bond. Biochemistry. 1973;12:4233–7. [DOI] [PubMed] [Google Scholar]
- [4].Cohen AS, Calkins E. THE ISOLATION OF AMYLOID FIBRILS AND A STUDY OF THE EFFECT OF COLLAGENASE AND HYALURONIDASE. The Journal of Cell Biology. 1964;21:481–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Shirahama T, Benson MD, Cohen AS, Tanaka A. Fibrillar Assemblage of Variable Segments of Immunoglobulin Light Chains: An Electron Microscopic Study. The Journal of Immunology. 1973;110:21–30. [PubMed] [Google Scholar]
- [6].Solomon A, Weiss DT, Murphy CL, Hrncic R, Wall JS, Schell M. Light chain-associated amyloid deposits comprised of a novel kappa constant domain. Proc Natl Acad Sci U S A. 1998;95:9547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Branden C, Tooze J. Introduction to protein structure. 2nd ed. New York: Garland Publishing, Inc.; 1999. [Google Scholar]
- [8].Abraham RS, Geyer SM, Ramirez-Alvarado M, Price-Troska TL, Gertz MA, Fonseca R. Analysis of somatic hypermutation and antigenic selection in the clonal B cell in immunoglobulin light chain amyloidosis (AL). J Clin Immunol. 2004;24:340–53. [DOI] [PubMed] [Google Scholar]
- [9].Poshusta TL, Sikkink LA, Leung N, Clark RJ, Dispenzieri A, Ramirez-Alvarado M. Mutations in specific structural regions of immunoglobulin light chains are associated with free light chain levels in patients with AL amyloidosis. PLoS One. 2009;4:e5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Katoh N, Poshusta TL, Manske MK, Dispenzieri A, Gertz MA, Abraham RS, et al. A reappraisal of immunoglobulin variable gene primers and its impact on assessing clonal relationships between PB B cells and BM plasma cells in AL amyloidosis. J Clin Immunol. 2011;31:1029–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kourelis TV, Dasari S, Theis JD, Ramirez-Alvarado M, Kurtin PJ, Gertz MA, et al. Clarifying immunoglobulin gene usage in systemic and localized immunoglobulin light-chain amyloidosis by mass spectrometry. Blood. 2017;129:299–306. [DOI] [PubMed] [Google Scholar]
- [12].Garay Sanchez SA, Rodriguez Alvarez FJ, Zavala-Padilla G, Mejia-Cristobal LM, Cruz-Rangel A, Costas M, et al. Stability and aggregation propensity do not fully account for the association of various germline variable domain gene segments with light chain amyloidosis. Biol Chem. 2017;398:477–89. [DOI] [PubMed] [Google Scholar]
- [13].del Pozo-Yauner L, Wall JS, Gonzalez Andrade M, Sanchez-Lopez R, Rodriguez-Ambriz SL, Perez Carreon JI, et al. The N-terminal strand modulates immunoglobulin light chain fibrillogenesis. Biochem Biophys Res Commun. 2014;443:495–9. [DOI] [PubMed] [Google Scholar]
- [14].del Pozo Yauner L, Ortiz E, Sanchez R, Sanchez-Lopez R, Guereca L, Murphy CL, et al. Influence of the germline sequence on the thermodynamic stability and fibrillogenicity of human lambda 6 light chains. Proteins. 2008;72:684–92. [DOI] [PubMed] [Google Scholar]
- [15].Kobayashi Y, Tsutsumi H, Abe T, Ikeda K, Tashiro Y, Unzai S, et al. Decreased amyloidogenicity caused by mutational modulation of surface properties of the immunoglobulin light chain BRE variable domain. Biochemistry. 2014;53:5162–73. [DOI] [PubMed] [Google Scholar]
- [16].Wall JS, Gupta V, Wilkerson M, Schell M, Loris R, Adams P, et al. Structural basis of light chain amyloidogenicity: comparison of the thermodynamic properties, fibrillogenic potential and tertiary structural features of four Vlambda6 proteins. J Mol Recognit. 2004;17:323–31. [DOI] [PubMed] [Google Scholar]
- [17].Arendt BK, Ramirez-Alvarado M, Sikkink LA, Keats JJ, Ahmann GJ, Dispenzieri A, et al. Biologic and genetic characterization of the novel amyloidogenic lambda light chain-secreting human cell lines, ALMC-1 and ALMC-2. Blood. 2008;112:1931–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pawlyn C, Morgan GJ. Evolutionary biology of high-risk multiple myeloma. Nat Rev Cancer. 2017;17:543–56. [DOI] [PubMed] [Google Scholar]
- [19].Schiffer M, Hardman KD, Wood MK, Edmundson AB, Hood ME, Ely KR, et al. A preliminary crystallographic investigation of human L-type Bence-Jones protein. J Biol Chem. 1970;245:728–30. [PubMed] [Google Scholar]
- [20].Bourne PC, Ramsland PA, Shan L, Fan ZC, DeWitt CR, Shultz BB, et al. Three-dimensional structure of an immunoglobulin light-chain dimer with amyloidogenic properties. Acta Crystallogr D Biol Crystallogr. 2002;58:815–23. [DOI] [PubMed] [Google Scholar]
- [21].Edmundson AB, Borrebaeck CA. Progress in programming antibody fragments to crystallize. Immunotechnology. 1998;3:309–17. [DOI] [PubMed] [Google Scholar]
- [22].Edmundson AB, Ely KR. Binding of N-formylated chemotactic peptides in crystals of the Mcg light chain dimer: similarities with neutrophil receptors. Mol Immunol. 1985;22:463–75. [DOI] [PubMed] [Google Scholar]
- [23].Edmundson AB, Ely KR, Abola EE. Conformational flexibility in immunoglobulins. Contemp Top Mol Immunol. 1978;7:95–118. [DOI] [PubMed] [Google Scholar]
- [24].Edmundson AB, Ely KR, Abola EE, Schiffer M, Panagiotopoulos N, Deutsch HF. Conformational isomerism, rotational allomerism, and divergent evolution in immunoglobulin light chains. Fed Proc. 1976;35:2119–23. [PubMed] [Google Scholar]
- [25].Edmundson AB, Ely KR, Girling RL, Abola EE, Schiffer M, Westholm FA, et al. Binding of 2,4-dinitrophenyl compounds and other small molecules to a crystalline lambda-type Bence-Jones dimer. Biochemistry. 1974;13:3816–27. [DOI] [PubMed] [Google Scholar]
- [26].Edmundson AB, Ely KR, He XM, Herron JN. Cocrystallization of an immunoglobulin light chain dimer with bis(dinitrophenyl) lysine: tandem binding of two ligands, one with and one without accompanying conformational changes in the protein. Mol Immunol. 1989;26:207–20. [DOI] [PubMed] [Google Scholar]
- [27].Edmundson AB, Ely KR, Herron JN. A search for site-filling ligands in the Mcg Bence-Jones dimer: crystal binding studies of fluorescent compounds. Mol Immunol. 1984;21:561–76. [DOI] [PubMed] [Google Scholar]
- [28].Edmundson AB, Ely KR, Herron JN, Cheson BD. The binding of opioid peptides to the Mcg light chain dimer: flexible keys and adjustable locks. Mol Immunol. 1987;24:915–35. [DOI] [PubMed] [Google Scholar]
- [29].Edmundson AB, Harris DL, Fan ZC, Guddat LW, Schley BT, Hanson BL, et al. Principles and pitfalls in designing site-directed peptide ligands. Proteins. 1993;16:246–67. [DOI] [PubMed] [Google Scholar]
- [30].Edmundson AB, Herron JN, Ely KR, He XM, Harris DL, Voss EW Jr, Synthetic site-directed ligands. Philos Trans R Soc Lond B Biol Sci. 1989;323:495–509. [DOI] [PubMed] [Google Scholar]
- [31].Edmundson AB, Schiffer M, Ely KR, Wood MK. Structure of a lambda-type Bence-Jones protein at 6-A resolution. Biochemistry. 1972;11:1822–7. [DOI] [PubMed] [Google Scholar]
- [32].Edmundson AB, Schiffer M, Wood MK, Hardman KD, Ely KR, Ainsworth CF. Crystallographic studies of an IgG immunoglobulin and the Bence-Jones protein from one patient. Cold Spring Harb Symp Quant Biol. 1972;36:427–32. [DOI] [PubMed] [Google Scholar]
- [33].Edmundson AB, Sheber FA, Ely KR, Simonds NB, Hutson NK, Rossiter JL. Characterization of human L type Bence-Jones proteins containing carbohydrate. Arch Biochem Biophys. 1968;127:725–40. [DOI] [PubMed] [Google Scholar]
- [34].Edmundson AB, Simonds NB, Sheber FA, Johnson T, Bangs B. Use of carboxypeptidase A for simultaneous assessment of purity and assignment of human Bence Jones proteins and light chains to K and L classes. Arch Biochem Biophys. 1969;132:502–8. [DOI] [PubMed] [Google Scholar]
- [35].Ely KR, Firca JR, Williams KJ, Abola EE, Fenton JM, Schiffer M, et al. Crystal properties as indicators of conformational changes during ligand binding or interconversion of Mcg light chain isomers. Biochemistry. 1978;17:158–67. [DOI] [PubMed] [Google Scholar]
- [36].Ely KR, Herron JN, Edmundson AB. Three-dimensional structure of a hybrid light chain dimer: protein engineering of a binding cavity. Mol Immunol. 1990;27:101–14. [DOI] [PubMed] [Google Scholar]
- [37].Ely KR, Herron JN, Harker M, Edmundson AB. Three-dimensional structure of a light chain dimer crystallized in water. Conformational flexibility of a molecule in two crystal forms. J Mol Biol. 1989;210:601–15. [DOI] [PubMed] [Google Scholar]
- [38].Ely KR, Peabody DS, Holm TR, Cheson BD, Edmundson AB. Accessible intrachain disulfide bonds in hybrids of light chains. Mol Immunol. 1985;22:85–92. [DOI] [PubMed] [Google Scholar]
- [39].Epp O, Lattman EE, Schiffer M, Huber R, Palm W. The molecular structure of a dimer composed of the variable portions of the Bence-Jones protein REI refined at 2.0-A resolution. Biochemistry. 1975;14:4943–52. [DOI] [PubMed] [Google Scholar]
- [40].Firca JR, Ely KR, Kremser P, Westholm FA, Dorrington KJ, Edmundson AB. Interconversion of conformational isomers of light chains in the Mcg immunoglobulins. Biochemistry. 1978;17:148–58. [DOI] [PubMed] [Google Scholar]
- [41].Hernandez-Santoyo A, del Pozo Yauner L, Fuentes-Silva D, Ortiz E, Rudino-Pinera E, Sanchez-Lopez R, et al. A single mutation at the sheet switch region results in conformational changes favoring lambda6 light-chain fibrillogenesis. J Mol Biol. 2010;396:280–92. [DOI] [PubMed] [Google Scholar]
- [42].Horne C, Klein M, Polidoulis I, Dorrington KJ. Noncovalent association of heavy and light chains of human immunoglobulins. III. Specific interactions between VH and VL. J Immunol. 1982;129:660–4. [PubMed] [Google Scholar]
- [43].Huang DB, Ainsworth C, Solomon A, Schiffer M. Pitfalls of molecular replacement: the structure determination of an immunoglobulin light-chain dimer. Acta Crystallogr D Biol Crystallogr. 1996;52:1058–66. [DOI] [PubMed] [Google Scholar]
- [44].Kolmar H, Frisch C, Kleemann G, Gotze K, Stevens FJ, Fritz HJ. Dimerization of Bence Jones proteins: linking the rate of transcription from an Escherichia coli promoter to the association constant of REIV. Biol Chem Hoppe Seyler. 1994;375:61–70. [DOI] [PubMed] [Google Scholar]
- [45].Novotny J, Haber E. Structural invariants of antigen binding: comparison of immunoglobulin VL-VH and VL-VL domain dimers. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:4592–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Roussel A, Spinelli S, Deret S, Navaza J, Aucouturier P, Cambillau C. The structure of an entire noncovalent immunoglobulin kappa light-chain dimer (Bence-Jones protein) reveals a weak and unusual constant domains association. Eur J Biochem. 1999;260:192–9. [DOI] [PubMed] [Google Scholar]
- [47].Schormann N, Murrell JR, Liepnieks JJ, Benson MD. Tertiary structure of an amyloid immunoglobulin light chain protein: a proposed model for amyloid fibril formation. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:9490–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Pokkuluri PR, Solomon A, Weiss DT, Stevens FJ, Schiffer M. Tertiary structure of human lambda 6 light chains. Amyloid. 1999;6:165–71. [DOI] [PubMed] [Google Scholar]
- [49].Huang DB, Ainsworth CF, Stevens FJ, Schiffer M. Three quaternary structures for a single protein. Proc Natl Acad Sci U S A. 1996;93:7017–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Oberti L, Rognoni P, Barbiroli A, Lavatelli F, Russo R, Maritan M, et al. Concurrent structural and biophysical traits link with immunoglobulin light chains amyloid propensity. Sci Rep. 2017;7:16809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Baden EM, Owen BA, Peterson FC, Volkman BF, Ramirez-Alvarado M, Thompson JR. Altered dimer interface decreases stability in an amyloidogenic protein. J Biol Chem. 2008;283:15853–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Randles EG, Thompson JR, Martin DJ, Ramirez-Alvarado M. Structural alterations within native amyloidogenic immunoglobulin light chains. J Mol Biol. 2009;389:199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Schiffer M, Ainsworth C, Xu ZB, Carperos W, Olsen K, Solomon A, et al. Structure of a second crystal form of Bence-Jones protein Loc: strikingly different domain associations in two crystal forms of a single protein. Biochemistry. 1989;28:4066–72. [DOI] [PubMed] [Google Scholar]
- [54].Baden EM, Randles EG, Aboagye AK, Thompson JR, Ramirez-Alvarado M. Structural insights into the role of mutations in amyloidogenesis. J Biol Chem. 2008;283:30950–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Peterson FC, Baden EM, Owen BA, Volkman BF, Ramirez-Alvarado M. A single mutation promotes amyloidogenicity through a highly promiscuous dimer interface. Structure. 2010;18:563–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Blancas-Mejia LM, Misra P, Ramirez-Alvarado M. Differences in Protein Concentration Dependence for Nucleation and Elongation in Light Chain Amyloid Formation. Biochemistry. 2017;56:757–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Richardson JS, Richardson DC. Natural β-sheet proteins use negative design to avoid edge-to-edge aggregation. Proceedings of the National Academy of Sciences. 2002;99:2754–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hurle MR, Helms LR, Li L, Chan W, Wetzel R. A role for destabilizing amino acid replacements in light-chain amyloidosis. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:5446–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wall J, Schell M, Murphy C, Hrncic R, Stevens FJ, Solomon A. Thermodynamic instability of human lambda 6 light chains: correlation with fibrillogenicity. Biochemistry. 1999;38:14101–8. [DOI] [PubMed] [Google Scholar]
- [60].Wetzel R Domain stability in immunoglobulin light chain deposition disorders. Adv Protein Chem. 1997;50:183–242. [DOI] [PubMed] [Google Scholar]
- [61].Chiti F, Dobson CM. Amyloid formation by globular proteins under native conditions. Nat Chem Biol. 2009;5:15–22. [DOI] [PubMed] [Google Scholar]
- [62].Ramirez-Alvarado M, Merkel JS, Regan L. A systematic exploration of the influence of the protein stability on amyloid fibril formation in vitro. Proc Natl Acad Sci U S A. 2000;97:8979–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Blancas-Mejia LM, Horn TJ, Marin-Argany M, Auton M, Tischer A, Ramirez-Alvarado M. Thermodynamic and fibril formation studies of full length immunoglobulin light chain AL-09 and its germline protein using scan rate dependent thermal unfolding. Biophys Chem. 2015;207:13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tischer A, Madde P, Blancas-Mejia LM, Auton M. A molten globule intermediate of the von Willebrand factor A1 domain firmly tethers platelets under shear flow. Proteins. 2014;82:867–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].McLaughlin RW, De Stigter JK, Sikkink LA, Baden EM, Ramirez-Alvarado M. The effects of sodium sulfate, glycosaminoglycans, and Congo red on the structure, stability, and amyloid formation of an immunoglobulin light-chain protein. Protein Sci. 2006;15:1710–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sikkink LA, Ramirez-Alvarado M. Salts enhance both protein stability and amyloid formation of an immunoglobulin light chain. Biophys Chem. 2008;135:25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Marin-Argany M, Guell-Bosch J, Blancas-Mejia LM, Villegas S, Ramirez-Alvarado M. Mutations can cause light chains to be too stable or too unstable to form amyloid fibrils. Protein Sci. 2015;24:1829–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Poshusta TL, Katoh N, Gertz MA, Dispenzieri A, Ramirez-Alvarado M. Thermal stability threshold for amyloid formation in light chain amyloidosis. Int J Mol Sci. 2013;14:22604–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Blancas-Mejia LM, Tischer A, Thompson JR, Tai J, Wang L, Auton M, et al. Kinetic control in protein folding for light chain amyloidosis and the differential effects of somatic mutations. J Mol Biol. 2014;426:347–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Blancas-Mejia LM, Martin EB, Williams A, Wall JS, Ramirez-Alvarado M. Kinetic stability and sequence/structure studies of urine-derived Bence-Jones proteins from multiple myeloma and light chain amyloidosis patients. Biophys Chem. 2017;230:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Enqvist S, Sletten K, Westermark P. Fibril protein fragmentation pattern in systemic AL-amyloidosis. J Pathol. 2009;219:473–80. [DOI] [PubMed] [Google Scholar]
- [72].Glenner GG, Cuatrecasas P, Isersky C, Bladen HA, Eanes ED. Physical and chemical properties of amyloid fibers. II. Isolation of a unique protein constituting the major component from human splenic amyloid fibril concentrates. J Histochem Cytochem. 1969;17:769–80. [DOI] [PubMed] [Google Scholar]
- [73].Vrana JA, Gamez JD, Madden BJ, Theis JD, Bergen HR, 3rd, Dogan A. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood. 2009;114:4957–9. [DOI] [PubMed] [Google Scholar]
- [74].Lavatelli F, Perlman DH, Spencer B, Prokaeva T, McComb ME, Theberge R, et al. Amyloidogenic and associated proteins in systemic amyloidosis proteome of adipose tissue. Mol Cell Proteomics. 2008;7:1570–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Yamamoto K, Yagi H, Lee YH, Kardos J, Hagihara Y, Naiki H, et al. The amyloid fibrils of the constant domain of immunoglobulin light chain. FEBS Lett. 2010;584:3348–53. [DOI] [PubMed] [Google Scholar]
- [76].Nokwe CN, Hora M, Zacharias M, Yagi H, John C, Reif B, et al. The Antibody Light-Chain Linker Is Important for Domain Stability and Amyloid Formation. J Mol Biol. 2015;427:3572–86. [DOI] [PubMed] [Google Scholar]
- [77].Klimtchuk ES, Gursky O, Patel RS, Laporte KL, Connors LH, Skinner M, et al. The critical role of the constant region in thermal stability and aggregation of amyloidogenic immunoglobulin light chain. Biochemistry. 2010;49:9848–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Kim Y, Wall JS, Meyer J, Murphy C, Randolph TW, Manning MC, et al. Thermodynamic modulation of light chain amyloid fibril formation. J Biol Chem. 2000;275:1570–4. [DOI] [PubMed] [Google Scholar]
- [79].Sikkink LA, Ramirez-Alvarado M. Biochemical and aggregation analysis of Bence Jones proteins from different light chain diseases. Amyloid. 2008;15:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Andrich K, Hegenbart U, Kimmich C, Kedia N, Bergen HR, 3rd, Schonland S, et al. Aggregation of Full-length Immunoglobulin Light Chains from Systemic Light Chain Amyloidosis (AL) Patients Is Remodeled by Epigallocatechin-3-gallate. J Biol Chem. 2017;292:2328–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Ramirez-Alvarado M Amyloid formation in light chain amyloidosis. Curr Top Med Chem. 2012;12:2523–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Blancas-Mejia LM, Hammernik J, Marin-Argany M, Ramirez-Alvarado M. Differential effects on light chain amyloid formation depend on mutations and type of glycosaminoglycans. J Biol Chem. 2015;290:4953–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Martin DJ, Ramirez-Alvarado M. Glycosaminoglycans promote fibril formation by amyloidogenic immunoglobulin light chains through a transient interaction. Biophys Chem. 2011;158:81–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Trinkaus-Randall V, Walsh MT, Steeves S, Monis G, Connors LH, Skinner M. Cellular response of cardiac fibroblasts to amyloidogenic light chains. Am J Pathol. 2005;166:197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Ren R, Hong Z, Gong H, Laporte K, Skinner M, Seldin DC, et al. Role of glycosaminoglycan sulfation in the formation of immunoglobulin light chain amyloid oligomers and fibrils. J Biol Chem. 2010;285:37672–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Wall JS, Richey T, Stuckey A, Donnell R, Macy S, Martin EB, et al. In vivo molecular imaging of peripheral amyloidosis using heparin-binding peptides. Proc Natl Acad Sci U S A. 2011;108:E586–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Foster JS, Williams AD, Macy S, Richey T, Stuckey A, Wooliver DC, et al. A Peptide-Fc Opsonin with Pan-Amyloid Reactivity. Front Immunol. 2017;8:1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Buxbaum JN. Animal models of human amyloidoses: are transgenic mice worth the time and trouble? FEBS Lett. 2009;583:2663–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Hrncic R, Wall J, Wolfenbarger DA, Murphy CL, Schell M, Weiss DT, et al. Antibody-mediated resolution of light chain-associated amyloid deposits. Am J Pathol. 2000;157:1239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].O’Nuallain B, Hrncic R, Wall JS, Weiss DT, Solomon A. Diagnostic and therapeutic potential of amyloid-reactive IgG antibodies contained in human sera. J Immunol. 2006;176:7071–8. [DOI] [PubMed] [Google Scholar]
- [91].Teng J, Turbat-Herrera EA, Herrera GA. An animal model of glomerular light-chain-associated amyloidogenesis depicts the crucial role of lysosomes. Kidney Int. 2014;86:738–46. [DOI] [PubMed] [Google Scholar]
- [92].Wall JS, Kennel SJ, Paulus M, Gregor J, Richey T, Avenell J, et al. Radioimaging of light chain amyloid with a fibril-reactive monoclonal antibody. J Nucl Med. 2006;47:2016–24. [PMC free article] [PubMed] [Google Scholar]
- [93].Ward JE, Ren R, Toraldo G, Soohoo P, Guan J, O’Hara C, et al. Doxycycline reduces fibril formation in a transgenic mouse model of AL amyloidosis. Blood. 2011;118:6610–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Brenner DA, Jain M, Pimentel DR, Wang B, Connors LH, Skinner M, et al. Human amyloidogenic light chains directly impair cardiomyocyte function through an increase in cellular oxidant stress. Circ Res. 2004;94:1008–10. [DOI] [PubMed] [Google Scholar]
- [95].Monis GF, Schultz C, Ren R, Eberhard J, Costello C, Connors L, et al. Role of endocytic inhibitory drugs on internalization of amyloidogenic light chains by cardiac fibroblasts. Am J Pathol. 2006;169:1939–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Sikkink LA, Ramirez-Alvarado M. Cytotoxicity of amyloidogenic immunoglobulin light chains in cell culture. Cell Death Dis. 2010;1:e98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Shi J, Guan J, Jiang B, Brenner DA, Del Monte F, Ward JE, et al. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38alpha MAPK pathway. Proc Natl Acad Sci U S A. 2010;107:4188–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Levinson RT, Olatoye OO, Randles EG, Howell KG, DiCostanzo AC, Ramirez-Alvarado M. Role of mutations in the cellular internalization of amyloidogenic light chains into cardiomyocytes. Sci Rep. 2013;3:1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Keeling J, Teng J, Herrera GA. AL-amyloidosis and light-chain deposition disease light chains induce divergent phenotypic transformations of human mesangial cells. Lab Invest. 2004;84:1322–38. [DOI] [PubMed] [Google Scholar]
- [100].Teng J, Russell WJ, Gu X, Cardelli J, Jones ML, Herrera GA. Different types of glomerulopathic light chains interact with mesangial cells using a common receptor but exhibit different intracellular trafficking patterns. Lab Invest. 2004;84:440–51. [DOI] [PubMed] [Google Scholar]
- [101].Teng J, Zhang PL, Russell WJ, Zheng LP, Jones ML, Herrera GA. Insights into mechanisms responsible for mesangial alterations associated with fibrogenic glomerulopathic light chains. Nephron Physiol. 2003;94:p28–38. [DOI] [PubMed] [Google Scholar]
- [102].McWilliams-Koeppen HP, Foster JS, Hackenbrack N, Ramirez-Alvarado M, Donohoe D, Williams A, et al. Light Chain Amyloid Fibrils Cause Metabolic Dysfunction in Human Cardiomyocytes. PLoS One. 2015;10:e0137716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Marin-Argany M, Lin Y, Misra P, Williams A, Wall JS, Howell KG, et al. Cell Damage in Light Chain Amyloidosis: FIBRIL INTERNALIZATION, TOXICITY AND CELL-MEDIATED SEEDING. J Biol Chem. 2016;291:19813–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Lin Y, Marin-Argany M, Dick CJ, Redhage KR, Blancas-Mejia LM, Bulur P, et al. Mesenchymal stromal cells protect human cardiomyocytes from amyloid fibril damage. Cytotherapy. 2017;19:1426–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Lavatelli F, Imperlini E, Orru S, Rognoni P, Sarnataro D, Palladini G, et al. Novel mitochondrial protein interactors of immunoglobulin light chains causing heart amyloidosis. FASEB J. 2015;29:4614–28. [DOI] [PubMed] [Google Scholar]
- [106].Diomede L, Romeo M, Rognoni P, Beeg M, Foray C, Ghibaudi E, et al. Cardiac Light Chain Amyloidosis: The Role of Metal Ions in Oxidative Stress and Mitochondrial Damage. Antioxid Redox Signal. 2017;27:567–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Diomede L, Rognoni P, Lavatelli F, Romeo M, di Fonzo A, Foray C, et al. Investigating heart-specific toxicity of amyloidogenic immunoglobulin light chains: A lesson from C. elegans. Worm. 2014;3:e965590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Diomede L, Rognoni P, Lavatelli F, Romeo M, del Favero E, Cantu L, et al. A Caenorhabditis elegans-based assay recognizes immunoglobulin light chains causing heart amyloidosis. Blood. 2014;123:3543–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Guan J, Mishra S, Qiu Y, Shi J, Trudeau K, Las G, et al. Lysosomal dysfunction and impaired autophagy underlie the pathogenesis of amyloidogenic light chain-mediated cardiotoxicity. EMBO Mol Med. 2014;6:1493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Guan J, Mishra S, Shi J, Plovie E, Qiu Y, Cao X, et al. Stanniocalcin1 is a key mediator of amyloidogenic light chain induced cardiotoxicity. Basic Res Cardiol. 2013;108:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Mishra S, Guan J, Plovie E, Seldin DC, Connors LH, Merlini G, et al. Human amyloidogenic light chain proteins result in cardiac dysfunction, cell death, and early mortality in zebrafish. Am J Physiol Heart Circ Physiol. 2013;305:H95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Blancas-Mejia LM, Ramirez-Alvarado M. Recruitment of Light Chains by Homologous and Heterologous Fibrils Shows Distinctive Kinetic and Conformational Specificity. Biochemistry. 2016;55:2967–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Martin DJ, Ramirez-Alvarado M. Comparison of amyloid fibril formation by two closely related immunoglobulin light chain variable domains. Amyloid. 2010;17:129–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Martin EB, Williams A, Wooliver C, Heidel RE, Adams S, Dunlap J, et al. Differential recruitment efficacy of patient-derived amyloidogenic and myeloma light chain proteins by synthetic fibrils-A metric for predicting amyloid propensity. PLoS One. 2017;12:e0174152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Pisitkun T, Shen RF, Knepper MA. Identification and proteomic profiling of exosomes in human urine. Proc Natl Acad Sci U S A. 2004;101:13368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Ramirez-Alvarado M, Ward CJ, Huang BQ, Gong X, Hogan MC, Madden BJ, et al. Differences in immunoglobulin light chain species found in urinary exosomes in light chain amyloidosis (Al). PLoS One. 2012;7:e38061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Ramirez-Alvarado M, Barnidge DR, Murray DL, Dispenzieri A, Marin-Argany M, Dick CJ, et al. Assessment of renal response with urinary exosomes in patients with AL amyloidosis: A proof of concept. Am J Hematol. 2017;92:536–41. [DOI] [PubMed] [Google Scholar]
- [118].Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci U S A. 2004;101:9683–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Porto-Carreiro I, Fevrier B, Paquet S, Vilette D, Raposo G. Prions and exosomes: from PrPc trafficking to PrPsc propagation. Blood Cells Mol Dis. 2005;35:143–8. [DOI] [PubMed] [Google Scholar]
- [120].Vella LJ, Sharples RA, Nisbet RM, Cappai R, Hill AF. The role of exosomes in the processing of proteins associated with neurodegenerative diseases. Eur Biophys J. 2008;37:323–32. [DOI] [PubMed] [Google Scholar]
- [121].Piehl DW, Blancas-Mejia LM, Ramirez-Alvarado M, Rienstra CM. Solid-state NMR chemical shift assignments for AL-09 VL immunoglobulin light chain fibrils. Biomol NMR Assign. 2017;11:45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Piehl DW, Blancas-Mejia LM, Wall JS, Kennel SJ, Ramirez-Alvarado M, Rienstra CM. Immunoglobulin Light Chains Form an Extensive and Highly Ordered Fibril Involving the N- and C-Termini. ACS Omega. 2017;2:712–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Murray DT, Kato M, Lin Y, Thurber KR, Hung I, McKnight SL, et al. Structure of FUS Protein Fibrils and Its Relevance to Self-Assembly and Phase Separation of Low-Complexity Domains. Cell. 2017;171:615–27 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, et al. Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat Struct Mol Biol. 2016;23:409–15. [DOI] [PMC free article] [PubMed] [Google Scholar]