

Abstract
Inositol hexakisphosphate kinases (IP6Ks) regulate a myriad of cellular processes, not only through their catalytic activity which synthesizes InsP7, a multifunctional inositol pyrophosphate signaling molecule, but also through protein-protein interactions. To further study the enzymatic function and distinguish between these different mechanisms, specific inhibitors that target IP6K catalytic activity are required. Only one IP6K inhibitor is commonly used: N2-(m-(trifluoromethyl)benzyl) N6-(p-nitrobenzyl)purine (TNP). However, TNP is compromised by weak potency, inability to distinguish between IP6K isoenzymes, off-target activities, and poor pharmacokinetic properties. Herein, we describe a new inhibitor discovery strategy, based on the high degree of structural conservation of the nucleotide binding sites of IP6Ks and protein kinases; we screened for novel IP6K2 inhibitors using a focused set of compounds with features known, or computationally predicted, to target nucleotide binding by protein kinases. We developed a time-resolved, fluorescence resonance energy transfer (TR-FRET) assay of ADP formation from ATP. Novel hit compounds for IP6K2 were identified and validated with dose-response curves and an orthogonal assay. None of these inhibitors affected another inositol pyrophosphate kinase, PPIP5K. Our screening strategy offers multiple IP6K2 inhibitors for future development and optimization. This approach will be applicable to inhibitor discovery campaigns for other inositol phosphate kinases.
Introduction
Inositol pyrophosphates, such as 5-diphosphoinositol pentakisphosphate (InsP7), regulate many cellular processes, although most attention is given to their actions at the interface of cell signaling and bioenergetic metabolism.1 InsP7 is synthesized by inositol hexakisphosphate kinases (IP6Ks); mammalian cells express three of these: types 1, 2, and 3. IP6K1 and IP6K2 are ubiquitously expressed, whereas IP6K3 expression is mainly restricted to the cerebellum and skeletal muscle.2 Genetic experiments in mice have revealed that IP6Ks have several non-overlapping functions. For example, only the IP6K1 knockout displays low body weight, low insulin levels, and male sterility3, protection from thrombotic challenge4, and lower weight gain on a high-fat diet5. These studies, and other work6–7, suggest that inhibition of IP6K1 could be of therapeutic benefit in treating diabetes, obesity and thrombosis. It has also been reported that IP6K2 promotes cancer cell migration, invasion, and tumor metastasis via inactivation of the tumor suppressor liver kinase B1 (LKB1).8 Therefore, inhibitors of IP6K2 offer promise as new cancer therapeutics.
Chemical probes that inhibit IP6Ks could also be used as research tools for functional characterization of their kinase activities, and also to distinguish this kinase-directed mechanism from separate, non-catalytic roles mediated by protein-protein interactions. Currently, only one IP6K inhibitor is in routine use: N2-(m-(trifluoromethyl)benzyl) N6-(p-nitrobenzyl)purine (TNP). However, this compound is compromised by weak (low micromolar) potency, inability to distinguish between different IP6K isoenzymes, and off-target liabilities.6
A recent study by Wormald et al.9 described how IP6K assays that monitor ATP consumption can be developed and used for compound screening campaigns. This group derived proof-of-principle of their approach using an annotated set of 1280 compounds, the Library of Pharmacologically Active Compounds (LOPAC). At least one pharmacological activity is known for each of these compounds and the main purpose of this library is to examine the performance of a high throughput assay, rather than to identify tractable inhibitors.9 However, a successful screening exercise depends upon identification of chemically tractable hit molecules. One approach to reaching this goal, in an efficient manner, lies in the curation and application of smaller, focused libraries with functionally and/or chemically related properties.10
To support our selection of focused compound sets, we describe here how we first compared the conserved core structure of an IP6K (from Entamoeba histolytica; PDB: 4O4F) with that of protein kinase A (PDB: 1L3R), and we found the nucleotide-binding sites to exhibit a substantial degree of similarity. Therefore, we reasoned that a focused screen using molecules known to have features of protein kinase inhibitors would be a potentially successful approach. Thus, we screened human IP6K2 with two focused compound sets: a 5K kinase library from The Center for Integrative Chemical Biology and Drug Discovery, University of North Carolina (UNC CICBDD)11 and the GSK Published Kinase Inhibitor Set (PKIS).12 We identified several novel hits for IP6K2, which showed specificity over PPIP5K, another inositol pyrophosphate kinase.
Materials and Methods
Protein expression and purification.
The catalytic domain of recombinant human PPIP5K213 and full length recombinant human IP6K214 were prepared as described previously. The purity of these proteins was estimated to be >90% as judged by SDS-PAGE. The purified proteins were concentrated to between 1 and 10 mg/ml and stored at −80ºC.
IP6K2 and PPIP5K TR-FRET assays for ADP-detection.
ATP-driven kinase activity was measured by detecting ADP formation from substrate phosphorylation using the Adapta™ Universal Kinase Assay (Thermo Fisher). The enzyme reaction was performed using optimized final conditions of 400 nM IP6K2, 10 μM ATP and 10 μM InsP6; and 500 nM PPIP5K, 10 μM ATP and 10 μM 5-InsP7. All assays were performed in 384-well plates in kinase reaction buffer: 50 mM HEPES pH 7.5, 0.01% Brij-35, 10 mM MgCl2, and 1 mM EGTA. Reaction conditions (additional details below) were selected such that displacement of the ADP tracer from the antibody was 70 – 80% (according to manufacturer guidelines).
Compound screening.
Compounds from a kinase-focused library of 4727 molecules (‘5K’ library) and from the Published Kinase Inhibitor Set (PKIS) of 843 molecules were screened at 10 µM and 1 µM, respectively, using a Mosquito (TTP Labtech) to dispense 50 nL compound (in DMSO) from the stock plates (1 mM for the 5K library and 0.1 mM for PKIS) in 384-well assay plates. A Multidrop Combi Reagent Dispenser (ThermoFisher) was used to dispense 2.5 µL of (2x) kinase to the assay plates and incubated 20 min at room temperature, followed by addition of 2.5 µL of (2x) ATP and substrate to a final concentration of 10 µM as stated above. The enzymatic reaction was performed for 30 min and the amount of ADP produced was detected by adding 2.5 µL of detection solution Adapta™ Eu-anti-ADP antibody, Alexa Fluor® 647 ADP tracer, and EDTA, for a final concentration of 2 nM, 12 nM, and 10 mM respectively. After an additional 30 minute equilibration period, the plate was read on EnVision (PerkinElmer) plate reader (excitation = 320 nm, emission = 665 nm and 615 nm). The HTRF signal was calculated as a ratio of the signals from the 665 nm (acceptor) and 615 nm (donor) channels.
IC50 determination.
Inhibitors (3-fold serial dilutions, 10 points) were incubated 10 to 20 min at room temperature with each kinase prior to addition of substrates at the concentrations and the enzymatic reaction was performed as described above. Percent inhibition was calculated on a scale of 0% (i.e., activity with DMSO vehicle only) to 100% (no enzyme added) using full column controls on each plate. The interquartile mean of 16 wells of each control was used per plate. ADP titration curves were routinely run on all compound screening and IC50 plates to assure that the enzyme assays produced near 20% maximum ATP turnover.
TNP Synthesis.
A suspension of 6-chloro-2-fluoro-9H-purine (200 mg, 1.0 mmol) in DMF (2.0 mL) was added 4-nitro benzylamine (300 mg, 1.0 mmol) and triethylamine (200 mg, 2.0 mmol). The reaction mixture was heated at 90°C under microwave irradiation for 20 min, quenched with water, and extracted with ethyl acetate. The organic layer was concentrated. The crude product was washed with 10 mL of MeOH and filtered to yield the desired product 2-fluoro-N-(4-nitrobenzyl)-9H-purin-6-amine (150 mg, 50%) which was used without further purification. A suspension of 2-fluoro-N-(4-nitrobenzyl)-9H-purin-6-amine (350 mg, 1.2 mmol) in (3-(trifluoromethyl)phenyl)methanamine (1.5 mL) was heated at 155°C for 2 hr. Silica was added to the reaction mixture. The mixture was loaded directly to an ISCO column and purified using a mixture of hexanes and ethyl acetate followed by another purification on pre-HPLC to provide the desired product, TNP (150 mg, 28%). 1H NMR (400 MHz, CD3OD) δ 8.11 (s, 1H), 8.08 – 7.98 (m, 2H), 7.55 – 7.34 (m, 6H), 4.84 (s, 2H), 4.68 (s, 2H).
HPLC assay of IP6K2 activity.
The base buffer for assaying IP6K2 activity was: 100 mM KCl, 16.5 mM NaCl, 20 mM HEPES pH 7.2, 8 mM MgCl2, 1 mM EDTA and 10 μM [3H]InsP6 (15,000 dpm). Two alternative concentrations of ATP were added, either 5 mM or 50 μM, with the quantity of IP6K2 adjusted to 30 nM or 100 nM, respectively. Assays were run for 15 min at 370C, and then quenched, neutralized, and assayed by strong anion exchange HPLC as previously described.14
Results and Discussion
A recent report describes the development of a high throughput bioluminescence assay (ADP-Glo; Promega) for IP6K activity.9 Here, we report the development of an alternative enzymatic assay for IP6K2 and PPIP5K. We employed a TR-FRET readout (Adapta™; Thermo Fisher) that detects ADP resulting from phosphate transfer to substrate from ATP; ADP is detected with a europium-labeled anti-ADP antibody which displaces an acceptor-labeled ADP (Figure 1A). The TR-FRET format protects against false positives due to the time-resolved nature of the fluorescence readout. In addition, the ratiometric readout for TR-FRET (acceptor/donor fluorescence) enhances assay robustness. Therefore, the TR-FRET assay is particularly well suited for screening campaigns and compound characterization.15
Figure 1.
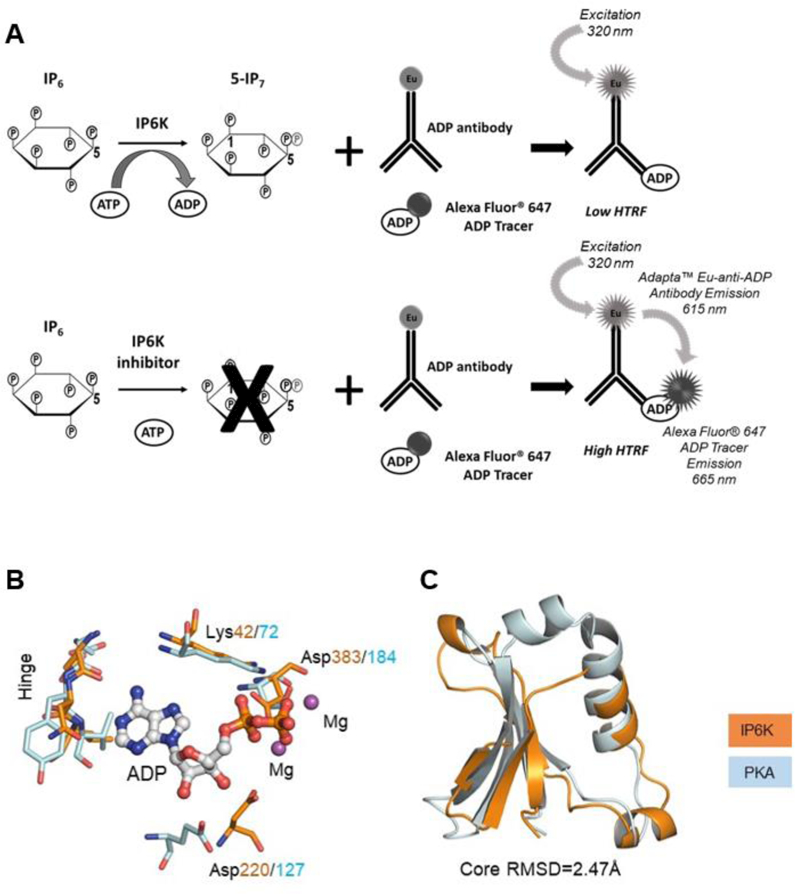
The IP6K2 screening strategy. (A) As shown in the schematic, IP6K2 was screened using the Adapta™ Universal Kinase Assay (Thermo Fisher). ADP is detected with a europium-labeled anti-ADP antibody which displaces an acceptor-labeled ADP. Inhibition of IP6K2 blocks ADP production and results in a high TR-FRET signal. (B) Superimposition19 of spatially conserved elements within the active sites of human PKA (PDB Accession Code 1L3R) and a model of IP6K2, in turn created by homology modeling20 using EhIP6KA (PDB Accession Code 4O4F) as the template. IP6K2 is denoted as orange stick; oxygen is colored red and nitrogen is colored blue. PKA is denoted as cyan stick. (C) Secondary structural superimposition19 of the N-lobe of a model of human IP6K2 (blue ribbon; residues 32–76 and 197–209) with human PKA (orange ribbon; residues 56–123).
We paid particular attention to the choice of a compound library for screening. Our decision was based on the following structural analysis: Using the structure of EhIP6K as a template, we constructed a homology model14, of the ATP-binding site of human IP6K2 which we compared with the corresponding region of a prototypical protein kinase (Protein Kinase A, PKA; Fig. 1B, C). There are several spatially conserved elements in these two proteins, including a hinge region between the N- and C-lobes of IP6K2 that corresponds to the hinge region in PKA, which figures prominently during the rational design of protein kinase inhibitors.16 The N-terminal lobes of the nucleotide-binding regions of IP6K2 and PKA are very similar (RMSD = 2.47Å). Therefore, we focused our screening campaign on compound sets directed toward kinase inhibitors: a protein kinase focused library (∼5000 compounds) from the UNC CICBDD11 and the GSK Published Kinase Inhibitor Set (PKIS; ∼800 compounds).12 The PKIS set contains high quality and previously profiled kinase inhibitors. Single shot kinase profiles using biochemical assays are publically available for compounds in this set. For the UNC CICBDD kinase directed set, compounds were computationally selected based on structural similarity to known kinase inhibitors as well as compounds having a hinge-binding motif (e.g. heterocycles with a high likelihood to bind the kinase hinge region conserved in nearly every kinase-small molecule X-ray structure).16
Our screening assay was designed to preferentially identify substrate competitive inhibitors by using substrate concentrations significantly lower than the Km value. While previous studies suggest the IPK Km for ATP is near 1 mM17, we chose an ATP concentration of 10 µM ATP. This low concentration also enabled us to reliably quantitate the amount of ADP formed in the enzyme reaction, given the affinity of the anti-ADP antibody and its degree of selectivity over ATP. We chose conditions that would allow for a maximum of approximately 20% conversion of ATP to ADP during the time course of the reaction. This low turnover assured that the reaction remained in the linear, initial velocity phase (data not shown).
We screened both IP6K2 and PPIP5K with the protein kinase focused sets and overall the screen performed well with an average signal/background ∼ 2 and average Z-prime value > 0.8 (Figure 2). No hits were identified for PPIP5K. For IP6K2, of the 5570 compounds screened, 96 compounds yielded >50% inhibition (1.7% hit rate) (Figure 2). We discarded 22 of these compounds because they altered europium donor emission by more than two standard deviations from the mean. That is, approximately 0.4% of the hits in the primary screen were removed due to quenching of donor fluorescence alone. The remaining 74 hits were retested with dose-response curves (see below).
Figure 2.
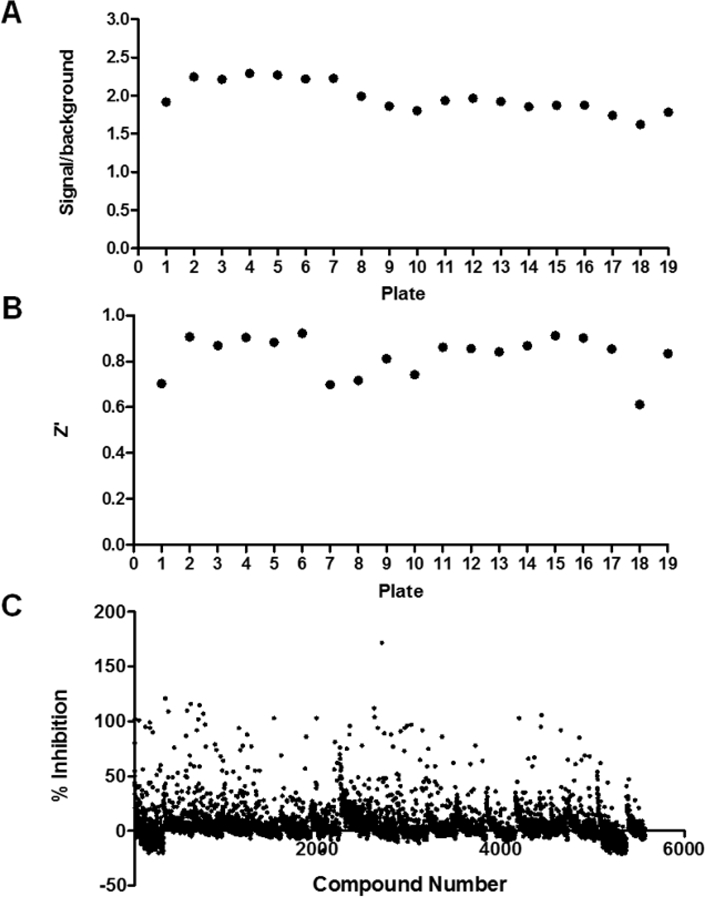
Assay performance for the IP6K2 screening campaign. The kinase focused library from the UNC CICBDD (5K) (plates 1–16) and the GSK Published Kinase Inhibitor Set (PKIS) (plates 17–19) were screened using the Adapta™ Universal Kinase Assay (Thermo Fisher). (A) Signal to background for each screening plate. Ratio represents the signal with substrates without enzyme, divided by the signal obtained with enzyme and substrates. Average signal/background = 1.98 ± 0.20. (B) Z’ for each screening plate. Average Z’ = 0.83 ± 0.09. (C) Scatter plot showing the data from the IP6K2 screening campaign. Single shot inhibition data are shown for compounds from the UNC CICBDD kinase-focused library that were screened at 10 µM and compounds from PKIS library that were screened at 1 µM.
We also evaluated the IP6K inhibitor, TNP, as a positive control during screening and dose-response follow up. It is worth noting that we found TNP to have an IC50 value of 7.36 µM for IP6K2 (Figure 3A). This is significantly weaker than the IC50 value of 0.43 µM (Ki value of 0.24 µM) previously reported for IP6K1.18 To confirm our result, we resynthesized TNP to produce a highly pure compound and obtained the same IC50 value as commercially obtained material. It is noteworthy that our IC50 value for TNP is close to that previously reported using the ADP-Glo Max assay.9
Figure 3.

Representative dose-response curves for inhibition of IP6K2 by (A) TNP (B) UNC10102221, (C) UNC10104261, (D) UNC10105760 and (E) UNC10225257. (F) Average IC50 values, standard deviation and number of experiments. The Adapta® Universal Kinase Assay (Thermo Fisher) was used to measure the amount of ADP produced by enzyme activity. Percent inhibition was calculated using controls with substrate alone (0% inhibition) and no enzyme (100% inhibition). Curve fits were performed using four parameter non-linear regression (GraphPad Prism).
We found that 46 of our initial hits were validated in secondary screening (i.e, IC50 < 5 µM). The dose-response curves for four illustrative hit compounds, plus TNP, are shown in Figure 3. One of these, UNC10225257 from the PKIS compound library, is a quite promiscuous inhibitor that targets >80 kinases with >90% inhibition at 1 µM12. Thus, it is unlikely this would be an optimum compound for further development. Reassuringly, none of the confirmed hits for IP6K2 inhibited PPIP5K. Generally, we did not observe significant structural similarities amongst the top 46 hits (other than their containing the kinase ‘hinge-binding’ motif16). Having a number of distinct chemotypes for future follow-up work is an encouraging outcome from our screening efforts.
To further validate our hits for IP6K2, we either repurified the compound (UNC10102221) or remade fresh solutions from solid stocks (UNC10104261 and UNC10105760) and performed an orthogonal assay; we used HPLC to monitor metabolism of radiolabeled InsP6 substrate. This is a laborious and time-intensive assay, so we only provide data with two inhibitor concentrations, 1 µM and 10 µM, and two ATP concentrations, 50 μM and 5 mM. As shown in Figure 4A and 4B, all three compounds were validated to yield significant inhibition in this assay. These hit compounds have chemically tractable features suitable for a future structure-guided medicinal chemistry optimization effort to improve potency and selectivity, particularly with regards to developing isoform-specific IP6K inhibitors. The latter may be a more challenging goal; the IC50 values for our top three IP6K2 inhibitors (Figure 3) are very similar to IC50 values we obtained in further experiments with IP6K1: UNC10102221, 1.3 μM; UNC10104261, 1.7 μM; UNC10105760, 0.5 μM. Further, we obtained selectivity data using a panel of 30 protein kinases that broadly represents the kinome, using a single compound concentration of 10 µM (Figure 4C). As shown, UNC10104261 (IC50 = 1.1 µM for IP6K2) only hits one protein kinase, ALK1, with inhibition >80% at the 10 µM dose. UNC10225257, a compound from the PKIS library, is broadly promiscuous which confirms publically available PKIS data.
Figure 4.
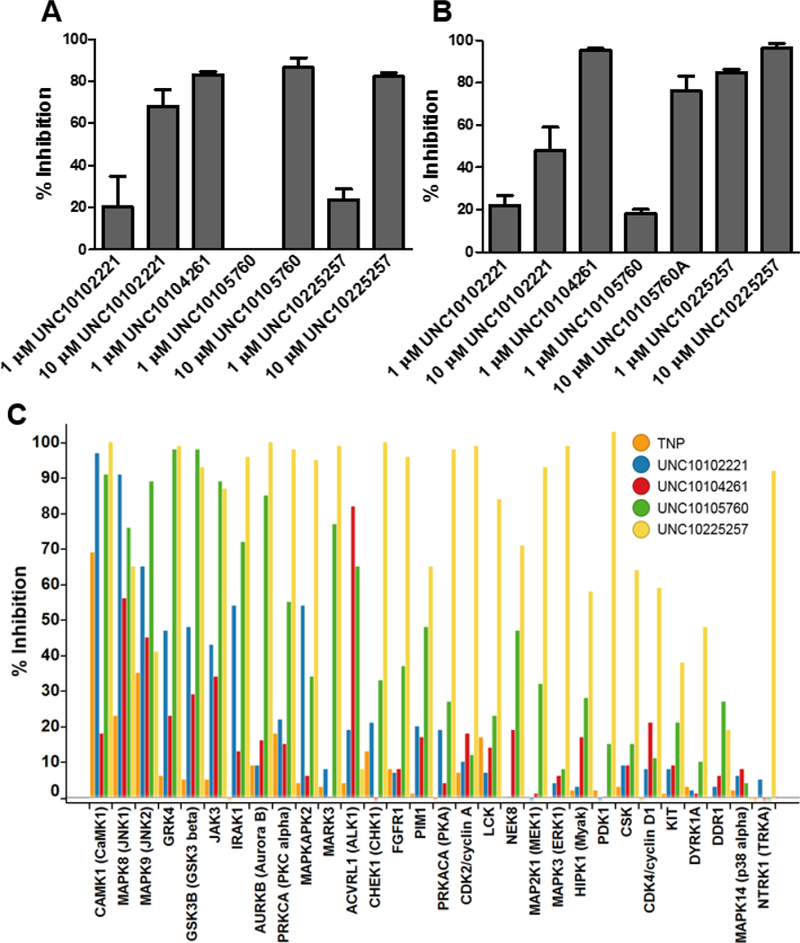
Evaluation of selected IP6K2 inhibitors by HPLC and protein kinase selectivity assays. Assays were performed as described in the Methods section using either (A) 5 mM ATP or (B) 50 μM ATP. (C) Selectivity testing using a panel of protein kinases that broadly cover the kinome was performed using the Thermo Fisher Scientific SelectScreen Kinase Profiling Service; compounds were screened using a single concentration of 10 µM with either the Adapta (ATP at Km), LanthaScreen, or ZLYTE (ATP at Km) assay format. The figure was generated using TIBCO Spotfire®.
A key development in this study has been our structure-based evidence that IP6K2 may be targeted by compounds resembling protein kinase inhibitors (Figure 1). This has allowed us to conduct an efficient screening campaign; we have used relatively small, focused libraries containing molecules that have features that are either known or computationally predicted to target a protein kinase nucleotide-binding site. This new approach is validated by our identification of several novel inhibitors of IP6K2 with IC50 values in the micromolar range. These results also suggest that commercially available kinase profiling panels, such as those supplied by CarnaBio, DiscoveRx, Eurofins, ReactionBiology, and Thermo SelectScreen, should include IP6Ks and perhaps other inositol phosphate kinases. Although most of these kinase panel screening services do contain inositol lipid kinases (such as PI3K, PI4K, and PIP5K1), these are a functionally and structurally different class of proteins from the inositol phosphate kinases. Overall, our targeted library approach could also prove useful for inhibitor discovery campaigns against other inositol phosphate kinases.
Acknowledgement
We thank Daowei Huang for help confirming TNP via NMR.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the National Institutes of Health under grant R01 DK101645.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Shears SB, Intimate connections: Inositol pyrophosphates at the interface of metabolic regulation and cell signaling. J Cell Physiol 2018, 233 (3), 1897–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thota SG; Bhandari R, The emerging roles of inositol pyrophosphates in eukaryotic cell physiology. J Biosci 2015, 40 (3), 593–605. [DOI] [PubMed] [Google Scholar]
- 3.Bhandari R; Juluri KR; Resnick AC; et al. Gene deletion of inositol hexakisphosphate kinase 1 reveals inositol pyrophosphate regulation of insulin secretion, growth, and spermiogenesis. Proc Natl Acad Sci U S A 2008, 105 (7), 2349–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghosh S; Shukla D; Suman K; et al. Inositol hexakisphosphate kinase 1 maintains hemostasis in mice by regulating platelet polyphosphate levels. Blood 2013, 122 (8), 1478–86. [DOI] [PubMed] [Google Scholar]
- 5.Chakraborty A; Koldobskiy MA; Bello NT; et al. Inositol pyrophosphates inhibit Akt signaling, thereby regulating insulin sensitivity and weight gain. Cell 2010, 143 (6), 897–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghoshal S; Zhu Q; Asteian A; et al. TNP [N2-(m-Trifluorobenzyl), N6-(p-nitrobenzyl)purine] ameliorates diet induced obesity and insulin resistance via inhibition of the IP6K1 pathway. Mol Metab 2016, 5 (10), 903–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu Q; Ghoshal S; Rodrigues A; et al. Adipocyte-specific deletion of Ip6k1 reduces diet-induced obesity by enhancing AMPK-mediated thermogenesis. J Clin Invest 2016, 126 (11), 4273–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rao F; Xu J; Fu C; et al. Inositol pyrophosphates promote tumor growth and metastasis by antagonizing liver kinase B1. Proc Natl Acad Sci U S A 2015, 112 (6), 1773–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wormald M; Liao G; Kimos M; et al. Development of a homogenous high-throughput assay for inositol hexakisphosphate kinase 1 activity. PLoS One 2017, 12 (11), e0188852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barnash KD; James LI; Frye SV, Target class drug discovery. Nat Chem Biol 2017, 13 (10), 1053–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hutti JE; Porter MA; Cheely AW; et al. Development of a high-throughput assay for identifying inhibitors of TBK1 and IKKepsilon. PLoS One 2012, 7 (7), e41494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elkins JM; Fedele V; Szklarz M; et al. Comprehensive characterization of the Published Kinase Inhibitor Set. Nat Biotechnol 2016, 34 (1), 95–103. [DOI] [PubMed] [Google Scholar]
- 13.Wang H; Falck JR; Hall TM; et al. Structural basis for an inositol pyrophosphate kinase surmounting phosphate crowding. Nat Chem Biol 2011, 8 (1), 111–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H; DeRose EF; London RE; et al. IP6K structure and the molecular determinants of catalytic specificity in an inositol phosphate kinase family. Nat Commun 2014, 5, 4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Degorce F; Card A; Soh S; et al. HTRF: A technology tailored for drug discovery - a review of theoretical aspects and recent applications. Curr Chem Genomics 2009, 3, 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xing L; Klug-Mcleod J; Rai B; et al. Kinase hinge binding scaffolds and their hydrogen bond patterns. Bioorg Med Chem 2015, 23 (19), 6520–7. [DOI] [PubMed] [Google Scholar]
- 17.Voglmaier SM; Bembenek ME; Kaplin AI; et al. Purified inositol hexakisphosphate kinase is an ATP synthase: diphosphoinositol pentakisphosphate as a high-energy phosphate donor. Proc Natl Acad Sci U S A 1996, 93 (9), 4305–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Padmanabhan U; Dollins DE; Fridy PC; et al. Characterization of a selective inhibitor of inositol hexakisphosphate kinases: use in defining biological roles and metabolic relationships of inositol pyrophosphates. J Biol Chem 2009, 284 (16), 10571–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emsley P; Cowtan K, Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004, 60 (Pt 12 Pt 1), 2126–32. [DOI] [PubMed] [Google Scholar]
- 20.Biasini M; Bienert S; Waterhouse A; et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 2014, 42 (Web Server issue), W252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]