

Abstract
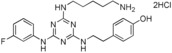
Low‐molecular‐weight synthetic molecules 1 with the general 2‐(fluorophenylamino)‐4,6‐disubstituted 1,3,5‐triazine structure and showing anti‐inflammatory and anticancer activities were explored. Structure–activity relationship studies demonstrated the importance of the aminopentyl chain, the 3‐ or 4‐fluorophenylaniline component, and the presence of at least one substituent, such as a tyramine moiety, attached directly to the triazine ring as essential for good activity. These compounds, represented by leads 4‐{2‐[4‐(5‐Aminopentylamino)‐6‐(3‐fluorophenylamino)‐1,3,5‐triazin‐2‐ylamino]ethyl}phenol (6) and 4‐{2‐[4‐(5‐Aminopentylamino)‐6‐(4‐fluorophenylamino)‐1,3,5‐triazin‐2‐ylamino]ethyl}phenol (10), displayed moderate and significant in vitro and in vivo dual activities, respectively, and address the molecular link between inflammation and cancer. Compound 10 demonstrated significant antitumor efficacy upon administration by the oral and intravenous routes in several animal models. This class of triazine compounds is new, safe, and nontoxic and offers a novel approach to the treatment of inflammation and cancer.
Keywords: autoimmune diseases, cancer, inflammation, structure–activity relationships, triazines
1. Introduction
Cancer is the second leading cause of death in the USA and many European countries.1 Whereas breast cancer is among the five most common types of cancers, the five‐year survival rate is one of the lowest of all cancers.2, 3, 4 Also, pancreatic cancer is currently one of the deadliest of the solid malignancies,5 and surgery remains the only option for cure.6 Another example of major cancer is melanoma.7 A need, therefore, exists for the discovery of novel compounds that are more effective but less toxic for the treatment of these cancers.
It has long been recognized that inflammation is related to cancer, and strong correlation between the presence of inflammation and the development of precancerous lesions has been established.8 One novel approach to the treatment of cancer lies in the discovery of new compounds with a new mechanism of action that are efficacious in reducing tumor size and/or the spread of metastasis and that can also reduce inflammation. These compounds, which simultaneously exhibit significant anticancer and anti‐inflammatory properties, offer a potential two‐pronged approach that targets both genetically unstable tumor cells (high mutation rate and subsequent resistance to chemotherapy) and genetically normal cells present in inflamed tissue. This two‐pronged approach to the treatment of cancer is made more compelling by the increasing awareness that a link exists between chronic inflammation and the subsequent development of cancer.9 It has been shown that chronic inflammation is associated with the development of numerous human cancers.10 For example, markers of a systemic inflammatory response have prognostic significance in advanced, inoperable pancreatic cancer,11 and inflammatory processes have emerged as key mediators of breast cancer development and progression.12 Inflammatory cytokines such as tumor necrosis factor (TNF‐α),13 IL‐1, IL‐6,14 and MCP‐115 have been shown to be involved in all stages of the malignant process. In a breast,16 prostate,17 and melanoma18 cancer study, both serum TNF‐α and IL‐6 correlated with the extent of the disease. Other inflammatory mediators, such as prostaglandins and leukotrienes, have also been reported to play a role in the development and progression of several cancers, including breast,19 melanoma,20 and pancreatic21 cancers.
1,3,5‐Triazine compounds have been studied extensively and are the subject of many reviews.22 The triazine scaffold has been exploited for the design of biologically relevant molecules with broad biomedical value as therapeutics.23 For example, these compounds possess potent antiprotozoa,24 antiviral,25 fungicidal,22 insecticidal, bactericidal,26 herbicidal,27 antimicrobial,28 and antimalarial29 activities. In addition, trisubstituted triazines have been shown to display protein A mimetic properties for the treatment of autoimmune diseases.30 As part of our autoimmune diseases program, small molecules were examined as potential anticancer agents. A recent review describes the versatile bioactivities of 1,3,5‐triazine derivatives as potential antitumor compounds.31 The reasons for selecting the 1,3,5‐triazine scaffold were threefold: 1) 1,3,5‐triazines are monocyclic, symmetrical molecules and have been utilized as promising scaffolds for their versatile biological potential;32 2) the presence of three nitrogen atoms in the 1,3,5‐triazine core can inherently impart polarity to the whole molecule, and the partition coefficient (c log P) values of the designed compounds can be about 2 or lower; 3) trifunctionalized 1,3,5‐triazines are ideal modular scaffolds for generating libraries. These molecules may demonstrate remarkable pharmacokinetics and oral bioavailability with additional modifications. In this paper, the synthesis and anticancer and anti‐inflammatory activities of trisubstituted triazines, as exemplified by the general structure 1, are reported.
2. Results and Discussion
As part of an effort towards the discovery of novel therapeutics for the treatment of autoimmune diseases, a search was initiated to discover small molecules that possess anti‐inflammatory activity. It was reported that fluorophenylaminotriazine derivatives possess anti‐inflammatory activity (kinase inhibitors,33 EGFR inhibitors,34 and glycosidase modulators35). Considering that we synthesized and tested in‐house many fluorophenylamino‐4,6‐disubstituted 1,3,5‐triazines28 as protein A mimetics and as antimicrobial agents, a number of these compounds were selected and screened on the basis of general structure 1.
The starting material for these compounds was cyanuric chloride. This is an inexpensive commercially available reagent, which makes its use attractive. The ease of displacement of the chlorine atoms in cyanuric chloride by various nucleophiles enhances the utility of this reagent for the preparation of mono‐, di‐, and trisubstituted 1,3,5‐triazines at controlled temperatures.36 The general synthetic sequence for the preparation of triazine compounds is outlined in Scheme 1. It illustrates the route employed for the preparation of trisubstituted triazine 5, for which the key intermediate was dichlorotriazine 2. Compound 5 was prepared by two different routes. The first route involved the reaction of cyanuric chloride with fluoroaniline at −10 °C in the presence of sodium bicarbonate to give dichlorotriazine intermediate 2. This reaction proceeded in excellent yield (>95 %) and was general for different aniline derivatives.30, 37 Aminoalkylamines were then added in the presence of a base to afford triazine 3 in high yield (90 %). This compound was then treated with different aryl‐ and aralkylamines to give products 5. Route 2 demonstrates the preparation of dichlorotriazine intermediate 2 as in route 1, followed first by the reaction with aryl‐ and aralkylamines to give 4 and then by the addition of aminoalkylamines at 130 °C for 10 min by using microwave irradiation to afford compound 5. The last step was the removal of the protecting groups.
Scheme 1.
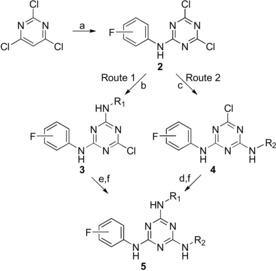
For intermediates 3–5, NHR1=4‐aminobutylamino or 5‐aminopentylamino; NHR2=substituted anilino or phenethylamino. Reagents and conditions: a) fluoroaniline, acetone/water, −10 °C to RT; b) 4‐(tert‐butoxycarbonylamino)butylamine or 5‐(tert‐butoxycarbonylamino)pentylamine, NaHCO3/H2O/THF/acetone, RT; c) R2NH2 (substituted anilino or phenethylamino); NaHCO3, acetone/H2O; d) 4‐aminobutylamine or 5‐aminopentylamine, THF/MeOH, 130 °C, 10 min, microwave; e) 4‐R‐phenylene‐(CH2)nNH2, Et3N, THF, 65 °C; f) removal of the protecting group (if applicable).
These compounds were evaluated for their anti‐inflammatory activity by using inhibition of TNF‐α production by lipopolysaccharide (LPS)‐stimulated J774A.1 cells (murine macrophages). A total of 15 compounds were chosen upon which to define a structure–activity relationship for a 2‐(3‐ or 4‐fluorophenylamino)‐4,6‐disubstituted 1,3,5‐triazine scaffold. In general, the analogues varied in their aminoalkyl chains [(CH2)mNH2, m=4 or 5] and tyramine units [4‐HOC6H4(CH2)nNH, 4‐H2NC6H4(CH2)nNH, 4‐H2NCOC6H4(CH2)nNH, 4‐H2NO2SC6H4(CH2)nNH, 4‐(CH3)2HNO2SC6H4(CH2)nNH, n=0–2], and the third group was held constant as a 3‐ or 4‐fluorophenylamine group. Table 1 illustrates the variation of these substituents and demonstrates in vitro the effect of analogues of 1 on tumor necrosis factor (TNF‐α) production, as measured by ELISA (enzyme‐linked immunosorbent assay) by using J774A.1 cells stimulated by LPS. J774A.1 cells were cultured in the presence or absence of LPS and the compound. The cells were pretreated with the compounds 1 h prior to LPS stimulation. The supernatants were collected to determine the concentration of TNF‐α by ELISA. The data were analyzed, and the concentration of compound that inhibited 50 % of TNF‐α production (IC50) was calculated. Prior work indicated the importance of the length of the carbon chain (n‐pentyl) between the amine groups. In fact, a decrease in the length of the chain by three carbon atoms (as in compound 12) or by one carbon atom (as in compounds 17 and 23) reduced the activity. However, homologation of the alkyl chain with a one‐carbon spacer chain length (as in compound 24) resulted in some activity but also some toxicity. Similarly, rigidifying the six‐carbon chain and keeping the terminal amine function (as in compound 25) gave some toxicity. However, introducing a heteroatom in the chain (as in compound 21) or increasing the rigidity of the chain by forming a ring with the primary amine function (as in compound 22) led to a drop in activity. It was observed that inhibition of TNF‐α was obtained with triazine compounds containing a tyramine moiety, for example, compounds 6, 10, and 11. However, substitution of the alkyl chain of the tyramine unit with a hydroxy group, as in compound 18, or a carboxylate group, as in compound 19, resulted in a loss of activity. Changing the tyramine moiety to an arylsulfonamide unit, as in 16, or a benzylamine, as in 20, also resulted in a loss of activity. Similarly, compounds 13, 9, and 8, in which the hydroxy group of the tyramine unit was replaced with an amide group, an amine, and a N,N‐dimethylsulfonamide, displayed lower activity. In contrast, corresponding carboxylic ester 14 and sulfonamides 7 and 15 showed weaker activity than compounds 6 and 10. Also, the correct acidity of the phenol group in the tyramine unit was important for the activity of the triazine compounds. For example, adding an ortho electron‐donating substituent such as a methoxy group to give compound 26 made the phenol less acidic and not active. In contrast, adding an ortho electron‐withdrawing substituent such as a chlorine atom to give compound 27 resulted in some activity but also some toxicity. In general, compound 10 in which the triazine ring is substituted with a 4‐fluoroaniline, tyramine, and a pentylamine chain resulted in good activity relative to that observed for corresponding 3‐fluoroanilines 6 and 11.
Table 1.
Effect of compounds on the inhibition of TNF‐α released by LPS induction from J774A.1 cells.
| Compound | Structure | TNF‐α inhibition IC50 [μm] |
|---|---|---|
| 6 |
|
29 |
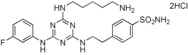
| 7 |
|
50 |
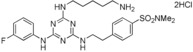
| 8 |
|
>90 |
| 9 |
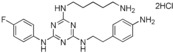
|
>90 |
| 10 |
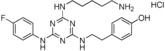
|
13 |
| 11 |
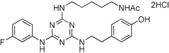
|
33 |
| 12 |
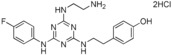
|
>90 |
| 13 |
|
>90 |
| 14 |
|
37 |
| 15 |
|
>90 |
| 16 |
|
>90 |
| 17 |
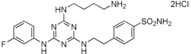
|
>90 |
| 18 |
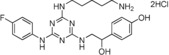
|
>90 |
| 19 |
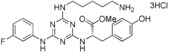
|
40 |
| 20 |
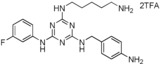
|
>90 |
| 21 |
|
>90 |
| 22 |
|
>90 |
| 23 |
|
>90 |
| 24 |
|
32[a] |
| 25 |
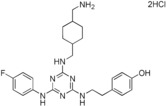
|
26[a] |
| 26 |
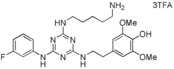
|
>90 |
| 27 |
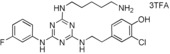
|
29[a] |
[a] Compound showed cytotoxicity at this concentration.
On the basis of the above results, three triazine compounds demonstrated inhibition of TNF‐α production. Compounds 6 and 7 showed moderate inhibition, whereas triazine 10 gave good TNF‐α inhibition. To confirm the anti‐inflammatory activity of triazine derivative 10, we studied the in vitro effect of this compound on the production of prostaglandin E2 (PGE2) in LPS‐stimulated J774A.1 cells. The results are shown in Figure 1. LPS‐stimulated J774A.1 cells produce PGE2, a bioactive lipid associated with inflammation and cancer. Compound 10 was demonstrated to inhibit PGE2 production in stimulated J774A.1 cells with an IC50 of 1.82 μm. It was next demonstrated that the in vitro activity of 7 and 10 would translate into moderate and significant in vivo activity, respectively, in a relevant model of inflammation. Therefore, compounds 7 and 10 were assessed in an LPS‐induced inflammation air‐pouch model in rats. This animal model has widely been used to study the anti‐inflammatory activity of test compounds in efficacy studies. Upon injection with LPS, the air‐pouch produces an inflammatory response characterized by the infiltration of inflammatory cells and the production of inflammation factors, such as TNF‐α and PGE2.
Figure 1.
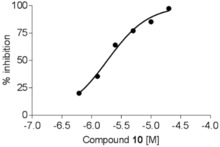
Effect of compound 10 on the inhibition of PGE2 released by LPS induction from J774A.1 cells.
Figure 2 represents the effect of the intravenous administration of the two compounds on TNF‐α production induced by LPS (2 h after induction) in the air‐pouch rat model. Compound 10 significantly inhibited TNF‐α production induced by LPS. As expected and on the basis of the above results, compound 7 had some effect on the concentration of TNF‐α in the exudates 2 h post‐LPS induction. Also, similar effects were obtained by intravenous administration of compounds 7 and 10 on TNF‐α production induced by LPS (12 h) in the air‐pouch rat model (Figure 3).
Figure 2.
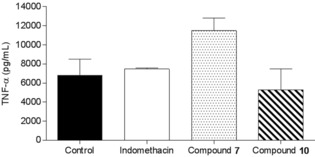
Effect of the intravenous administration of compound 7 or 10 on TNF‐α production induced by LPS (2 h) in an air‐pouch rat model.
Figure 3.
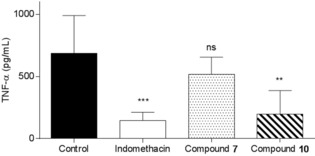
Effect of the intravenous administration of compound 7 or 10 on TNF‐α production induced by LPS (12 h) in an air‐pouch rat model. **: p<0.01, ***: p<0.001: ns: not significant.
Another experiment was performed to confirm the anti‐inflammatory activity of compound 10 compared to that of compound 7. Figure 4 represents the effect of the intravenous administration of compound 7 or 10 on PGE2 production induced by LPS (12 h after induction) in an air‐pouch rat model. Compound 10 and indomethacin significantly inhibited PGE2 production induced by LPS. However, compound 7 demonstrated a weak and nonsignificant effect on PGE2 production.
Figure 4.
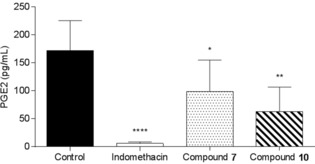
Effect of the intravenous administration of compound 7 and 10 on PGE2 production induced by LPS (12 h) in an air‐pouch rat model. *: p<0.05, **: p<0.01: ****: p<0.0001.
The next step was to examine the anticancer activity of the two best compounds, 6 and the lead 10, in in vitro and in vivo models. Cell proliferation, enhanced cell motility, cell adhesion, proteolytic degradation of the extracellular matrix, and cell migration are inter‐related processes that are responsible for the invasion and metastasis of cancer. In prostate cancer, androgen independence and bone metastasis are lethal complications in patients. The first in vitro experiment was to evaluate the effect of compound 10 on PC‐3 prostate cancer cell line proliferation by using DNA synthesis by 3H‐thymidine incorporation and the cell cycle. This compound reduced PC‐3 cell proliferation with an IC50 of 43.3 μm at 24 h (Figure 5) and 72 h but exhibited no effect on the cell cycle below 20 μm. We demonstrated, from cell‐cycle experiments (Figure 5), that there was cell‐cycle arrest in the G0/G1 phase. No increase in the sub G0/G1 phase was observed, and hence, no apoptosis occurred at compound concentrations of 40 and 80 μm. Therefore, compound 10 at a concentration of 10 μm was used for the remaining in vitro experiments. In a second experiment, it was desired to demonstrate the effect of the compounds on epidermal growth factor (EGF)‐induced migration of the PC‐3 prostate cancer cell line in an in vitro scratch wound healing assay. A migration assay was used to assess cell mobility in two dimensions. Confluent cells were quiesced by starvation and mitomycin C treatment to prevent the confounding issue of cell proliferation. Cells were incubated in the presence or absence of EGF and 6 or 10 for 24 h. Photographs were taken at 24 h. Figure 6 represents the effects of EGF and compounds 6 and 10 on PC‐3 cell migration or invasion. EGF promotes the migration or invasion of PC‐3 cells treated with mitomycin compared to a control (without growth factor). The addition of different concentrations of 10 to the cell culture resulted in inhibition of the EGF‐induced PC‐3 migration or invasion (Figure 6). However, as expected, compound 6 performed less well than compound 10 (Figure 6). The addition of different concentrations of compound 6 to the cell culture produced partial inhibition of the EGF‐induced PC‐3 migration or invasion after culture for 24 h.
Figure 5.
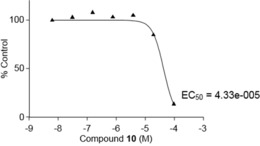
Cytotoxicity effect of compound 10 on the PC‐3 cell line (24 h). EC50=median effective concentration.
Figure 6.
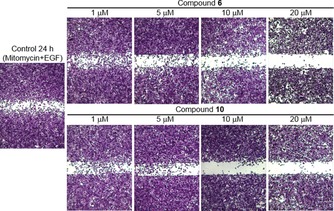
Effect of compounds 6 and 10 on EGF‐induced PC‐3 cell migration or invasion (typical experiment of three replicates).
Compound 10 showed in vitro activity on PC‐3 prostate cancer, which is a model that involves inflammation. The next step was to determine if this activity translated into anticancer/anti‐inflammatory mouse models. The first experiment was to study the antitumor effects of compound 10 on primary tumor P815 cells. This cell line has been used extensively as a cancer model to establish a relationship between the tumor‐induced local, regional, and systemic increase of proinflammatory mediators and progression of tumors in vivo.38 Figure 7 shows the effect of the oral administration (PO) of triazine derivative 10 (50 mg kg−1) and acetylsalicylic acid (positive control, 50 mg kg−1) on primary tumor P815 cells. Compound 10 induced a significant reduction T/C (mean relative tumor volume of treated group/mean relative tumor volume of control group) between 40 and 50 % of tumor growth. Furthermore, the efficacy of this compound was comparable to that of the gold standard, soluble acetylsalicylic acid (lysine salt).
Figure 7.
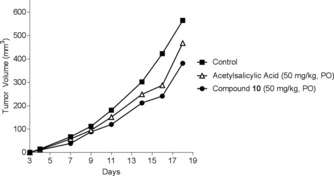
Antitumor efficacy of compound 10 and acetylsalicylic acid on P815 primary tumor.
To validate this observation, another in vivo experiment was performed to demonstrate the effect of the oral administration of compound 10 (25 mg kg−1) on primary melanoma tumor B16F10 cells. Triazine 10 induced a significant reduction (T/C<40 %, p=0.001) in the tumor volume compared to the control, Cytoxan (cyclophosphamide, 100 mg kg−1) (Figure 8).
Figure 8.
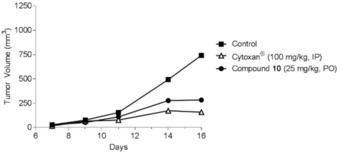
Antitumor efficacy of compound 10 (25 mg kg−1) and Cytoxan (cyclophosphamide, 100 mg kg−1) on B16F10 primary tumor.
Also, additional experiments were undertaken with other types of cancer. Figure 9 shows the antitumor efficacy of compound 10 on pancreatic cancer (PAN02 primary tumor), which is one of the most malignant tumors of the gastrointestinal tract. To evaluate long‐term tumor growth, the study lasted 35 days. The syngeneic tumor PAN02 cells were grown in RPMI‐1640 with 10 % fetal bovine serum (FBS). At day 0, 50 μL of 7.5×105 viable PAN02 cells were intradermally injected to produce localized tumors in 6‐to‐8‐week‐old C57BL/6 mice. Mice were treated daily with oral administration of vehicle (negative control) or compound 10 (50 mg kg−1) and with intraperitoneal injection of gemcitabine (50 mg kg−1) at day 6 and day 12. Mice were sacrificed at day 35. Figure 9 shows the effect of oral administration of compound 10 and gemcitabine (positive control) on primary tumor PAN02 cells. The effect of 10 was comparable to that of the gold standard, gemcitabine (T/C between 52 and 77 %), which is used for pancreatic cancer therapy.
Figure 9.
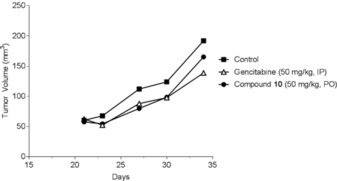
Antitumor activity of compound 10 on pancreatic cancer.
The fact that compound 10 showed similar anticancer and anti‐inflammatory activities suggests that there is a direct relationship between the two diseases. Indeed, it was reported that a few small molecules39 and several natural products40 exhibited a strong correlation between anti‐inflammatory and anticancer activities.
An additional in vivo experiment was performed by testing the activity of compound 10 (good anti‐inflammatory activity) on a metastatic tumor model. Mice were treated with intravenous administration (5 mg kg−1, Figure 10) or by oral administration (50 mg kg−1, Figure 11) of compound 10 with intraperitoneal injection (IP) of Cytoxan (cyclophosphamide) on primary DA‐3 breast tumor. IP injection of cyclophosphamide (100 mg kg−1) was undertaken at days 11 and 18 and with intravenous administration (5 mg kg−1) or oral treatment (50 mg kg−1) of compound 10 at days 11, 12, 13, 15, 18, and 20. Tumors were palpable 7 to 10 days postinoculation. In both types of administration, compound 10 induced significant (p<0.05) inhibition of tumor volume with T/C between 35 and 57 %, which is comparable to cyclophosphamide, which induces significant (p<0.04) inhibition of tumor volume with T/C between 33 and 59 %.
Figure 10.
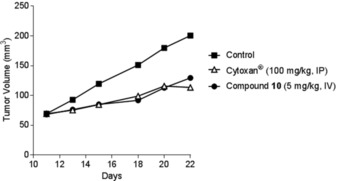
Antitumor effects of compound 10 (IV) on a primary DA‐3 breast cancer.
Figure 11.
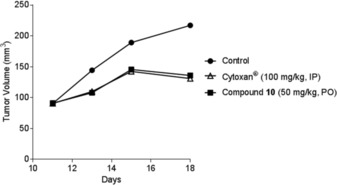
Antitumor effects of compound 10 (PO) on a primary DA‐3 breast cancer.
We then sought to demonstrate if compound 10 could inhibit other types of cancer, especially those that spread (metastasize) to other parts of the body. Figure 12 shows the antitumor efficacy of the oral administration of compound 10 (50 mg kg−1) or cyclophosphamide [100 mg kg−1, intravenous (IV) administration] on xenograft human prostate PC‐3 tumor. Compound 10 significantly inhibited (p<0.05) tumor volume with T/C between 14 and 40 %. Cyclophosphamide induced significant inhibition (p<0.05) of tumor volume with T/C between 1 and 39 %. Injection of Cytoxan was undertaken at days 29 and 36.
Figure 12.
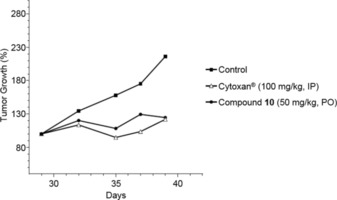
Antitumor efficacy of compound 10 and Cytoxan (cyclophosphamide) on xenograft human prostate PC‐3 tumor.
The mechanism by which most‐active compound 10 demonstrates anti‐inflammatory and anticancer properties in vivo is under investigation. To address this issue, different experiments were undertaken to identify the target. Compound 10 inhibits TNF‐α synthesis and activity without affecting TNF‐α binding to its receptors (TNF‐RI and TNF‐RII). It inhibits PGE2 production41 with no effect on Cox‐1 and Cox‐2 activity. It has weak in vitro cytotoxicity (IC50>10 μm) on NHDF (normal human dermal fibroblasts), HUVEC (human umbilical vein endothelial cells), PC‐3, K562, P815, and resting PBML (peripheral blood mononuclear leukocytes) (Table 2). Even though compound 10 showed some toxicity in vitro on normal cells, the in vivo air‐pouch rat model showed that this compound inhibited both TNF‐α and PGE2. These results indicate the effect of compound 10 on proinflammatory cytokines that can affect tumor growth. In fact, in all types of cancers studied in vivo, we observed no regression of tumors but a slow down of growth, which is an indication that compounds 6, 7, and 10 affect cancer cell proliferation but do not kill the cells directly. It is most likely scaffold linked/dependent. Furthermore, compound 10 had no effect on PLA2, 5‐Lox, and 15‐Lox enzymatic activities or modulation of iNOS expression. On PC‐3 prostate cancer cells, compound 10 (20 μm) induced cell‐cycle block in G1. Compound 10 had no effect on microtubule polymerization.
Table 2.
In vitro cytoxicity of compound 10.
| Time [h] | IC50 [μm] | |||||
|---|---|---|---|---|---|---|
| NHDF | HUVEC | PC‐3 | K562 | P815 | Resting PBML | |
| 24 | 78.6 | 31.8 | 43.3 | 36.0 | 23.5 | >10 |
| 72 | 45.8 | 51 | 26.8 | 15.5 | 7.5 | nd[a] |
[a] nd: not determined.
3. Conclusions
Low‐molecular‐weight synthetic molecules 1 with a general 2‐(fluorophenylamino)‐4,6‐disubstituted 1,3,5‐triazine structure and showing anti‐inflammatory and anticancer activities were described. Basic structure–activity relationship studies demonstrated the importance of the 3‐ or 4‐fluorophenylaniline group attached to the triazine ring, which played a role in the activity of these compounds. Two lead triazines, 6 and 10, displayed both in vitro and in vivo dual activity at moderate and significant levels, respectively, in anticancer/anti‐inflammatory models. This addresses the molecular link between inflammation and cancer. Compound 10 demonstrated significant antitumor efficacy in several animal models, such as the breast DA‐3 adenocarcinoma and human xenogeneic prostate PC‐3 cancer models. This was confirmed by the ability of compound 10 to inhibit PGE2 in vitro in stimulated J774 cell (murine macrophage) and in vivo (LPS‐induced air‐pouch assay). Also, compound 10 reduced PC‐3 cell proliferation with an IC50 of 20 μm by inducing cell‐cycle arrest at the G0/G1 phase. In an in vitro model of wound healing, compound 10 inhibited PC‐3 cell migration/invasion. This class of triazine compounds is new, safe,42 and nontoxic43 and offers a novel approach to the treatment of inflammation and cancer.
Experimental Section
General Methods
All HPLC chromatograms and mass spectra were recorded with an HP 1100 LC‐MS Agilent instrument by using a diode array detector. An analytical C18 column (75×4.6 mm, 5 μm) with a gradient of 1–40 % acetonitrile/water containing 0.01 % trifluoroacetic acid (TFA) in 6 min and a flow of 2 mL min−1 (method 1), an analytical C18 column (75×4.6 mm, 5 μm) with a gradient of 15–99 % acetonitrile/water containing 0.01 % TFA in 6 min and a flow of 2 mL min−1 (method 2), an analytical C18 column (75×4.6 mm, 5 μm) with a gradient of 0.1–20 % acetonitrile/water containing 0.01 % TFA in 5 min and a flow of 1 mL min−1 (method 3), or an analytical C18 column (75×4.6 mm, 5 μm) with a gradient of 1–50 % acetonitrile/water containing 0.01 % TFA in 5 min and a flow of 1 mL min−1 (method 4) was used.
Syntheses
1. ‐{2‐[4‐(5‐Aminopentylamino)‐6‐(3‐fluorophenylamino)‐1,3,5‐triazin‐2‐ylamino]ethyl}phenol Hydrochloride Salt (6)
Cyanuric chloride (10.0 g, 54.2 mmol) was added in small portions to a cooled (−10 °C) mixture of water (50 mL) and acetone (50 mL). A solution of 3‐fluoroaniline (5.2 mL, 54.2 mmol) in acetone (50 mL) was added slowly over 50 min, maintaining the temperature of the reaction below −5 °C. The mixture was then stirred at ambient temperature for 1 h. The pH of the mixture was adjusted from 2 to 8 with saturated aqueous sodium bicarbonate (200 mL), and stirring was continued for another 30 min. The precipitated solid was collected by filtration, washed with water, and dried in vacuo. This gave 2,4‐dichloro‐3‐fluorophenylamino‐1,3,5‐triazine as a white solid (13.3 g, 94 %): 1H NMR (400 MHz, [D6]DMSO): δ=6.97–7.01 (1 H, m), 7.38–7.43 (2 H, m), 7.52–7.55 (1 H, m), 11.25 ppm (1 H, br); LRMS (ESI): m/z: 259 [M+H]+; HPLC (method 2): t R=4.1 min. The product was used in the next step without further purification. This dichlorotriazine derivative (6.4 g, 24.7 mmol) was dissolved in THF (70 mL) at RT and was treated with a solution of 5‐(tert‐butoxycarbonylamino)pentylamine (7.5 g, 37.0 mmol) in a mixture of acetone (50 mL) and water (50 mL). The resulting solution was then treated with saturated aqueous sodium bicarbonate (70 mL). The mixture was stirred at RT for 2.5–3 h. The mixture was then concentrated in vacuo, and the residue was diluted with water and extracted with ethyl acetate. The combined organic extract was washed with saturated aqueous sodium chloride, 2 m aqueous HCl, saturated sodium chloride, saturated sodium bicarbonate, and saturated sodium chloride. The solution was then dried (magnesium sulfate/charcoal), filtered through Celite diatomaceous earth, and concentrated in vacuo to 200 mL. This solution was poured, with stirring, into hexane (1.2 L), and the precipitate was collected by filtration, washed with hexane, and dried in vacuo to yield the monochloro‐1,3,5‐triazine derivative as a white solid (6.6 g, 63 %): 1H NMR (400 MHz, [D6]DMSO): δ=1.23–1.30 (2 H, m), 1.31–1.56 (2 H, m), 1.34 (9 H, s), 1.44–1.56 (2 H, m), 2.85–2.91 (2 H, m), 3.20–3.30 (2 H, m), 6.70–6.77 (1 H, m), 6.79–6.85 (1 H, m), 7.25–7.33 (1 H, m), 7.38–7.43 (1 H, m), 7.67–7.75 & 7.76–7.85 (1 H, br), 8.14–8.21 & 8.22–8.30 (1 H, br), 10.05–10.11 & 10.15–10.26 ppm (1 H, br); LRMS (ESI): m/z: 425 [M+H]+, 447 [M+H+Na]+; HPLC (method 2): t R=4.5 min. A solution of the monochlorotriazine (6.6 g, 15.6 mmol) in THF (300 mL) was treated with tyramine (6.4 g, 46.7 mmol) and triethylamine (77.7 mmol, 10.9 mL). The mixture was heated at 65–70 °C for 16–60 h, cooled to ambient temperature, and concentrated in vacuo. The residue was extracted with ethyl acetate and filtered. The filtrate was washed with 1 m aqueous HCl, saturated sodium chloride, saturated aqueous sodium bicarbonate, and saturated sodium chloride; dried (magnesium sulfate/charcoal); filtered through Celite diatomaceous earth; and concentrated in vacuo. The residue was then dissolved in ether (150 mL), and this solution was added dropwise to hexane (1.4 L) with vigorous stirring. The precipitated solid was collected by filtration and dried in vacuo to yield the tri(amino‐substituted) 1,3,5‐triazine derivative as an off‐white solid (6.5 g, 80 %): 1H NMR (400 MHz, [D6]DMSO): δ=1.21–1.29 (2 H, m), 1.32–1.41 (2 H, m), 1.34 (9 H, s), 1.44–1.54 (2 H, m), 2.65–2.71 (2 H, m), 2.88 (2 H, dt, J=6.5, 6.5 Hz), 3.15–3.27 (2 H, m), 3.33–3.42 (2 H, m), 6.61–6.70 (1 H, m), 6.67 (2 H, d, J=8.5 Hz), 6.71–6.76 (1 H, m), 6.84–7.02 (1 H, m), 7.01 (2 H, d, J=8.5 Hz), 7.16–7.23 (1 H, m), 7.39–7.47 (1 H, m), 7.87–7.91 (1 H, m), 8.92–8.94 & 9.00–9.06 (1 H, 2×br), 9.13 ppm (1 H, s); LRMS (ESI): m/z: 526 [M+H]+, 548 [M+H+Na]+; HPLC (method 2): t R=2.9 min. A solution of the tert‐butoxycarbonyl (Boc)‐protected compound (6.5 g, 12.4 mmol) in 4 m HCl/1,4‐dioxane (100 mL) and water (10 mL) was stirred at RT for 2 h. The solvents and the excess amount of acid were evaporated in vacuo, and the trace amounts of water were removed by co‐evaporation (2×) with 2‐propanol (25 mL). The dried residue was dissolved in 2‐propanol (25 mL), and the solution was added dropwise to ether (450 mL) with vigorous stirring. The precipitated solid was collected by filtration, dried in vacuo, and then dissolved in pyrogen‐free water (800 mL), filtered (0.22 μm), and lyophilized to give deprotected compound 6 as an off‐white solid (5.5 g, 89 %): 1H NMR (400 MHz, [D6]DMSO): δ=1.26–1.35 (2 H, m), 1.47–1.57 (4 H, m), 2.64–2.73 (4 H, m), 3.24–3.31 (2 H, m), 3.32–3.55 (5 H, m, CH2+NH3 +), 6.63 (2 H, d, J=8.5 Hz), 6.82–6.89 (1 H, m), 6.93–7.06 (2 H, m), 7.24–7.39 (2 H, m), 7.61–7.73 (1 H, m), 7.81–7.93 (3 H, m), 8.15–8.25, 8.40–8.60, 9.10–9.30, 10.25–10.40 & 10.55–10.65 ppm (2 H, br); 19F NMR (376.5 MHz, CD3OD): δ=−114.5 to −113.8 ppm (1 F, m); LRMS (ESI): m/z: 426 [M+H]+, 448 [M+H+Na]+; HPLC (method 2): t R=1.6 min.
2. ‐{2‐[4‐(5‐Aminopentylamino)‐6‐(3‐fluorophenylamino)‐1,3,5‐triazin‐2‐ylamino]ethyl}benzenesulfonamide Dihydrochloride Salt (7)
This compound was prepared by using the same method as that outlined for compound 6 by using 4‐(2‐aminoethyl)benzenesulfonamide instead of tyramine. White solid; 77 % yield; m.p. 145–147 °C; 1H NMR (400 MHz, D2O): δ=1.14–1.26 (2 H, m), 1.33–1.44 (2 H, m), 1.46–1.55 (2 H, m), 2.64–2.84 (4 H, m), 3.04–3.15 (2 H, m), 3.33–3.56 (2 H, m), 6.68–6.84 (1 H, m), 6.88–6.99 (1 H, m), 7.09–7.32 (4 H, m), 7.44–7.63 ppm (2 H, m); 19F NMR (376.5 MHz, CD3OD): δ=−114.5 to −113.8 ppm (1 F, m); LRMS (ESI): m/z: 489 [M+H]+; HPLC (method 2): t R=1.6 min.
3. ‐{2‐[4‐(5‐Aminopentylamino)‐6‐(4‐fluorophenylamino)‐1,3,5‐triazin‐2‐ylamino]ethyl}phenol Hydrochloride Salt (10)
2,4‐Dichloro‐4‐fluorophenylamino‐1,3,5‐triazine was prepared by using the same method as that outlined for compound 6 by using 4‐fluoroaniline (18 mL, 190 mmol) instead of 3‐fluoroaniline to yield a white solid (44.3 g, 90): LRMS (ESI): m/z: 259 [M+H]+ HPLC (method 2): t R=4.0 min. The dichlorotriazine (44.2 g, 0.2 mole) was coupled with tyramine (35.1 g, 0.3 mole) according to compound 6 by using tyramine instead of 5‐(tert‐butoxycarbonylamino)pentylamine to yield a white solid (56.1 g, 91 %): LRMS (ESI): m/z: 360 [M+H]+, 382 [M+H+Na]+; HPLC (method 2): t R=3.7 min. A solution of the monochlorotriazine (15.0 g, 41.8 mmol) and 1,5‐diaminopentane (24.5 mL, 209 mmol) in tetrahydrofuran (125 mL) and methanol (60 mL) was divided into nine portions. Each portion was heated in a chemistry microwave apparatus at 130 °C for 10 min. The portions were then recombined and concentrated in vacuo, and the residue was dissolved in ethyl acetate. The ethyl acetate solution was washed with water and saturated sodium chloride and then extracted with 2 m aqueous HCl. The aqueous extract was treated with ethyl acetate and then saturated aqueous sodium bicarbonate. The precipitate was extracted with ethyl acetate, and the combined extract was washed with saturated sodium chloride, dried (magnesium sulfate/charcoal), filtered through Celite diatomaceous earth, and concentrated in vacuo. The residue was dissolved in methanol (300 mL), and the solution was treated with 1 m HCl in ether (60 mL). The solution was then concentrated in vacuo. The residue was dissolved in hot 2‐propanol (150 mL), and this solution was added dropwise to ether (1.5 L) with vigorous stirring. The precipitated solid was collected by filtration, dried in vacuo, and then dissolved in pyrogen‐free water (1.6 L), filtered (0.22 μm), and lyophilized to give compound 10 as the hydrochloride salt (14.9 g, 72 %): m.p. 130–133 °C; 1H NMR (400 MHz, D2O): δ=1.16–1.27 (2 H, m), 1.37–1.54 (4 H, m), 2.53–2.64 (2 H, m), 2.76–2.83 (2 H, m), 3.09–3.17 (2 H, m), 3.21–3.48 (2 H, m), 6.56–6.64 (2 H, m), 6.85–7.02 (4 H, m), 7.16–7.27 ppm (2 H, m); 19F NMR (376.5 MHz, CD3OD): δ=−118.1 to −116.0 ppm (1 F, m); LRMS (ESI): m/z: 426 [M+H]+; HPLC (method 2): t R=1.6 min.
4. ‐{2‐[4‐(5‐Aminopentylamino)‐6‐(3‐fluorophenylamino)‐1,3,5‐triazin‐2‐ylamino]ethyl}‐N,N‐dimethylbenzenesulfonamide Dihydrochloride Salt (8)
This compound was prepared according to the procedure outlined for compound 10 by using N,N‐dimethyl‐4‐[2‐aminoethyl]benzenesulfonamide instead of tyramine. N,N‐Dimethyl‐4‐(2‐aminoethyl)benzenesulfonamide was synthesized as follows: A solution of 4‐(2‐aminoethyl)benzenesulfonamide (26.5 g, 0.1 mole) in anhydrous DMF (120 mL) was treated with phthalic anhydride (23.5 g, 0.2 mole), and the mixture was heated at 70 °C for 4 h. The mixture was cooled to ambient temperature and 1,1′‐carbonyldiimidazole (21.5 g, 0.1 mole) was added in small portions; the mixture was stirred at ambient temperature overnight. The solvent was evaporated in vacuo, and the residue was washed with water, dried, and triturated with ethyl acetate to give the phthaloyl‐protected compound as a white solid (38.1 g, 89): 1H NMR (400 MHz, [D6]DMSO): δ=2.98 (2 H, t, J=7.0 Hz), 3.82 (2 H, t, J=7.0 Hz), 7.29 (2 H, s), 7.38 (2 H, d, J=8.0 Hz), 7.69 (2 H, t, J=8.0 Hz), 7.76–7.84 ppm (4 H, m); LRMS (ESI): m/z: 331 [M+H]+, 348 [M+H+Na]+; HPLC (method 2): t R=2.9 min. A solution of the phthaloyl‐protected compound (12.7 g, 38.6 mmol) in anhydrous DMF (120 mL) was treated with NaH (60 % dispersion in oil; 3.5 g, 88.8 mmol) in small portions under a N2 atmosphere at 0 °C over 15 min, and the mixture was stirred under a N2 atmosphere at 0 °C for 1 h. Iodomethane (4.8 mL, 77.2 mmol) was then added dropwise over 15 min, and the mixture was stirred under a N2 atmosphere at 0 °C to RT overnight. The resultant yellow suspension was poured onto ice/water (1.4 L) and was stirred for 30 min. The precipitate was collected by filtration; washed sequentially with water, hexane, and ether; and then dried in vacuo to give the N,N‐dimethylbenzenesulfonamide derivative as a white solid (11.3 g, 81 %): LRMS (ESI): m/z: 359 [M+H]+, 381 [M+H+Na]+; HPLC (method 2): t R=3.7 min. A solution of the phthaloyl‐protected, N,N‐dimethyl compound (11.3 g, 31.5 mmol) and hydrazine hydrate (4.6 mL, 44.6 mmol) in 95 % ethanol (125 mL) was heated at reflux for 2 h. The white solid that formed was removed by filtration and was washed with ethanol. The filtrate and washings were combined, and the solution was concentrated in vacuo. The solid that formed was removed by filtration and was washed with ethanol. This procedure was repeated (3×), and the final filtrate was concentrated to dryness in vacuo. The solid was extracted with ethyl acetate. The extracts were concentrated in vacuo to give the free amine as a yellow oil (4.8 g, 67 %): 1H NMR (400 MHz, CD3OD): δ=2.65 (6 H, s), 2.84–2.92 (4 H, m), 7.47 (2 H, d, J=8.5 Hz), 7.71 ppm (2 H, d, J=8.5 Hz); LRMS (ESI): m/z: 229 [M+H]+, 251 [M+H+Na]+; HPLC (method 2): t R=2.3 min. This compound was treated with dichlorotriazine followed by the alkylamine and was then deprotected to give the final product as a white solid (2.2 g, 92 %): m.p. 143–146 °C; 1H NMR (400 MHz, CD3OD): δ=1.42–1.53 (2 H, m), 1.64–1.78 (4 H, m), 2.60 & 2.64 (6 H, 2 x s), 2.92–2.99 (2 H, m), 3.01–3.07 (2 H, m), 3.39–3.48 (2 H, m), 3.68–3.78 (2 H, m), 6.83–6.92 (1 H, m), 7.24–7.37 (2 H, m), 7.42–7.71 ppm (5 H, m); LRMS (ESI): m/z: 517 [M+H]+, 539 [M+H+Na]+; HPLC (method 1): t R=4.3 min.
N‐(5‐Aminopentyl)‐N′‐[2‐(4‐aminophenyl)ethyl]‐N′′‐(4‐fluorophenyl)‐1,3,5‐triazine‐2,4,6‐triamine Dihydrochloride Salt (9)
This compound was prepared according to the procedure outlined for compound 6 by using 2‐(4‐aminophenyl)ethylamine instead of tyramine. Yellow solid (97 %): m.p. 155–158 °C; 1H NMR (400 MHz, D2O): δ=1.42–1.53 (2 H, m), 1.63–1.76 (4 H, m), 2.87–3.02 (4 H, m), 3.40–3.48 (2 H, m), 3.62–3.77 (2 H, m), 7.07–7.15 (2 H, m), 7.28–7.38 (3 H, m), 7.40–7.49 (1 H, m), 7.52–7.63 ppm (2 H, m); LRMS (ESI): m/z: 425 [M+H]+, 447 [M+H+Na]+; HPLC (method 3): t R=1.9 min.
N‐{5‐[4‐(3‐Fluorophenylamino)‐6‐(4‐hydroxyphenethylamino)‐1,3,5‐triazin‐2‐ylamino]pentyl}acetamide Hydrochloride Salt (11)
This compound was prepared according to the procedure outlined for compound 6 by using N‐(5‐aminopentyl)acetamide instead of 5‐(tert‐butoxycarbonylamino)pentylamine. Orange solid (37 mg): 1H NMR (400 MHz, CD3OD): δ=1.34–1.41 (2 H, m), 1.46–1.62 (4 H, m), 1.90 (3 H, s), 2.76 (2 H, t, J=7.1 Hz), 3.14 (2 H, t, J=7.1 Hz), 3.26–3.40 (2 H, m), 3.48–3.56 (2 H, m), 6.62–6.71 (1 H, m), 6.70 (2 H, d, J=8.4 Hz), 7.05 (2 H, d, J=8.2 Hz), 7.18–7.30 (1 H, m), 7.75–7.83 ppm (1 H, m); LRMS (ESI): m/z: 468 [M+H]+; HPLC (method 2): t R=2.3 min.
5. ‐{2‐[4‐(2‐Aminoethylamino)‐6‐(4‐fluorophenylamino)‐1,3,5‐ triazin‐2‐ylamino]ethyl}phenol Dihydrochloride Salt (12)
This compound was prepared according to the procedure outlined for compound 10 by using ethylenediamine instead of 1,5‐diaminopentane. White solid (566 mg): 1H NMR (400 MHz, CD3OD): δ=2.77–2.82 (2 H, m), 3.14–3.20 (2 H, m), 3.54–3.70 (4 H, m), 6.70 (2 H, d, J=7.8 Hz), 6.98–7.13 (4 H, m), 7.52–7.62 ppm (2 H, m); LRMS (ESI): m/z: 384 [M+H]+; HPLC (method 1): t R=1.6 min.
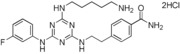
6. ‐{2‐[4‐(5‐Aminopentylamino)‐6‐(3‐fluorophenylamino)‐1,3,5‐triazin‐2‐ylamino]ethyl}benzamide Dihydrochloride Salt (13)
This compound was prepared according to the procedure outlined for compound 10 by using 4‐(2‐aminoethyl)benzamide and 3‐fluoroaniline instead of tyramine and 4‐fluoroaniline, respectively. 4‐(2‐Aminoethyl)benzamide was prepared as follows: A suspension of 4‐(2‐aminoethyl)benzoic acid hydrochloride (5.0 g, 24.8 mmol) in methanol (200 mL) was treated with a 4 m solution of HCl in 1,4‐dioxane (10 mL, 40 mmol), and the mixture was heated at reflux overnight. The solvents and the excess amount of acid were removed in vacuo. The residue was triturated with ether and dried in vacuo to give the ester as a white solid (5.5 g, quant.): 1H NMR (400 MHz, CD3OD): δ=3.04 (2 H, t, J=7.0 Hz), 3.21 (2 H, td, J=7.0, 0.5 Hz), 3.89 (3 H, s), 7.41 (2 H, dd, J=8.0, 0.5 Hz), 8.00 ppm (2 H, d, J=8.0 Hz). A suspension of this hydrochloride salt (5.4 g, 24.8 mmol) in tetrahydrofuran (60 mL) and methanol (30 mL) was treated with diisopropylethylamine (4.8 mL, 27.3 mmol) and di‐tert‐butyl dicarbonate (8.1 g, 37.2 mmol). The mixture was stirred at ambient temperature under a N2 atmosphere for 5 h. The solvents were evaporated in vacuo, and the residue was dissolved in ethyl acetate. The solution was washed with water and saturated sodium chloride and was then dried (magnesium sulfate), filtered, and concentrated in vacuo. The residue was triturated with cold ether and dried in vacuo to give the protected compound as a white solid (5.6 g, 81 %): LRMS (ESI): m/z: 192 [M+H]+, 302 [M+H+Na]+; HPLC (method 2): t R=3.9 min. A solution of the ester (5.6 g, 20.0 mmol) in 1,4‐dioxane (36 mL) was treated with saturated aqueous ammonia (36 mL). The mixture was heated in a sealed tube at 100 °C overnight. After cooling, the precipitated solid was collected by filtration, washed with water, and dried in vacuo to give the amide as a white solid (4.4 g, 82 %): LRMS (ESI): m/z: 287 [M+H+Na]+; HPLC (method 2): t R=2.6 min. Deprotection of the tert‐butoxycarbonyl compound (4.4 g, 16.5 mmol) was undertaken by modifying the procedure outlined for compound 6, in that the water co‐solvent was omitted and the solid was dried in vacuo rather than lyophilized to yield a white solid (3.3 g, quant.): LRMS (ESI): m/z: 165 [M+H]+, 187 [M+H+Na]+; HPLC (method 2): t R=0.3 min. This compound was treated with dichlorotriazine and the alkylamine and was then deprotected to give the final product as a white solid (1.0 g, 20 %): m.p. 190–192 °C; 1H NMR (400 MHz, D2O): δ=1.13–1.26 (2 H, m), 1.31–1.55 (4 H, m), 2.54–2.84 (4 H, m), 2.99–3.12 (2 H, m), 3.23–3.49 (2 H, m), 6.66–6.82 (1 H, m), 6.86–7.14 (3 H, m), 7.16–7.25 (2 H, m), 7.36–7.57 ppm (2 H, m); LRMS (ESI): m/z: 453 [M+H]+; HPLC (method 2): t R=1.5 min.
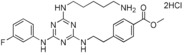
Methyl 4‐{2‐[4‐(5‐aminopentylamino)‐6‐(3‐fluorophenylamino)‐1,3,5‐triazin‐2‐ylamino]ethyl}benzoate Dihydrochloride Salt (14)
This compound was prepared according to the procedure outlined for compound 6 by using methyl 4‐(2‐aminoethyl)benzoate instead of tyramine. Off‐white solid (88 mg): 1H NMR (400 MHz, CD3OD): δ=1.44–1.53 (2 H, m), 1.65–1.76 (4 H, m), 2.93–3.04 (4 H, m), 3.43–3.50 (2 H, m), 3.69–3.76 (2 H, m), 3.84 (3 H, s), 6.87–6.96 (1 H, m), 7.24–7.43 (4 H, m), 7.57–7.68 (1 H, m), 7.88–7.96 ppm (2 H, m); LRMS (ESI): m/z: 468 [M+H]+; HPLC (method 2): t R=1.9 min.
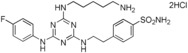
7. ‐{2‐[4‐(5‐Aminopentylamino)‐6‐(4‐fluorophenylamino)‐1,3,5‐triazin‐2‐ylamino]ethyl}benzenesulfonamide Dihydrochloride Salt (15)
This compound was prepared according to the procedure outlined for compound 7 by using 4‐fluoroaniline in place of 3‐fluoroaniline. The final Boc‐protected intermediate (4.8 mmol) was deprotected by using a variation of the procedure outlined for compound 6. In this case, 4 m HCl/1,4‐dioxane (36 mL) in CH2Cl2 (30 mL) was used at 0 °C to ambient temperature to yield a low‐density, white solid (87 %): m.p. 165–168 °C; 1H NMR (400 MHz, CD3OD): δ=1.40–1.51 (2 H, m), 1.62–1.74 (4 H, m), 2.87–3.04 (4 H, m), 2.93 (2 H, t, J=7.5 Hz), 3.01 (2 H, t, J=6.5 Hz), 3.41 (2 H, t, J=7.5 Hz), 3.64–3.77 (2 H, m), 7.07–7.16 (2 H, m), 7.29–7.48 (2 H, m), 7.50–7.62 (2 H, m), 7.77–7.86 ppm (2 H, m); 19F NMR (376.5 MHz, CD3OD): δ=−120.2 to −119.8 ppm (1 F, m); LRMS (ESI): m/z: 245 [M+H]+, 489 [M+H+Na]+; HPLC (method 1): t R=3.6 min.
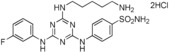
8. ‐[4‐(5‐Aminopentylamino)‐6‐(3‐fluorophenylamino)‐1,3,5‐tri‐ azin‐2‐ylamino]benzenesulfonamide Dihydrochloride Salt (16)
This compound was prepared according to the procedure outlined for compound 10 by using 4‐aminobenzenesulfonamide and 3‐fluoroaniline instead of tyramine and 4‐fluoroaniline, respectively. Pale‐beige solid (95 % yield): m.p. 162–163 °C; 1H NMR (400 MHz, CD3OD): δ=1.47–1.56 (2 H, m), 1.67–1.78 (4 H, m), 2.95 (2 H, t, J=7.5 Hz), 3.52 (2 H, t, J=7.0 Hz), 6.92–7.00 (1 H, m), 7.29–7.42 (2 H, m), 7.60–7.78 (1 H, m), 7.82–7.95 ppm (4 H, m); 19F NMR (376.5 MHz, CD3OD): δ=−114.2 to −113.7 ppm (1 F, m); LRMS (ESI): m/z: 461 [M+H]+, 483 [M+H+Na]+; HPLC (method 4): t R=3.7 min.
9. ‐{2‐[4‐(4‐Aminobutylamino)‐6‐(3‐fluorophenylamino)‐1,3,5‐triazin‐2‐ylamino]ethyl}benzenesulfonamide Dihydrochloride Salt (17)
This compound was prepared according to the procedure outlined for compound 10 by using 4‐(2‐aminoethyl)benzenesulfonamide and 4‐aminobutylamine instead of tyramine and 5‐aminopentylamine, respectively. White solid (74 %): m.p. 181–184 °C; 1H NMR (400 MHz, CD3OD): δ=1.65–1.77 (4 H, m), 2.93–3.04 (4 H, m), 3.42–3.54 (2 H, m), 3.68–3.78 (2 H, m), 6.86–6.95 (1 H, m), 7.24–7.50 (4 H, m), 7.57–7.66 (1 H, m), 7.78–7.86 ppm (2 H, m); 19F NMR (376.5 MHz, CD3OD): δ=−116.10 to −115.43 ppm (1 F, m); LRMS (ESI): m/z: 475 [M+H]+, 497 [M+H+Na]+; HPLC (method 1): t R=3.6 min.
(RS)‐4‐{2‐[4‐(5‐Aminopentylamino)‐6‐(4‐fluorophenylamino)‐1,3,5‐triazin‐2‐ylamino]‐1‐hydroxyethyl}phenol Dihydrochloride Salt (18)
This compound was prepared according to the procedure outlined for compound 10 by using [±]‐octopamine instead of tyramine. White solid (36 %): m.p. 122–125 °C; 1H NMR (400 MHz, CD3OD): δ=1.36–1.43 (2 H, m), 1.50 (2 H, tt, J=7.0, 7.0 Hz), 1.57–1.63 (2 H, m), 2.62 (2 H, t, J=7.0 Hz), 3.28–3.40 (2 H, m), 3.45 (1 H, J=13.5, 8.0 Hz), 3.54–3.63 (1 H, m), 4.67–4.75 (1 H, m), 6.69 (2 H, d, J=8.5 Hz), 6.96–7.00 (2 H, m), 7.13 (2 H, d, J=8.5 Hz), 7.56–7.67 ppm (2 H, m); LRMS (ESI): m/z: 442 [M+H]+; HPLC (method 1): t R=3.3 min.
Methyl (S)‐2‐[4‐(5‐aminopentylamino)‐6‐(3‐fluorophenylamino)‐1,3,5‐triazin‐2‐ylamino]‐3‐(4‐hydroxyphenyl)propanoate Dihydrochloride Salt (19)
This compound was prepared according to the procedure outlined for compound 6 by using methyl (S)‐tyrosine instead of tyramine. Colorless residue (1 mg): LRMS (ESI): m/z: 484 [M+H]+; HPLC (method 2): t R=1.8 min.
N‐(4‐Aminobenzyl)‐N′‐(5‐aminopentyl)‐N′′‐(3‐fluorophenyl)‐1,3,5‐triazine‐2,4,6‐triamine Trihydrochloride Salt (20)
This compound was prepared according to the procedure outlined for compound 6 by using 4‐aminobenzylamine instead of tyramine/triethylamine in step 3. Orange solid (quant.): m.p. 197–199 °C; 1H NMR (400 MHz, CD3OD): δ=7.59–7.73 (m, 1 H), 7.53 (d, J=7.9 Hz, 2 H), 7.37 (d, J=7.9 Hz, 2 H), 7.09–7.33 (m, 2 H), 6.82–6.96 (m, 1 H), 4.69 (s, 2 H), 3.44–3.53 (m, 2 H), 2.90–2.97 (m, 2 H), 1.62–1.76 (m, 4 H), 1.42–1.55 ppm (m, 2 H); 13C NMR (101 MHz, CD3OD): δ=complex rotamers; 19F NMR (377 MHz, CD3OD): δ=−114.5 to −113.9 (1 F, m); LRMS (ESI): m/z (%): 411.2 (100) [M+H]+; HPLC (10–99 % 5 min): t R=0.89 min.
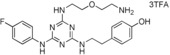
10. ‐(2‐{4‐[2‐(2‐Aminoethoxy)ethylamino]‐6‐(4‐fluorophenylamino)‐1,3,5‐triazin‐2‐ylamino}ethyl)phenol bis(Trifluoroacetate) Salt (21)
This compound was prepared according to the procedure outlined for compound 10 by using 2,2′‐oxydiethylamine dihydrochloride instead of 1,5‐diaminopentane. White solid (33 mg): 1H NMR (400 MHz, CD3OD): δ=2.75–2.83 (2 H, m), 3.12–3.17 (2 H, m), 3.54–3.73 (8 H, m), 6.67–6.72 (2 H, m), 6.96–7.14 (4 H, m), 7.55–7.60 ppm (2 H, m); LRMS (ESI): m/z: 428 [M+H]+; HPLC (method 4): t R=3.2 min.
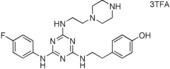
11. ‐(2‐{4‐(4‐Fluorophenylamino)‐6‐[2‐(piperazin‐1‐yl)ethylamino]‐1,3,5‐triazin‐2‐ylamino}ethyl)phenol Trifluoroacetate Salt (22)
This compound was prepared according to the procedure outlined for compound 10 by using 2‐[4‐(tert‐butoxycarbonyl)piperazin‐1‐yl]ethylamine instead of 1,5‐diaminopentane with a final deprotection step as for compound 6. White solid (15 mg): 1H NMR (400 MHz, CD3OD): δ=2.60–2.82 (6 H, m), 2.81 (2 H, t, J=7.0 Hz), 3.08–3.17 (2 H, m), 3.64 (2 H, t, J=7.0 Hz), 3.82–3.98 (4 H, m), 6.71 (2 H, d, J=8.4 Hz), 7.05 (2 H, d, J=8.4 Hz), 7.10–7.17 (2 H, m), 7.46–7.57 ppm (2 H, m); LRMS (ESI): m/z: 453 [M+H]+; HPLC (method 4): t R=3.1 min.
12. ‐{2‐[4‐(4‐Aminobutylamino)‐6‐(4‐fluorophenylamino)‐1,3,5‐triazin‐2‐ylamino]ethyl}phenol Trifluoroacetate Salt (23)
This compound was prepared according to the procedure outlined for compound 10 by using 1,4‐diaminobutane instead of 1,5‐diaminopentane. White solid (1.9 mg): LRMS (ESI): m/z: 412 [M+H]+; HPLC (method 2): t R=4.1 min.
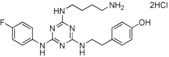
13. ‐{2‐[4‐(6‐Aminohexylamino)‐6‐(4‐fluorophenylamino)‐1,3,5‐triazin‐2‐ylamino]ethyl}phenol Dihydrochloride Salt (24)
This compound was prepared according to the procedure outlined for compound 10 by using 1,6‐diaminohexane instead of 1,5‐diaminopentane. White solid (30 mg): 1H NMR (400 MHz, CD3OD): δ=1.39–1.47 (4 H, m), 1.61–1.70 (4 H, m), 2.76–2.84 (2 H, m), 2.88–2.94 (2 H, m), 3.37–3.48 (2 H, m), 3.56–3.67 (2 H, m), 6.71 (2 H, d, J=7.8 Hz), 6.97–7.13 (4 H, m), 7.52–7.62 ppm (2 H, m); LRMS (ESI): m/z: 440 [M+H]+; HPLC (method 4): t R=4.0 min.
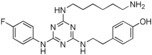
14. ‐{2‐[4‐{[4‐(Aminomethyl)cyclohexyl]methylamino}‐6‐(4‐fluorophenylamino)‐1,3,5‐triazin‐2‐ylamino]ethyl}phenol (25)
This compound was prepared according to the procedure outlined for compound 10 by using 1,4‐bis(aminomethyl)cyclohexane instead of 1,5‐diaminopentane. Pale‐pink solid (13 mg): 1H NMR (400 MHz, CD3OD): δ=0.86–1.02 (2 H, m), 1.33–1.58 (6 H, m), 1.68–1.90 (2 H, m), 2.47–2.60 (2 H, m), 2.69–2.79 (2 H, m), 3.16–3.24 (2 H, m), 3.45–3.53 (2 H, m), 6.70 (2 H, d, J=8.2 Hz), 6.95–7.03 (4 H, m), 7.53–7.68 ppm (2 H, m); 19F (377 MHz, CD3OD): δ=−124.1 to −123.7 ppm (1 F, m); LRMS (ESI): m/z: 466 [M+H]+; HPLC (method 4): t R=3.5 min.
15. ‐{2‐[4‐(5‐Aminopentylamino)‐6‐(3‐fluorophenylamino)‐1,3,5‐triazin‐2‐ylamino]ethyl}‐2,6‐dimethoxyphenol Dihydrochloride Salt (26)
This compound was prepared according to the procedure outlined for compound 6 by using 4‐(2‐aminoethyl)‐2,6‐dimethoxyphenol instead of tyramine. Pink solid (22 mg): 1H NMR (400 MHz, CD3OD): δ=1.45–1.53 (2 H, m), 1.64–1.77 (4 H, m), 2.82–2.88 (2 H, m), 2.90–2.98 (2 H, m), 3.39–3.50 (2 H, m), 3.66–3.74 (2 H, m), 3.76–3.82 (2 H, m), 3.78 (3 H, s), 3.81 (3 H, s), 6.47–6.60 (2 H, m), 6.84–6.93 (1 H, m), 7.26–7.37 (2 H, m), 7.60–7.67 ppm (1 H, m); 19F (377 MHz, CD3OD): δ=−114.5 to −113.8 ppm (1 F, m); LRMS (ESI): m/z: 486 [M+H]+; HPLC (method 4): t R=3.1 min.
16. ‐{2‐[4‐(5‐Aminopentylamino)‐6‐(3‐fluorophenylamino)‐1,3,5‐triazin‐2‐ylamino]ethyl}‐2‐chlorophenol, tris(trifluoroacetate) Salt (27)
This compound was prepared according to the procedure outlined for compound 6 by using 4‐(2‐aminoethyl)‐2‐chlorophenol instead of tyramine. White solid (18 mg): 1H NMR (400 MHz, CD3OD): δ=1.43–1.52 (2 H, m), 1.65–1.74 (4 H, m), 2.77–2.83 (2 H, m), 2.90–2.96 (2 H, m), 3.43–3.49 (2 H, m), 3.58–3.65 (2 H, m), 6.78–6.91 (2 H, m), 6.94–7.03 (1 H, m), 7.13–7.21 (1 H, m), 7.30–7.36 (2 H, m), 7.54–7.69 ppm (1 H, m); 19F (377 MHz, CD3OD): δ=−114.6 to −113.9 ppm (1 F, m); LRMS (ESI): m/z: 460 [M+H]+; HPLC (method 4): t R=3.7 min.
Anticancer Activity
In Vitro Effect of Compounds on the Inhibition of TNF‐α Released by LPS Induction from J774A.1 Cells
J774A.1 cells were cultured in the presence or absence of LPS and compounds. Cells were cultured at 37 °C for 24 h, and thereafter, the supernatants were collected for determination of the concentration of PGE2 by ELISA, as recommended by the manufacturer (GE Healthcare). Data were analyzed in Microsoft Excel software, and the concentration of compound that inhibited 50 % of PGE2 production (IC50) was calculated by using Prism software.
In Vitro Effect of Compounds on PC‐3 Cell Migration or Invasion
An in vitro migration assay was used to assess cell mobility in two dimensions. PC‐3 cells were plated on a 12‐well plate and were grown to confluence in RPMI+10 % FBS. A rubber policeman was used to create a denuded area. Confluent cells were quiesced by mitomycin C treatment (0.5 μm) at the concentration used to prevent the confounding issue of cell proliferation and protein synthesis. These cells were also incubated in the presence or absence of endothelial growth factor (EGF) and the compound for 24 h, and they were then photographed.
In Vitro Effect of Compounds on the Production of PGE2 in LPS‐Stimulated J774A.1
J774A.1 cells were cultured in the presence or absence of LPS and the compounds. Cells were cultured at 37 °C for 24 h, and thereafter, the supernatants were collected for determination of the concentration of PGE2 by ELISA, as recommended by the manufacturer (GE Healthcare). Data were analyzed in Microsoft Excel software, and the concentration of compound that inhibited 50 % of PGE2 production (IC50) was calculated by using Prism software.
In Vivo Experiments
All animal studies were reviewed and approved by the animal care and use committee of the National Institute of Scientific Research, INRS‐Institut‐Armand‐Frappier Center (Laval, QC, Canada).
Antitumor Effects of Compounds on a Primary P815 Mastocytoma Tumor
The syngeneic tumor P815 is a DBA/2 (H‐2d)‐derived mastocytoma obtained from ATCC (TIB64). P815 cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10 % fetal bovine serum. At day 0, 5×105 viable P815 cells (50 μL) were intradermally injected to produce localized tumors in 6‐to‐8‐week‐old DBA/2 mice. The animals were then serially monitored by manual palpation for evidence of tumor. Mice were then treated every day with oral administration of vehicle (negative control), acetylsalicylic acid (positive control, 50 mg kg−1), or compound (50 mg kg−1). Mice were sacrificed at day 23. Serial tumor volume was obtained by bi‐dimensional diameter measurements with calipers by using Equation (1):
| (1) |
in which a is the major tumor diameter and b is the minor perpendicular diameter. Tumors were palpable, in general, 3 days to 5 days postinoculation.
Antitumor Effects of Compounds on Pancreatic PAN02
The syngeneic tumor PAN02 is a pancreatic tumor cell line obtained from NCI (0507232). PAN02 cells were grown in RPMI‐1640 containing 10 % fetal bovine serum. At day 0, 7.5×105 viable PAN02 cells (50 μL) were intradermally injected to produce localized tumors in 6‐to‐8‐week‐old C57BL/6 mice. The animals were then serially monitored by manual palpation for evidence of tumor. Mice were then treated every day with oral administration of vehicle (negative control) or compound (50 mg kg−1) and with intraperitoneal injection of gemcitabine (50 mg kg−1) at day 6 and day 12. Mice were sacrificed at day 35. Serial tumor volume was obtained by bi‐dimensional diameter measurements with calipers by using Equation (1). Tumors were palpable, in general, 3 days to 5 days postinoculation.
Antitumor Effects of Compounds on a Primary DA‐3 Breast Tumor
The syngeneic tumor DMBA3 (DA‐3, breast carcinoma model) arose from a preneoplastic lesion treated with 7,12‐dimethylbenzanthracene in female BALB/c mice. DA‐3 cells were grown as monolayer cultures in plastic flasks in RPMI‐1640 containing 0.1 mm nonessential amino acids, 0.1 μm sodium pyruvate, and 2 mm l‐glutamine. This was further supplemented with 50 μm 2‐mercaptoethanol and 10 % fetal bovine serum. The DA‐3 tumors were serially passaged in vivo by intradermal inoculation of 5×105 viable tumor cells (50 μL) to produce localized tumors in 6‐to‐8‐week‐old BALB/c mice. The animals were then serially monitored by manual palpation for evidence of tumor. Mice were treated at days 11 and 18 with cyclophosphamide (100 mg kg−1, IV injection) and by intravenous treatment at days 11, 12, 13, 15, 18, and 20 with compound. Mice were sacrificed at day 18. Serial tumor volume was obtained by bi‐dimensional diameter measurements with calipers by using Equation (1). Tumors were palpable, in general, 7 days to 10 days postinoculation.
Antitumor Effects of Compounds on Xenograft Human Prostate PC‐3 Tumor
The xenogenic human prostate tumor PC‐3 was obtained from ATCC (CRL1435). PC‐3 cells were grown in RPMI‐1640 containing 10 % fetal bovine serum. At day 0, viable PC‐3 (1.5 to 2×106, 50 μL) cells were intradermally injected to produce localized tumors in 6‐to‐8‐week‐old male CD1 nu/nu mice. The animals were then serially monitored by manual palpation for evidence of tumor. When the tumors reached a satisfactory volume, mice were randomized and then treated four, three, and three times a week for the first, second, and third weeks, respectively, with intravenous injection of vehicle (negative control), cyclophosphamide (positive control, 100 mg kg−1), or oral administration of compound (50 mg kg−1). Mice were sacrificed at day 50. Serial tumor volume was obtained by bi‐dimensional diameter measurements with calipers by using Equation (1).
Conflict of interest
Authors are employees of Prometic
Acknowledgements
The authors thank Lyne Marcil for preparation of this manuscript.
B. Zacharie, S. D. Abbott, J.-S. Duceppe, L. Gagnon, B. Grouix, L. Geerts, L. Gervais, F. Sarra-Bournet, V. Perron, N. Wilb, C. L. Penney, P. Laurin, ChemistryOpen 2018, 7, 737.
References
- 1. Hoelder S., Clarke P. A., Workman P., Mol. Oncol. 2012, 6, 155–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Al-Hajj M., Wicha M. S., Benito-Hernandez A., Morrison S. J., Clarke M. F., Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dai X., Li T., Bai Z., Yang Y., Liu X., Zhan J., Shi B., Am. J. Cancer Res. 2015, 5, 2929–2943. [PMC free article] [PubMed] [Google Scholar]
- 4. Hutchinson L., Nat. Rev. Clin. Oncol. 2010, 7, 669–670. [DOI] [PubMed] [Google Scholar]
- 5. Wolfgang C. L., Herman J. M., Laheru D. A., Klein A. P., Erdek M. A., Fishman E. K., Hruban R. H., CA Cancer J. Clin. 2013, 63, 318–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Makohon A., Jacobuzio-Donahue C. A., Nat. Rev. Cancer 2016, 16, 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Foletto M. C., Haas S. E., An. Bras. Dermatol. 2014, 89, 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rayburn E. R., Ezell S. J., Zhang R., Mol. Cell. Pharmacol. 2009, 1, 29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Grivennikov S., Greten F. R., Karin M., Cell 2010, 140, 883–899; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Mohan A., Narayanan S., Sethuraman S., Krishnan U. M., Anti-cancer Agents Med. Chem. 2013, 13, 281–295. [DOI] [PubMed] [Google Scholar]
- 10. Coussens L. M., Werb Z., Nature 2002, 420, 860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McKay C. J., Glen P., McMillan D. C., Best Pract. Res. Clin. Gastroenterol. 2008, 22, 65–73. [DOI] [PubMed] [Google Scholar]
- 12. Tobias D. K., Akinkuolie A. O., Chandler P. D., Lawler P. R., Manson J. E., Buring J. E., Ridker P. M., Wang L., Lee I.-M., Mora S., Am. J. Epidemiol. 2018, 187, 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Balkwill F., Cancer Metastasis Rev. 2006, 25, 409–416. [DOI] [PubMed] [Google Scholar]
- 14. Paule B., Terry S., Kheuang P., Soyeux F., Vacherot A., Taille A. de la, World J. Urol. 2007, 25, 477–489. [DOI] [PubMed] [Google Scholar]
- 15. Lu Y., Cai Z., Galson D. L., Xiao G., Liu Y., George D. E., Melhem M. F., Yao Z., Zhang J., The Prostate 2006, 66, 1311–1318. [DOI] [PubMed] [Google Scholar]
- 16. Tripsianis G., Papadopoulou E., Anagnostopoulos K., Botaitis S., Katotomichelakis M., Romanidis K., Kontomanolis E., Tentes I., Kortsaris A., Neoplasma 2014, 61, 205–212. [DOI] [PubMed] [Google Scholar]
- 17. Michalaki V., Syrigos K., Charles P., Waxman J., Br. J. Cancer 2004, 90, 2312–2316 (Correction: [DOI] [PMC free article] [PubMed] [Google Scholar]; Michalaki V., Odontiadis M., Syrigos K., Charles P., Waxman J., Br. J. Cancer 2004, 91, 1227). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Colombo M. P., Maccalli C., Mattei S., Melani C., Radrizzani M., Parmiani G., Melanoma Res. 1992, 2, 181–189. [DOI] [PubMed] [Google Scholar]
- 19. Hammamieh R., Sumaida D., Zhang X. Y., Das R., Jett M., BMC Cancer 2007, 7, 138; http://www.biomedcentral.com/1471-2407/7/138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Torres D., Paget C., Fontaine J., Mallevaey T., Matsuoka T., Maruyama T., Narumiya S., Capron M., Gosset P., Faveeuw C., Trottein F., J. Immunol. 2008, 180, 783–792. [DOI] [PubMed] [Google Scholar]
- 21. Greene E. R., Huang S., Serhan C. N., Panigrahy D., Prostaglandins Other Lipid Mediators 2011, 96, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Basedia D. K., Dubey B. K., Shrivastava B., Am. J. Pharmtech Res. 2011, 1, 174–193 [Google Scholar]
- 23.D. R. Shah, R. P. Modh, K. H. Chikhalia, Future Med. Chem 2014, https://www.future-science.com/doi/pdf/10.4155/fmc.13.212. [DOI] [PubMed]
- 24. Baliani A., Bueno G. J., Stewart M. L., Yardlev V., Brun R., Barrett M. P., Gilbert I. H., J. Med. Chem. 2005, 48, 5570–5579. [DOI] [PubMed] [Google Scholar]
- 25. Xiong Y.-Z., Chen F.-E., Balzarini J., De Clercq E., Pannecouque C., Eur. J. Med. Chem. 2008, 43, 1230–1236 and references cited therein. [DOI] [PubMed] [Google Scholar]
- 26. Yuefen Z., Zhongxiang S., Jamie M. F., Bioorg. Med. Chem. Lett. 2006, 16, 5451–5456.16890435 [Google Scholar]
- 27. Wackett L. P., Sadowsky M. J., Martinez B., Shapir N., Appl. Microbiol. Biotechnol. 2002, 58, 39–45. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Saleh M., Abbott S., Perron V., Lauzon C., Penney C., Zacharie B., Bioorg. Med. Chem. Lett. 2010, 20, 945–949; [DOI] [PubMed] [Google Scholar]
- 28b. Zhou C., Min J., Liu Z., Young Z., Deshazer H., Gao T., Chang Y.-T., Kallenbach R., Bioorg. Med. Chem. Lett. 2008, 18, 1308–1311; [DOI] [PubMed] [Google Scholar]
- 28c. Srinivas K., Srinivas U., Bhanuprakash K., Harakishore K., Murthy U. S. N., Jayathirtha Rao V., Eur. J. Med. Chem. 2006, 41, 1240–1246. [DOI] [PubMed] [Google Scholar]
- 29. Melato S., Prosperi D., Coghi P., Basilico B., Monti D., ChemMedChem 2008, 3, 873–876 and references cited therein. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Zacharie B., Abbott S., Bienvenu J.-F., Cameron A. D., Cloutier J., Duceppe J.-S., Ezzitouni A., Fortin D., Houde K., Lauzon C., Moreau N., Perron V., Wilb N., Asselin M., Doucet A., Fafard M.-E., Gaudreau D., Grouix B., Sarra-Bournet F., St-Amant N., Gagnon L., Penney C. L.. J. Med. Chem. 2010, 53, 1138–1145; [DOI] [PubMed] [Google Scholar]
- 30b. Zacharie B., Fortin D., Wilb N., Bienvenu J.-F., Asselin M., Grouix B., Penney C., Bioorg. Med. Chem. Lett. 2009, 19, 242–246. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Liu B., Sun T., Zhou Z., Du L., Med. Chem. 2015, 5, 131–148; [Google Scholar]
- 31b. Fraczyk J., Kolesinska B., Swiontek M., Lipinski W., Drozdowska D., Kaminski Z. J., Anti-Cancer Agents Med. Chem. 2016, 16, 1435–1444. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Venkatesan A. M., Dehnhardt C. M., Santos E. D., Chen Z., Dos Santos O., Ayral-Kaloustian S., Khafizova G., Brooijmans N., Mallon R., Hollander I., Feldberg L., Lucas J., Yu K., Gibbons J., Abraham R. T., Chaudhary I., Mansour T. S., J. Med. Chem. 2010, 53, 2636–2645; [DOI] [PubMed] [Google Scholar]
- 32b. Singla P., Luxami V., Paul K., Eur. J. Med. Chem. 2015, 102, 39–57. [DOI] [PubMed] [Google Scholar]
- 33. Hunt J. T., Mitt T., Borzilleri R., Gullo-Brown J., Fargnoli J., Fink B., Han W.-C., Mortillo S., Vite G., Wautlet B., Wong T., Yu C., Zheng X., Bhide R., J. Med. Chem. 2004, 47, 4054–4059. [DOI] [PubMed] [Google Scholar]
- 34.L. Hong et al., Chinese Patent 1970552 (A), 2007.
- 35.R. T. Timmer, C. W. Alexander, S. Pillarisetti, U. Saxena, K. R. Yeleswarapu, M. Pal, J. T. Reddy, V. V. Reddy, M. K. Rama, B. S. Sridevi, P. R. Kumar, G. O. Reddy, International Patent 2004/026844, 2004.
- 36. Blotny G., Tetrahedron 2006, 62, 9507–9522 and references cited therein. [Google Scholar]
- 37. Baldaniya B. B., Patel P. K., E-J. Chem. 2009, 6, 673–680. [Google Scholar]
- 38. Kamate C., Baloul S., Grootenboer S., Pessis E., Chevrot A., Tulliez M., Marchiol C., Viguier M., Fradelizi D., Int. J. Cancer 2002, 100, 571–579. [DOI] [PubMed] [Google Scholar]
- 39. Gao H., Sun W., Zhao J., Wu X., Lu J.-J., Chen X., Xu Q.-M., Khan I. A., Yang S., Sci. Rep. 2016, 6, 33720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fantozzi A., Christofori G., Breast Cancer Res. 2006, 8, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nakanishi M., Rosenberg D. W., Semin. Immunopathol. 2013, 35, 123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.For the safety profile of triazine compounds, see: Beaufils F., Cmiljanovic N., Cmiljanovic V., Bohnacker T., Melone A., Marone R., Jackson E., Zhang X., Sele A., Borsari C., Mestan J., Hebeisen P., Hillmann P., Giese B., Zvelebil M., Fabbro D., Williams R. L., Rageot D., Wymann M. P., J. Med. Chem. 2017, 60, 7524–7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.For the nontoxic profile of triazine compounds, see: Prashant G., Surajit K. G., J. Enzyme Inhib. Med. Chem. 2012, 27, 281–293.21657948 [Google Scholar]