

Abstract
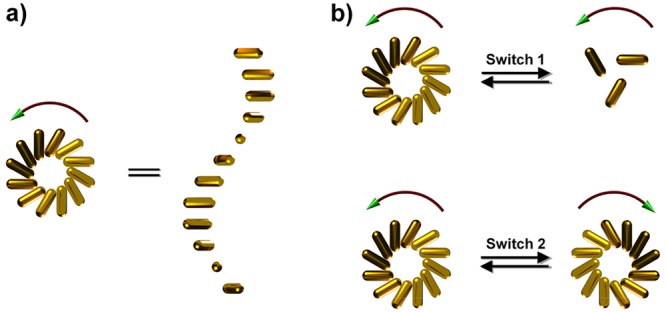
The ability to dynamically tune the self-assembled structures of nanoparticles is of significant interest in the fields of chemistry and material studies. However, it continues to be challenging to dynamically tune the chiral superstructures of nanoparticles and actively switch the chiral optical properties thereof. Here, we dynamically controlled a gold nanorod 3D chiral plasmonic superstructure (a stair helix with a pinwheel end view) templated by a DNA origami supramolecular polymer, using DNA-toehold-mediated conformational change in the DNA template. The gold nanorod chiral plasmonic helix was controllably reconfigured between a tightly folded state (with a small inter-rod angle) and an extended state (with a wide inter-rod angle) of the same handedness, or between two mirror-image-like structures of opposite handedness. As a result, the chiral plasmonic properties of the gold nanorod helix superstructures, in terms of the circular dichroism amplitude, peak response frequency, and signature of chirality, were actively switched upon the DNA-guided structural reconfiguration. We envision that the strategy demonstrated here will boost the advancement of reconfigurable chiral materials with increased complexity for active light control applications through rational molecular design and predictable self-assembly procedures.
Introduction
With the rapid increase in the sophistication of nanomaterials, reconfigurable materials and integrated devices have opened exciting new opportunities because of the emerging capability to dynamically tune and switch their functionalities.1−3 Noticeably, reconfigurable chiral materials enable us to actively control the light polarization,4 thus creating the possibility of various potential applications, for example, in smart sensing, tunable refractions, active color display, optical communication, and others. Fundamentally, chiral nanoparticle superstructures provide a framework for studying the collective optical interactions between nanoparticles. Their dynamic reconfiguration will offer a new pathway for active manipulation of the chiral-symmetry-breaking couplings with light.
To date, much effort has been dedicated to the fabrication of chiral nanostructures through wet synthesis and colloidal self-assembly, featured by parallel productions, and precise morphological and compositional control. For instance, chiral biomolecules are used to control the chiral crystalline structure of gold nanoparticles,5 semiconductor nanocrystals,6 and magnetic nanoparticles7 by influencing the growth of the nanocrystals. Also, various templates, such as nematic liquid crystal,8 fibers,9−11 peptides,12,13 DNA nanostructures,14−22 and others,23 have been used to guide the in situ synthesis or self-assembly of nanoparticles into chiral arrangements with tunable chirality. However, most of the developed chiral systems are static and display nonswitchable optical responses once formed.
Although there exists a variety of responsive/reconfigurable colloidal systems that are driven by molecular triggers,24−27 temperature,28,29 and light,30,31 for example, reconfigurable DNA-linked gold nanoparticle clusters32 and superlattice crystals,33,34 only scarce examples of reconfigurable chiral colloidal materials are available.35,36 Liu et al. created a chiral dimer of gold nanorod (AuNR) with a DNA origami template that can change the conformation in response to DNA triggers35 or light exposure.37 Kotov et al. reported chiral soft composites that can be reversibly twisted upon external mechanical tension by the deposition of gold nanoparticles layer-by-layer onto a polymer substrate.36 In sharp contrast to the strength in fabricating static chiral systems, the ability to dynamically reconfigure chiral nanostructures and actively switch the chiral optical properties is still largely limited. Especially, the dynamic tuning of the chiral nanoparticle superstructures through controllable and predictable nanoparticle self-assembly, with features engineered on the scale of tens of or hundreds of nanometers, remains a significant challenge so far.
A helix is one of the prototypical forms of chirality in nature, which can be found in α-helix peptides, DNA double helices, and plants. Therefore, it is fundamentally interesting to understand and tune the chirality of helices under various scenarios. Here, we created controllable reconfigurations of AuNR chiral plasmonic helixes that were templated by self-assembled chiral superstructures of DNA origami. DNA strand displacement reactions were rationally designed to dynamically tune the geometry of the DNA origami monomer. For example, with the dynamic modification of the interarm angle of a V-shaped DNA origami monomer, the AuNR chiral superstructures were transformed between a tightly folded state with a small inter-rod dihedral angle and an extended state with a larger inter-rod angle. The plasmonic chiroptical properties in terms of the chiral signal magnitude and frequency were reversibly switched upon the structural reconfiguration. Also, by dynamically changing the DNA origami monomer into its mirror-image structure, the AuNR chiral superstructure was transformed into its mirror image as well. This led to the inversion of the plasmonic chiral signal. To our knowledge, these results are the first demonstration of a reconfigurable chiral colloidal superstructure with controllably switched chirality through a DNA guided transformation process.
Principle
Figure 1 shows the principle of the DNA-guided self-assembly and reconfiguration of the AuNR superstructures. A new V-shaped DNA origami is designed, which consists of 3 layers of parallel DNA helixes in each arm and a 2 × 4 or 2 × 2 helix bundle switchable DNA spacer between the two arms (Figure 1A and B), or a fixed 4 helix bundle as the spacer (Figure 1C). Each DNA origami is decorated with one AuNR on one of the arms through complementary DNA hybridization. DNA origami monomers can be hierarchically connected in the left-handed direction, forming a helical supramolecular structure, through hybridization with a group of connector strands (38 strands in total, 13 nt or 14 nt each) at the corresponding helical ends of the two arms (shown as dashed lines in Figure 1A). Consequently, AuNRs functionalized with DNA origami are self-assembled into a left-handed chiral plasmonic superstructure (a stair helix with a pinwheel bottom-view shown in Figure 1A right side). Following the dynamic reconfiguration of the DNA origami at the spacer region (Figure 1B) or the hinge region (Figure 1C) via toe-hold-mediated strand displacement, the interarm angle of the V-shaped DNA origami is changed, so that the AuNR chiral superstructures are controllably transformed between the tightly folded and the extended states (Figure 1B), or between the left-handed and the right-handed states (Figure 1C).
Figure 1.

Schematic illustration of the principle of self-assembly and structural reconfiguration of the chiral plasmonic superstructures of AuNR. (A) AuNRs (12 nm × 38 nm) functionalized with V-shaped DNA origami are hierarchically self-assembled into a left-handed chiral superstructure (a stair helix with a pinwheel bottom-view), through DNA hybridization with connector strands at corresponding locations as indicated by the dashed lines. (B) DNA origami is dynamically converted between a tightly folded state and an extended state to make the reconfiguration of AuNR superstructure possible. The reconfiguration between these two states occurs via a toe-hold-mediated strand displacement reaction. Key strands and lock strands [(K1 and L2) or (K2 and L1)] are added in an alternating fashion to create the structural transformation. Each arm of the DNA origami is 36 nm in width, 24 nm along the helical axis, and 7.8 nm in thickness. The DNA spacer in the tightly folded state (left) is a 2 × 4 helix bundle with the DNA helical axis parallel to the helical direction of the arms that separates the two arms at an angle of approximately 30°. In the extended state (right) the spacer is structurally switched into a 2 × 2 helical bundle with the helical axis perpendicular to the helical direction of the arms that separates the two arms at an angle of ∼120°. (C) The DNA origami is designed to allow the dynamic conversion between the two mirror-image like structures, which enables the handedness inversion of the AuNR superstructure. The design of the DNA origami is similar to that in (B), but with a fixed 4 helix bundle in the middle as the spacer. The hinge of the V-shape is closed by a zipping hybridization of the two DNA handles (there are actually two pairs; only one pair is shown here) and opened by a toe-hold-mediated strand displacement. A combination of block strands and release strands [(B1 and R2) or (B2 and R1)] are added in an alternating fashion to make the structural transformation. The interarm angle is approximately 40° in both cases, taking into account the flexibility of the double helix connections at the hinges. A slightly larger angle for the right V-shape is expected as the 4 helix bundle linker between the two arms is not in the exact center position.
More precisely, to convert the AuNR superstructure from the tightly folded state to the extended state, and vice versa, the spacer of the DNA origami is dynamically modified upon DNA strand displacements, which makes the interarm angle of DNA origami reversibly change between ∼30° and ∼120°. As shown in Figure 1B, two sets of DNA helper strands (in gray) each contain a 12-nucleotide (nt)-long DNA toehold (as highlighted in blue or purple). These strands are the locking strands L1 and L2, respectively, which can hybridize with the scaffold to form two different DNA spacers necessary for the folded state and the extended state, respectively. During the conversion of the DNA origami from the folded state to the extended state, both of the key strands K1 and the locking strands L2 are added. The K1 strands are fully complementary to the L1 strands, and K1–L1 hybridization causes the 2 × 4 helical-bundle spacer of the folded state to decompose through a branch migration process.38 In the presence of the L2 strands, the new 2 × 2 helical-bundle spacer for the extended state is formed, and the interarm angle is increased. Similarly, the addition of the key strands K2 and the locking strands L1 enables the DNA origami to be reverted into the folded state that has the smaller interarm angle. With repeated alternative additions of the key strands and the locking strands, the DNA origami polymer and AuNR superstructure can be converted between the tightly folded state and the extended state.
To achieve the chiral inversion of the AuNR superstructure, an H-shaped DNA origami with a fixed DNA spacer is designed, wherein the two arms could be alternatively connected at one end or the other by complementary DNA hybridization to form a right V- or a left V-shaped structure (Figure 1C). Pairing of the touching strands extended from the edge helixes forms the hinge. The two arms are opened at one end with the addition of the blocking strands (B1 or B2) to deactivate the corresponding touching strands. With the addition of the release strands (R2 or R1), the blocking strands initially bound at the opposite end are dissociated from the DNA origami, and the corresponding touching strands are reactivated to pair with each other. For example, starting from the left side V-shape, the addition of B1 blocking strands (black), which have longer sequences complementary to the touching strands (red) on the left side, opens the closed hinge on the left through a branch migration process mediated by the cyan toehold (10-nt-long). The R2 release strands that have longer sequences complementary to the B2 (gray) strands initially bound on the right side can release the B2 strands, producing double-stranded B2:R2 wastes. This release will reactivate the touching strands (green) to enable the base-pairing and thus formation of the hinge on the right. Similarly, the R1 release strands release the complementary B1 blocking strands (black) bound on the left and reactivate the formation of the hinge on the left via hybridization of the red segment of the touching strands. Therefore, with a cycled activation of the touching strands on one end and a simultaneous deactivation on the other end, the connection of the DNA arms is destroyed and reproduced repeatedly, which causes the reversible conversion of the V-shaped DNA origami between two mirror-image-like structures.
Results and Discussion
We first used transmission electron microscopy (TEM) to characterize the DNA-guided self-assembly of AuNRs into a chiral plasmonic helix superstructure (Figure 2), and the subsequent reconfiguration of the superstructures between the folded state and the extended state. The DNA-capped AuNRs were site-specifically attached to the DNA origami monomer through DNA hybridization (DNA sequences are listed in the Supporting Information). When the DNA connector strands were added, the AuNRs functionalized with the DNA origami hierarchically self-assembled into a left-handed chiral plasmonic helix superstructure, as shown in Figure 2A. Because of the dry conditions necessary for the TEM imaging and the strong adhesion of the AuNRs to the grid surface, the flattening and the distortion of the three-dimensional superstructure was expected. The quasi-periodic arrangement of the AuNRs and long-range ordering of the superstructure are still evident in Figure 2. This result is in agreement with the model that is based solely on the structural and dimensional considerations of the DNA template. The black arrows in Figure 2A show deviations from the observation from the model, which can be explained by the adhesion of the DNA origami and the AuNRs on the TEM grid surface at the ends of superstructures. The red arrows show that when approaching the twisting point of the superstructure, AuNRs tend to stand up on the surface, due to the global restriction from the long-range ordering. The statistics in Figure 2B reveals that the assembled superstructures consist mainly of 6–12 AuNRs, which form helical superstructures of 0.5–1 turn.
Figure 2.

TEM characterization of the self-assembled AuNR chiral superstructures. (A) Different views of the 3D model of the AuNR superstructure (i), observed objects after flattening, and corresponding models that are traced from the observation (ii–i). The black arrows point to the adhesion of AuNRs to the surface, while the red arrows show the twist point of the superstructure. (B) The statistics of number of AuNRs in the assembled superstructures. 500 superstructures were counted.
To convert the DNA origami template between the folded state and the extended state, the molar ratio of both the locking strands and the key strands to DNA origami was set to (40 × N):1 in each step reaction, where N is the step number. Successful reconfigurations of DNA origami on the level of both monomers and supramolecular polymers were confirmed (Figure 3). The transformation efficiency of the DNA origami monomers was above 80%. It is noteworthy that the DNA origami may land on the TEM grid surface at different orientations because of their three-dimensional structure. However, the dominant attachment direction on the surface was relatively controlled through the appropriate modification of the hydrophilicity of the grid surface (Supporting Information, note S4). As illustrated in Figure 3A and C, the DNA origami of the folded state, before reconfiguration and after recovery, were made to have a differential preference of attachment to the surface with either the sides or the ends of the DNA helix of the arm, controlled by adjusting the hydrophilicity of the grid surface (Figure 3B and Figure S4). As such, we verified the correct formation/transformation of the DNA origami from viewing the origami under TEM imaging under different views.
Figure 3.

Characterization of the reconfiguration of the DNA origami template between the folded state and extended state. (A–C) A full cycle of successive transformation of the DNA origami monomer between the folded state and extended state. Zoomed-in images of a DNA origami monomer and models with corresponding attachment directions on the surface are presented. The scale bars in the zoomed-in and zoomed-out images are 25 and 50 nm, respectively. (D–F) A full cycle of successive transformation of the DNA origami supramolecular polymer between the folded state and extended state. The top and side views of the supra-structure model are presented. The scale bars in the zoomed-in and zoomed-out images are 50 and 100 nm, respectively.
Figure 3 shows the successive conversion of both the monomeric and the polymeric DNA origami from a folded state to an extended state and then back to the folded state. Clear morphological changes were observed after each step of the transformation. In the close-up view of the monomeric DNA origami, the observed objects matched well with the designed model, with a preferred corresponding attachment orientation on the surface (Figure 3A–C). The polymeric DNA origami of the folded state showed a clearly left-handed hierarchical superstructure (Figure 3D and F), while the extended-state polymeric structures displayed a quasi-one-dimensional arrangement of DNA arms with alternative units standing-up, which also match well with those predicted from the design (Figure 3E). The high structural fidelity before and after a full cycle of structural conversions of both DNA origami monomers and polymers manifests a highly reversible structural reconfiguration.
The optical behaviors after the structural conversion of the AuNR chiral superstructures between the folded state and the extended state were examined. Circular dichroism (CD), which denotes the difference between absorptions for left-handed and right-handed circularly polarized light (LCP and RCP) (Figure 4A), was used to characterize the optical responses after each step of the structural transformation of the chiral superstructures. As illustrated in Figure 4B, the AuNR superstructure of the folded state gave rise to a peak-dip bisignated CD line-shape due to the left-handed conformation. The relatively weak CD intensity is due to the low concentration of the superstructures typically in the range of 0.01–0.05 nM in the CD measurement. However, the dissymmetry factor was about 0.01, which is comparable to that of previously reported self-assembled chiral superstructures.9,15 When the AuNR superstructure was converted into the extended state, the peaked CD value decreased, partially due to the highly repeating structures (3 units per turn) of the extended state, which causes a decrease in the chiral anisotropy. The peak wavelength was red-shifted, because the dominant plasmon resonances between the AuNRs changed from antibonding modes for the folded state to bonding modes for the extended state.39Figure 4C demonstrates the reversible oscillations of the CD peak value and peak wavelengths with the cycling of the structural transformation.
Figure 4.

Optical switching of AuNR chiral superstructures due to structural reconfigurations between a folded state and an extended state. (A) Illustration of interactions between the incident circularly polarized light and the AuNR chiral superstructure. (B) Experimentally measured CD spectra of the superstructure from the folded state (black) and after its conversion to an extended state (red). (C) Cycling of the CD peak value and peak wavelength with a reversible reconfiguration of the superstructure. (D) Theoretically calculated absorption spectra of the superstructure in the folded state (30°) under LCP and RCP, respectively. Only the absorptions with light illumination along the axis of the superstructure (Z direction) were shown. The inset shows a chiral field profile of one individual AuNR in the superstructure, excited under LCP at 670 nm (CD peak wavelength) (Figure S14). (E) Calculated CD spectra of the superstructure in the folded state (30°), in the extended state (120°), and in an intermediate state (51°). The CD calculation is averaged over X, Y, and Z directions. (F) The statistics of the interarm angle of the DNA origami for a one-step conversion. The folded state and the extended state show a peaked interarm angle at 30° and 120°, respectively. 500 and 1000 particles were counted for the folded state and the extended state, respectively. After reconfiguration, an intermediate state shows a small peak at the 50° interarm angle.
We further verified our experimental observations through theoretical calculations with a finite-difference time-domain (FDTD) method (see Supporting Information for details). The calculated absorption spectra of the superstructure under LCP exhibited a blue shift as compared to that under RCP (Figure 4D). A power absorption map and field profile of one individual AuNR (e.g., the inset in Figure 4D) within the superstructure was also calculated; the results further confirmed the absorption differences and mode generation under LCP and RCP (Figures S13 and 14). The calculated CD spectra for different inter-rod angles all displayed a peak-dip CD line shape (Figure 4E). The physical origin of this CD line shape is in the plasmon–plasmon interactions between the gold nanorods that are arranged in a helical fashion.40 The AuNR superstructure in the extended state (120°) exhibited a lower CD intensity and red-shifted CD line shape as compared to the folded state (30°) in the calculated results (Figure 4E), which is qualitatively consistent with the experimental observations (Figure 4B). However, the quantitative results of the intensity differences and the degrees of spectral shift in the experimental CD spectra were both smaller than those in the calculated results. This was because the experimental transformation efficiency of the superstructure between the two states was less than 100% and some intermediate conformations of the superstructure were present. From the statistical analysis of the interarm angle of the DNA origami monomer for a one-step reconfiguration (Figure 4F), the intermediate conformations exhibited an interarm angle of around 50°. We also calculated the CD spectrum of an intermediate AuNR superstructure consisting of 7 nanorods with an interarm angle of ∼51°. The calculated CD spectrum was between those of 30° and 120° (Figure 4E). This result explains the deviation of the experimental CD signals from the calculations after the reconfiguration mainly due to the less than 100% structural conversion yield (Figure 4B and E).
Our use of self-assembled chiral plasmonic superstructures not only allows us to dynamically tune the optical responses in terms of chiral signal intensity and frequency, but we can also actively switch the intrinsic optical chirality. As shown in Figure 5, we further demonstrated the dynamic control of a V-shape DNA origami template between two mirror-image structures for the chirality inversion of AuNR superstructures. The original H-shape was in a relaxed state whereby pairing of the touching strands was deactivated by the blocking strands (B1 and B2). However, this H-shape could be converted into a locked V-shape with interarm connections on the left side or right side (Figure 5A) when the R1 or R2 release strands are added, respectively. The TEM images validated the successful conversions (Figure 5B). Experimentally, the release strands were added to the DNA origami in a molar ratio of 100:1 to increase the transition efficiency. The efficiency of the structural transformation (from H to V) was ∼70% as evidenced by the wide field images (Figure S8). The self-assembly of the V-shaped DNA origami monomers into the polymeric superstructures is similar to that presented above.
Figure 5.

Reconfiguration of a DNA origami to create a chiral inversion of an AuNR superstructure. (A) The mechanism of the conversion reaction from an H shape into two mirror-image V-shaped structures. (B) TEM images to confirm the structural conversion of the DNA origami.
We explored the optical chirality inversion of the AuNR superstructures through both experimental CD measurements and theoretical simulations. Figure 6A schematically illustrates the dynamic reconfiguration of AuNR superstructures between the ideally relaxed state (middle) and the targeted chiral conformation of opposite handedness (left and right). The relaxed state of AuNR superstructures was obtained from the polymerization of AuNRs functionalized with H-shaped DNA origami monomers (Figure 6B). After the transformation of the AuNR superstructures from the relaxed state into a left-handed or right-handed conformation, the experimentally measured CD spectra displayed vertically mirrored shapes (Figure 6C). With the reconfiguration of the superstructures from the left-handed to the right-handed shapes, a reversed CD spectrum was also observed (Figure 6D). The structural conversion was reversible; however, an even lower recovery efficiency was observed in this case. One possible reason was due to the leakage of the strand displacement reactions in the presence of numerous toehold-carrying strands, which included interactions among the blocking strands, the release strands, and the touching strands. This may interfere with the hybridization of the appropriate pair of touching strands and thus result in a decrease in the conversion efficiency of the DNA origami. Furthermore, different from the scaffold-involved formation of a rigid spacer for controlling the interarm angle of the DNA origami as discussed above, only two pairs of touching strands were used here to connect the DNA arms at each end. This gave rise to more structural flexibility, and made the DNA origami more vulnerable to strand interferences in the multi-DNA system. To further improve the efficiency of handedness switching, more optimization of the structural characteristics of the superstructures are needed, such as the interarm angle of the DNA origami, flexibility of the scaffold and helper links between the DNA arm and the DNA spacer, DNA sequences of the blocking strands and release strands, and even the gold nanorod sizes that will influence their charge repulsions. Nonetheless, we demonstrated the working principle for the structural switching, the AuNR superstructures could be converted between the two enantiomeric conformations as reflected by the inversion of the optical chirality. As expected, theoretical calculations of the left-handed and right-handed superstructures exhibited reversed CD line shapes in accordance with the experiments (Figure 6E).
Figure 6.

Optical chirality inversion of AuNR superstructures. (A) Mechanistic illustration of the conversion of AuNR superstructures into two mirror-image states. (B) TEM images of the relaxed state of the DNA origami superstructures (i) and AuNR superstructures (ii), respectively. Different views of the superstructures are presented. The scale bar is 50 nm. (C) Experimentally measured CD spectra of the two mirror-imaged states converted from the relaxed state. (D) CD spectra for a full cycle of conversion between the left-handed and right-handed superstructures. The dotted spectrum shows a low recovery efficiency after a cycle of reconfigurations. (E) Theoretically calculated CD spectra of a left-handed and a right-handed superstructure. An interarm angle of 30° was used for the left-handed structure in the calculation, and 51° for the right-handed structure. These parameters were obtained from analyzing the TEM images of the DNA origami template for each superstructure (images similar to those shown in Figure 5B).
Conclusion
We presented the first example of a reconfigurable chiral nanoparticle helix superstructure with fully switchable chirality. Through DNA-based self-assembly of AuNR chiral helix superstructures and rationally designed DNA strand displacement reactions, the active transformation of the assembled superstructures was achieved. The superstructures were transformed between a tightly folded state and an extended state of the same handedness, or between two mirror-image states of opposite handedness, following the dynamic control of the underlying DNA template. The chiral plasmonic responses of the superstructures were actively switched upon the DNA-guided structural reconfiguration. We envision that our developed general framework of nanoparticle helix will bring new possibilities of chiral optical tuning and multifunctionality of chiral materials when different functional building blocks are utilized for the self-assembly, such as molecular quantum emitters, semiconductor quantum dots, or even magnetic nanoparticles. We expect that the explorations of the hybrid nanoparticle chiral superstructures will enhance our understanding of a plethora of physical phenomena, such as magnetochirality, chiral quantum optics, etc., and their dynamic engineering will add a new dimension for various active light control applications. The biomolecule-triggered responsiveness of the chiral nanoparticle superstructures also shows the potential for building up new artificial smart biophotonic systems with increased complexity, transformable tuning, and optical switching.
Acknowledgments
This work was supported by grants from the Army Research Office, National Institute of Health, Office of Naval Research, and National Science Foundation to H.Y. and Y.L. We acknowledge Dr. Shuoxing Jiang and Dr. Fei Zhang for helpful discussions. This work was also supported by Volkswagen Foundation (A.O.G.) and via the 1000-talent Award of Sichuan, China (A.O.G.). A.O.G. holds Chang Jiang (Yangtze River) Chair Professorship in China. T.L. is financially supported by the Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b06526.
Methods, DNA origami design, optical calculation, and supplementary figures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Song J.; Li Z.; Wang P. F.; Meyer T.; Mao C. D.; Ke Y. G. Science 2017, 357, eaan3377. 10.1126/science.aan3377. [DOI] [PubMed] [Google Scholar]
- Zheludev N. I.; Plum E. Nat. Nanotechnol. 2016, 11, 16–22. 10.1038/nnano.2015.302. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Xiao J.; Zhu H. Y.; Li Y.; Alsaid Y.; Fong K. Y.; Zhou Y.; Wang S. Q.; Shi W.; Wang Y.; Zettl A.; Reed E. J.; Zhang X. Nature 2017, 550, 487–491. 10.1038/nature24043. [DOI] [PubMed] [Google Scholar]
- Lee D.; Han S. E. Nat. Mater. 2016, 15, 377–378. 10.1038/nmat4605. [DOI] [PubMed] [Google Scholar]
- Lee H. E.; Ahn H. Y.; Mun J.; Lee Y. Y.; Kim M.; Cho N. H.; Chang K.; Kim W. S.; Rho J.; Nam K. T. Nature 2018, 556, 360–365. 10.1038/s41586-018-0034-1. [DOI] [PubMed] [Google Scholar]
- Ben-Moshe A.; Wolf S. G.; Bar Sadan M.; Houben L.; Fan Z. Y.; Govorov A. O.; Markovich G. Nat. Commun. 2014, 5, 4302. 10.1038/ncomms5302. [DOI] [PubMed] [Google Scholar]
- Yeom J.; Santos U. S.; Chekini M.; Cha M.; de Moura A. F.; Kotov N. A. Science 2018, 359, 309–314. 10.1126/science.aao7172. [DOI] [PubMed] [Google Scholar]
- Querejeta-Fernandez A.; Chauve G.; Methot M.; Bouchard J.; Kumacheva E. J. Am. Chem. Soc. 2014, 136, 4788–4793. 10.1021/ja501642p. [DOI] [PubMed] [Google Scholar]
- Guerrero-Martinez A.; Auguie B.; Alonso-Gomez J. L.; Dzolic Z.; Gomez-Grana S.; Zinic M.; Cid M. M.; Liz-Marzan L. M. Angew. Chem., Int. Ed. 2011, 50, 5499–5503. 10.1002/anie.201007536. [DOI] [PubMed] [Google Scholar]
- Lin Y. Y.; Pashuck E. T.; Thomas M. R.; Amdursky N.; Wang S. T.; Chow L. W.; Stevens M. M. Angew. Chem., Int. Ed. 2017, 56, 2361–2365. 10.1002/anie.201610976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa M.; Kawai T. J. Am. Chem. Soc. 2018, 140, 4991–4994. 10.1021/jacs.8b00910. [DOI] [PubMed] [Google Scholar]
- Mokashi-Punekar S.; Merg A. D.; Rosi N. L. J. Am. Chem. Soc. 2017, 139, 15043–15048. 10.1021/jacs.7b07143. [DOI] [PubMed] [Google Scholar]
- Merg A. D.; Boatz J. C.; Mandal A.; Zhao G.; Mokashi-Punekar S.; Liu C.; Wang X.; Zhang P.; van der Wel P. C.; Rosi N. L. J. Am. Chem. Soc. 2016, 138, 13655–13663. 10.1021/jacs.6b07322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan X.; Chen Z.; Dai G.; Lu X.; Ni W.; Wang Q. J. Am. Chem. Soc. 2013, 135, 11441–11444. 10.1021/ja404354c. [DOI] [PubMed] [Google Scholar]
- Lan X.; Lu X. X.; Shen C. Q.; Ke Y. G.; Ni W. H.; Wang Q. B. J. Am. Chem. Soc. 2015, 137, 457–462. 10.1021/ja511333q. [DOI] [PubMed] [Google Scholar]
- Mastroianni A. J.; Claridge S. A.; Alivisatos A. P. J. Am. Chem. Soc. 2009, 131, 8455–8459. 10.1021/ja808570g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzyk A.; Schreiber R.; Fan Z.; Pardatscher G.; Roller E. M.; Hogele A.; Simmel F. C.; Govorov A. O.; Liedl T. Nature 2012, 483, 311–314. 10.1038/nature10889. [DOI] [PubMed] [Google Scholar]
- Cecconello A.; Kahn J. S.; Lu C. H.; Khorashad L. K.; Govorov A. O.; Willner I. J. Am. Chem. Soc. 2016, 138, 9895–9901. 10.1021/jacs.6b04096. [DOI] [PubMed] [Google Scholar]
- Urban M. J.; Dutta P. K.; Wang P. F.; Duan X. Y.; Shen X. B.; Ding B. Q.; Ke Y. G.; Liu N. J. Am. Chem. Soc. 2016, 138, 5495–5498. 10.1021/jacs.6b00958. [DOI] [PubMed] [Google Scholar]
- Yan W. J.; Xu L. G.; Xu C. L.; Ma W.; Kuang H.; Wang L. B.; Kotov N. A. J. Am. Chem. Soc. 2012, 134, 15114–15121. 10.1021/ja3066336. [DOI] [PubMed] [Google Scholar]
- Shen X.; Song C.; Wang J.; Shi D.; Wang Z.; Liu N.; Ding B. J. Am. Chem. Soc. 2012, 134, 146–149. 10.1021/ja209861x. [DOI] [PubMed] [Google Scholar]
- Lan X.; Su Z.; Zhou Y.; Meyer T.; Ke Y.; Wang Q.; Chiu W.; Liu N.; Zou S.; Yan H.; Liu Y. Angew. Chem., Int. Ed. 2017, 56, 14632–14636. 10.1002/anie.201709775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B.; Zhu Z. N.; Li Z. T.; Zhang W.; Tang Z. Y. J. Am. Chem. Soc. 2014, 136, 16104–16107. 10.1021/ja506790w. [DOI] [PubMed] [Google Scholar]
- Shim T. S.; Estephan Z. G.; Qian Z. X.; Prosser J. H.; Lee S. Y.; Chenoweth D. M.; Lee D.; Park S. J.; Crocker J. C. Nat. Nanotechnol. 2017, 12, 41–47. 10.1038/nnano.2016.192. [DOI] [PubMed] [Google Scholar]
- Ohta S.; Glancy D.; Chan W. C. W. Science 2016, 351, 841–845. 10.1126/science.aad4925. [DOI] [PubMed] [Google Scholar]
- Nie Z.; Fava D.; Kumacheva E.; Zou S.; Walker G. C.; Rubinstein M. Nat. Mater. 2007, 6, 609–614. 10.1038/nmat1954. [DOI] [PubMed] [Google Scholar]
- He J.; Liu Y. J.; Babu T.; Wei Z. J.; Nie Z. H. J. Am. Chem. Soc. 2012, 134, 11342–11345. 10.1021/ja3032295. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Han X.; He L.; Yin Y. Angew. Chem., Int. Ed. 2012, 51, 6373–6377. 10.1002/anie.201201816. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Santos P. J.; Gabrys P. A.; Lee S.; Liu C.; Macfarlane R. J. J. Am. Chem. Soc. 2016, 138, 16228–16231. 10.1021/jacs.6b11052. [DOI] [PubMed] [Google Scholar]
- Ding T.; Valev V. K.; Salmon A. R.; Forman C. J.; Smoukov S. K.; Scherman O. A.; Frenkel D.; Baumberg J. J. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 5503–5507. 10.1073/pnas.1524209113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y.; Chen J. I.; Ginger D. S. Nano Lett. 2012, 12, 2530–2536. 10.1021/nl300739n. [DOI] [PubMed] [Google Scholar]
- Aldaye F. A.; Sleiman H. F. J. Am. Chem. Soc. 2007, 129, 4130–4131. 10.1021/ja070017i. [DOI] [PubMed] [Google Scholar]
- Mason J. A.; Laramy C. R.; Lai C. T.; O’Brien M. N.; Lin Q. Y.; Dravid V. P.; Schatz G. C.; Mirkin C. A. J. Am. Chem. Soc. 2016, 138, 8722–8725. 10.1021/jacs.6b05430. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Macfarlane R. J.; Mirkin C. A. J. Am. Chem. Soc. 2013, 135, 10342–10345. 10.1021/ja405988r. [DOI] [PubMed] [Google Scholar]
- Kuzyk A.; Schreiber R.; Zhang H.; Govorov A. O.; Liedl T.; Liu N. Nat. Mater. 2014, 13, 862–866. 10.1038/nmat4031. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Yeom B.; Arteaga O.; Yoo S. J.; Lee S. G.; Kim J. G.; Kotov N. A. Nat. Mater. 2016, 15, 461–468. 10.1038/nmat4525. [DOI] [PubMed] [Google Scholar]
- Kuzyk A.; Yang Y. Y.; Duan X.; Stoll S.; Govorov A. O.; Sugiyama H.; Endo M.; Liu N. Nat. Commun. 2016, 7, 10591. 10.1038/ncomms10591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D. Y.; Seelig G. Nat. Chem. 2011, 3, 103–113. 10.1038/nchem.957. [DOI] [PubMed] [Google Scholar]
- Prodan E.; Radloff C.; Halas N. J.; Nordlander P. Science 2003, 302, 419–422. 10.1126/science.1089171. [DOI] [PubMed] [Google Scholar]
- Fan Z. Y.; Govorov A. O. Nano Lett. 2010, 10, 2580–2587. 10.1021/nl101231b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.