

Abstract
Bioorthogonal reactions are selective transformations that are not affected by any biological functional group and are widely used for chemical modification of biomolecules. Recently, we reported that vinylboronic acids (VBAs) gave exceptionally high reaction rates in the bioorthogonal inverse electron-demand Diels–Alder (iEDDA) reaction with tetrazines bearing a boron-coordinating pyridyl moiety compared to tetrazines lacking such a substituent. In this integrated experimental and theoretical study, we show how the reaction rate of the VBA-tetrazine ligation can be accelerated by shifting the equilibrium from boronic acid to the boronate anion in the reaction mixture. Quantum chemical activation strain analyses reveal that this rate enhancement is a direct consequence of the excellent electron-donating capability of the boronate anion in which the π HOMO is pushed to a higher energy due to the net negative potential of this species. We have explored the second-order rate constants of several tetrazines containing potential VBA-coordinating hydroxyl substituents. We observed an increase in rate constants of several orders of magnitude compared to the tetrazines lacking a hydroxyl substituent. Furthermore, we find the hydroxyl-substituted tetrazines to be more selective toward VBAs than toward the commonly used bioorthogonal reactant norbornene, and more stable in aqueous environment than the previously studied tetrazines containing a pyridyl substituent.
Introduction
The development of bioorthogonal reactions has advanced tremendously as it allows selective modification of biomolecules without interfering with any naturally occurring biochemical functionality.1−4 The tetrazine ligation is one of the most popular bioorthogonal reactions due to its selectivity and high reaction rates.5−8 So far, several bioorthogonal reactants have been developed for this inverse electron-demand Diels–Alder (iEDDA) reaction, such as strained alkynes (e.g., bicyclo[6.1.0]nonyne (BCN)),9,10 strained alkenes (e.g., trans-cyclooctene (TCO),11,12 norbornene,13 and cyclopropene),14,15 and nonstrained alkenes (e.g., primary alkenes).16,17 During our efforts to improve the reaction rates of the slow-reacting nonstrained alkenes, we found that vinylboronic acids (VBAs) show impressive second-order rate constants (k2) with 3,6-dipyridiyl-s-tetrazines of up to 27 M–1 s–1 in 5% MeOH/PBS (pH = 7.4).18 The VBAs are easily accessible, biocompatible with cellular components, and suitable for protein modification. Recently, we showed that vinylboronic acids are also suitable for a bioorthogonal application within the living cell.19
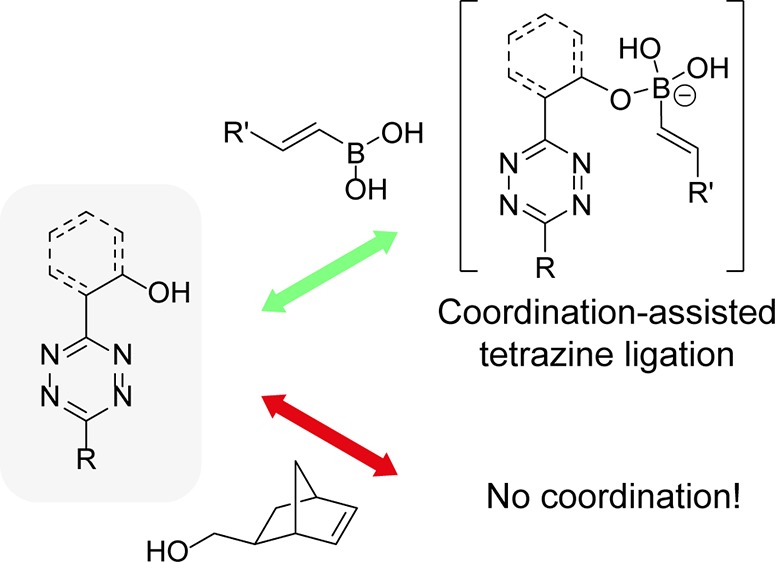
Boronic acids possess a vacant p orbital and are mild organic Lewis acids that form the more electron-rich boronate complex after coordination by a Lewis base. In aqueous media, the boronic acids are in equilibrium with their negatively charged boronate anion, although the VBAs have high pKa values and therefore reside mainly in the trivalent boronic acid form at physiological pH (pH = 7.4) (Figure 1A).20 Recently, we reported that VBAs are much more reactive toward pyridyl-substituted tetrazines than toward tetrazines lacking this Lewis basic substituent (Figure 1B).18,21 We observed that the high reaction rates are caused by coordination of the nitrogen of the pyridyl ring to the boron of the VBA, which favors the reaction due to the induced proximity and the inductive effect. We used this unique reactivity of the VBA to develop two orthogonal tetrazine ligations and perform a simultaneous dual labeling of two proteins containing a VBA and a norbornene moiety.21
Figure 1.

(A) Equilibrium between boronic acid 1a and boronate anion 1b in aqueous environment. (B) Coordination of the nitrogen of the pyridyl ring of tetrazine 2a to the boronic acid of VBA 1, yielding dihydropyridazine 3 as a single isomer.
Herein, we explore the reactivity of the boronic acids and their tetravalent boronate counterparts toward tetrazines in more detail in a combined theoretical and experimental study, based on density functional theory (DFT) and rate measurements as a function of pH. In addition, we discuss the reactivity of a set of tetrazines bearing conceivable boron-coordinating hydroxyl substituents. We hypothesized that the hydroxyl moiety would favor coordination to the VBA and increase the rate constants of the tetrazine-VBA ligation. Furthermore, we anticipate that orthogonality of these tetrazines toward VBAs could be established as more electron-rich tetrazines react less favorably in the general iEDDA reaction with unsubstituted (strained) alkenes.
Results and Discussion
DFT Calculations of the Tetrazine Ligation with VBAs
To provide more insight into the reactivity of the VBA and the boronate anion in the iEDDA reaction with tetrazines, we have explored suitable model reactions using DFT calculations as implemented in the Amsterdam Density Functional (ADF) 2013 software.22 For all calculations, the generalized gradient approximation (GGA) was used at the OLYP level.23−25 As a basis set, the TZ2P level was used.22 Vibrational analyses confirmed zero imaginary frequencies for equilibrium structures and a single imaginary frequency associated with normal mode along the reaction coordinate for transition states.
We first determined the optimized geometries, reaction energies, and transition states of the reaction of vinylboronic acid 6a with tetrazine 2b which bears no substituents and cannot coordinate to the boronic acid of the VBA, until the transition state (TS) (Table 1). We compare this reaction to the tetrazine ligations with a nonsubstituted alkene, ethene 4, and with an alkene containing an electron-donating substituent, methylvinylether 5. The tetrazine ligation involving methylvinylether 5 has a transition state barrier (ΔETS) that is comparable to that for ethene 4, although both have a lower ΔETS than VBA 6a (Table 1). Also, the reaction energy (Er) with ethene 4 is more favorable than with VBA 6a, although all reactions are pronouncedly exothermic, with values of −17, −8.0, and −12.8 kcal mol–1 for 4, 5, and 6a, respectively.
Table 1. Quantum-Chemically Computed Key Parameters of the Ligation of Tetrazine 2b with Alkenes 4–6a.
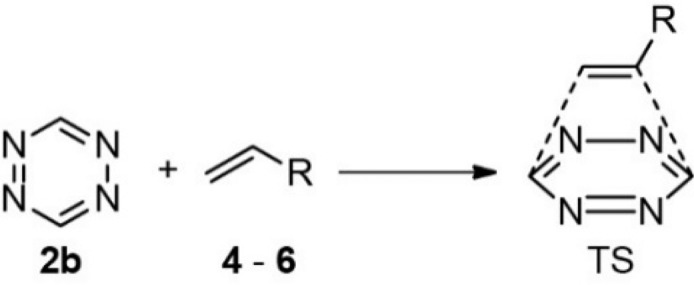

Computed at OLYP/TZ2P. Reaction energy (Er) and change in transition state (ΔETS), interaction (ΔEint), and strain energies (ΔEstrain) in kcal mol–1 with ΔETS = ΔEstrain – ΔEint. HOMO and LUMO energies in eV. [a] Energy of the HOMO–2, which is the occupied π orbital.
Next, we compared the tetrazine ligations of tetrazine 2b with vinylboronic acid 6a and the tetracoordinated boronate anion 6b (Table 1). Both ΔETS and Er are much lower for boronate 6b than for boronic acid 6a, with ΔETS + 1.6 kcal mol–1 and Er of −29.1 kcal mol–1 for 6b, and only ΔETS of +18.2 kcal mol–1 and Er of −12.8 kcal mol–1 for 6a.
To trace the origin of the enhanced reactivity of 6b relative to 6a, we have carried out activation-strain analyses (also known as distortion/interaction analyses),26 in combination with an analysis of the bonding mechanism between the reactants in the TS based on Kohn–Sham MO theory.27 The lower barrier for the boronate anion compared to vinylboronic acid reacting with tetrazine is mainly caused by a more stabilizing interaction energy ΔEint in the case of the former. Next, we analyzed the orbital electronic structure of the alkenes 4–6 and tetrazine 2b (Tables 1 and S3). Figure 2 is a schematic representation of the frontier molecular orbital interactions between VBA 6a and its boronate anion 6b with the tetrazine as they emerge from our quantitative Kohn–Sham MO analyses. The dominant bonding mechanism is a donor–acceptor orbital interaction between the occupied C–C π bonding orbital of the alkene and the LUMO+1 acceptor orbital of tetrazine 2b. In the case of vinylboronic acid 6a, the electron-donating π orbital is the HOMO whereas for vinylboronate 6b, it is the HOMO–2.
Figure 2.
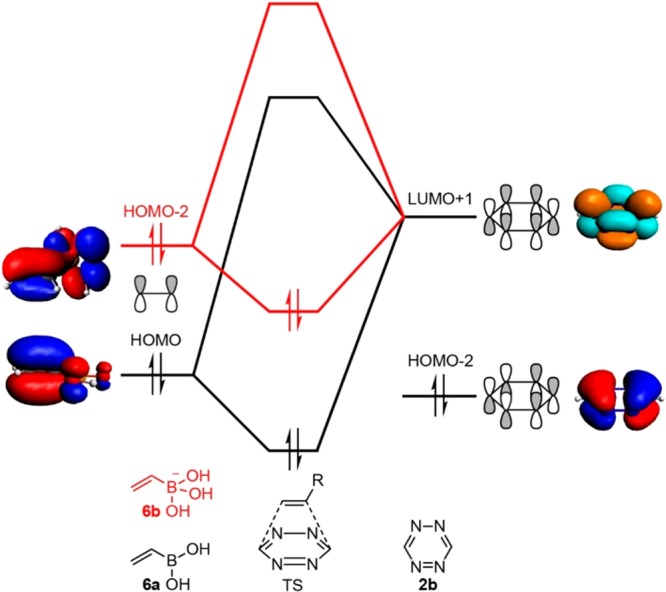
Frontier orbital interactions in the tetrazine ligation. Schematic representation of the frontier molecular orbital interactions of 1,2,4,5-tetrazine 2b and vinylboronic acid 6a (black) or vinylboronate 6b (red) from our quantitative Kohn–Sham MO analyses.
Our analyses reveal that the π orbital in boronate 6b is at higher orbital energy than that in boronic acid 6a, because of the more negative potential in the former, anionic reactant. Consequently, the orbital-energy gap with the tetrazine acceptor orbital is smaller and the interaction ΔEint more stabilizing for the boronate anion 6b than for its boronic acid analogue 6a. Eventually, as pointed out above, this leads to the lower barrier for the reaction involving 6b. Alkenes 4 and 5 have a similar or slightly higher HOMO than vinylboronic acid 6a (Table 1). Therefore, 4 and 5 should also undergo an iEDDA reaction with tetrazine 2b (vide infra). Our quantum chemical results agree with our previous finding21 that only tetrazines bearing the pyridyl substituent gave high rate constants with VBAs compared to tetrazines lacking a Lewis basic substituent.
pH Effect on the Tetrazine Ligation with VBAs
Next, we investigated whether the lower reaction energy of the boronate anion toward tetrazines could be validated in an experimental setup (Figure 3A). As formation with the boronate anion is favored upon increasing pH, we expected that this would also advance the tetrazine ligation. We examined the reaction of (E)-phenylvinylboronic acid 1 with 3-phenyl-s-tetrazine 2c, lacking a boron-coordinating atom, between pH 8 and 11. Indeed, the rate of the reaction of VBA 1 with tetrazine 2c increased at higher pH, indicating that the boronate anion is more reactive than the boronic acid, in accordance with the DFT calculations (Figure 3B). As a control experiment, we examined the reaction of tetrazine 2c with norbornene 5 in the same buffers and observed that the rate is independent of the pH, as expected. In the absence of an alkene, tetrazine 2c slowly degrades, especially in the basic aqueous solutions (Figure S1).
Figure 3.
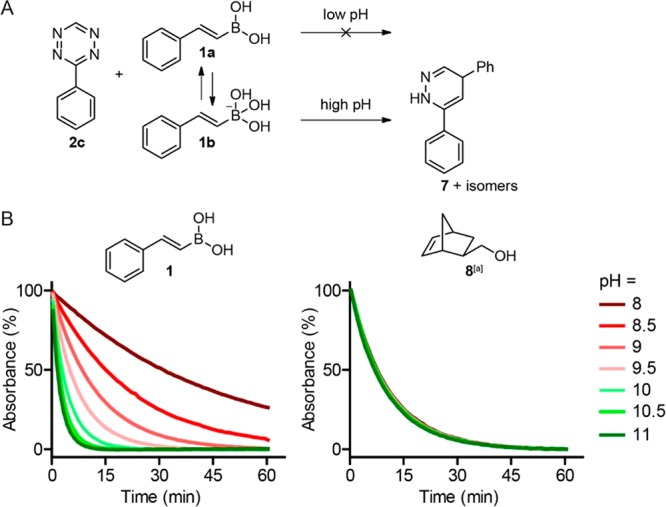
(A) Schematic overview of the pH effect on the reaction of tetrazine 2c with VBA 1 yielding dihydropyridazine 7. (B) Reactions of tetrazine 2c with VBA 1 (left) and norbornene 8 (right) in 50% MeOH and 50% Na2B4O7 buffer, ranging from pH 8 until 11. The reactions were measured at 20 °C by following the decay of the tetrazine at 540 nm. [a] Endo/exo 2:1.
Second-Order Rate Constants of VBAs with Tetrazines Bearing a Hydroxyl-Substituent
As we established that the tetrazine ligation proceeds faster when the VBA adopts the boronate anion configuration, we next explored the reactivity of a set of tetrazines bearing hydroxyl substituents. The coordination of the hydroxyl to the VBA could at physiological pH promote the rate of the iEDDA reaction by making the VBA more reactive and favor the cycloaddition due to the induced proximity. In addition, as the tested hydroxyl substituents are more electron-rich than the pyridine substituent, we expect that the hydroxyl-tetrazines react less favorably in the iEDDA reaction with unsubstituted alkenes, such as norbornene, and thereby become more selective for reaction with VBAs.
We synthesized several tetrazines possessing a hydroxyl substituent, and compared the k2 values of the tetrazine ligations of VBA 1 with that of norbornene 8 (Figure 4, Table S1). As previously shown, VBA 1 and norbornene 8 have a comparable rate constant of 1.4 M–1 s–1 with dipyridyl-s-tetrazine 2a in 50% MeOH/PBS at 20 °C.18 Methyl pyridyl tetrazine 2e still possesses a boron-coordinating atom, although a 10-fold lower k2 value was found for VBA 1 than for norbornene 8. Phenyl tetrazine 2c also showed a lower rate constant for VBA 1 than for norbornene 8. Phenyl methyl tetrazine 2h, the derivative of 2e lacking a coordination atom, gives a 100-fold lower rate constant for VBA 1 than for norbornene 8. Of note, we have previously observed that pyrimidine tetrazines 2f and 2g also react poorly with VBAs, which we reasonably ascribe to the lower basicity of the pyrimidine compared to the pyridine substituent.21
Figure 4.

(A) Second-order rate constants of tetrazines 2a,c–n with VBA 1 and norbornene 8 measured in 1:1 MeOH/PBS (pH = 7.4) at 20 °C, shown on a logarithmic scale. (B) Product formation of the reaction of tetrazine 2l with VBA 1 and subsequent oxidation with chloranil, yielding pyridazine 9 as a single isomer. [a] Previously reported.18 [b] Previously reported.21 [c] Endo/exo = 2:1.
Introducing an aliphatic hydroxyl substituent, as in tetrazine 2i, resulted in a tremendous increase in reaction rate compared to the nonhydroxyl-containing tetrazine 2h, giving a k2 value of VBA 1 with 2i that is almost 500-fold higher than with 2h. The rate constant of norbornene 8 with tetrazine 2i slightly dropped compared to the constant with 2h and was 10-fold lower than the k2 of 2i with VBA, demonstrating that this hydroxylated tetrazine is more selective toward vinylboronic acids. The reaction of VBA 1 with disubstituted hydroxyethyl-substituted tetrazine 2j and the hydroxyethyl-methyl-substituted tetrazine 2k showed lower rate constants than with tetrazine 2i, possibly due to lack of an aromatic substituent on the tetrazine for stacking. Norbornene 8 gave a slightly lower k2 value with both tetrazines 2j and 2k compared to the more electron deficient tetrazine 2e, as expected.
Next, we investigated tetrazines bearing a hydroxyl substituent on the phenyl ring in the reaction with VBA and norbornene. The o-hydroxyphenyl methyl tetrazine 2l gave a high rate constant of 0.28 M–1 s–1 for VBA 1, almost 1000-fold higher than the k2 of phenyl methyl tetrazine 2h. Moreover, the reactivity of 2l with norbornene 8 was 23-fold lower, making this tetrazine more selective for VBA. In contrast, m-hydroxyphenyl tetrazine 2m gave a more than 3 orders of magnitude decrease in reaction rate compared to the o-hydroxy-substituted 2l, possibly due to unfavorable positioning of the hydroxyl for coordination. Important to emphasize is that the hydroxyl substitution pattern is not relevant for the tetrazine ligations with norbornene, as m-hydroxy-substituted tetrazine 2m gave a comparable rate constant as the o-hydroxy-substituted tetrazine 2l. Furthermore, o-hydroxyphenyl pyridyl tetrazine 2m additionally increased the reaction rate more than 30-fold to 9.3 M–1 s–1 for VBA and thereby exceeding the rate constant of dipyridyl-s-tetrazine 2a with about 1 order of magnitude. We additionally attempted to synthesize and measure the rate constants for disubstituted o-hydroxyphenyl tetrazine; however, the insolubility of this tetrazine in methanol unfortunately hampered our measurements.
The results above indicates that coordination of a hydroxyl-substituent on the tetrazine to the boronic acid has a positive influence on the rate of the iEDDA cycloaddition. Furthermore, the hydroxyl-substituted tetrazines are much more selective for VBA than for norbornene. Whereas dipyridyl-s-tetrazine 2a gives comparable rate constants for both alkenes, tetrazine 2n gives, for example, a 52-fold higher k2 for VBA 1 than for norbornene 8. To validate that the increase in reactivity of the VBAs toward the hydroxyl-substituted tetrazines is indeed caused by coordination, we isolated the product of VBA 1 and tetrazine 2l (Figure 4B). Since the reaction of VBA 1 with tetrazine 2l gave several tautomers of the dihydropyridazine, we oxidized the product to the corresponding pyridazine to facilitate characterization of the product. This two-step reaction gave pyridazine 9 as a single isomer in nearly quantitative yield with the phenyl substituent on the 5-position of the pyridazine ring, demonstrating the coordination of the boronic acid to the o-hydroxyphenyl ring of tetrazine 2l. The reaction of VBA 1 with tetrazine 2n also resulted in a single product after oxidation; however, the exact regioisomer could not be established due to the absence of a clear NOESY signal in 2D 1H NMR.
Stability of Tetrazines in Aqueous Environment
The bioorthogonal application of tetrazines requires that these moieties are stable in aqueous solution or biological environment. However, some tetrazines slowly decompose in aqueous environment with especially electron-withdrawing substituents destabilizing the aromatic ring.28,29 As we expect that the hydroxyl-containing tetrazines described above are more electron-rich than the pyridyl- or pyrimidyl-substituted tetrazines, we predicted that the former also have superior stability. Therefore, we tested the stability of tetrazines 2a,c–n in aqueous environment at 37 °C by measuring the decrease in absorbance of the tetrazines at 540 nm (Figure 5, Table S2).
Figure 5.

Decrease of absorbance at 540 nm of tetrazines 2a,c–n in 1:9 DMSO/PBS (pH = 7.4) at 37 °C. The normalized mean with SD is plotted. [a] Measured in 1:1 DMSO/PBS (pH = 7.4) as tetrazines 2d and 2m were insoluble in 1:9 DMSO/PBS.
As expected, we observed that dipyridyl-s-tetrazines 2a as well as pyrimidyl-substituted tetrazines 2f and 2g were rather unstable, with 60–85% of the tetrazines being degraded after 12 h in 1:9 DMSO/PBS. In contrast, the more electron-rich pyridyl tetrazines 2d and 2e and phenyl tetrazines 2c and 2h were more stable, with at least 75% of the tetrazines remaining after 12 h. To our delight, all hydroxyl-substituted tetrazines 2i–n were similarly stable and only marginal degradation was visible after 12 h.
Conclusions
To conclude, we set out here to gain more insight in the notable reactivity of VBAs toward tetrazines containing a pyridine substituent, of which the nitrogen coordinates to the boronic acid of the VBA. We compared the reactivity of vinylboronic acids to their negatively charged boronate anion in the tetrazine ligation using DFT calculations and an experimental setup, where we changed the pH of the reaction buffer, and observed that the more electron-rich boronate anion gave a faster reaction with tetrazines. We synthesized several hydroxyl-substituted tetrazines and showed that the hydroxyl substituent coordinates to the boronic acid and increases the rate constants of the tetrazine ligation with VBAs of several orders of magnitude. Moreover, these new tetrazines were more electron-rich than the pyridyl-substituted tetrazines giving lower rate constants with norbornene and favoring reactivity toward VBA due to coordination. Furthermore, the hydroxyl-substituted tetrazines were found to be more stable in aqueous media and therefore more suitable for a bioorthogonal application requiring long incubation times compared to the pyridyl-substituted tetrazines. The developed hydroxyl-substituted tetrazines are a valuable asset for bioorthogonal conjugation due to their small size, stability, and hydrophilic character. Furthermore, the selectivity of the hydroxyl tetrazines toward VBAs renders them potential candidates for use in orthogonal bioorthogonal tetrazine ligations for dual labeling of biomolecules, when combined with the reaction of a strained alkene and a tetrazine lacking a boron-coordinating substituent.
Acknowledgments
Selma Eising and Kimberly M. Bonger thank The Netherlands Research Institute for Chemical Biology (NRSCB) and the Institute of Molecules and Materials (IMM) of the Radboud University in Nijmegen for financial support. F. Matthias Bickelhaupt acknowledges financial support from The Netherlands Organization for Scientific Research (NWO).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.8b00439.
Experimental details, full spectroscopic data for all new compounds, Cartesian coordinates of all species occurring in our model DFT computations, kinetics for rate constants determination, and additional figures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Shih H.-W.; Kamber D. N.; Prescher J. A. (2014) Building better bioorthogonal reactions. Curr. Opin. Chem. Biol. 21, 103–111. 10.1016/j.cbpa.2014.07.002. [DOI] [PubMed] [Google Scholar]
- Shieh P.; Bertozzi C. R. (2014) Design Strategies for Bioorthogonal Smart Probes. Org. Biomol. Chem. 12, 9307–9320. 10.1039/C4OB01632G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M.; Wagner A. (2014) Developments in the field of bioorthogonal bond forming reactions - past and present trends. Bioconjugate Chem. 25, 825–839. 10.1021/bc500028d. [DOI] [PubMed] [Google Scholar]
- Chen X.; Wu Y.-W. (2016) Selective chemical labeling of proteins. Org. Biomol. Chem. 14, 5417–5439. 10.1039/C6OB00126B. [DOI] [PubMed] [Google Scholar]
- Šečkutė J.; Devaraj N. K. (2013) Expanding room for tetrazine ligations in the in vivo chemistry toolbox. Curr. Opin. Chem. Biol. 17, 761–767. 10.1016/j.cbpa.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knall A.-C.; Slugovc C. (2013) Inverse electron demand Diels-Alder (iEDDA)-initiated conjugation: a (high) potential click chemistry scheme. Chem. Soc. Rev. 42, 5131–5142. 10.1039/c3cs60049a. [DOI] [PubMed] [Google Scholar]
- Kozma E.; Demeter O.; Kele P. (2017) Bioorthogonal Fluorescent Labelling of Biopolymers via Inverse Electron Demand Diels-Alder Reactions. ChemBioChem 18, 486–501. 10.1002/cbic.201600607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira B. L.; Guo Z.; Bernardes G. J. L. (2017) Inverse electron demand Diels-Alder reactions in chemical biology. Chem. Soc. Rev. 46, 4895–4950. 10.1039/C7CS00184C. [DOI] [PubMed] [Google Scholar]
- Borrmann A.; Milles S.; Plass T.; Dommerholt J.; Verkade J. M. M.; Wiessler M.; Schultz C.; van Hest J. C. M.; van Delft F. L.; Lemke E. A. (2012) Genetic Encoding of a Bicyclo[6.1.0]nonyne-Charged Amino Acid Enables Fast Cellular Protein Imaging by Metal-Free Ligation. ChemBioChem 13, 2094–2099. 10.1002/cbic.201200407. [DOI] [PubMed] [Google Scholar]
- Lang K.; Davis L.; Wallace S.; Mahesh M.; Cox D. J.; Blackman M. L.; Fox J. M.; Chin J. W. (2012) Genetic encoding of bicyclononynes and trans-cyclooctenes for site-specific protein labeling in vitro and in live mammalian cells via rapid fluorogenic diels-alder reactions. J. Am. Chem. Soc. 134, 10317–10320. 10.1021/ja302832g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman M. L.; Royzen M.; Fox J. M. (2008) Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels-Alder Reactivity. J. Am. Chem. Soc. 130, 13518–13519. 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj R.; Fox J. M. (2013) trans-cyclooctene - a stable, voracious dienophile for bioorthogonal labeling. Curr. Opin. Chem. Biol. 17, 753–760. 10.1016/j.cbpa.2013.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj N. K.; Weissleder R.; Hilderbrand S. A. (2008) Tetrazine-based cycloadditions: application to pretargeted live cell imaging. Bioconjugate Chem. 19, 2297–2299. 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson D. M.; Nazarova L. A.; Xie B.; Kamber D. N.; Prescher J. A. (2012) Functionalized cyclopropenes as bioorthogonal chemical reporters. J. Am. Chem. Soc. 134, 18638–18643. 10.1021/ja3060436. [DOI] [PubMed] [Google Scholar]
- Yang J.; Šečkutė J.; Cole C. M.; Devaraj N. K. (2012) Live-cell imaging of cyclopropene tags with fluorogenic tetrazine cycloadditions. Angew. Chem., Int. Ed. 51, 7476–7479. 10.1002/anie.201202122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederwieser A.; Späte A.-K.; Nguyen L. D.; Jüngst C.; Reutter W.; Wittmann V. (2013) Two-color glycan labeling of live cells by a combination of diels-alder and click chemistry. Angew. Chem., Int. Ed. 52, 4265–4268. 10.1002/anie.201208991. [DOI] [PubMed] [Google Scholar]
- Lee Y.-J.; Kurra Y.; Yang Y.; Torres-Kolbus J.; Deiters A.; Liu W. R. (2014) Genetically encoded unstrained olefins for live cell labelling with tetrazine dyes. Chem. Commun. 50, 13085–13088. 10.1039/C4CC06435F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eising S.; Lelivelt F.; Bonger K. M. (2016) Vinylboronic Acids as Fast Reacting, Synthetically Accessible, and Stable Bioorthogonal Reactants in the Carboni–Lindsey Reaction. Angew. Chem., Int. Ed. 55, 12243–12247. 10.1002/anie.201605271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eising S.; van der Linden N. G. A.; Kleinpenning F.; Bonger K. M. (2018) Vinylboronic Acids as Efficient Bioorthogonal Reactant for Tetrazine Labeling in Living Cells. Bioconjugate Chem. 29, 982–986. 10.1021/acs.bioconjchem.7b00796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall D. G. (2011) Boronic acids, Preparation and Applications in Organic Synthesis, Medicine and Materials, WILEY-VCH, Weinheim. [Google Scholar]
- Eising S.; Xin B. T.; Kleinpenning F.; Heming J.; Florea B. I.; Overkleeft H. S.; Bonger K. M. (2018) Coordination-Assisted Bioorthogonal Chemistry: Orthogonal Tetrazine Ligations with Vinylboronic Acid and a Strained Alkene. ChemBioChem 19, 1648. 10.1002/cbic.201800275. [DOI] [PubMed] [Google Scholar]
- te Velde G.; Bickelhaupt F. M.; Baerends E. J.; Fonseca Guerra C.; van Gisbergen S. J. A.; Snijders J. G.; and Ziegler T. (2001) Chemistry with ADF. J. Comput. Chem. 22, 931–967. 10.1002/jcc.1056. [DOI] [Google Scholar]
- Baker J.; Pulay P. (2002) Assessment of the Handy-Cohen optimized exchange density functional for organic reactions. J. Chem. Phys. 117, 1441–1449. 10.1063/1.1485723. [DOI] [Google Scholar]
- Handy N. C.; Cohen A. J. (2001) Left-right correlation energy. Mol. Phys. 99, 403–412. 10.1080/00268970010018431. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Bickelhaupt F. M.; Houk K. N. (2017) Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem., Int. Ed. 56, 10070–10086. 10.1002/anie.201701486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickelhaupt F. M., and Baerends E. J. (2000) Reviews in Computational Chemistry (Lipkowitz K. B., and Boyd D. B.) Wiley-VCH, New York. [Google Scholar]
- Versteegen R. M.; Rossin R.; Ten Hoeve W.; Janssen H. M.; Robillard M. S. (2013) Click to release: Instantaneous doxorubicin elimination upon tetrazine ligation. Angew. Chem., Int. Ed. 52, 14112–14116. 10.1002/anie.201305969. [DOI] [PubMed] [Google Scholar]
- Karver M. R.; Weissleder R.; Hilderbrand S. A. (2011) Synthesis and evaluation of a series of 1,2,4,5-tetrazines for bioorthogonal conjugation. Bioconjugate Chem. 22, 2263–2270. 10.1021/bc200295y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.