

Abstract
Complementary phenylacetylene oligomers equipped with phenol and phosphine oxide recognition sites form stable multiply H-bonded duplexes in toluene solution. Oligomers were prepared by Sonogashira coupling of diiodobenzene and bis-acetylene building blocks in the presence of monoacetylene chain terminators. The product mixtures were separated by reverse phase preparative high-pressure liquid chromatography to give a series of pure oligomers up to seven recognition units in length. Duplex formation between length complementary homo-oligomers was demonstrated by 31P NMR denaturation experiments using dimethyl sulfoxide as a competing H-bond acceptor. The denaturation experiments were used to determine the association constants for duplex formation, which increase by nearly 2 orders of magnitude for every phenol-phosphine oxide base-pair added. These experiments show that the phenylacetylene backbone supports formation of extended duplexes with multiple cooperative intermolecular H-bonding interactions, and together with previous studies on the mixed sequence phenylacetylene 2-mer, suggest that this supramolecular architecture is a promising candidate for the development of synthetic information molecules that parallel the properties of nucleic acids.
Introduction
Nucleic acids encode information in the sequence of monomer units, and this structure provided the molecular basis for the evolution of biological life. The information is read through sequence-selective duplex formation and copied through template synthesis.1 The sequence of monomer units also defines the three-dimensional structures and functional properties of single-stranded nucleic acids.2 Modified analogues of nucleic acids have been reported,3 where the sugar,4 the phosphate linker,6 or the base pairing system7 have been replaced and the ability of forming duplexes was not affected, suggesting that fully synthetic information molecules may be able to have some or all the functions of nucleic acids. There are some examples of synthetic oligomers that form duplexes using metal coordination, salt bridges, or H-bonding as the base-pairing interactions.8−12Figure 1a shows a blueprint for duplex forming oligomers that is currently being investigated in our laboratory. The modular nature of this design allows the three different components to be explored independently, providing insights into the key requirements for architectures that will lead to sequence-selective duplex formation.
Figure 1.
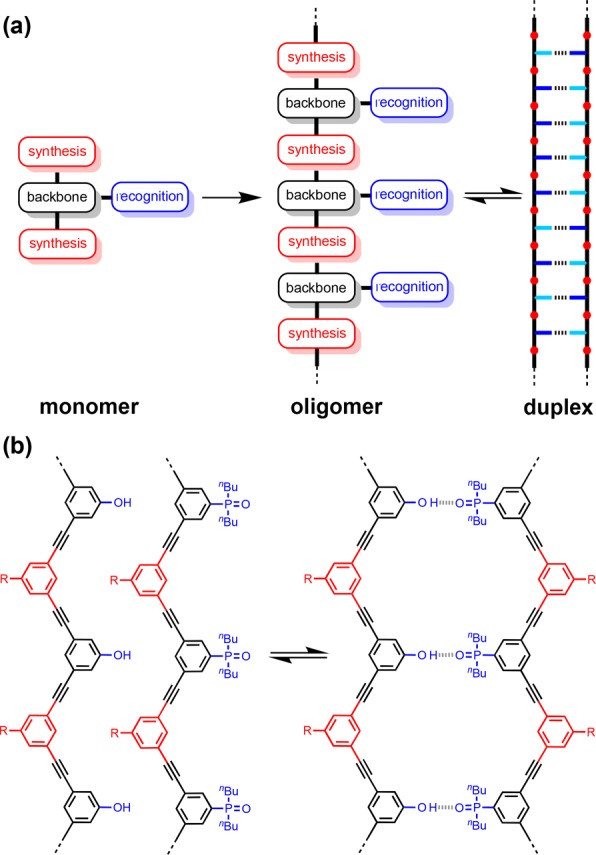
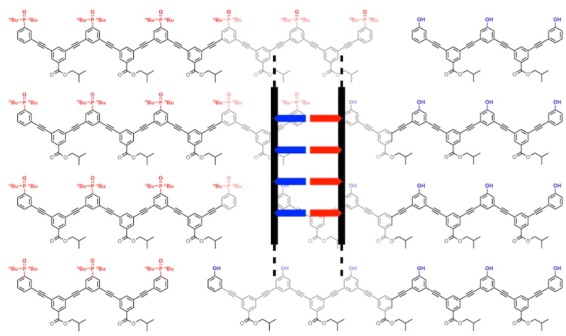
(a) A blueprint for duplex forming molecules. There are three key design elements: the coupling chemistry used for the synthesis of oligomers (red), the recognition module, which controls intermolecular binding (blue), and the backbone module, which links these components together (black). (b) Proposed duplex formed by phenylacetylene oligomers equipped with phenol and phosphine oxide recognition modules (R is a solubilizing group).
We have shown that oligomers equipped with single H-bond recognition modules form stable duplexes in nonpolar solvents.13a−13g It is possible to interchange different H-bond donor–acceptor recognition motifs (D·A) on the same backbone and maintain the duplex forming properties of the oligomers.13c Oligomers have been prepared using reductive amination chemistry and using thiol–ene coupling, and both types of backbone lead to the formation of stable duplexes. Experiments on three isomeric reductive amination backbones indicate that an important requirement for duplex formation is a backbone with sufficient flexibility to adopt to a compatible conformation.13b,13g
However, when mixed donor–acceptor sequences were prepared, it became clear that the conformational properties of the backbone also play a key role in discriminating between intramolecular H-bonding interactions that lead to folding and intermolecular interactions that lead to duplex formation. Highly flexible backbones lead to folding that precludes effective sequence-selective duplex formation in mixed sequence oligomers.13e These studies have identified two backbones that have promising conformational properties, and we recently showed that one of these systems supports sequence-selective duplex formation for mixed sequence 3-mers.13fFigure 1b shows the other backbone that is sufficiently rigid to preclude intramolecular folding in the mixed sequence AD 2-mer.13e This phenylacetylene backbone is particularly attractive for a number of reasons. Phenylacetylene oligomers have been extensively studied by Moore,14 who developed solid phase synthesis methods that could be used to make oligomers with controlled length and sequence.15 Oligomers longer than about eight repeat units fold into helical structures in acetonitrile solution, due to stacking interactions,16 so the single-stranded forms may have interesting properties. Formation of phenylacetylene duplexes based on dynamic covalent base-pairs was investigated previously, but slow kinetics limited the fidelity.17 Here we describe the synthesis of families of recognition-encoded oligomers based on the phenylacetylene backbone and show that this molecular architecture leads to the formation of very stable H-bonded duplexes in toluene.
Results and Discussion
Stepwise synthesis of oligomeric molecules requires multiple deprotection-coupling cycles and is labor intensive. Here we investigate a one-step process, statistically controlled oligomerization, to rapidly access families of oligomers of varying length (Figure 2). The average oligomer length can be controlled by adding monofunctional chain stoppers to the reaction mixture, and the resulting product mixture can be separated by preparative HPLC. Sonogashira coupling of bis-acetylene recognition modules with diiodo-linkers in the presence of the corresponding monoacetylene recognition modules was therefore investigated as a simple route to homo-oligomers.
Figure 2.
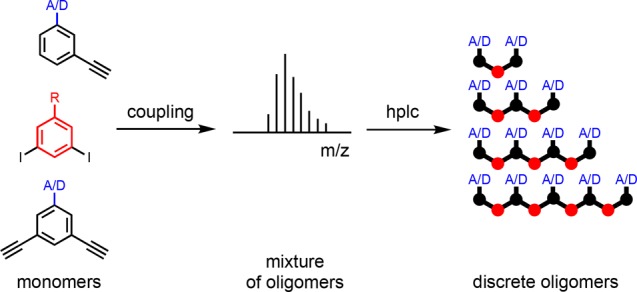
Oligomer synthesis using statistical coupling of mono- and bifunctional monomers to produce mixtures that can be separated by chromatography. H-bond donor (D) or acceptor (A) recognition groups can be used, and R is a solubilizing group.
Synthesis of Building Blocks
The monoacetylene donor recognition module (1) was commercially available, and the monoacetylene acceptor recognition module (2) was synthesized as previously described (Scheme 1).13e Recognition modules bearing two acetylene moieties were synthesized using the route shown in Scheme 1. Treatment of tribromobenzene (3) with nBuLi followed by reaction with iodine gave dibromoiodobenzene (4). The difference in reactivity of halides allowed for selective P-arylation with di-n-butylphosphine oxide to give 5. Sonogashira coupling with TMSA yielded 6, and TBAF mediated deprotection yielded the bis-acetylene acceptor recognition module (7). Acetylation of 3,5-dibromophenol (8) with acetic anhydride yielded the acetate derivative (9). Acetylation was required to increase the reactivity in the subsequent Sonogashira coupling with TMSA to obtain 10. Simultaneous base-mediated deprotection of the phenol and acetylenes yielded the bis-acetylene donor recognition module (11). The diiodo-linker (13) was synthesized by an EDC-mediated esterification of 3,5-diiodobenzoic acid (12) with iBuOH (Scheme 2).
Scheme 1. Mono-Acetylene Recognition Modules, and Synthesis of Bis-Acetylene Recognition Modules.

Scheme 2. Synthesis of Diiodo-Linker.
Oligomer Synthesis
Donor homo-oligomers were synthesized by oligomerization of the phenol recognition modules (1 and 11) with linker 13 under Sonogashira coupling conditions in toluene (Scheme 3).13e,18 The ratio of starting materials was chosen to target the DDD 3-mer as the major product (1:11:13 = 2:1:2). Although the crude reaction mixture contained a large amount of insoluble material, it could be dissolved in DMSO. LCMS analysis of the DMSO solution showed the presence of oligomers up to the 6-mer (Figure 3a). Preparative HPLC was used to separate the products (Figure 2b), and samples of donor homo-oligomers from the 3-mer to 6-mer were isolated by this method (Table 1).19 The overall yield with respect to the diiodo-linker 13 was 15%, due to the low solubility of the oligomeric products. The most abundant oligomer isolated was the DDD 3-mer, as expected from the stoichiometry of the two recognition modules used in the reaction.
Scheme 3. Oligomerization of Phenol Recognition Modules to Yield Donor Homo-Oligomers (n = 1–4 Corresponding to the 3-mer to 6-mer Donor Homo-Oligomers).
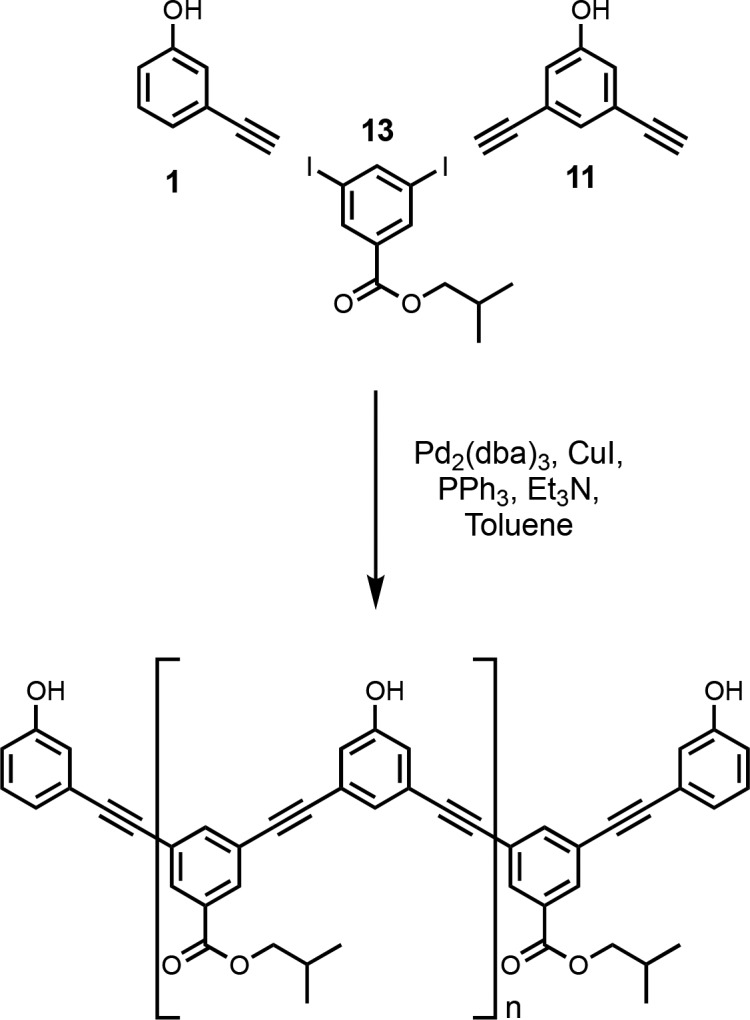
Figure 3.

Product distribution from the donor oligomerization reaction shown in Scheme 3. (a) LCMS analysis of the crude reaction mixture using a Hichrom C8C18 column with a water/THF (0.1% formic acid) solvent system. (b) Preparative HPLC separation of donor homo-oligomers using a HIRBP-6988 prep column with a water/THF solvent system. Samples were prepared in DMSO, UV/vis absorption was measured at 290 nm, and the peaks identified by MS are labeled with retention time in minutes. dba = dibenzylideneacetone.
Table 1. Donor homo-oligomers isolated from the oligomerization reaction in Scheme 3.
| product | sequence | % mol fraction | % yield |
|---|---|---|---|
| 14 (3-mer) | DDD | 40 | 6 |
| 15 (4-mer) | DDDD | 27 | 4 |
| 16 (5-mer) | DDDDD | 21 | 3 |
| 17 (6-mer) | DDDDDD | 12 | 2 |
Acceptor homo-oligomers were synthesized by oligomerization of the phosphine oxide recognition modules (2 and 7) with linker 13 under Sonogashira conditions in toluene (Scheme 4). The ratio of starting materials was chosen to target the AAA 3-mer as the major product (2:7:13 = 2:1:2). The reaction mixture was dissolved in ethanol and analyzed by LCMS (Figure 3a). Oligomers up to the 7-mer were observed. Preparative HPLC was used to separate the products (Figure 4b), and samples of the acceptor homo-oligomers from the 3-mer to 7-mer were isolated (Table 2). The overall yield with respect to the diiodo-linker 13 was 33%. The most abundant oligomer in the mixture was the AAA 3-mer, as expected from the stoichiometry of the two recognition modules used in the reaction.
Scheme 4. Oligomerization of Phosphine Oxide Recognition Modules to Yield Acceptor Homo-Oligomers (n = 1–5 Corresponding to the 3-mer to 7-mer Acceptor Homo-Oligomers).

Figure 4.

Product distribution from the acceptor oligomerization reaction shown in Scheme 4. (a) LCMS analysis of the crude reaction mixture using a Hichrom C8C18 column with a water/THF (0.1% formic acid) solvent system. (b) Preparative HPLC separation of acceptor homo-oligomers using a HIRBP-6988 prep column with a water/THF solvent system. Samples were prepared in EtOH, UV/vis absorption was measured at 290 nm, and the peaks identified by MS are labeled with retention time in minutes. dba = dibenzylideneacetone.
Table 2. Acceptor Homo-Oligomers Isolated from the Oligomerization Reaction in Scheme 4.
| product | sequence | % mol fraction | % yield |
|---|---|---|---|
| 18 (3-mer) | AAA | 58 | 19 |
| 19 (4-mer) | AAAA | 27 | 9 |
| 20 (5-mer) | AAAAA | 10 | 3 |
| 21 (6-mer) | AAAAAA | 4 | 1 |
| 22 (7-mer) | AAAAAAA | 1 | <1 |
Oligomer Characterization
Mass spectrometry and 1H NMR spectroscopy were used to confirm the structures of the oligomers. The terminal recognition modules, the internal recognition modules, and the linkers all give rise to distinct signals in the 1H NMR spectra, and the ratios of the integrals of these signals were used to confirm oligomer length. For the donor oligomers, the ratio of the integrals of the 1H NMR signals due to the terminal linkers (blue and orange) compared with the signals due to the internal linkers (red and green) allowed quantification of number of linkers and therefore oligomer length (Figure 5). Similarly, for the acceptor oligomers, the ratio of the integrals of the 1H NMR signals due to the terminal linkers (blue) compared with the signals due to the internal linkers (red) allowed quantification of the number of linkers and therefore oligomer length. These results were confirmed using the ratio of integrals of the 31P NMR signals due to the terminal recognition modules (blue) compared with the signals due to the internal recognition modules (red) (Figure 6).
Figure 5.

(a) Chemical structures of the donor homo-oligomers. (b) Partial 500 MHz 1H NMR spectra in THF-d6. The signals in the 1H NMR spectra are assigned to the chemical structures using color coding. The 4-mer contains traces of an impurity that could not be removed by HPLC.
Figure 6.

(a) Chemical structures of the acceptor homo-oligomers. (b) Partial 500 MHz 1H NMR spectra in CDCl3. (c) 162 MHz 31P NMR spectra in CDCl3. The signals in the 1H and 31P NMR spectra are assigned to the chemical structures using color coding.
For both the donor homo-oligomers in THF and the acceptor homo-oligomers in chloroform, the chemical shifts of the signals due to equivalent protons from equivalent building blocks do not change as the length of the oligomers increases. Moore and co-workers have studied the properties of a related family of phenylacetylene oligomers in acetonitrile, and they observed substantial upfield changes in chemical shift (0.5 ppm) for the signals due to the aromatic protons as the length of the oligomers increased.15 These chemical shift changes are indicative of folding into a helical conformation with stacking of the aromatic rings. The 1H NMR data in Figures 5 and 6 show that the oligomers described in this work do not fold in chloroform or THF, but the solubility is not sufficient to record NMR spectra in acetonitrile.
Binding Studies
Previous studies of duplex assembly used toluene as the solvent, because toluene is a very weak H-bond donor and acceptor, which maximizes the strength of the phenol-phosphine oxide interactions. The intrinsic strength of this base-pairing interaction is important for maximizing K EM, the parameter that governs chelate cooperativity in zipping up the duplex.13,20 The poor solubility of the donor homo-oligomers in toluene described here made it impossible to measure association constants for duplex formation by titration experiments. However, in the presence of the complementary acceptor homo-oligomer, both DDD and DDDD are soluble at millimolar concentrations in toluene. The longer donor homo-oligomers do not dissolve in toluene even in the presence of the complementary acceptor homo-oligomer.
The 31P NMR spectrum of a 1:1 mixture of AAA and DDD in toluene-d8 shows one broad signal at 40–41 ppm. In contrast in the 31P NMR spectrum of AAA in toluene-d8, there are two signals at 35.9 ppm. These differences in 31P chemical shift are characteristic of phosphine oxide groups that are fully H-bonded to phenol in the mixture of AAA and DDD. In the 1H NMR spectrum of this mixture, the two signals due to the phenol OH groups appear at 11 ppm, which indicates that the three phenol groups of DDD are fully H-bonded to phosphine oxide groups on AAA. These data provide good evidence that at a concentration of 1 mM in toluene-d8 the AAA·DDD duplex is fully assembled with three intermolecular phenol-phosphine oxide H-bonds. Similarly for a 1:1 mixture of AAAA and DDDD in toluene-d8, the 31P NMR spectrum has two signals at 40 ppm, and the 1H NMR spectrum has two signals due to the phenol OH groups at 11 ppm. Thus, the AAAA·DDDD duplex is also fully assembled with four intermolecular phenol-phosphine oxide H-bonds at a concentration of 1 mM in toluene-d8. In all cases, equilibration was rapid, and no slow exchange processes were observed.
The stabilities of the AAA·DDD and AAAA·DDDD duplexes were characterized by denaturation experiments with DMSO. DMSO can H-bond to the phenol groups on the donor homo-oligomers and thus dissociate the duplexes. If the association constant for the phenol-DMSO interaction is known (Kd), it is possible to use the denaturation isotherm to deduce the association constant for duplex formation between two complementary N-mers (KN). To test the validity of this approach, the 1-mers and 2-mers, which were reported previously,13e were used to directly compare the results from a titration and a denaturation experiment (Figure 7).
Figure 7.
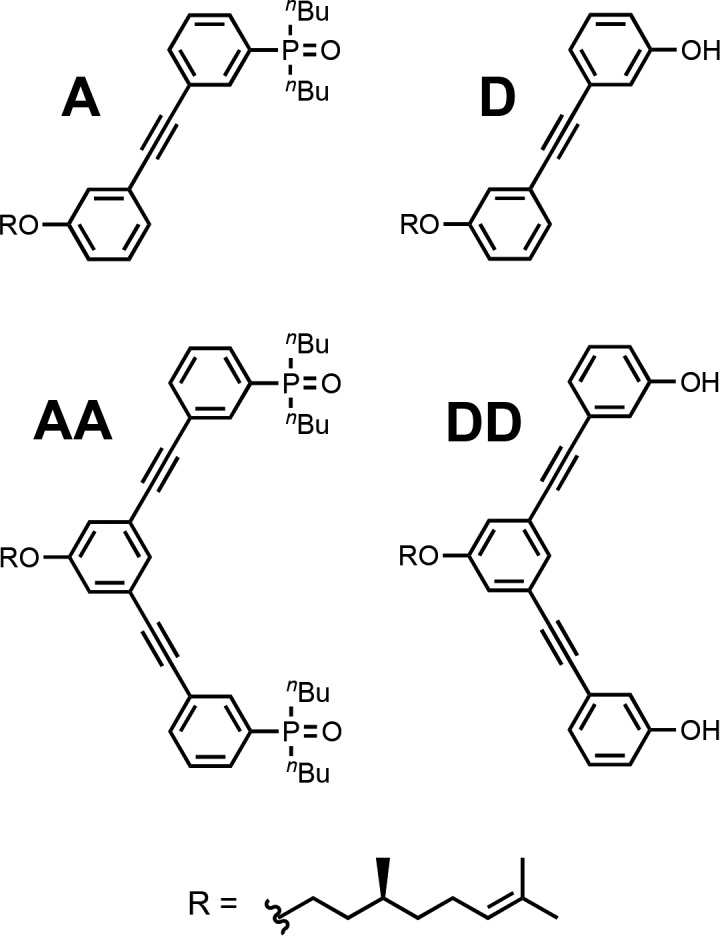
1-mers and 2-mers used to validate the DMSO denaturation experiment.
Association constants for formation of the A·D and AA·DD complexes in toluene-d8 were determined using 31P NMR titration experiments. The data fit well to 1:1 binding isotherms (see the Supporting Information, SI), and the results are summarized in Table 3. The large downfield changes in 31P NMR chemical shift (+6–7 ppm) are indicative of H-bond formation, and the bound chemical shifts are very similar to those observed for the AAA·DDD and AAAA·DDDD duplexes described above. The association constant for the AA·DD complex (K2 = 44 000 M–1) is 2 orders of magnitude higher than the association constant for the A·D complex (K1 = 760 M–1) indicating that the duplex is fully assembled with two cooperative intermolecular phenol-phosphine oxide H-bonds.
Table 3. Association Constants for Duplex Formation (KN), Effective Molarities (EM), and 31P NMR Chemical Shifts (δ) Measured by NMR Titration and DMSO Denaturation Experiments in Toluene-d8 at 298 Ka.
| method | complex | KN (M–1) | EM (mM) | δf (ppm) | δb (ppm) |
|---|---|---|---|---|---|
| titration | A·D | 760 | 35.3 | 42.1 | |
| titration | AA·DD | 44 000 | 38 | 35.3 | 41.3 |
| denaturation | AA·DD | 83 000 | 72 | 34.8 | 42.1 |
| denaturation | AAA·DDD | 2 300 000 | 52 | 34.8 | 40.5 |
| denaturation | AAAA·DDDD | 130 000 000 | 59 | 35.3 | 40.1 |
Errors are estimated at ±30% based on two repeats of the experiment.
The value of Kd was measured by 1H NMR titration of DMSO into the D 1-mer in toluene (Kd = 150 M–1, see SI). For denaturation of the AA·DD duplex, an equimolar 1 mM solution of AA and DD was prepared in toluene-d8. At this concentration, AA is 90% bound to DD, and the 31P signal was accordingly observed close to the bound chemical shift at 41 ppm. Addition of DMSO-d6 lead to a decrease in the chemical shift of the 31P NMR signal to 36 ppm, due to disruption of the H-bonding interactions (Figure 8b). However, the final chemical shift is 1 ppm higher than the free chemical shift from the titration experiment (35 ppm), and Figure 8b shows that the 31P chemical shift starts to increase at very high concentrations of DMSO at the end of the denaturation experiment. This observation is presumably due to a change in the nature of the solvent for DMSO concentrations of the order 1 M. DMSO-d6 was therefore titrated into a 1 mM sample of the A 1-mer in toluene-d8 where no H-bonding interactions are present (Figure 8a). An increase of about 1 ppm was observed in the 31P chemical shift at very high DMSO concentrations, confirming that the unusual shape of the AA·DD denaturation data is due to nonspecific effects of the change in solvent. The 31P chemical shift data for the A 1-mer were therefore used to correct the 31P chemical shifts from the AA·DD denaturation experiment for the effects of DMSO on the chemical shift of the unbound phosphine oxide signal. The proportion of duplex present at any point in the denaturation experiment is therefore given by eq 1.
| 1 |
where δobs is the observed chemical shift for the 1:1 mixture at a specific concentration of denaturant, δA is the observed chemical shift of the A 1-mer at the same concentration of denaturant, δf is the free chemical shift of the acceptor homo-oligomer in pure toluene-d8, and δb is the bound chemical shift of the duplex.
Figure 8.

202 MHz 31P NMR spectra for titration of DMSO-d6 into a) A (1 mM), and DMSO-d6 denaturation of equimolar 1 mM solutions of b) DD and AA, c) DDD and AAA, and d) DDDD and AAAA in toluene-d8 at 298 K.
The data from the AA·DD denaturation experiment did not fit to a simple two-state, all-or-nothing denaturation isotherm (see SI), which suggests that partially denatured species must also be considered. Figure 9 shows all possible complexes present in the AA·DD denaturation experiment. Both the fully bound duplex and unbound species are present at the start of the experiment. Binding of one molecule of DMSO to the duplex leads to the partially denatured AA·DD·DMSO complex. It is possible to estimate the equilibrium constant for the formation of this termolecular complex as the product of Kd (the D·DMSO association constant) and K1 (the A·D association constant), multiplied by a statistical factor of 4. The unbound DD 2-mer can bind one or two molecules of DMSO, and the equilibrium constants for formation of the DD·DMSO and DD·(DMSO)2 complexes can be estimated using the D·DMSO association constant as 2Kd and Kd2 respectively.
Figure 9.
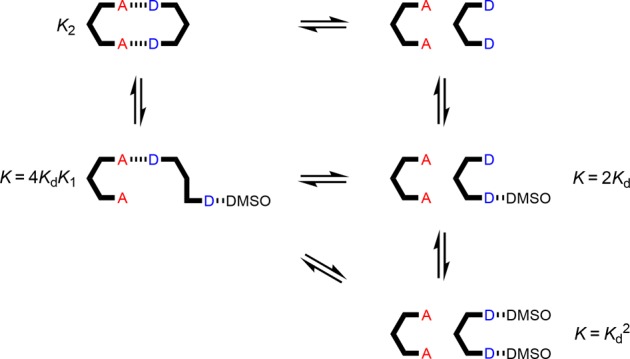
Equilibria involved in denaturation of AA·DD. Kd is the D·DMSO association constant, and KN is the association constant for duplex formation between homo-oligomers of length N in toluene-d8.
The data for denaturation of the AA·DD duplex fit well to a denaturation isotherm that allows for all of these species, and this allowed determination of K2 as the only unknown equilibrium constant. The results are shown in Table 3. The value of K2 determined in the denaturation experiment is comparable to the value determined in the corresponding titration experiment in toluene, which indicates that the denaturation methodology provides a robust method for determining duplex stability.
The 31P NMR DMSO denaturation experiments for the AAA·DDD and AAAA·DDDD duplexes are shown in Figure 8c and d, respectively, and the results are similar to those obtained for AA·DD. Fitting the denaturation data for the longer oligomers requires consideration of a larger number of partially denatured species (Figures 10 and 11). The equilibrium constants for formation of most of these species can also be estimated from the values of Kd and K1, but some partially denatured duplexes have no direct analogues. For example, a number of different structures are possible for the AAA·DDD·DMSO complex, and we do not know whether complexes with DMSO bound to one of the terminal recognition units have similar stability to the complex with DMSO bound to the central recognition unit. By analogy with the AA·DD·DMSO complex, one might assume that the association constant for AAA·DDD·DMSO complex, KP, will be the product of Kd and K2, multiplied by a statistical factor, but to avoid any bias in the fitting, KP was optimized as a variable along with K3 in analysis of the AAA·DDD denaturation data. The results of fitting the proportion of duplex present determined using eq 1 to the denaturation isotherm are shown in Figure 12, and the resulting association constant for duplex formation is reported in Table 3. The optimized value of KP from the fitting is equal to 3.5 KdK2. If the duplex preferentially denatures from the ends, so that the central H-bond is intact in the partially denatured AAA·DDD·DMSO complex, the statistical factor would be 4, which is close to the optimized value of 3.5. The AAA·DDD duplex is more than an order of magnitude more stable than the AA·DD duplex, which confirms that the fully assembled triply H-bonded duplex is formed in toluene.
Figure 10.
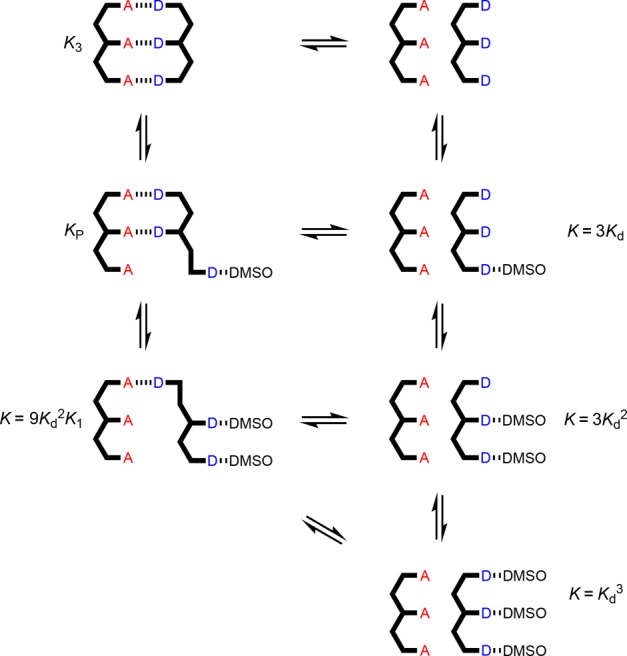
Equilibria involved in denaturation of AAA·DDD. Kd is the D·DMSO association constant, and KN is the association constant for duplex formation between homo-oligomers of length N in toluene-d8. For some of the complexes, isomeric arrangements are possible (not shown).
Figure 11.
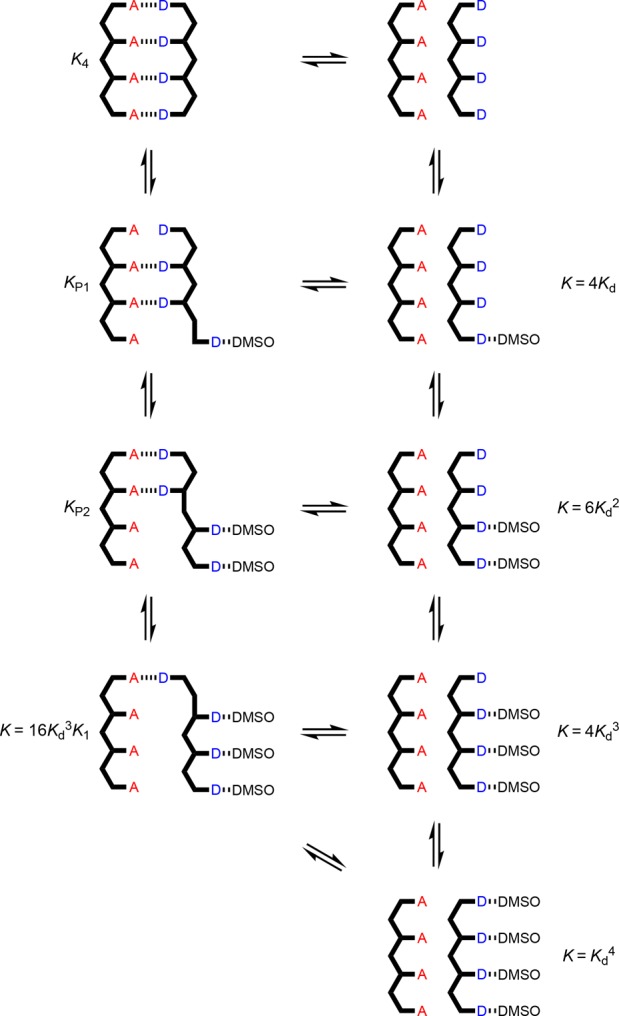
Equilibria involved in denaturation of AAAA·DDDD. Kd is the D·DMSO association constant, and KN is the association constant for duplex formation between homo-oligomers of length N in toluene-d8. For some of the complexes, isomeric arrangements are possible (not shown).
Figure 12.
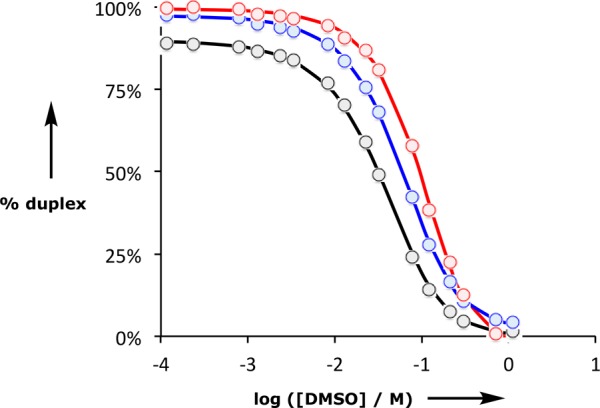
Duplex denaturation data plotted as a function of DMSO-d6 concentration in toluene-d8 at 298 K for AA·DD (black), AAA·DDD (blue), and AAAA·DDDD (red). The dots represent the experimental values obtained using eq 1, and the lines are the calculated denaturation isotherms.
For denaturation of the AAAA·DDDD duplex, there are two partially denatured complexes with unknown stability, AAAA·DDDD·DMSO and AAAA·DDDD·(DMSO)2 (Figure 11). Therefore, fitting of the denaturation data for this system involved optimization of three equilibrium constants, K4, KP1, and KP2. The results are shown in Figure 12 and Table 3. The AAAA·DDDD is more than an order of magnitude more stable than the AAA·DDD duplex, which confirms that the fully assembled duplex with all four H-bonds is formed in toluene. The value of KP1 is equal to 7.5 KdK3. If the duplex preferentially denatures from the ends, so that the central H-bonds are both intact in the partially denatured AAAA·DDDD·DMSO complex, then the statistical factor would be 4, which is smaller than the optimized value of 7.5. This result suggests that there are some additional partially denatured states where the central H-bonds are broken. The value of KP2 is equal to 8.0 Kd2K2. If the duplex preferentially denatures from the ends, then the statistical factor would be 9, which is close to the optimized value.
Figure 13 shows the speciation of different complexes calculated from the denaturation isotherms. In the denaturation of all three duplexes, the first step is breaking of one duplex H-bonds to form a phenol-DMSO H-bond, and this complex is the major partially denatured state in all cases (blue lines in Figure 13a–c). For the AA·DD duplex, this partially denatured complex is not very stable, so the DD·DMSO complex is also significantly populated (pink line in Figure 13a). At high concentrations of DMSO, the fully denatured state dominates in all cases (red lines in Figure 13a–c).
Figure 13.
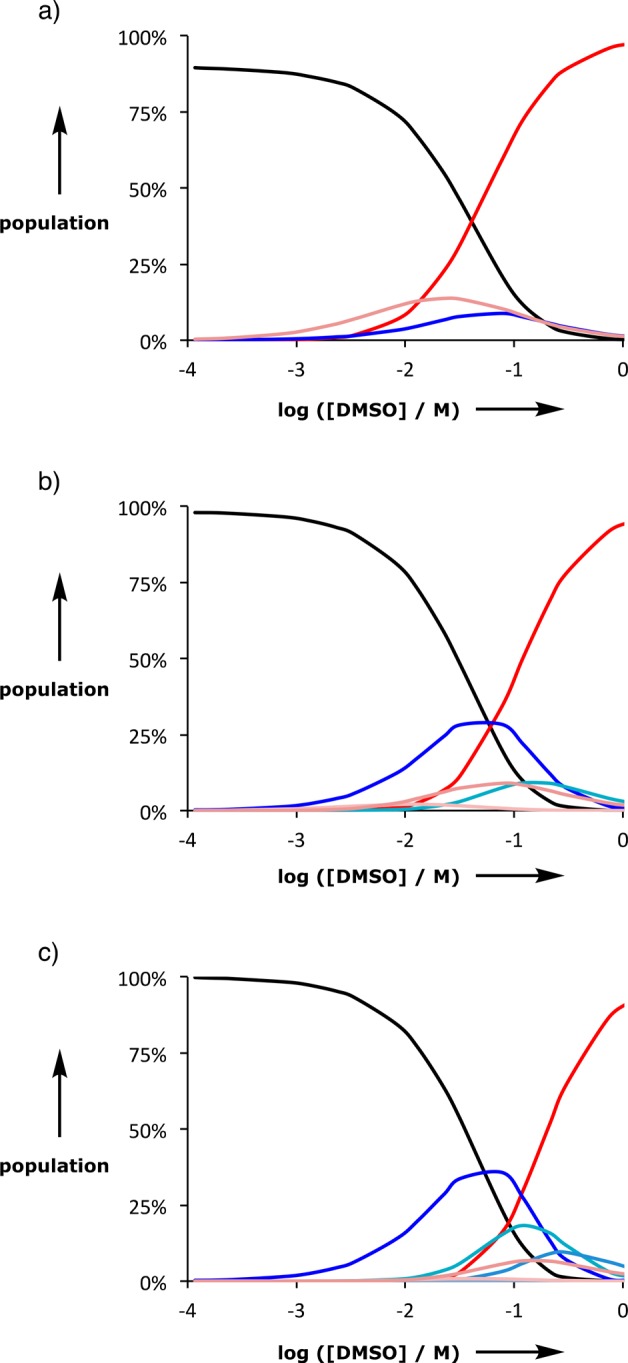
Speciation from DMSO denaturation experiments on length complementary homo-oligomer duplexes. a) The 2-mer duplex: AA·DD (black), DD·DMSO (pink), AA·DD·DMSO (blue), and DD·(DMSO)2 (red). b) The 3-mer duplex: AAA·DDD (black), AAA·DDD·DMSO (blue), DDD·(DMSO)2 (pink), AAA·DDD·(DMSO)2 (green), and DDD·(DMSO)3 (red). c) The 4-mer duplex: AAAA·DDDD (black), AAAA·DDDD·DMSO (blue), AAAA·DDDD·(DMSO)2 (cyan), DDDD·(DMSO)3 (pink), and DDDD·(DMSO)4 (red). Free acceptor homo-oligomer populations are not shown, and all other possible complexes are not populated to a significant extent.
Denaturation of the duplexes can also be monitored by 1H NMR spectroscopy (Figure 14). The signals due to the aromatic protons of the oligomers move by 0.1–0.6 ppm on denaturation. For the AA·DD duplex, the signal due to the phenol OH proton starts at 6.2 ppm, suggesting that the duplex is not fully bound, and moves to 10 ppm when bound to DMSO (Figure 14a). For both the AAA·DDD and AAAA·DDDD duplexes, the signals due to the phenol OH protons start at about 11 ppm, suggesting that the duplexes are fully bound, and move to about 10 ppm when bound to DMSO (Figure 14b and c). As observed for the 31P NMR spectra, there are differences at very high concentrations of DMSO-d6 due to the change in the nature of the solvent.
Figure 14.

500 MHz 1H NMR spectra for DMSO-d6 denaturation of equimolar 1 mM solutions of a) A, b) DD·AA, c) DDD·AAA, and d) DDDD·AAAA in toluene-d8 at 298 K.
The association constants for duplex formation, the average effective molarities (EM) for intramolecular H-bonding interactions in the duplexes, and the free and bound chemical shifts are shown in Table 3. The consistency of the values suggests that the results obtained from the denaturation experiments are accurate. The association constants increase with each additional H-bond, and the value of EM does not vary significantly between duplexes.
Figure 15 shows the value of log KN plotted as a function of the number of recognition modules in the oligomers (N). There is a uniform increase of 1.7 log units for every additional H-bond in the duplex. The results presented here can be compared with previous experiments on different backbone stuctures equipped with the same phenol-phosphine oxide recognition modules. The data in Figure 15 show that the phenylacetylene backbone (red) gives the most stable duplexes, presumably because these oligomers are more rigid than the aniline (green) and thioether (blue) backbones. Although the increased preorganization and geometric complementarity of the phenylacetylene backbone leads to higher values of EM for formation of the intramolecular H-bonds that zip up the duplex, the increase is actually quite small compared with the more flexible backbones (50 mM compared with 20 mM and 30 mM for the green and blue backbones, respectively, in Figure 15). However, small differences in H-bond strength and EM are cumulative along an oligomer, so that the phenylacetylene 4-mer duplex is 2 orders of magnitude more stable than the corresponding duplex with the aniline backbone.
Figure 15.
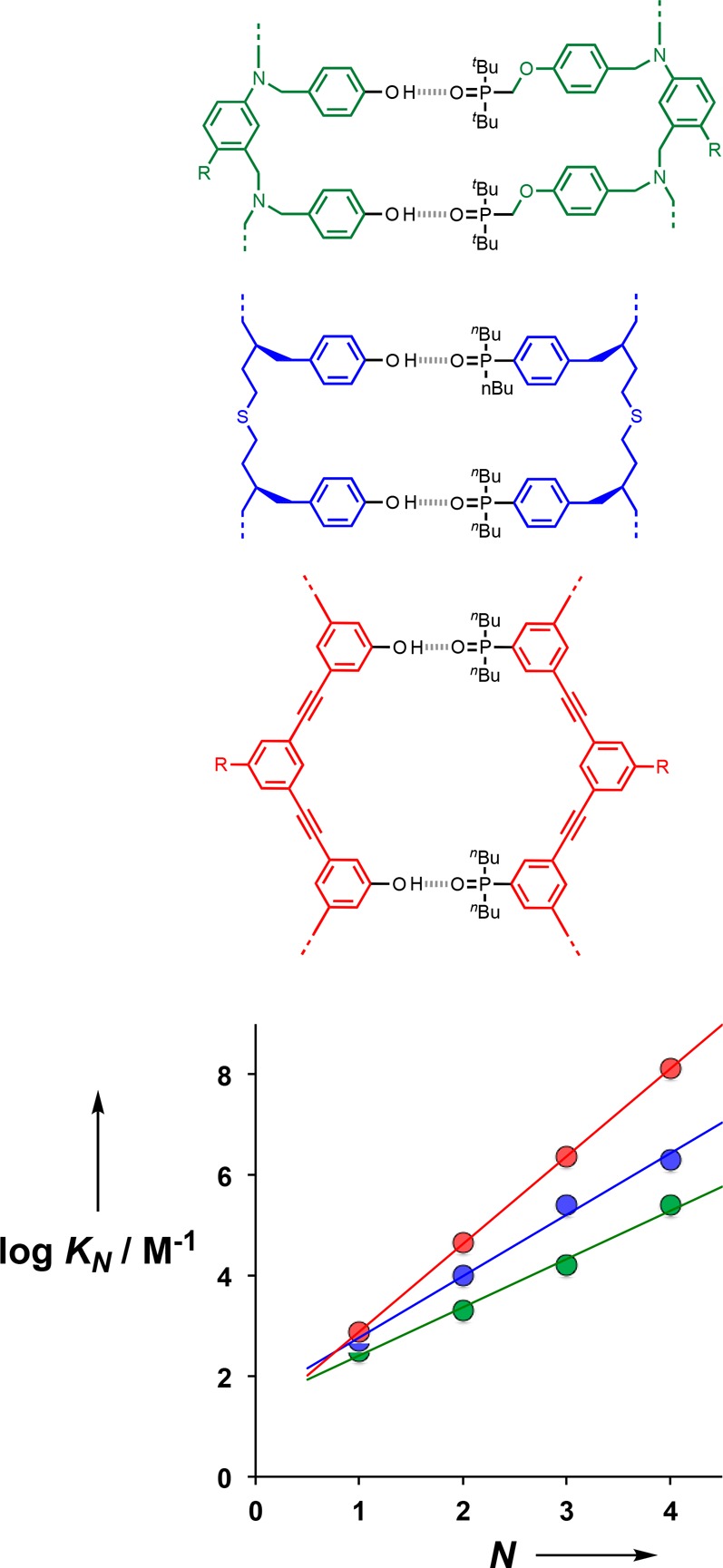
Relationship between the association constant for duplex formation between length-complementary oligomers in toluene-d8 at 298 K (KN) and the number of intermolecular H-bonds formed (N). Data are shown for three different backbones (color coded), each equipped with phenol-phosphine oxide recognition modules. The lines of best fit are shown: red log KN = 1.7N + 1.3; blue log KN = 1.2N + 1.6; green log KN = 1.0N + 1.5. R represents a solubilizing group.
Conclusions
Oligomerization of bifunctional building blocks in the presence of monofunctional chain terminators provides a straightforward approach to access families of oligomeric molecules with minimal synthetic effort. Here we describe the use of this strategy to prepare phenylacetylene oligomers equipped with phenol and phosphine oxide recognition sites. Pure samples of homo-oligomers up to seven recognition units in length were obtained by preparative hplc of the crude reaction mixtures. The phenol oligomers form stable duplexes with length complementary phosphine oxide oligomers via intermolecular phenol-phosphine oxide H-bonding interactions in toluene solution. NMR spectroscopy indicates that all possible base-pairing interactions between recognition sites are fully bound in the duplexes.
Addition of a competing H-bond acceptor, DMSO, dissociated the duplexes, and DMSO denaturation experiments were used to measure the association constants for duplex formation. The association constants increase by almost 2 orders of magnitude for every additional H-bonded base-pair added to the duplex, reaching 108 M–1 for AAAA·DDDD. The average effective molarity for the intramolecular H-bonding interactions responsible for zipping up the duplex is EM = 50 mM, and the corresponding value of the intramolecular equilibrium constant that quantifies chelate cooperativity in these systems is K EM = 40 (the product of the association constant for formation of one intermolecular phenol-phosphine oxide H-bond and the effective molarity). A value of 40 for K EM means that in the duplex the recognition sites are 98% bound, which is consistent with the NMR data. The uniform increase in duplex stability with oligomer length suggests that this supramolecular architecture is likely to be compatible with the formation extended H-bonded duplexes in longer oligomers. Given that the mixed sequence AD 2-mer does not exhibit any intramolecular folding, the recognition-encoded phenylacetylene oligomer system described here appears to be a promising candidate for the development of synthetic information molecules with sequence-selective duplex-forming properties that resemble nucleic acids.
Acknowledgments
We thank the Engineering and Physical Sciences Research Council (EP/J008044/2) and European Research Council (ERC-2012-AdG 320539-duplex) for funding.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b08087.
Detailed experimental procedures and 1H and 13C of all compounds, NMR titration and dilution spectra, details of fitting the binding isotherm, and detailed HPLC methods used for separation of oligomers (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Crick F. H. C.; Watson J. D. Nature 1953, 171, 964. 10.1038/171964b0. [DOI] [PubMed] [Google Scholar]; b Kyogoku Y.; Lord R. C.; Rich A. Science 1966, 154, 518. 10.1126/science.154.3748.518. [DOI] [PubMed] [Google Scholar]; c Kyogoku Y.; Lord R. C.; Rich A. J. Am. Chem. Soc. 1967, 89, 496. 10.1021/ja00979a005. [DOI] [PubMed] [Google Scholar]; d Newmark R. A.; Cantor C. R. J. Am. Chem. Soc. 1968, 90, 5010. 10.1021/ja01020a041. [DOI] [PubMed] [Google Scholar]; e Porschke D.Biopolymers 1971, 10, 1989. [Google Scholar]; f Crick F. Nature 1970, 227, 561. 10.1038/227561a0. [DOI] [PubMed] [Google Scholar]
- a Ellington A. D.; Szostak J. W. Nature 1990, 346, 818. 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]; b Rothemund P. W. K. Nature 2006, 440, 297. 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]; c Aldaye F. A.; Palmer A. L.; Sleiman H. F. Science 2008, 321, 1795. 10.1126/science.1154533. [DOI] [PubMed] [Google Scholar]; d Seeman N. C. Nano Lett. 2010, 10, 1971. 10.1021/nl101262u. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Ke Y.; Ong L. L.; Shih W. M.; Yin P. Science 2012, 338, 1177–1183. 10.1126/science.1227268. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Pinheiro A. V.; Han D.; Shih W. M.; Yan H. Nat. Nanotechnol. 2011, 6, 763. 10.1038/nnano.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Zhang F.; Nangreave J.; Liu Y.; Yan H. J. Am. Chem. Soc. 2014, 136, 11198. 10.1021/ja505101a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Eschenmoser A. Science 1999, 284, 2118. 10.1126/science.284.5423.2118. [DOI] [PubMed] [Google Scholar]; b Benner S. A. Acc. Chem. Res. 2004, 37, 784. 10.1021/ar040004z. [DOI] [PubMed] [Google Scholar]; c Benner S. A.; Chen F.; Yang Z.. Chemical Synthetic Biology; Luisi P. L., Chiarabelli C., Eds.; John Wiley & Sons, Ltd: Chichester, UK, 2011; pp 69–106. [Google Scholar]; d Benner S. A. Biological Theory 2013, 8, 357. 10.1007/s13752-013-0142-y. [DOI] [Google Scholar]; e Wilson C.; Keefe A. D. Curr. Opin. Chem. Biol. 2006, 10, 607. 10.1016/j.cbpa.2006.10.001. [DOI] [PubMed] [Google Scholar]; f Appella D. H. Curr. Opin. Chem. Biol. 2009, 13, 687. 10.1016/j.cbpa.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Kool E. T. Curr. Opin. Chem. Biol. 2000, 4, 602. 10.1016/S1367-5931(00)00141-1. [DOI] [PubMed] [Google Scholar]
- Sugar modifications of nucleic acids; a Schöning K.; Scholz P.; Guntha S.; Wu X.; Krishnamurthy R.; Eschenmoser A. Science 2000, 290, 1347. 10.1126/science.290.5495.1347. [DOI] [PubMed] [Google Scholar]; b van Aerschot A.; Verheggen I.; Hendrix C; Herdewijn P.. Angew. Chem. Int. Ed. Engl. 1995, 34, 1338. [Google Scholar]; c Renneberg D.; Leumann C. J. J. Am. Chem. Soc. 2002, 124, 5993. 10.1021/ja025569+. [DOI] [PubMed] [Google Scholar]; d Braasch D. A.; Corey D. R. Chem. Biol. 2001, 8, 1. 10.1016/S1074-5521(00)00058-2. [DOI] [PubMed] [Google Scholar]; e Singh S. K.; Koshkin A. A.; Wengel J.; Nielsen P. Chem. Commun. 1998, 455. 10.1039/a708608c. [DOI] [Google Scholar]; f Nguyen H. V.; Zhao Z. Y.; Sallustrau A.; Horswell S. L.; Male L.; Mulas A.; Tucker J. H. R. Chem. Commun. 2012, 48, 12165. 10.1039/c2cc36428j. [DOI] [PubMed] [Google Scholar]; g Zhang L.; Peritz A.; Meggers E. J. Am. Chem. Soc. 2005, 127, 4174. 10.1021/ja042564z. [DOI] [PubMed] [Google Scholar]; h Schlegel M. K.; Peritz A. E.; Kittigowittana K.; Zhang L.; Meggers E. ChemBioChem 2007, 8, 927. 10.1002/cbic.200600435. [DOI] [PubMed] [Google Scholar]; i Karri P.; Punna V.; Kim K.; Krishnamurthy R. Angew. Chem., Int. Ed. 2013, 52, 5840. 10.1002/anie.201300795. [DOI] [PubMed] [Google Scholar]
- Phosphate modifications of nucleic acids:; a Benner S. A.; Hutter D. Bioorg. Chem. 2002, 30, 62. 10.1006/bioo.2001.1232. [DOI] [PubMed] [Google Scholar]; b Huang Z.; Schneider K. C.; Benner S. A. J. Org. Chem. 1991, 56, 3869. 10.1021/jo00012a018. [DOI] [Google Scholar]; c Huang Z.; Benner S. A. J. Org. Chem. 2002, 67, 3996. 10.1021/jo0003910. [DOI] [PubMed] [Google Scholar]; d Richert C.; Roughton A. L.; Benner S. A. J. Am. Chem. Soc. 1996, 118, 4518. 10.1021/ja952322m. [DOI] [Google Scholar]; e Li P.; Sergueeva Z. A.; Dobrikov M.; Shaw B. R. Chem. Rev. 2007, 107, 4746. 10.1021/cr050009p. [DOI] [PubMed] [Google Scholar]; f Isobe H.; Fujino T.; Yamazaki N.; Guillot-Nieckowski M.; Nakamura E. Org. Lett. 2008, 10, 3729. 10.1021/ol801230k. [DOI] [PubMed] [Google Scholar]; g Eriksson M.; Nielsen P. E. Q. Q. Rev. Biophys. 1996, 29, 369. 10.1017/S0033583500005886. [DOI] [PubMed] [Google Scholar]; h Nielsen P. E. Chem. Biodiversity 2010, 7, 786. 10.1002/cbdv.201000005. [DOI] [PubMed] [Google Scholar]; i Nielsen P. E.; Egholm M. Curr. Issues Mol. Biol. 1999, 1, 89. [PubMed] [Google Scholar]; j Nielsen P. E.; Haaima G. Chem. Soc. Rev. 1997, 26, 73. 10.1039/cs9972600073. [DOI] [Google Scholar]; k Ura Y.; Beierle J. M.; Leman L. J.; Orgel L. E.; Ghadiri M. R. Science 2009, 325, 73. 10.1126/science.1174577. [DOI] [PubMed] [Google Scholar]
- Bases modification of nucleic acids:; a Piccirilli J. A.; Krauch T.; Moroney S. E.; Benner S. A. Nature 1990, 343, 33. 10.1038/343033a0. [DOI] [PubMed] [Google Scholar]; b Yang Z.; Hutter D.; Sheng P.; Sismour A. M.; Benner S. A. Nucleic Acids Res. 2006, 34, 6095. 10.1093/nar/gkl633. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wojciechowski F.; Leumann C. J. Chem. Soc. Rev. 2011, 40, 5669. 10.1039/c1cs15027h. [DOI] [PubMed] [Google Scholar]; d Benner S. A. Curr. Opin. Chem. Biol. 2012, 16, 581. 10.1016/j.cbpa.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Liu H.; Gao J.; Lynch S. R.; Saito Y. D.; Maynard L.; Kool E. T. Science 2003, 302, 868. 10.1126/science.1088334. [DOI] [PubMed] [Google Scholar]; f Kool E. T. Acc. Chem. Res. 2002, 35, 936. 10.1021/ar000183u. [DOI] [PubMed] [Google Scholar]; g Kool E. T.; Lu H.; Kim S. J.; Tan S.; Wilson J. N.; Gao J.; Liu H. Nucleic Acids Symp. Ser. 2006, 50, 15. 10.1093/nass/nrl008. [DOI] [PubMed] [Google Scholar]; h Wilson J. N.; Kool E. T. Org. Biomol. Chem. 2006, 4, 4265. 10.1039/b612284c. [DOI] [PubMed] [Google Scholar]
- a Kramer R.; Lehn J.-M.; Marquis-Rigault A. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 5394. 10.1073/pnas.90.12.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Marquis A.; Smith V.; Harrowfield J.; Lehn J.-M.; Herschbach H.; Sanvito R.; Leize-Wagner E.; van Dorsselaer A. Chem. - Eur. J. 2006, 12, 5632. 10.1002/chem.200600143. [DOI] [PubMed] [Google Scholar]
- a Gong B.; Yan Y.; Zeng H.; Skrzypczak-Jankunn E.; Kim Y. W.; Zhu J.; Ickes H. J. Am. Chem. Soc. 1999, 121, 5607. 10.1021/ja990904o. [DOI] [Google Scholar]; b Gong B. Synlett 2001, 2001, 0582. 10.1055/s-2001-13350. [DOI] [Google Scholar]; c Zeng H.; Miller R. S.; Flowers R. A.; Gong B. J. Am. Chem. Soc. 2000, 122, 2635. 10.1021/ja9942742. [DOI] [Google Scholar]; d Zeng H.; Ickes H.; Flowers R. A.; Gong B. J. Org. Chem. 2001, 66, 3574. 10.1021/jo010250d. [DOI] [PubMed] [Google Scholar]; e Gong B. Polym. Int. 2007, 56, 436. 10.1002/pi.2175. [DOI] [Google Scholar]; f Gong B. Acc. Chem. Res. 2012, 45, 2077. 10.1021/ar300007k. [DOI] [PubMed] [Google Scholar]; g Yang Y.; Yang Z. Y.; Yi Y. P.; Xiang J. F.; Chen C. F.; Wan L. J.; Shuai Z. G. J. Org. Chem. 2007, 72, 4936. 10.1021/jo070525a. [DOI] [PubMed] [Google Scholar]
- a Tanaka Y.; Katagiri H.; Furusho Y.; Yashima E. Angew. Chem. 2005, 117, 3935. 10.1002/ange.200501028. [DOI] [PubMed] [Google Scholar]; b Ito H.; Furusho Y.; Hasegawa T.; Yashima E. J. Am. Chem. Soc. 2008, 130, 14008. 10.1021/ja806194e. [DOI] [PubMed] [Google Scholar]; c Yamada H.; Furusho Y.; Ito H.; Yashima E. Chem. Commun. 2010, 46, 3487. 10.1039/c002170a. [DOI] [PubMed] [Google Scholar]; d Yashima E.; Ousaka N.; Taura D.; Shimomura K.; Ikai T.; Maeda K. Chem. Rev. 2016, 116, 13752. 10.1021/acs.chemrev.6b00354. [DOI] [PubMed] [Google Scholar]
- a Anderson H. L. Inorg. Chem. 1994, 33, 972. 10.1021/ic00083a022. [DOI] [Google Scholar]; b Taylor P. N.; Anderson H. L. J. Am. Chem. Soc. 1999, 121, 11538. 10.1021/ja992821d. [DOI] [Google Scholar]; c Berl V.; Huc I.; Khoury R. G.; Krische M. J.; Lehn J.-M. Nature 2000, 407, 720. 10.1038/35037545. [DOI] [PubMed] [Google Scholar]; d Berl V.; Huc I.; Khoury R. G.; Lehn J.-M. Chem. - Eur. J. 2001, 7, 2810.. [DOI] [PubMed] [Google Scholar]; e Sánchez-Quesada J.; Seel C.; Prados P.; de Mendoza J.; Dalcol I.; Giralt E. J. Am. Chem. Soc. 1996, 118, 277. 10.1021/ja953243d. [DOI] [Google Scholar]; f Bisson A. P.; Carver F. J.; Eggleston D. S.; Haltiwanger R. C.; Hunter C. A.; Livingstone D. L.; McCabe J. F.; Rotger C.; Rowan A. E. J. Am. Chem. Soc. 2000, 122, 8856. 10.1021/ja0012671. [DOI] [Google Scholar]; g Bisson A. P.; Hunter C. A. Chem. Commun. 1996, 1723. 10.1039/cc9960001723. [DOI] [Google Scholar]; h Yang Y.; Yang Z.-Y.; Yi Y.-P.; Xiang J.-F.; Chen C.-F.; Wan L.-J.; Shuai Z.-G. J. Org. Chem. 2007, 72, 4936. 10.1021/jo070525a. [DOI] [PubMed] [Google Scholar]; i Chu W.-J.; Yang Y.; Chen C.-F. Org. Lett. 2010, 12, 3156. 10.1021/ol101068n. [DOI] [PubMed] [Google Scholar]; j Chu W.-J.; Chen J.; Chen C.-F.; Yang Y.; Shuai Z. J. Org. Chem. 2012, 77, 7815. 10.1021/jo301434a. [DOI] [PubMed] [Google Scholar]; k Archer E. A.; Krische M. J. J. Am. Chem. Soc. 2002, 124, 5074. 10.1021/ja012696h. [DOI] [PubMed] [Google Scholar]; l Gong H.; Krische M. J. J. Am. Chem. Soc. 2005, 127, 1719. 10.1021/ja044566p. [DOI] [PubMed] [Google Scholar]
- a Dervan P. B.; Burli R. W. Curr. Opin. Chem. Biol. 1999, 3, 688. 10.1016/S1367-5931(99)00027-7. [DOI] [PubMed] [Google Scholar]; b Renneberg D.; Dervan P. B. J. J. Am. Chem. Soc. 2003, 125, 5707. 10.1021/ja0300158. [DOI] [PubMed] [Google Scholar]; c Doss R. M.; Marques M. A.; Foister S.; Chenoweth D. M.; Dervan P. B. J. Am. Chem. Soc. 2006, 128, 9074. 10.1021/ja0621795. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Meier J. L.; Yu A. S.; Korf I.; Segal D. J.; Dervan P. B. J. Am. Chem. Soc. 2012, 134, 17814. 10.1021/ja308888c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Stross A. E.; Iadevaia G.; Hunter C. A. Chem. Sci. 2016, 7, 94. 10.1039/C5SC03414K. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Iadevaia G.; Stross A. E.; Neumann A.; Hunter C. A. Chem. Sci. 2016, 7, 1760. 10.1039/C5SC04467G. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Stross A. E.; Iadevaia G.; Hunter C. A. Chem. Sci. 2016, 7, 5686. 10.1039/C6SC01884J. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Núñez-Villanueva D.; Hunter C. A. Chem. Sci. 2017, 8, 206. 10.1039/C6SC02995G. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Núñez-Villanueva D.; Iadevaia G.; Stross A. E.; Jinks M. A.; Swain J. A.; Hunter C. A. J. Am. Chem. Soc. 2017, 139, 6654. 10.1021/jacs.7b01357. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Stross A. E.; Iadevaia G.; Núñez-Villanueva D.; Hunter C. A. J. Am. Chem. Soc. 2017, 139, 12655. 10.1021/jacs.7b06619. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Iadevaia G.; Núñez-Villanueva D.; Stross A. E.; Hunter C. A. Org. Biomol. Chem. 2018, 16, 4183. 10.1039/C8OB00819A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhang J.; Moore J. S.; Xu Z.; Aguirre R. A. J. Am. Chem. Soc. 1992, 114, 2273–2274. 10.1021/ja00032a060. [DOI] [Google Scholar]; b Wu Z.; Lee S.; Moore J. S. J. Am. Chem. Soc. 1992, 114, 8730–8732. 10.1021/ja00048a073. [DOI] [Google Scholar]; c Xu Z.; Kahr M.; Walker K. L.; Wilkins C. L.; Moore J. S. J. Am. Chem. Soc. 1994, 116, 4537–4550. 10.1021/ja00090a002. [DOI] [Google Scholar]; d Lahiri S.; Thompson J. L.; Moore J. S. J. Am. Chem. Soc. 2000, 122, 11315–11319. 10.1021/ja002129e. [DOI] [Google Scholar]; e Prince R. B.; Brunsveld L.; Meijer E. W.; Moore J. S. Angew. Chem., Int. Ed. 2000, 39, 228–230. . [DOI] [PubMed] [Google Scholar]; f Prince R. B.; Moore J. S.; Brunsveld L.; Meijer E. W. Chem. - Eur. J. 2001, 7, 4150–4154. . [DOI] [PubMed] [Google Scholar]; g Tanatani A.; Mio M. J.; Moore J. S. J. Am. Chem. Soc. 2001, 123, 1792–1793. 10.1021/ja003678n. [DOI] [PubMed] [Google Scholar]; h Brunsveld L.; Meijer E. W.; Prince R. B.; Moore J. S. J. Am. Chem. Soc. 2001, 123, 7978–7984. 10.1021/ja010751g. [DOI] [PubMed] [Google Scholar]; i Cary J. M.; Moore J. S. Org. Lett. 2002, 4, 4663–4666. 10.1021/ol0270982. [DOI] [PubMed] [Google Scholar]; j Zhao D.; Moore J. S. Chem. Commun. 2003, 807–818. 10.1039/b207442g. [DOI] [PubMed] [Google Scholar]; k Stone M. T.; Moore J. S. Org. Lett. 2004, 6, 469. 10.1021/ol036238k. [DOI] [PubMed] [Google Scholar]
- a Young J. K.; Nelson J. C.; Moore J. S. J. Am. Chem. Soc. 1994, 116, 10841–10842. 10.1021/ja00102a082. [DOI] [Google Scholar]; b Nelson J. C.; Young J. K.; Moore J. S. J. Org. Chem. 1996, 61, 8160–8168. 10.1021/jo961250u. [DOI] [PubMed] [Google Scholar]
- a Prince R. B.; Saven J. G.; Wolynes P. G.; Moore J. S. J. Am. Chem. Soc. 1999, 121, 3114. 10.1021/ja983995i. [DOI] [Google Scholar]; b Prince R. B.; Barnes S. A.; Moore J. S. J. Am. Chem. Soc. 2000, 122, 2758. 10.1021/ja993830p. [DOI] [Google Scholar]; c Prince R. B.; Okada T.; Moore J. S. Angew. Chem., Int. Ed. 1999, 38, 233–236. . [DOI] [Google Scholar]; d Hill D. J.; Mio M. J.; Prince R. B.; Hughes T. S.; Moore J. S. Chem. Rev. 2001, 101, 3893. 10.1021/cr990120t. [DOI] [PubMed] [Google Scholar]
- a Hartley C. S.; Elliott E. L.; Moore J. S. J. Am. Chem. Soc. 2007, 129, 4512. 10.1021/ja0690013. [DOI] [PubMed] [Google Scholar]; b Elliott E. L.; Hartley C. S.; Moore J. S. Chem. Commun. 2011, 47, 5028–5030. 10.1039/c1cc11242b. [DOI] [PubMed] [Google Scholar]
- Sonogashira K. J. J. Organomet. Chem. 2002, 653, 46–49. 10.1016/S0022-328X(02)01158-0. [DOI] [Google Scholar]
- a Lawrence J.; Lee S.-H.; Abdilla A.; Nothling M. D.; Ren J. M.; Knight A. S.; Fleischmann C.; Li Y.; Abrams A. S.; Schmidt B. V. K. J.; Hawker M. C.; Connal L. A.; McGrath A. J.; Clark P. G.; Gutekunst W. R.; Hawker C. J. J. Am. Chem. Soc. 2016, 138, 6306. 10.1021/jacs.6b03127. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lawrence J.; Goto E.; Ren J. M.; McDearmon B.; Kim D. S.; Ochiai Y.; Clark P. G.; Laitar D. S.; Higashihara T.; Hawker C. J. J. Am. Chem. Soc. 2017, 139, 13735. 10.1021/jacs.7b05299. [DOI] [PubMed] [Google Scholar]
- Hunter C. A.; Anderson H. L. Angew. Chem., Int. Ed. 2009, 48, 7488–7499. 10.1002/anie.200902490. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.