

Conspectus
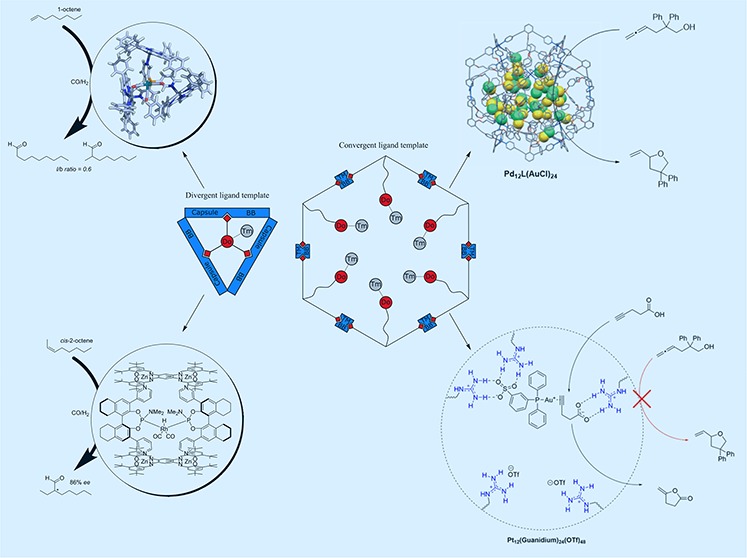
Binding of molecules in molecular cages based on self-assembled concave building blocks has been of great interest to scientists for decades. The binding of static molecular fragments inside cage-like molecular structures is generally based on complementarity of host and guest in terms of shape and interactions. The encapsulation of homogeneous catalysts in molecular cages is of interest as activity, selectivity, and stability can be controlled by the cage as second coordination sphere, reminiscent of how enzymes control chemical reactivity. Homogeneous catalysts, however, are not static guest molecules as catalysts change in shape, charge, and polarity during the catalytic cycle, representing the challenges involved in cage controlled catalysis. To address these issues, we developed a new strategy that we coined the “ligand template approach for catalyst encapsulation”. This strategy relies on ligand building blocks that contain multiple orthogonal binding sites: the central ligand (mostly phosphorus) is bound to the transition metal required for catalysis, while other binding sites are used to construct a cage structure around the transition metal atom through self-assembly. By design, the catalyst is inside the capsule during the catalytic cycle, as the central ligand is coordinated to the catalyst. As the approach is based on a self-assembly process of building blocks, the catalyst properties can be easily modulated by modification of building blocks involved.
In this Account, we elaborate on template ligand strategies for single catalyst encapsulation, based on divergent ligand templates and the extension to nanospheres with multiple metal complexes, which are formed by assembly of convergent ligand templates. Using the mononuclear approach, a variety of encapsulated catalysts can be generated, which have led to highly (enantio)selective hydroformylation reactions for encapsulated rhodium atoms. Besides the successes of encapsulated rhodium catalysts in hydroformylation, mononuclear ligand template capsules have been applied in asymmetric hydrogenation, the Heck reaction, copolymerization, gold catalyzed cyclization reactions, and hydrosilylation reactions. By changing the capsule building blocks the electronic and steric properties around the transition metal atom have successfully been modified, which translates to changes in catalyst properties. Using the convergent ligand templates, nanospheres have been generated with up to 24 complexes inside the sphere, leading to very high local concentrations of the transition metal. The effect of local concentrations was explored in gold catalyzed cyclization reactions and ruthenium catalyzed water oxidation, and for both reactions, spectacular reaction rate enhancements have been observed. This Account shows that the template ligand approach to provide catalyst in well-defined specific environments is very versatile and leads to catalyst properties that are not achievable with traditional approaches.
Introduction
For decades chemists have been fascinated by the properties of molecules, and the ever increasing knowledge of synthetic methodologies facilitates the exploration of a diverse set of molecules with different sizes and shapes. The generation of concave 3-D shaped molecules provided the first opportunities to study the binding of guest molecules in the cavities and cages of such structures, and this initiated a new scientific field that is now well-known as supramolecular chemistry.1−3 Active research by many scientists in the field has resulted in many spectacular examples of molecular constructs, often based on self-assembly of smaller components, that specifically bind guest molecules that are complementary in shape. The interactions and entropic contributions relevant for these binding events are well-understood.4,5 A variety of applications for these new molecular materials have been proposed, including their use in the field of catalysis. Molecular cages have been used as catalysts to promote several organic transformations mostly through preorganization of the substrates and stabilizing the crucial transition state by interactions with the cage environment, leading to rate acceleration.6−11
Transition metal catalysis can also be carried out in molecular cages.11−16 Traditionally, the key properties of transition metal catalysts are controlled by ligand effects. Electronic properties, steric environment, and coordination mode can all be fine-tuned by ligand modifications, and in turn this has large influence on the catalytic properties of the metal complex. Placing such catalytically active complexes in confined spaces provides additional tools to control the crucial parameters of the catalytic system. Compared to using cages themselves as catalysts, this strategy is more challenging. First of all, the metal complex and the substrate need to be in the cage at the same time, even though excess of substrate is present in solution, as otherwise the number of catalytic cycles the catalyst can perform will remain low. Second, the transition metal complex generally changes during a catalytic cycle along the different steps such as oxidative addition, reductive elimination, ligand dissociation, or substrate insertion. This means that the complex will change in shape, size, and electronic properties throughout the catalytic cycle, and yet it should stay in the molecular cage, coencapsulated with different substrates at different stages of the reaction. This sketches the complication when transition metal catalysis is attempted in molecular cages.
Despite the challenge, several examples have been reported in which metal complexes are encapsulated with catalyst properties that are substantially different from the unencapsulated analogue.12 Encapsulation strategies include covalent attachment of ligands to the cage or binding of metal complexes in cages via ionic interaction, which so far is limited to cationic metal complexes.5−16 The capsule size is often found detrimental for encapsulation of metal complexes, and optimization may involve painstaking synthesis for systems with the catalyst covalently attached to the capsule.
We have developed a novel strategy that we coined “ligand template strategy for catalyst encapsulation”.17 In this strategy, we prepare relatively simple building blocks that by a self-assembly process lead to transition metal catalysts in cage structures (Figure 1). A ligand is utilized with functional groups, to which other building blocks can bind, and as such this ligand functions as a template for encapsulation. This Account highlights the progress in template ligand strategies for catalyst encapsulation. We will show that crucial catalyst parameters, such as activity and selectivity, can be controlled by second coordination sphere effects. Changing the catalyst is relative simple, as it involves just mixing of different building blocks. A diverse set of reactions have been explored and shown to be compatible with the approach. Recently, we have extended the approach from mononuclear complexes in cages, to template ligand strategies that lead to multinuclear complexes in nanospheres. This will be discussed in the second part of this Account.
Figure 1.

Schematic representation of the two distinct ligand template approaches for encapsulation of catalysts: for the mononuclear approach, a divergent ligand template is encapsulated by capsule building blocks, forming a confined space around a single transition metal atom. For the multinuclear approach, multiple convergent ligand template molecules self-assemble into a nanosphere with multiple metal complexes in confined space, leading to very high local concentration of transition metal atoms.
Template Ligand Strategies for Encapsulation of Single Catalysts
We started our template ligand approach with trispyridylphosphine ligand 1, in combination with simple zinc(II)tetraphenylporphyrin (Zn(II)TPP) building blocks. Mixing 3 equiv of Zn(II)TPP with 1 leads to the self-assembled structure 2 with the phosphorus ligand located in the middle of the cage structure, as established by titration studies in solution18,19 and later by X-ray analysis (Figure 2).20
Figure 2.

Encapsulation of ligand 1 with Zn(II)TPP leads to self-assembled structure 2. The crystal structure of 2 is displayed with CH−π interactions highlighted in green. Catalyst 3 is formed under syngas pressure in the presence of a rhodium precursor (DFT optimized structure depicted; for the chemdraw structure, see Figure 5).
From the X-ray structure, it is clear that the structure is based on pyridyl–zinc coordination, as well as CH−π interactions between adjacent Zn(II)TPP building blocks. The latter also explains why cooperative binding is observed, the third porphyrin is bound 5 times more strongly to 1 than the first (K ≈ 103–104; Figure 2).19,21 The strength of the template ligand approach for encapsulation was first demonstrated by application of capsule 2 (1(Zn(II)TPP)3) in the rhodium catalyzed hydroformylation of 1-octene (Figure 3). Catalyst 3 favors formation of the branched aldehyde product in the hydroformylation of terminal alkenes, which usually is the minor product. Alongside the unprecedented selectivity, the encapsulated catalyst was ten times more active than the rhodium catalysts formed by ligand 1 in the absence of Zn(II)TPP. The higher rates observed are partly explained by the formation of rhodium complexes with only one phosphine coordinated, as a result of the encapsulation of the template ligand.19
Figure 3.

Unique selectivity achieved in 1-octene and trans-2-octene hydroformylation with encapsulated catalyst 3 (for the chemdraw structure of 3, see Figure 5).
Hydroformylation of internal alkenes is challenging because of the inherent low reactivity of the disubstituted double bond. It is very difficult to control the regioselectivity, as the two carbon atoms are very similar in terms of sterics and electronics, and as such traditional catalysts generally produce both isomers in equal amounts. Interestingly, high selectivity was observed when catalyst 3 was applied in hydroformylation of internal alkenes, with the innermost carbon atom being the favored position for CO insertion, leading to the 3-aldehyde formation from 2-alkenes with a selectivity of up to 91%.20,22 Kinetic studies, in situ spectroscopy, and computational studies show that hydride migration is selectivity determining in these reactions. For trans-2-octene, the migration leading to the formation of the aldehyde at C2 requires significant structural rearrangement of the capsule, whereas the hydride migration leading to the 3-aldehyde requires no rearrangement of the second coordination sphere, explaining how these capsules induce the selectivity in this reaction.20 Next to this high selectivity, encapsulation also leads to a rate enhancement in the hydroformylation of trans-2-octene. Similar effects are observed for a variety of internal alkenes.20 Catalyst 3 was also applied at higher, industrially more relevant, temperatures (up to 75 °C), and 1-octene was converted with high rates and characteristic branched selectivity, but only at high partial CO pressure (60 bar). At elevated temperature and 10 bar of CO (and 10 bar hydrogen), the complex no longer shows the monophosphine coordination that is typical for the encapsulated catalysts, but bisphosphine coordination complexes formed instead. The equilibrium shifts back to monocoordination at higher pressure and with that also the selectivity of the catalyst.23
The use of zinc(II)tetraphenylporpholactone, formed by oxidation of Zn(II)TPP, increases the association constant as the porphyrin is more electron poor. Cooperative binding of the third porphyrin moiety is still possible as the porphyrin structure does not change much (Figure 4).21 The cage based on Zn(II)porpholactone 4 was applied in the hydroformylation of propene, where the higher association constant facilitated the formation of a stable capsule in polar solvents, leading to higher activity and a better branched selectivity than the related catalyst 3. In fact, the encapsulated catalyst is one of the most selective catalysts reported to date, favoring formation of the branched aldehyde, which is difficult to form using traditional ligand systems.21 Another way to make the binding of the porphyrin to the ligand template stronger is by changing the metal in the porphyrin building block. For example, the binding of Ru(II)(CO)TPP to ligand template 1 is ten times stronger than for Zn(II)TPP, yet the shape of the cage formed is identical.19 In addition, the ruthenium–pyridyl bond is less dynamic. Indeed, the rhodium catalyst based on the capsule formed by template ligand 1 and Ru(II)(CO)tetraphenylporphyrin 6 results in higher selectivity for the branched product (l/b = 0.4) in the hydroformylation of 1-octene at room temperature.19
Figure 4.

Capsule building block variations: Porpholactone 4 binds more strongly to ligand template molecules. Zn(II)porphyrins 5 can be substituted on the phenyl rings, leading to different electronic properties. To retain the cooperative binding, meta substituents should be used. Ruthenium porphyrin 6 binds much more strongly to ligand templates. Phthallocyanin 7 generates a larger capsule around ligand templates. Zn(II)salphens (8)24,25 and Zn(II)bisthiosemicarbazonates (9)26 generally bind more strongly to ligand template molecules and are highly modular but much smaller than the Zn(II)TPP.
Several different ligand templates and capsule building block combinations have been studied in the hydroformylation of terminal and internal unfunctionalized alkenes (Figure 4). The porphyrin capsule building blocks can be modified with a single meta substituent on each phenyl ring, allowing the CH−π interaction with the other porphyrins and thereby forming a tight capsule by cooperative binding.20 Substitution of the Zn(II)TPP on ortho, para, or multiple positions of the phenyl ring changes the orientation of the phenyl ring and disturbs the CH−π interactions between the porphyrins, leading to a weaker coordination of the third porphyrin.19 To demonstrate the importance of the CH−π interactions, the Zn(II)TPP-d20 porphyrin with deuterium at all phenyl rings was used to form a close analogue catalyst 3, and this caged catalyst displayed a slightly lower selectivity than 3 (70% vs 91% 3-aldehyde). In addition, steric interactions between the porphyrins with substituents also change the shape of the cavity around the catalyst, and as a result the product distribution can be controlled from 91% for the 3-aldehyde to 60% for the 2-aldehyde. An encapsulated catalyst based on trispyridylphosphine ligand 1 and zinc(II)phthalocyanine 7 was also used for hydroformylation of internal alkenes, and modeling shows that the cavity created around the rhodium catalyst is much larger when Zn(II)phthalocyanine is used as building block (Figure 5). Interestingly, the phthalocyanine based capsular catalyst favors the formation of the 2-aldehyde in hydroformylation of trans-2-octene (70%). These experiments show that by only changing the second coordination sphere the selectivity can be controlled as the catalyst itself is identical to that of catalyst 3, and this catalyst with the smaller cage formed predominantly the 3-aldehyde product.20 These cages formed by the ligand template approach resembles strategies encountered in nature as in enzymes the selectivity is largely controlled by the second coordination sphere and not by the active site itself, and in the currently example, a selectivity issue in the hydroformylation of internal alkenes was addressed that is not easily solved by ligand design.
Figure 5.

Product selectivity in the hydroformylation of trans-2-octene can be controlled by changing only the cavity size of the capsule, which can be modified using different building blocks for the assembly of the catalyst: space-filling models of rhodium catalyst 3 (left, DFT calculated structure) and the catalyst based on phthallocyanin building block 7 (right, DFT calculated structure) showing that the cavity size around the rhodium atom is 5 times larger.
The shape and size of the cavity formed can be modified by changing the capsule building block, as discussed above, but also by using different ligand templates. For example, application of ligand template 10 was explored in 1-octene hydroformylation (Figure 6).19,27 Because of the different position of the pyridine group on the ligand, the capsule formed is larger, allowing for two ligand template molecules to be coordinated to rhodium in the presence of capsule building blocks. In situ spectroscopy and the results in catalysis confirmed the formation of a bisphosphorus coordinated rhodium species under catalytic conditions. As the Zn(II)TPP building blocks are coordinated to the pyridine groups, the capsule formed is significantly larger than for the parent complex 3. The selectivity of the catalyst is typical for bisphosphorus rhodium catalysts, and the cage has only an effect on the activity of the catalyst, which depends on the substituents on the phenyl groups of the porphyrin building block. This indicates electronic communication between the phosphine and the 4-pyridyl, which is not observed for the 3-pyridyl; electron withdrawing substituents give rise to faster catalysis, and electron donating substituents decrease the catalytic rate.27
Figure 6.

Encapsulation of tris-4-pyridylphosphine (10) leads to the formation of a bisphosphorus coordinated rhodium catalyst that displays linear selective hydroformylation of 1-octene; changing the substituents on the porphyrin can be used to tune the activity of the catalyst.
Trispyridylphosphite 11 is a slightly larger ligand template than 1, and as such it can accommodate larger porphyrins. In addition, the electronic properties of the phosphorus ligand are different: 1 is more electron donating, while 11 is a more π-accepting ligand (Figure 7).19 Application of Zn(II)TPP with ligand template 11 in 1-octene hydroformylation leads to slow catalysis but with a clear change in selectivity; the branched aldehyde is preferably formed, while in absence of Zn(II)TPP, the linear aldehyde is formed (l/b = 6.9). Application of bulkier porphyrin 4a increases the rate of the reaction, but the product selectivity is lost. When porphyrin 4b is used to form the encapsulated hydroformylation catalyst, the rate compared to Zn(II)TPP is doubled but the typical encapsulated selectivity is retained.19
Figure 7.

Ligand template 11 can form an encapsulated catalyst with bulky porphyrins; the exact steric environment around the catalyst has large effects on the activity and selectivity of the catalyst in 1-octene hydroformylation.
Encapsulated catalysts based on the ligand template approach were successfully applied in the asymmetric hydroformylation of internal alkenes (Figure 8).28,29 Analysis of the active species revealed that only one ligand template is attached to rhodium during the catalysis. In absence of Zn(II)TPP, the phosphorus is coordinated in the equatorial plane, and in the presence of Zn(II)TPP, an unusual coordination complex is formed with both the phosphorus and the hydride at the axial positions. The monocoordinated complexes result in relatively high rates, and the change in coordination geometry upon capsule formation results in increased selectivity as the ee of the product is a factor 2 higher. For comparison, catalysis with the benchmark (R,S)-BINAPHOS ligand yielded a racemic product for trans-2-octene, showing the potential of these encapsulated monodentate ligand structures.28 Several ligand variations of ligand template 12 were developed, and the effect of the coordination of different capsule building blocks showed that the electronic properties of different Zn(II)porphyrins and Zn(II)salphens influence the electron density at the rhodium atom, influencing the activity of the catalyst, whereas the selectivity was about the same for all encapsulated catalysts.29
Figure 8.

Encapsulation of ligand template 12·RhH(CO)3 changes the geometry around the rhodium atom, increasing the enantiomeric excess (ee) of the aldehyde product formed.
In an attempt to further improve the catalyst selectivity, ligand 14 was encapsulated in more rigid supramolecular cage 13, consisting of two porphyrin moieties held together by pillar like structures (Figure 9).30 In situ spectroscopy demonstrated that also in this complex the coordination geometry is such that the hydride and the phosphorus are coordinated at the axial position, but embedding the same catalyst in this cage did not give any selectivity in the asymmetric hydroformylation of 2-octene. In contrast, when catalyst 15 was applied in the hydroformylation of styrene derivatives, there was a very clear effect of catalyst encapsulation on the selectivity as the enantiomeric excess increased from 9% to 74%, and also higher turnover numbers were obtained compared to 14·RhH(CO)3 and 14·RhH(CO)3·(Zn(II)TPP)2.30
Figure 9.

Formation of supramolecular porphyrin cage 13, encapsulation of ligand template 14·RhH(CO)3 in cage 13 forms catalyst 15; the increase in confined space leads to an increase in ee from 9% for the nonencapsulated catalyst (14·RhH(CO)3) to 74% for the encapsulated catalyst (15).
To expand the ligand template approach to bidentate ligands, ligand template 14 was combined with bis-zinc(II)salphen 16 leading to the construction of a box-like capsule in which bidentate coordination is facilitated (Figure 10).31 The self-assembled ligand templated capsule 17 was applied in the hydroformylation of both cis- and trans-2-octene, leading to high enantioselectivity in both cases and with good regioselectivity for the formation of the 3-aldehyde, in contrast to benchmark bidentate ligands applied in the same reaction.31 The substrate scope was extended to shorter and longer internal alkenes, for which ligand template capsule 17 gave similar results.
Figure 10.

Application of bis-zinc(II)salphen complex 16 to generate bidentate ligand template capsule 17 leads to up to 86% ee in the enantioselective hydroformylation of internal alkenes.
In a different strategy to arrive at encapsulated bidentate ligands, a bidentate analogue of 12, ligand template 18 (Figure 11), was designed to probe the influence of encapsulation on such bidentate complexes.32 Coordination studies under catalytic conditions reveal that the phosphine is coordinated trans to the hydride at the axial position, and the phosphoramidite is coordinated in the equatorial plane, regardless of the presence of Zn(II)TPP. When these systems were applied in the hydroformylation of styrene derivatives, the highest selectivities were obtained when Ru(II)(CO)TPP was used for encapsulation, showing that the bidentate ligand template requires more strongly coordinating capsule building blocks to obtain the optimal effect. The effect of catalyst encapsulation on the improvement of the enantioselectivity was again substantial, producing the aldehyde in 82% ee compared to 18% in the absence of the porphyrin template.
Figure 11.

Bidentate phosphine–phosphoramidite ligand template 18 requires encapsulation with strongly coordinating Ru(II)(CO)TPP 6 for optimal chiral induction.
Ligand Template Encapsulated Catalysts As a General Approach in Homogeneous Catalysis
The previous section highlighted the strength of the ligand template approach to catalyst encapsulation to solve challenges in hydroformylation catalysis, and in this section, we will demonstrate that this approach can be extended to a variety of other metal catalyzed reactions. Ligand 1 was found to be applicable in the palladium catalyzed Heck reaction, where the encapsulation of the ligand template with Zn(II)TPP coordinated led to faster initiation of the catalyst.18 Encapsulation facilitates the dissociation of phosphine ligands from the palladium(0) species, required to enter the catalytic cycle.
Ligand template 18 was applied in the asymmetric hydrogenation, leading to very high conversion and enantioselectivity when encapsulated by strongly binding building blocks (Figure 12).33 Most significantly, when the rhodium complex of ligand template 18 was applied in the asymmetric hydrogenation of dimethyl itaconate, the product was generated in racemic form, whereas the encapsulated analogue, with either strongly binding Zn(II)salphens or Ru(II)(CO)porphyrins, resulted in formation of the enantiopure product.
Figure 12.

Asymmetric hydrogenation of dimethyl itaconate proceeds with high enantioselectivity when strongly binding porphyrin building blocks are used to form the capsule, whereas in absence of these building blocks the products are generated in racemic form.
BIAN based catalyst 19 was applied in the copolymerization of 4-tert-butylstyrene and carbon monoxide (Figure 13).34 Encapsulation of this catalyst was achieved using Zn(II)salphen building blocks, and encapsulation increases the activity of the catalyst and produces a syndiotactic copolymer, instead of the atactic copolymer formed in absence of capsule building blocks.
Figure 13.

Palladium–BIAN complex 19 encapsulated by Zn(II)salphens results in increase of the activity of the catalyst and changes the stereoregularity of the polymer product.
Ligand template 12 was successfully applied in gold catalyzed ring-closing reactions (Figure 14).35 When the phenyl substituted analog of 12 was applied, a mixture of the five- and the six-membered ring product were formed. The gold complex inside the cage formed by template ligand 12 and two zinc(II)salphens was demonstrated to be an active catalyst that resulted in the exclusive formation of the five-membered ring. Although a stereogenic carbon atom is formed in this reaction, the chiral induction by the complexes is poor, leading to low ee values in all cases.
Figure 14.

Gold catalyzed ring-closing reaction selectively produces the five-membered ring product when an encapsulated catalyst is applied.
1,3,5-Triaza-7-phosphaadamantane (PTA) has three nitrogen donor atoms, just like trispyridylphosphine 1, and as such its use as a ligand template was studied (Figure 15).36 Coordination studies revealed that two or three zinc(II)salphen building blocks can coordinate to the nitrogen atoms of the ligand template, depending on the size of the zinc(II)salphen building blocks used, as confirmed by X-ray crystallography. The encapsulated PTA ligand was applied in the rhodium catalyzed hydrosilylation of 1-hexene, and the results indicated that the encapsulated PTA ligand behaves similar to bulky phosphine ligands when encapsulated. The steric bulk could be tuned by the introduction of different zinc(II)salphen capsule building blocks.
Figure 15.

Number of capsule building blocks encapsulating the PTA ligand template depends on the size of the Zn(II)salphen building block, as proved from the X-ray structures.
Ligand Template Assembly To Generate Multinuclear Complexes in Nanospheres
In the first part of this Account, the (divergent) ligand template encapsulation strategy presented aimed for modulation of the second coordination sphere around a single catalyst site, and with that it provides a new tool to control crucial properties such as activity and product selectivity. In the final part of this Account, we present our (convergent) ligand template strategies to form multinuclear complexes in well-defined nanospheres. Fujita introduced the self-assembly of dipyridyl-type building blocks into MnL2n nanospheres upon coordination of the pyridine moieties to Pd or Pt (M) atoms (Figure 16). These structures allow both exo- and endo-functionalization, by simple modification of the building block, and they contain large apertures to allow easy diffusion of substrates and products.37 When the ligand is endo-functionalized with either a ligand or a metal complex, the template ligand approach leads to formation of M12L24 nanospheres that contain up to 24 complexes. Interestingly, the local concentration of metal complexes in the nanosphere is around one molar (1 M), a concentration regime that is virtually unexplored, and the effect of these extremely high local catalyst concentrations can nicely be investigated.
Figure 16.

Formation of a M12L24 nanosphere with endo-functionalization (3D model; Au atom in yellow and Cl atom in green are highlighted) and its application in the cyclization of allenol substrate.
The influence of the local concentration of phosphine gold chloride complexes on their reactivity was investigated using gold building block L(AuCl) that self-assembles into the nanosphere Pd12(L(AuCl))24 in the presence of a palladium precursor. The local concentration could be further tuned by generating spheres based on mixtures of L(AuCl) and a building block that contains a methoxy group instead of the gold complex. By changing the ratio of these building blocks in the preparation of the nanospheres, the local concentration of gold in the spheres could be modulated from 0.05 to 1.07 M.38 The reactivity of the gold complex inside the sphere was investigated in gold-catalyzed cyclization of allenol, and surprisingly, at high local concentration no chloride abstraction was required in order to observe product formation. This effect was attributed to aurophilic interactions between the gold complexes inside the nanospheres observed by UV–vis spectroscopy, suggesting that dinuclear (or multinuclear) complexes are responsible for the activity. Importantly, the activity (higher TOF and TON) increases with the local concentration of gold (Figure 17).
Figure 17.
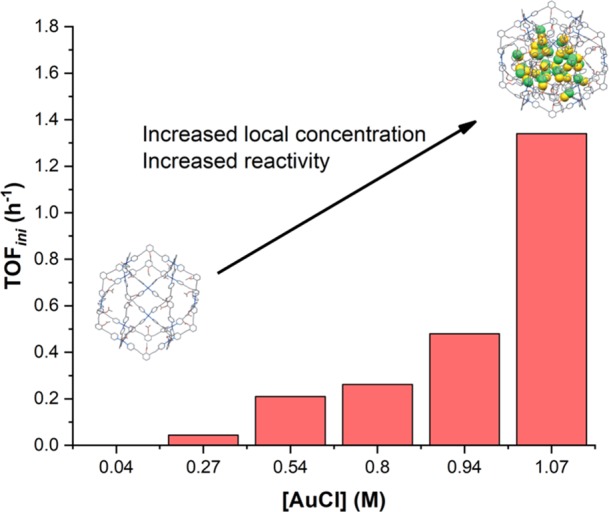
Correlation between the local concentration of gold chloride inside the nanosphere and the reactivity in the cyclization of allenol.
To further increase the reactivity and extend the substrate scope, formation of cationic gold complex inside the nanosphere was probed using silver hexafluorophosphate, but this resulted in nanopshere precipitation and decomposition.39 In order to circumvent this problem, more stable platinum nanospheres were synthesized, and these are stable upon chloride abstraction. These nanosphere catalysts have been applied in various gold-catalyzed cyclizations, and again the higher local concentration of gold complexes in the nanosphere resulted in higher reactivity and in different selectivity for several reactions (Figure 18).
Figure 18.

Extension of the reactivity of the Pt12L(AuCl)24 by chloride abstraction. The main product observed with the nanosphere is indicated in red with the corresponding ratio and those for PPh3AuCl between brackets.
While these examples demonstrate that high local concentration of metal complex can lead to better catalyst performance, substrate preorganization is not taken into account. Additionally, for each catalyst a separate building block has to be prepared. To address these issues, a more general strategy to template the formation of multinuclear complexes in nanospheres has been developed using a guanidinium functionalized nanosphere (Figure 19).40 Guanidine functional groups can strongly bind sulfonate functionalized catalysts through cooperative hydrogen-bonding and at the same time preorganize carboxylate functionalized substrate molecules, which bind more weakly but significantly, leading to subtle control of the local concentration of both the catalyst and the substrate inside nanosphere. The concept was demonstrated by evaluation of the gold catalyzed cyclization of 4-pentynoic acid. Performing this reaction inside the guanidinium nanosphere, coined the nanoconcentrator, using 4 equivalents of sulfonated gold phosphine catalyst resulted in increased TOF and TON up to 50-fold compared to the reaction in the absence of sphere, as a result of substrate preorganization nearby the gold catalyst that is bound to guanidinium moieties. Substrate preorganization can also be used for substrate selective catalysis. In a competition experiment between 4-pentynoic acid and allenol, only the 4-pentynoic acid that is bound to the guanidine group is converted into product when the reaction is performed in the presence of the nanoconcentrator, in contrast to the control experiment in the absence of the nanoconcentrator in which case both products are formed in equal amounts.
Figure 19.

Preparation of the nanoconcentrator based on a guanidinium ligand and schematic representation of the preorganization effect of the nanoconcentrator on the cyclization of 4-pentynoic acid. Substrate selectivity is based on recognition.
Next, the power of catalyst preconcentration using the guanidinium functionalized M12L24 nanosphere was demonstrated in electrochemical water oxidation using a ruthenium catalyst (Figure 20).41 Water oxidation catalysis can proceed via an oxyl radical mechanism (I2M), as well as a water nucleophilic attack (WNA) mechanism, the former generally leading to higher rates at milder overpotentials. The phenanthroline based complex is reported to proceed via WNA, whereas the bipyridine analogue can proceed via I2M, but only when the concentration of catalyst is sufficient to allow the coupling of two metal–oxo moieties (Figure 20). Electrochemical experiments performed at the usual low concentration (10–5 M) in the presence of various amounts of guanidinium functionalized M12L24 nanosphere show that the reaction rate can be increased by a factor 140 by only increasing the local concentration of sulfonated ruthenium catalyst. In contrast, the effect of local concentration on the catalyst that proceeds via WNA (i.e., the mononuclear pathway) is negligible. Bulk electrolysis demonstrated the stability of the cage as well as the catalyst, which was confirmed by NMR analysis of the sample after the experiment.
Figure 20.

Ru-catalyzed electrochemical water oxidation proceeding via water nucleophilic attack (WNA) or (dinuclear) oxyl radical mechanism (I2M) depending on the ligand. Relative rate for the electrochemical water oxidation (compared to free catalyst) as a function of the local ruthenium concentration; the reaction that proceeds via I2M is accelerated by the nanoconcentrator.
Conclusion and Outlook
The field of transition metal catalysis has been dominated by ligand design strategies in the past five decades, and only more recently the value of the second coordination sphere is appreciated. Whereas for simple transformations it is relatively easy to perform reactions in molecular cages, controlling the reaction process by the second coordination sphere, this is relatively difficult for transition metal catalysts as the complex changes substantially in the various steps of the catalytic cycle. Over the past two decades, we have demonstrated that the ligand template assembly strategy for encapsulation of transition metal catalysts is a highly effective and general strategy. Various catalytic reactions can now be controlled via the second coordination sphere as we have a general route for encapsulation of transition metal catalysts. Several different ligand templates have been developed and numerous building blocks that vary in steric properties, electronics, and binding strength have been studied, also allowing the application of self-assembled encapsulated catalysts under industrially relevant conditions.21,23 Encapsulated rhodium catalysts gave branched selective hydroformylation of 1-octene,18,19 and also the first catalysts for regioselective hydroformylation of internal unfunctionalized alkenes with especially high selectivity for hydroformylation of the inner carbon atom were achieved for the first time.20,22 In addition, unprecedented high enantioselectivity in the asymmetric hydroformylation of internal unfunctionalized alkenes28,29,31 has been achieved. The ligand template strategy was also applied in the Heck reaction,18 polymerization,34 hydrogenation,33 hydrosilylation,36 and gold catalysis.35 Divergent ligand templates have been used to generate nanospheres with multiple complexes in confined space. For example, based on the endo-functionalized nanospheres reported by Fujita, a ligand template strategy for encapsulation of 24 gold complexes was developed leading to high local concentrations of gold. This preorganization was extended to a supramolecular approach, in which catalysts were bound through binding of sulfonated ligands to the guanidinium moieties inside the nanosphere. Beneficial effects of the high local concentration were found in gold-catalyzed cyclization and electrochemical water oxidation. As many reactions benefit from high concentrations, this may be a general powerful approach for optimization of transition metal catalysts.
Acknowledgments
We thank the European Research Council (ERC-AG 339786-NAT_CAT) for financial support.
Biographies
Lukas Jongkind obtained his M.Sc. degree in chemistry cum laude from the University of Amsterdam in 2014, after which he joined the group of Prof. J. N. H. Reek to pursue a Ph.D. on the development of new supramolecular systems and their applications in homogeneous catalysis.
Xavier Caumes obtained his Ph.D. from the Université Pierre et Marie Curie in 2016 under the supervision of Dr. L. Bouteiller and Dr. M. Raynal. He joined the group of Prof. J. N. H. Reek in 2017 to pursue research on chirality amplification in supramolecular cages and their application in transition metal catalysis.
Arnout Hartendorp received his M.Sc. in chemistry at the University of Amsterdam and is a Ph.D. candidate under supervision of Prof. J. N. H. Reek and Prof. J. van Maarseveen at this same university. His research focuses on peptide cyclizations in supramolecular assemblies.
Joost Reek obtained his Ph.D. at the University of Nijmegen under supervision of Prof. R. J. M. Nolte in the area of supramolecular chemistry. After a postdoctoral appointment with Prof. Crossley in Sydney, he moved to the University of Amsterdam in 1998, where he was promoted to full professor in 2006, and faculty professor in 2017. His research interests include homogeneous catalysis and supramolecular chemistry, and he is exploring new research on the border of these research topics. In addition, he has a research program on catalysis for green energy applications, aiming at solar to fuel devices based on molecular components.
The authors declare no competing financial interest.
Special Issue
Published as part of the Accounts of Chemical Research special issue “Supramolecular Chemistry in Confined Space and Organized Assemblies”.
References
- Lehn J.-M. Supramolecular Chemistry—Scope and Perspectives Molecules, Supermolecules, and Molecular Devices. Angew. Chem., Int. Ed. Engl. 1988, 27, 89–112. 10.1002/anie.198800891. [DOI] [Google Scholar]
- Cram D. J. The Design of Molecular Hosts, Guests, and Their Complexes. Angew. Chem., Int. Ed. Engl. 1988, 27, 1009–1112. 10.1002/anie.198810093. [DOI] [PubMed] [Google Scholar]
- Pedersen C. J. The Discovery of Crown Ethers. Angew. Chem. 1988, 100, 1053–1059. 10.1002/ange.19881000805. [DOI] [PubMed] [Google Scholar]
- Kang J.; Rebek J. Entropically Driven Binding in a Self-Assembling Molecular Capsule. Nature 1996, 382, 239–241. 10.1038/382239a0. [DOI] [PubMed] [Google Scholar]
- Koblenz T. S.; Wassenaar J.; Reek J. N. H. Reactivity within a Confined Self-Assembled Nanospace. Chem. Soc. Rev. 2008, 37, 247–262. 10.1039/B614961H. [DOI] [PubMed] [Google Scholar]
- Catti L.; Zhang Q.; Tiefenbacher K. Advantages of Catalysis in Self-Assembled Molecular Capsules. Chem. - Eur. J. 2016, 22, 9060–9066. 10.1002/chem.201600726. [DOI] [PubMed] [Google Scholar]
- Yoshizawa M.; Klosterman J. K.; Fujita M. Functional Molecular Flasks: New Properties and Reactions within Discrete, Self-Assembled Hosts. Angew. Chem., Int. Ed. 2009, 48, 3418–3438. 10.1002/anie.200805340. [DOI] [PubMed] [Google Scholar]
- Vriezema D. M.; Comellas Aragonès M. C.; Elemans J. A. A. W.; Cornelissen J. J. L. M.; Rowan A. E.; Nolte R. J. M. Self-Assembled Nanoreactors. Chem. Rev. 2005, 105, 1445–1490. 10.1021/cr0300688. [DOI] [PubMed] [Google Scholar]
- Dong Z.; Luo Q.; Liu J. Artificial Enzymes Based on Supramolecular Scaffolds. Chem. Soc. Rev. 2012, 41, 7890. 10.1039/c2cs35207a. [DOI] [PubMed] [Google Scholar]
- Zarra S.; Wood D. M.; Roberts D. A.; Nitschke J. R. Molecular Containers in Complex Chemical Systems. Chem. Soc. Rev. 2015, 44, 419–432. 10.1039/C4CS00165F. [DOI] [PubMed] [Google Scholar]
- Brown C. J.; Toste F. D.; Bergman R. G.; Raymond K. N. Supramolecular Catalysis in Metal-Ligand Cluster Hosts. Chem. Rev. 2015, 115, 3012–3035. 10.1021/cr4001226. [DOI] [PubMed] [Google Scholar]
- Leenders S. H. A. M.; Gramage-Doria R.; de Bruin B.; Reek J. N. H. Transition Metal Catalysis in Confined Spaces. Chem. Soc. Rev. 2015, 44, 433–448. 10.1039/C4CS00192C. [DOI] [PubMed] [Google Scholar]
- Raynal M.; Ballester P.; Vidal-Ferran A.; Van Leeuwen P. W. N. M. Supramolecular Catalysis. Part 1: Non-Covalent Interactions as a Tool for Building and Modifying Homogeneous Catalysts. Chem. Soc. Rev. 2014, 43, 1660–1733. 10.1039/C3CS60027K. [DOI] [PubMed] [Google Scholar]
- Raynal M.; Ballester P.; Vidal-Ferran A.; van Leeuwen P. W. N. M. Supramolecular Catalysis. Part 2: Artificial Enzyme Mimics. Chem. Soc. Rev. 2014, 43, 1734–1787. 10.1039/C3CS60037H. [DOI] [PubMed] [Google Scholar]
- Wiester M. J.; Ulmann P. A.; Mirkin C. A. Enzyme Mimics Based upon Supramolecular Coordination Chemistry. Angew. Chem., Int. Ed. 2011, 50, 114–137. 10.1002/anie.201000380. [DOI] [PubMed] [Google Scholar]
- Otte M. Size-Selective Molecular Flasks. ACS Catal. 2016, 6, 6491–6510. 10.1021/acscatal.6b01776. [DOI] [Google Scholar]
- Kleij A. W.; Reek J. N. H. Ligand-Template Directed Assembly: An Efficient Approach for the Supramolecular Encapsulation of Transition-Metal Catalysts. Chem. - Eur. J. 2006, 12, 4218–4227. 10.1002/chem.200500875. [DOI] [PubMed] [Google Scholar]
- Slagt V. F.; Reek J. N. H.; Kamer P. C. J.; Van Leeuwen P. W. N. M. Assembly of Encapsulated Transition Metal Catalysts. Angew. Chem., Int. Ed. 2001, 40, 4271–4274. 10.1002/1521-3773(20011119)40:22<4271::AID-ANIE4271>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Slagt V. F.; Kamer P. C. J.; Van Leeuwen P. W. N. M.; Reek J. N. H. Encapsulation of Transition Metal Catalysts by Ligand-Template Directed Assembly. J. Am. Chem. Soc. 2004, 126, 1526–1536. 10.1021/ja0386795. [DOI] [PubMed] [Google Scholar]
- Bocokić V.; Kalkan A.; Lutz M.; Spek A. L.; Gryko D. T.; Reek J. N. H. Capsule-Controlled Selectivity of a Rhodium Hydroformylation Catalyst. Nat. Commun. 2013, 4, 2670. 10.1038/ncomms3670. [DOI] [PubMed] [Google Scholar]
- Wang X.; Nurttila S. S.; Dzik W. I.; Becker R.; Rodgers J.; Reek J. N. H. Tuning the Porphyrin Building Block in Self-Assembled Cages for Branched-Selective Hydroformylation of Propene. Chem. - Eur. J. 2017, 23, 14769–14777. 10.1002/chem.201702113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuil M.; Soltner T.; Van Leeuwen P. W. N. M.; Reek J. N. H. High-Precision Catalysts: Regioselective Hydroformylation of Internal Alkenes by Encapsulated Rhodium Complexes. J. Am. Chem. Soc. 2006, 128, 11344–11345. 10.1021/ja063294i. [DOI] [PubMed] [Google Scholar]
- Besset T.; Norman D. W.; Reek J. N. H. Supramolecular Encapsulated Rhodium Catalysts for Branched Selective Hydroformylation of Alkenes at High Temperature. Adv. Synth. Catal. 2013, 355, 348–352. 10.1002/adsc.201200790. [DOI] [Google Scholar]
- Jacobs I.; van Duin A. C. T.; Kleij A. W.; Kuil M.; Tooke D. M.; Spek A. L.; Reek J. N. H. Conformational Studies of Ligand-Template Assemblies and the Consequences for Encapsulation of Rhodium Complexes and Hydroformylation Catalysis. Catal. Sci. Technol. 2013, 3, 1955. 10.1039/c3cy20665c. [DOI] [Google Scholar]
- Kleij A. W.; Lutz M.; Spek A. L.; van Leeuwen P. W. N. M.; Reek J. N. H. Encapsulated Transition Metal Catalysts Comprising Peripheral Zn(Ii)Salen Building Blocks: Template-Controlled Reactivity and Selectivity in Hydroformylation Catalysis. Chem. Commun. 2005, (29), 3661. 10.1039/b503708e. [DOI] [PubMed] [Google Scholar]
- Bocokić V.; Lutz M.; Spek A. L.; Reek J. N. H. Bis-(Thiosemicarbazonato) Zn(II) Complexes as Building Blocks for Construction of Supramolecular Catalysts. Dalton Trans. 2012, 41, 3740. 10.1039/c2dt12096h. [DOI] [PubMed] [Google Scholar]
- Kleij A. W.; Kuil M.; Tooke D. M.; Spek A. L.; Reek J. N. H. Template-Assisted Ligand Encapsulation; the Impact of an Unusual Coordination Geometry on a Supramolecular Pyridylphosphine-Zn(II)Porphyrin Assembly. Inorg. Chem. 2005, 44, 7696–7698. 10.1021/ic050858v. [DOI] [PubMed] [Google Scholar]
- Bellini R.; Chikkali S. H.; Berthon-Gelloz G.; Reek J. N. H. Supramolecular Control of Ligand Coordination and Implications in Hydroformylation Reactions. Angew. Chem., Int. Ed. 2011, 50, 7342–7345. 10.1002/anie.201101653. [DOI] [PubMed] [Google Scholar]
- Bellini R.; Reek J. N. H. Coordination Studies on Supramolecular Chiral Ligands and Application in Asymmetric Hydroformylation. Chem. - Eur. J. 2012, 18, 7091–7099. 10.1002/chem.201200225. [DOI] [PubMed] [Google Scholar]
- García-Simón C.; Gramage-Doria R.; Raoufmoghaddam S.; Parella T.; Costas M.; Ribas X.; Reek J. N. H. Enantioselective Hydroformylation by a Rh-Catalyst Entrapped in a Supramolecular Metallocage. J. Am. Chem. Soc. 2015, 137, 2680–2687. 10.1021/ja512637k. [DOI] [PubMed] [Google Scholar]
- Gadzikwa T.; Bellini R.; Dekker H. L.; Reek J. N. H. Self-Assembly of a Confined Rhodium Catalyst for Asymmetric Hydroformylation of Unfunctionalized Internal Alkenes. J. Am. Chem. Soc. 2012, 134, 2860–2863. 10.1021/ja211455j. [DOI] [PubMed] [Google Scholar]
- Bellini R.; Reek J. N. H. Application of Supramolecular Bidentate Hybrid Ligands in Asymmetric Hydroformylation. Chem. - Eur. J. 2012, 18, 13510–13519. 10.1002/chem.201202044. [DOI] [PubMed] [Google Scholar]
- Bellini R.; Reek J. N. H. Supramolecular Hybrid Bidentate Ligands in Asymmetric Hydrogenation. Eur. J. Inorg. Chem. 2012, 2012, 4684–4693. 10.1002/ejic.201200549. [DOI] [Google Scholar]
- Flapper J.; Reek J. N. H. Templated Encapsulation of Pyridyl-Bian Palladium Complexes: Tunable Catalysts for CO/4-Tert-Butylstyrene Copolymerization. Angew. Chem., Int. Ed. 2007, 46, 8590–8592. 10.1002/anie.200703294. [DOI] [PubMed] [Google Scholar]
- Gramage-Doria R.; Bellini R.; Rintjema J.; Reek J. N. H. Supramolecular Ligands in Gold(I) Catalysis. ChemCatChem 2013, 5, 1084–1087. 10.1002/cctc.201200541. [DOI] [Google Scholar]
- Anselmo D.; Gramage-Doria R.; Besset T.; Escárcega-Bobadilla M. V.; Salassa G.; Escudero-Adán E. C.; Martínez Belmonte M.; Martin E.; Reek J. N. H.; Kleij A. W. Supramolecular Bulky Phosphines Comprising 1,3,5-Triaza-7-Phosphaadamantane and Zn(Salphen)s: Structural Features and Application in Hydrosilylation Catalysis. Dalton Trans. 2013, 42, 7595. 10.1039/c3dt00078h. [DOI] [PubMed] [Google Scholar]
- Harris K.; Fujita D.; Fujita M. Giant Hollow M(n)L(2n) Spherical Complexes: Structure, Functionalisation and Applications. Chem. Commun. 2013, 49, 6703–6712. 10.1039/c3cc43191f. [DOI] [PubMed] [Google Scholar]
- Gramage-Doria R.; Hessels J.; Leenders S. H. A. M.; Tröppner O.; Dürr M.; Ivanović-Burmazović I.; Reek J. N. H. Gold(I) Catalysis at Extreme Concentrations inside Self-Assembled Nanospheres. Angew. Chem., Int. Ed. 2014, 53, 13380–13384. 10.1002/anie.201406415. [DOI] [PubMed] [Google Scholar]
- Leenders S. H. A. M.; Dürr M.; Ivanovic-Burmazovic I.; Reek J. N. H. Gold Functionalized Platinum M12L24-Nanospheres and Their Application in Cyclization Reactions. Adv. Synth. Catal. 2016, 358, 1509–1518. 10.1002/adsc.201600071. [DOI] [Google Scholar]
- Wang Q. Q.; Gonell S.; Leenders S. H. A. M.; Dürr M.; Ivanovic-Burmazovic I.; Reek J. N. H. Self-Assembled Nanospheres with Multiple Endohedral Binding Sites Pre-Organize Catalysts and Substrates for Highly Efficient Reactions. Nat. Chem. 2016, 8, 225–230. 10.1038/nchem.2425. [DOI] [PubMed] [Google Scholar]
- Yu F.; Poole D. III; Mathew S.; Yan N.; Hessels J.; Orth N.; Ivanović-Burmazović I.; Reek J. N. H. Control over Electrochemical Water Oxidation Catalysis by Preorganization of Molecular Ruthenium Catalysts in Self-Assembled Nanospheres. Angew. Chem., Int. Ed. 2018, 10.1002/anie.201805244. [DOI] [PMC free article] [PubMed] [Google Scholar]