

Peptidoglycan biosynthesis is a dynamic and well-controlled pathway. The molecular assembly of PG and the regulatory pathways ensuring its maintenance are still not well understood. Here we studied the newly discovered Escherichia coli factor ElyC, which is important for PG biosynthesis at low temperatures. We revealed an important protein-folding defect in the ΔelyC mutant and showed that overproduction of the periplasmic chaperone DsbG or Spy was sufficient to correct the protein-folding defect and restore PG biosynthesis. These results show that the PG defect in the absence of ElyC is caused, at least in part, by a protein-folding problem in the cell envelope. Furthermore, we showed, for the first time, that the periplasmic protein DsbG has chaperone activity in vivo.
KEYWORDS: chaperones, peptidoglycan, ElyC, protein folding, protein chaperone
ABSTRACT
Peptidoglycan (PG) is the main structural component of bacterial envelopes. It protects bacterial cells against variations in osmotic pressure and cell lysis. The newly discovered Escherichia coli factor ElyC has been shown to be important for peptidoglycan biosynthesis at low temperatures. PG production in ΔelyC mutant cells is totally blocked after a few hours of growth at 21°C, triggering cell lysis. In this study, we took a candidate approach to identify genetic suppressors of the ΔelyC mutant cell lysis phenotype. We identified the periplasmic proteins DsbG and Spy as multicopy suppressors and showed that their overproduction restores PG biosynthesis in the ΔelyC mutant. Interestingly, we found that DsbG acts by a novel mechanism, which is independent of its known reductase activity and substrates. DsbG, like Spy, acts as a chaperone to reduce the amounts of protein aggregates in the envelopes of ΔelyC cells. In fact, we found that the amount of protein aggregates was greater in the ΔelyC mutant than in the wild type. Taken together, our results show a protein-folding defect in the envelope compartments of ΔelyC cells that blocks PG production, and they reveal a new physiological activity of DsbG.
IMPORTANCE Peptidoglycan biosynthesis is a dynamic and well-controlled pathway. The molecular assembly of PG and the regulatory pathways ensuring its maintenance are still not well understood. Here we studied the newly discovered Escherichia coli factor ElyC, which is important for PG biosynthesis at low temperatures. We revealed an important protein-folding defect in the ΔelyC mutant and showed that overproduction of the periplasmic chaperone DsbG or Spy was sufficient to correct the protein-folding defect and restore PG biosynthesis. These results show that the PG defect in the absence of ElyC is caused, at least in part, by a protein-folding problem in the cell envelope. Furthermore, we showed, for the first time, that the periplasmic protein DsbG has chaperone activity in vivo.
INTRODUCTION
The bacterial cell envelope is a complex and dynamic multilayered structure. It is the first and the major line of defense against threats from hostile external environments. In Gram-negative bacteria, such as Escherichia coli, the envelope is composed of the outer membrane (OM) and the inner membrane (IM), with a thin layer of peptidoglycan (PG) in between, in the periplasmic space (1, 2). PG is a unique and essential structural component of the bacterial envelope. It preserves cell integrity by providing mechanical strength to withstand variations in osmotic pressure, maintains the cell shape, and serves as a platform for the attachment of cell envelope proteins and polysaccharides (3–5). A defect in the PG biosynthesis pathway causes cell lysis, rendering this pathway one of the best targets for antibacterials.
PG is composed of linear glycan strands cross-linked by short peptides. The biosynthesis of this macromolecular structure occurs in three principal stages. First, the nucleotide precursors UDP-N-acetylglucosamine (UDP-GlcNAc) and UDP-N-acetylmuramyl pentapeptide (UDP-MurNAc) are synthesized in the cytoplasm by the action of several ligases. The next stage occurs in the cytoplasmic leaflet of the IM, where the nucleotide precursors are associated with the lipid carrier undecaprenyl phosphate (Und-P) to form the subunit of PG (lipid II). Thereafter, lipid II is flipped across the IM. In the final stage, lipid II is polymerized into glycan strands and cross-linked to the PG layer (6, 7). This stage is more complex than the earlier stages and is also much less understood. It is a dynamic and well-controlled process involving the spatial and temporal coordination of multiple enzymatic complexes, including synthetic complexes, which build the nascent PG and attach it to the existent PG, and hydrolytic complexes, which cut and remove the preexistent PG, allowing the insertion of new material.
The ElyC factor was identified recently by Paradis-Bleau et al. (8) following a genetic screen for bacterial envelope biogenesis mutants of E. coli. Phenotypic characterization of ΔelyC mutant cells showed that they grow normally at low temperatures (21 to 22°C) until they reach the late-exponential phase, when they start to lyse via a membrane-bulging mechanism. Cell lysis was shown to be a consequence of PG biosynthesis arrest. Indeed, direct measurement of PG biosynthesis in the cells by monitoring the incorporation of the radiolabeled PG precursor meso-[3H]diaminopimelic acid showed complete blockage of PG biosynthesis in the ΔelyC mutant after 5.5 h of growth at low temperatures. Because the cells started to lyse after 8 h of growth, it was suggested that cell lysis is initiated shortly after one mass doubling of the culture, following the expression of the PG-defective phenotype (8). It was also shown that overproduction of the PG biosynthesis enzymes controlling the major transitional point of the process (MurA, UppS, and penicillin-binding protein 1b [PBP1b]) fully suppresses the lysis phenotype of ΔelyC cells. MurA catalyzes the first step of nucleotide precursor biosynthesis in the cytoplasm (9). UppS produces the lipid carrier Und-P (10), which links to the PG precursor and allows its attachment to the inner membrane. PBP1b carries out the final polymerization and cross-linking reaction of the PG precursors in the periplasm (11). Interestingly, Paradis-Bleau et al. (8) showed that overproduction of UppS, unlike that of MurA or PBP1b, does not suppress the lysis phenotype of the PG-defective mutant inactivated for PBP1 (ΔmrcB). The fact that UppS could correct the PG defect in the ΔelyC mutant but not in the ΔmrcB mutant suggests that PG biosynthesis in the ΔelyC mutant is blocked at the level of the inner membrane steps of the pathway. Supporting this hypothesis is the observation that blocking the biosynthesis pathway of enterobacterial common antigen (ECA) can suppress the lysis phenotype of the ΔelyC mutant. Indeed, the production of ECA, like that of PG, requires the lipid carrier Und-P, and thus, ECA production competes with PG synthesis for the available Und-P pool (8).
ElyC is predicted to be an inner membrane protein with two transmembrane domains and a large domain of unknown function (DUF 218) in the periplasm (12). It is thus more likely that ElyC plays a role in the periplasm or the inner membrane than in the cytoplasm, which would also support the hypothesis that the PG biosynthesis pathway is blocked at the level of the envelope. The DUF 218 domain is highly conserved in bacteria, but little is known about the function(s) of DUF 218 domain-containing proteins (12). This large family of proteins possesses several conserved charged amino acids, suggesting that it plays an evolutionarily conserved role (12, 13). In E. coli, four proteins, ElyC, SanA, YgjQ, and YdcF, each carry a DUF 218 domain. However, the precise function(s) of these proteins is still unknown. SanA has been shown to play a role in vancomycin resistance, suggesting a possible implication of this protein in envelope biogenesis (14, 15).
Recent results in our laboratory have shown that ΔelyC mutant cells are sensitive to oxygen. Indeed, when the mutant grows under anaerobic conditions, it does not have a lysis phenotype. This suggests that oxygen is, at least in part, involved in ΔelyC cell lysis, likely by creating oxidative cell damage. Indeed, during aerobic respiration, the gain of a single electron by oxygen leads to the formation of harmful reactive oxygen species (ROS) (16, 17). Under normal physiological conditions, the ROS are neutralized by different enzymes. However, under stress conditions, the ROS damage cell components, including DNA, lipids, and proteins. Almost 70% of the molecules damaged by ROS are proteins (18). Thus, we hypothesize that proteins may be damaged in ΔelyC mutant cells. Starting from this hypothesis, we developed a candidate approach to identify genetic suppressors of the ΔelyC cell lysis phenotype. We identified the periplasmic proteins DsbG and Spy as multicopy suppressors of ΔelyC cell lysis. We showed that DsbG suppresses mutant cell lysis independently of its reductase activity and its known substrates. Moreover, DsbG was shown, for the first time, to possess a chaperone function in vivo. Unlike DsbG, Spy is already known to have a chaperone function; thus, the two suppressors identified act by the same mechanism in the ΔelyC mutant. Furthermore, analysis of protein aggregation showed that the ΔelyC mutant contains a protein-folding defect that is overcome by the overproduction of DsbG or Spy. As expected, we found that overproducing DsbG or Spy corrected the PG biosynthesis defect of ΔelyC cells. Taken together, these results reveal that a defect in the folding of periplasmic proteins is, at least in part, responsible for PG biosynthesis arrest and cell death in the absence of the ElyC factor.
RESULTS
The periplasmic reductase DsbG is a multicopy suppressor of the ΔelyC mutant lysis phenotype.
The amino acid cysteine is particularly sensitive to oxidation, since it processes an electron-rich sulfur atom in its side chain. Cysteine oxidation could lead to an irreversible modification that inactivates the proteins. Thus, oxidized cysteine should be reduced, or should interact with another cysteine of the same protein to form a native disulfide bond, so as to avoid protein inactivation. Based on our hypothesis that the sensitivity of the ΔelyC mutant to oxygen (see Fig. S1 in the supplemental material) could be the consequence of oxidative damage, we looked at the effect of oxidoreductase overexpression in the ΔelyC mutant. We tested periplasmic oxidoreductases from the Dsb (disulfide bond) family that are involved in the formation of a disulfide bond between two cysteines (DsbA, DsbB, DsbC) and in cysteine reduction (DsbC, DsbD, DsbG) (19). We used plasmids from a mobile plasmid library that consists of multicopy plasmids carrying different open reading frames (ORFs) of the E. coli genome (20). Plasmids carrying an ORF corresponding to a Dsb protein were transformed individually into ΔelyC mutant cells, and expression was induced by adding isopropyl-β-d-thiogalactopyranoside (IPTG). The transformed cells were tested using the chlorophenol red-β-d-galactopyranoside (CPRG) phenotypic assay as described previously (8). CPRG is a β-galactosidase (LacZ) substrate that cannot enter cells with an intact envelope. Mutants with a permeable envelope or a lysis phenotype produce bright-pink spots on plates (CPRG+ phenotype) because of CPRG cleavage by LacZ. The results of the CPRG phenotypic assay following overnight incubation at 21°C are shown in Fig. 1A. As expected, the control plasmid carrying the ElyC factor coding sequence suppressed the CPRG+ phenotype of the ΔelyC mutant. Interestingly, among the five different Dsb proteins tested, only DsbG could suppress the CPRG+ phenotype due to ΔelyC cell lysis.
FIG 1.

Overexpression of the periplasmic reductase DsbG suppresses the ΔelyC mutant lysis phenotype. (A) Screening for ΔelyC genetic suppressors with the CPRG assay. Cells from the E. coli wild-type strain MG1655 (WT), its ΔelyC mutant (strain EM9), and the ΔelyC mutant carrying a multicopy plasmid containing the indicated gene were patched onto LB agar supplemented with CPRG (20 μg/ml) and IPTG (100 μM) and were grown overnight at 21°C. (B) Testing the specificity of the DsbG suppression effect with the CPRG assay. ΔmrcB mutant (strain MM39) cells with or without a multicopy plasmid carrying the mrcB or dsbG gene were grown on a CPRG plate and were incubated overnight at 21°C. (C) Testing the DsbG suppression effect in LB liquid medium. Shown are growth curves of WT cells, ΔelyC cells, and ΔelyC cells with a plasmid carrying dsbG. The experiment was performed in triplicate. Data points and error bars represent average values and standard errors, respectively.
To determine whether the suppression of the CPRG+ phenotype by DsbG overproduction is specific to the ΔelyC mutant, we tested the ability of the DsbG-overproducing plasmid to correct the CPRG+ phenotype of the ΔmrcB mutant. This mutant lacks penicillin-binding protein 1b (PBP1b), an enzyme involved in the polymerization step of PG biosynthesis (21). We found that overproducing DsbG did not correct the CPRG+ phenotype of the ΔmrcB mutant. This result suggests that DsbG overproduction specifically corrects the problems leading to ΔelyC cell lysis.
As predicted, we found that overproducing DsbG totally restored the growth of ΔelyC cells in liquid LB medium at 21°C. In this experiment, DsbG expression was induced by adding 100 μM of IPTG to the medium. The ΔelyC mutant cells overexpressing DsbG grew as well as the wild-type (WT) strain and did not lyse as the mutant does (Fig. 1C). Indeed, as reported previously (8), ΔelyC mutant cells started to lyse at the late-exponential phase (optical density at 600 nm [OD600], ≈0.6). Taken together, our results show that the periplasmic reductase DsbG can act as a specific suppressor of the ΔelyC mutant lysis phenotype.
DsbG suppresses the lysis phenotype of ΔelyC mutant cells independently of its l,d-transpeptidase substrates.
DsbG is a V-shaped dimeric protein with a thioredoxin fold and a CXXC motif (Fig. 2A). The cysteines of DsbG are kept reduced by the inner membrane protein DsbD. Reduced DsbG, in turn, reduces the cysteine residue of proteins containing only one cysteine to protect it from oxidation (Fig. 2A). DsbG has been shown to reduce the cysteine residue of the l,d-transpeptidase YbiS and to interact with two other l,d-transpeptidases, YnhG and ErfK, suggesting that DsbG also reduces their single cysteines (22, 23). Reduced l,d-transpeptidases catalyze reactions of transpeptidation between two PG peptide chains to form PG cross-links, as well as between PG peptide chains and Braun's lipoprotein (Lpp) in the outer membrane (23, 24). It is thus possible that DsbG corrects the ΔelyC lysis phenotype by acting on its l,d-transpeptidase substrates. To investigate this possibility, we tested the ability of DsbG to suppress the lysis of an ΔelyC mutant in the absence of its substrates. First, we constructed a mutant with deletions of the genes encoding the three l,d-transpeptidases and named it the Δ3 mutant. This mutant was then tested on CPRG plates to ensure that it does not present an envelope or cell lysis defect (see Fig. S2 in the supplemental material). Starting from the Δ3 mutant, which has a CPRG− phenotype, we constructed the Δ4 mutant by deleting the ElyC coding sequence. Unlike the Δ3 mutant, the Δ4 mutant presents a CPRG+ phenotype, indicating that this phenotype is induced by the elyC gene deletion (Fig. 2B). Importantly, DsbG overproduction in the Δ4 mutant was still able to suppress the CPRG+ phenotype induced by the absence of ElyC. Furthermore, deletion of the two additional genes ycbB and ycfS, coding for l,d-transpeptidases that are not known to be substrates of DsbG (resulting in the Δ6 mutant), did not eliminate the ability of DsbG overproduction to suppress the CPRG+ phenotype of ΔelyC cells. Taken together, these results show that DsbG corrects the lysis phenotype of ΔelyC cells independently of its known l,d-transpeptidase substrates, and they suggest that DsbG acts on other proteins.
FIG 2.

DsbG does not act on the l,d-transpeptidases to suppress the ΔelyC lysis phenotype. (A) Catalytic mechanism of DsbG reductase. DsbG is translocated to the periplasm, where it forms a V-shaped dimeric protein with a thioredoxin fold and a CXXC motif. The two cysteines of the active site form a disulfide bond (S—S), inactivating DsbG. DsbD, which is an inner membrane reductase, maintains the cysteines of DsbG in a reduced state (SH—SH). Active DsbG, in turn, reduces single-cysteine-containing proteins, such as the l,d-transpeptidase YbiS, to protect this catalytic cysteine from the oxidizing ROS and activates the protein. Arrows represent the reduction mechanism. (B) CPRG assays of the ΔelyC and Δ4 (ΔelyC ΔybiS ΔynhG ΔerfK) (strain IK6) mutants with or without a plasmid encoding DsbG. (C) CPRG assays of the ΔelyC and Δ6 (ΔelyC ΔybiS ΔynhG ΔerfK ΔycbB ΔycfS) (strain IK15) mutants with or without a plasmid encoding DsbG.
DsbG suppresses the lysis phenotype of ΔelyC mutant cells independently of its reductase activity.
Since DsbG does not act on its known substrates to correct the lysis of ΔelyC cells, we hypothesized that DsbG might use a novel activity to suppress the ΔelyC mutant lysis phenotype. The group of James Bardwell showed that DsbG has an in vitro chaperone activity that is independent of its active site and its reduced state (25). As shown in Fig. 2A, the reductase activity of DsbG requires the reduction of its two catalytic cysteine residues in the active site (CXXC) by the inner membrane protein DsbD (22, 26). To assess if DsbG requires its reductase activity in order to suppress the lysis of ΔelyC cells, we used two different approaches. First, we tested the ability of DsbG overproduction to correct the lysis phenotype of ΔelyC cells lacking dsbD. We found that DsbG was still able to correct the phenotype in this double mutant. Then we replaced the catalytic cysteine residues of DsbG with alanine residues [DsbG(AXXA)] and tested the ability of this modified DsbG to suppress the lysis phenotype of the ΔelyC mutant. This catalytic mutant of DsbG suppressed the ΔelyC lysis phenotype (Fig. 3A and B). In addition, overproduction of the catalytic mutant of DsbG [DsbG(AXXA)] suppressed the growth defect of the ΔelyC mutant in liquid LB medium (Fig. 3C). These results demonstrate that the known reductase activity of DsbG is not required for suppression of the ΔelyC lysis phenotype.
FIG 3.

DsbG does not act by its reductase activity. (A) CPRG assays of the ΔelyC and ΔelyC ΔdsbD (Δ2) mutants (strains EM9 and IK16, respectively) with or without a plasmid encoding DsbG. (B) CPRG assays of the ΔelyC mutant alone and the ΔelyC mutant carrying a plasmid encoding either the native form of DsbG [DsbG(CXXC)] or its catalytic mutant [DsbG(AXXA)]. (C) Growth curves in LB liquid medium of the WT strain (MG1655), the ΔelyC mutant, and the ΔelyC mutant carrying a multicopy plasmid coding for the DsbG(AXXA) catalytic mutant. The experiment was performed in triplicate; data points and error bars indicate average values and standard errors, respectively.
Overexpression of the periplasmic chaperone Spy can also correct the ΔelyC mutant defects.
At this stage, we suspected that the described chaperone activity of DsbG might be required to correct the ΔelyC phenotypes. The envelope compartment contains a variety of molecular chaperones and folding catalysts that assist in protein folding and prevent the aggregation and degradation of stress-damaged proteins. Because chaperones have a broad range and overlapping substrates, DsbG might not be the only chaperone that could suppress the ΔelyC defect. We used a candidate approach to test this hypothesis. We individually overproduced several periplasmic chaperones (Skp, SurA, DegP, and Spy) and folding catalysts (FkpA, PpiA, and PpiD) (27) in the ΔelyC mutant. The suppression effect of each candidate was tested on CPRG plates supplemented with 100 μM IPTG. Overproduction of the periplasmic chaperone Spy fully corrected the CPRG+ phenotype of the ΔelyC mutant (Fig. 4A). However, this was not the case for the other chaperones or folding catalysts. Furthermore, the effect of Spy is specific to the ΔelyC mutant, since its overexpression did not correct the CPRG+ phenotype of the ΔmrcB mutant (Fig. 4B). As is the case for DsbG, Spy also corrected the growth defect of the ΔelyC mutant in liquid LB medium (Fig. 4C). Thus, these results demonstrate that the chaperone activity of Spy can correct the phenotypes of the ΔelyC mutant, and they support our hypothesis that the chaperone activity of DsbG is required to correct these phenotypes.
FIG 4.

Overexpression of the periplasmic chaperone Spy suppresses the ΔelyC mutant lysis defect at 21°C. (A) CPRG screen for periplasmic protein-folding helpers as ΔelyC genetic suppressors. ΔelyC cells alone or cells of the ΔelyC mutant carrying the indicated multicopy plasmid from the mobile plasmid library were patched onto CPRG indicator plates. (B) CPRG assay of the specificity of the Spy suppression effect. ΔmrcB cells with or without a multicopy plasmid carrying mrcB or spy were grown on LB agar with CPRG and IPTG. (C) Growth curves in LB broth cultures of the WT strain (MG1655), the ΔelyC mutant (EM9), and the ΔelyC mutant with a spy-carrying plasmid. The experiment was performed in triplicate. Data points and error bars indicate average values and standard errors, respectively.
Overexpression of DsbG and Spy restores peptidoglycan biosynthesis in the ΔelyC mutant.
Previous studies have shown that ΔelyC cells lyse due to a defect at the level of peptidoglycan biosynthesis (8). We therefore tested the ability of DsbG and Spy overproduction to correct the PG biosynthesis defect. PG was analyzed by using ultraperformance liquid chromatography (UPLC). The analysis was done for three biological replicates (each) of the wild type, the ΔelyC mutant, and the ΔelyC mutant individually overproducing native DsbG, the catalytic mutant of DsbG, nonreduced DsbG (DsbG expressed in the absence of its reductase, DsbD), or Spy. Cells growing at 21°C were harvested at an OD600 of 0.5, and the PG was isolated as described previously (28). The sacculi were digested with muramidase, and the resulting soluble muropeptides were analyzed by UPLC, followed by statistical analysis of the data. As expected, overproduction of Spy or DsbG restored peptidoglycan biosynthesis in the ΔelyC mutant (Fig. 5A). The catalytic mutant of DsbG was as effective as the native form of DsbG in correcting the PG defect, thus confirming that DsbG most likely corrected the PG biosynthesis defect only via its chaperone activity. We also found that Spy totally restores PG levels in the ΔelyC mutant.
FIG 5.

Overexpression of DsbG or Spy restores peptidoglycan biosynthesis in the ΔelyC mutant. (A) Relative amounts of PG in WT cells, ΔelyC cells, ΔelyC cells carrying a plasmid encoding DsbG(CXXC), DsbG(AXXA), or Spy, or ΔelyC ΔdsbD cells carrying a plasmid encoding DsbG(CXXC). The cells were grown in LB broth at 21°C to an OD600 of 0.5, and PG was isolated and prepared for UPLC analysis as described in Materials and Methods. The total amount of PG was calculated from the total intensity detected by UPLC, and the relative amounts of PG were then determined by using the wild-type strain as the reference. Data are means ± standard errors of the means. The asterisk indicates a significant difference from the WT (P < 0.05). (B) Representative chromatograms of the muramidase-digested sacculi of WT cells, ΔelyC cells, and ΔelyC cells carrying a plasmid encoding DsbG(AXXA) or Spy. The peaks are numbered, and the corresponding muropeptides are identified in Table S4 in the supplemental material.
We also performed compositional analysis of the PG to determine whether the overproduction of DsbG or Spy leads to structural or compositional modifications of the PG in the ΔelyC mutant. However, we observed no significant difference in muropeptide composition or relative abundance (Fig. 5B; see also Tables S4 and S5 in the supplemental material). Furthermore, measurement of the percentages of cross-linking, the average glycan strand length, and the level of PG-OM links revealed no significant differences from the PG from wild-type cells (see Table S6 in the supplemental material). Taken together, these results show that overproduction of the chaperones DsbG and Spy can correct the PG biosynthesis defect of the ΔelyC mutant without altering the normal composition or structure of the PG.
The ΔelyC mutant has a protein-folding defect, which is overcome by overproducing DsbG(AXXA) or Spy.
Molecular chaperones help proteins to fold and prevent the aggregation of misfolded and damaged proteins (29, 30). The fact that overproduction of periplasmic chaperones corrects the phenotype of ΔelyC cells suggests a protein-folding problem in the periplasm of these cells. It is therefore possible that DsbG or Spy overproduction acts by helping proteins to fold or by preventing the aggregation of damaged proteins.
To investigate this possibility, we isolated protein aggregates by using the 2001 protocol of Tomoyasu et al. (29), with some changes as described in Materials and Methods. This protocol allows the isolation of total cellular protein aggregates. Wild-type and ΔelyC cells, as well as ΔelyC cells overexpressing DsbG(AXXA) or Spy, were grown at 21°C until one generation time before cell lysis; the cells were then harvested, and the aggregated proteins were isolated. The levels of total and aggregated proteins were measured, and statistical analysis was performed in six different experiments. The level of aggregated proteins was approximately 2-fold higher in the ΔelyC mutant than in the wild type, corresponding to 10% and 5% of the total-protein level, respectively (Fig. 6). Furthermore, overproduction of DsbG(AXXA) or Spy in ΔelyC cells reduced the level of aggregated proteins in the mutant, bringing it close to the level seen in wild-type cells. DsbG(AXXA) was found to be slightly more effective than Spy. Thus, these results demonstrate the occurrence of excess protein aggregation in ΔelyC cells. Since it is corrected by overproducing periplasmic chaperones, the problem is likely at the level of the envelope compartment. Moreover, these results confirm our conclusion that both DsbG and Spy act via their chaperone activities.
FIG 6.
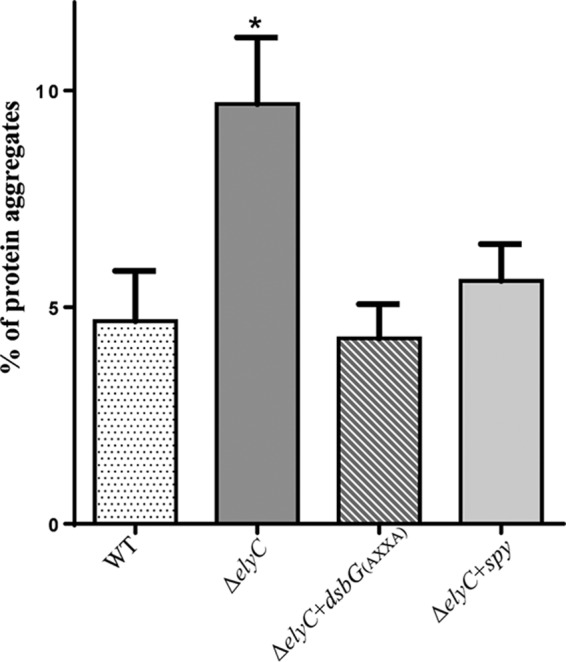
Relative amounts of protein aggregates. Data are average proportions of protein aggregates calculated from six independent experiments. The protein aggregates were isolated from WT cells, ΔelyC cells, and ΔelyC cells carrying a plasmid encoding DsbG(AXXA) or Spy, as described in Materials and Methods. The amount of aggregated proteins was measured by a Bradford assay, and the percentage of aggregation was calculated from the total-protein concentration. Statistical analysis of the data was carried out in comparison to WT cells. The asterisk indicates a significant difference (P < 0.05).
DISCUSSION
A previous study has identified a new Escherichia coli factor, named ElyC, that is important for peptidoglycan biosynthesis at low temperatures. The same study showed that the ΔelyC mutant cells lyse at the end of the exponential phase of growth at 21°C. This clearly established that cell lysis is caused by a blockage in the PG biosynthesis pathway, which occurs just before one doubling of the culture (8). In the present study, we aimed to broaden our knowledge about the function of ElyC by identification of genetic suppressors of the ΔelyC cell lysis phenotype and by characterization of the suppression mechanism.
In this study, we identified two multicopy suppressors of the ΔelyC cell lysis phenotype: the periplasmic reductase DsbG and the periplasmic chaperone Spy. We showed that these proteins proceed by a chaperone activity to restore ΔelyC cell growth. Characterization of the suppression mechanism revealed that the ΔelyC mutant presents a defect in protein folding that leads to an important increase in the level of protein aggregates. Furthermore, we showed that overexpression of either DsbG or Spy significantly reduces the amount of protein aggregates and totally suppresses the PG biosynthesis defect.
DsbG is known as a periplasmic reductase that protects single-cysteine-containing proteins from oxidation (22). However, in this study, we showed that DsbG does not act through this activity to suppress ΔelyC cell lysis. In fact, the two catalytic cysteines of DsbG were not essential for alleviating the major phenotypes of the ΔelyC mutant. Rather, our study strongly suggests that the chaperone activity of DsbG is most likely involved in the suppression of the ΔelyC mutant phenotype. Shao et al. showed in 2000 that DsbG has a general chaperone activity in vitro, which does not require the two catalytic cysteines (25). Other results in this study further support the fact that DsbG acts as a chaperone. First, Spy, a periplasmic chaperone, can also act as a multicopy suppressor of ΔelyC cell lysis. This demonstrates that the ΔelyC cell lysis defect can be overcome by overexpressing specific periplasmic chaperones. Second, an important level of aggregated proteins was found in ElyC-defective cells. Furthermore, this defect could be alleviated by the assistance of molecular chaperones. Shao et al. suggested in 2000 that the ability of DsbG to chaperone protein folding is likely to increase the effectiveness of its catalytic activity (25). However, in this study, we identified an experimental condition under which DsbG acts totally as a chaperone protein. We also showed that DsbG does not act on the substrates of its reductase activity. These results revealed that DsbG has two independent physiological functions: a chaperone activity and a reductase activity. It is possible that the chaperone activity of DsbG is found only under specific physiological conditions. DsbG might exhibit chaperone activity at low temperatures, as does the periplasmic protein DegP. Indeed, DegP acts as a protease at 42°C and as a chaperone at temperatures below 28°C (31). Another possibility is that DsbG acts as a chaperone when it is expressed at high levels in the periplasm, which is the case for the chaperone Spy (32). Further studies must be carried out to investigate these hypotheses.
Spy has recently been identified as a periplasmic chaperone (32). The spy gene is massively and rapidly induced following the cell's exposure to protein-unfolding agents, such as butanol, ethanol, and tannic acid (32–34). Furthermore, Spy overproduction has been shown to be necessary and sufficient for enhancing the folding level of the unstable protein Im7 in the periplasm of E. coli (32). Spy expression is under the control of the Bae and Cpx envelope stress response systems (35). These two systems are known to respond to stress conditions that induce protein-folding defects in the envelope compartment (36–38). Thus, it is clearly established that Spy is a general periplasmic chaperone that acts effectively to assist protein folding and prevent protein aggregation under envelope stress conditions. The identification of Spy and DsbG as multicopy suppressors of ΔelyC cell lysis strongly supports the notion that the protein-folding defect is localized in the cell envelope. Furthermore, both DsbG overproduction and Spy overproduction reduce the level of protein aggregation in the ΔelyC cell to a wild-type level, further supporting the hypothesis that the protein-folding defect is present in the envelope compartment.
Unlike the general chaperones DsbG and Spy, SurA, Skp, DegP, and FkpA are specific chaperones involved in outer membrane protein folding (39, 40). Their overexpression in the ΔelyC mutant does not suppress the lysis phenotype, suggesting that either outer membrane proteins are not damaged or, at least, they are not the only group of proteins misfolded in the ΔelyC mutant. DsbG and Spy are predicted to interact with a wide variety of substrates. Spy has a cradle-shaped structure that is very flexible and could allow the attachment of various proteins (32). DsbG is a V-shaped homodimeric protein with a large cleft that allows it to hold either small or large proteins (41). The high levels of DsbG or Spy required for the suppression effect, their general chaperone activities, and their wide ranges of substrate specificity lead us to suggest that the folding of several envelope proteins may be affected by the absence of ElyC. DsbG and Spy, unlike the chaperones of the outer membrane proteins, might bind and coat the surfaces of a wide range of damaged proteins in the periplasm of ΔelyC mutant cells to inhibit their aggregation and promote their refolding.
PBP1b is an inner membrane protein that carries out the final PG biosynthesis steps, consisting of the polymerization and cross-linking of the glycan chains (11). Cells lacking PBP1b are viable but have reduced PG surface density (42). However, at low temperatures, PBP1b mutant cells showed a lysis phenotype (8). The cold-sensitive phenotype observed in both ΔmrcB and ΔelyC mutants suggests that the PG biosynthesis pathway may present at least one cold-sensitive step. The inner membrane steps of the PG biosynthesis pathway could be more sensitive to cold temperatures due to the phospholipid compositional changes that occur in the inner membrane at low temperatures. These changes could make the PG biosynthesis machinery less effective and therefore more sensitive to mutations that affect the inner membrane steps of the process. The protein export machinery is a good example of a cold-sensitive mechanism induced by inner membrane compositional changes. Indeed, mutations affecting several components of this machinery result in a cold-sensitive phenotype (43–45). The cold-sensitive phenotype of the ΔelyC mutants strongly suggests that PG biosynthesis is blocked at the level of the inner membrane steps.
Although the ΔmrcB and ΔelyC mutants showed great similarity in their growth phenotypes, overexpression of the chaperone DsbG or Spy did not correct the ΔmrcB mutant defect. Therefore, we infer that the PG defect in the absence of PBP1b and the PG defect in the absence of ElyC are not caused by the same mechanisms. In the ΔmrcB mutant, the polymerization and cross-linking of the glycan chains may be blocked or slowed down due to the absence of PBP1b combined with the cold temperature effect. However, we showed that PG biosynthesis arrest in the ΔelyC mutant is caused, at least in part, by a problem in envelope protein folding.
MATERIALS AND METHODS
Media.
Cells were grown in Luria-Bertani broth (LB) (1% tryptone, 0.5% yeast extract, 1% NaCl), with the addition of 1.5% agar for plates. Antibiotics were used at 50, 20, and 10 μg/ml for ampicillin, kanamycin, and chloramphenicol, respectively. Chlorophenol red-β-d-galactopyranoside (CPRG) was used at 20 μg/ml and IPTG at 100 μM.
Strains and plasmids.
Strains, plasmids, and primers are listed in Tables S1, S2, and S3 in the supplemental material, respectively. All E. coli strains used in this study are derivatives of MG1655. Gene deletions in the strains listed in Table S1 were introduced by P1 transduction. The kanamycin resistance cassette was eliminated using FLP recombinase produced from pCP20 (46). All strains were confirmed by PCR using the primers listed in Table S3.
Plasmids from the multicopy ORF library (20) consist of ColE1-derived vectors that encode untagged proteins expressed under the control of the tac promoter. The plasmid-borne copy of dsbG was mutated in the two cysteine residues of the active site of DsbG by using Q5 Hot Start High-Fidelity DNA polymerase (New England BioLabs) and primer pair DsbG(AXXA)_FW/DsbG(AXXA)_RV, resulting in plasmid pIK4 (see Table S2 in the supplemental material).
Identification of multicopy suppressors.
Multicopy plasmids carrying specific ORFs from E. coli MG1655 were transformed individually into ΔelyC mutant cells. The transformed cells were streaked onto LB agar supplemented with ampicillin. The control cells, wild type and ΔelyC mutant, were streaked onto LB agar without ampicillin. Single colonies from the fresh-streaked LB agar were patched onto CPRG plates containing LB agar supplemented with 20 μg/ml of CPRG and 100 μM IPTG. The plates were incubated for 14 to 16 h at 21°C and were photographed. Cultures with strong pink colors (CPRG+) indicate strains with the lysis phenotype, while white cultures (CPRG−) correspond to cells with normal growth. (See the description of the CPRG assay in Results.)
Assessment of growth in liquid medium.
To monitor the growth of the various cell cultures in liquid medium, overnight cultures of each strain were grown at 37°C in LB medium, and cultures with multicopy plasmids were supplemented with ampicillin. These cultures were diluted to an optical density at 600 nm (OD600) of 0.02 in fresh LB medium, and 100 μM IPTG was added to cultures carrying multicopy plasmids to induce gene expression. The diluted cultures were then incubated at 21°C with an agitation of 250 rpm, and the OD600 was measured every 2 h.
Peptidoglycan analysis by UPLC.
Overnight cultures were diluted to an OD600 of 0.02 in 500 ml of LB medium, and cell cultures with multicopy plasmids were supplemented with 100 μM IPTG for gene expression. The cultures were grown at 21°C to an OD600 of 0.5, and the PG was isolated using the boiling SDS method (47, 48). In brief, samples were boiled in 5% SDS for 2 h, and sacculi were repeatedly washed with Milli-Q water by ultracentrifugation (110,000 rpm, 10 min, 20°C) until detergent was no longer detected (no bubble formation). The isolated sacculi were then treated with pronase E at 100 μg/ml to remove covalently bound Braun's lipoprotein. After the addition of SDS to a final concentration of 1% (wt/vol), reactions were heat inactivated, and detergent was removed by additional washing steps as described above. The washed samples were digested with 100 μg/ml muramidase for 16 h at 37°C, and the reaction was stopped by boiling. The coagulated proteins were removed by centrifugation at 14,000 rpm for 10 min. The supernatants were first adjusted to pH 8.5 to 9.0 with sodium borate buffer, and then sodium borohydride was added to a final concentration of 10 mg/ml. After reduction for 30 min at room temperature, the pH of the samples was finally adjusted to 3.5 with orthophosphoric acid.
UPLC analyses of muropeptides were performed on a Waters UPLC system (Waters Corporation, USA) equipped with an Acquity UPLC ethylene bridged hybrid (BEH) C18 column (130 Å, 1.7 μm, 2.1 mm by 150 mm; Waters, USA) and a dual-wavelength absorbance detector. Elution of muropeptides was detected at 204 nm. Muropeptides were separated at 37°C using a linear gradient from buffer A (50 mM phosphate buffer [pH 4.35]) to buffer B (50 mM phosphate buffer [pH 4.95], 15% methanol [vol/vol]) in a 20-min run, with a 0.25-ml/min flow rate. Relative PG amounts were calculated by comparison of the total intensities of the chromatograms (total area) from three biological replicates normalized to the same OD600 and extracted with the same volumes, since there is a direct correlation between muropeptide content and detected intensity. Muropeptides were identified by comparison with standard chromatograms of known composition, and their relative abundances were calculated as the relative area of the corresponding peak compared to the total area of the normalized chromatogram. The injection volume was optimized to ensure that the signal was within the detection limits of the system.
Isolation of aggregated proteins.
Aggregated proteins were isolated as described in reference 29 with the following modification. Bacterial cultures were grown in 25 ml of LB medium at 21°C, and the cultures carrying multicopy plasmids were supplemented with 100 μM IPTG. At an OD600 of 0.3, the cells were harvested by centrifugation. The pellets were resuspended in 40 μl of buffer A (10 mM potassium phosphate buffer [pH 6.5], 1 mM EDTA, 20% [wt/vol] sucrose, 1 mg/ml of lysozyme, and 1.4 mM phenylmethylsulfonyl fluoride [PMSF]), followed by 30 min of incubation on ice. Cells were then supplemented with 360 μl of buffer B (10 mM potassium phosphate buffer [pH 6.5], 1 mM EDTA, and 1.4 mM PMSF), mixed, and sonicated under cooling with a Sonics Materials VCX 400 sonifier (microtip, level 2, 50% duty, 10 cycles). Intact cells were removed by centrifugation at 2,000 × g for 15 min at 4°C. The insoluble fraction containing membrane and aggregated proteins was isolated by centrifugation at 15,000 × g for 20 min at 4°C. The pellet fractions were frozen, resuspended in 1 ml of buffer B by brief sonication, and then centrifuged at 15,000 × g for 20 min at 4°C. This washing procedure was repeated. Afterwards, 200 μl of 10% (wt/vol) NP-40 was added, and the mixture was incubated for 4 h at 4°C with agitation. The aggregated proteins were isolated by centrifugation at 15,000 × g for 30 min at 4°C. The soluble fractions containing membrane proteins were conserved, and the insoluble pellets (containing aggregates) were resuspended in 200 μl of buffer B. Protein quantification was performed using the Bradford method (49).
Statistical analysis.
The Prism software program, version 6.0 (GraphPad Inc., San Diego, CA), was used for all statistical analyses. To determine the significance of the data, the t test (unpaired) was performed for three independent replicates.
Supplementary Material
ACKNOWLEDGMENTS
We thank Marc Drolet and Rim Marrakchi for critical reading of the manuscript and advice. We also thank Gabrielle Gendron Lepage for assisting in obtaining CPRG images and helping with the isolation of aggregated proteins.
Research in the Paradis-Bleau lab was funded by the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Université de Montréal. Research in the Cava lab was funded by the Knut and Alice Wallenberg Foundation (KAW), the Laboratory of Molecular Infection Medicine Sweden (MIMS), the Swedish Research Council, and the Kempe Foundation.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00245-18.
REFERENCES
- 1.Silhavy TJ, Kahne D, Walker S. 2010. The bacterial cell envelope. Cold Spring Harb Perspect Biol 2:a000414. doi: 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sutcliffe IC. 2010. A phylum level perspective on bacterial cell envelope architecture. Trends Microbiol 18:464–470. doi: 10.1016/j.tim.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 3.Braun V, Sieglin U. 1970. The covalent murein-lipoprotein structure of the Escherichia coli cell wall. The attachment site of the lipoprotein on the murein. Eur J Biochem 13:336–346. [DOI] [PubMed] [Google Scholar]
- 4.Neuhaus FC, Baddiley J. 2003. A continuum of anionic charge: structures and functions of d-alanyl-teichoic acids in gram-positive bacteria. Microbiol Mol Biol Rev 67:686–723. doi: 10.1128/MMBR.67.4.686-723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vollmer W, Blanot D, de Pedro MA. 2008. Peptidoglycan structure and architecture. FEMS Microbiol Rev 32:149–167. doi: 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]
- 6.Bouhss A, Trunkfield AE, Bugg TD, Mengin-Lecreulx D. 2008. The biosynthesis of peptidoglycan lipid-linked intermediates. FEMS Microbiol Rev 32:208–233. doi: 10.1111/j.1574-6976.2007.00089.x. [DOI] [PubMed] [Google Scholar]
- 7.van Heijenoort J. 2001. Recent advances in the formation of the bacterial peptidoglycan monomer unit. Nat Prod Rep 18:503–519. doi: 10.1039/a804532a. [DOI] [PubMed] [Google Scholar]
- 8.Paradis-Bleau C, Kritikos G, Orlova K, Typas A, Bernhardt TG. 2014. A genome-wide screen for bacterial envelope biogenesis mutants identifies a novel factor involved in cell wall precursor metabolism. PLoS Genet 10:e1004056. doi: 10.1371/journal.pgen.1004056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown ED, Vivas EI, Walsh CT, Kolter R. 1995. MurA (MurZ), the enzyme that catalyzes the first committed step in peptidoglycan biosynthesis, is essential in Escherichia coli. J Bacteriol 177:4194–4197. doi: 10.1128/jb.177.14.4194-4197.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barreteau H, Magnet S, El Ghachi M, Touze T, Arthur M, Mengin-Lecreulx D, Blanot D. 2009. Quantitative high-performance liquid chromatography analysis of the pool levels of undecaprenyl phosphate and its derivatives in bacterial membranes. J Chromatogr B Analyt Technol Biomed Life Sci 877:213–220. doi: 10.1016/j.jchromb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 11.Vollmer W, Bertsche U. 2008. Murein (peptidoglycan) structure, architecture and biosynthesis in Escherichia coli. Biochim Biophys Acta 1778:1714–1734. doi: 10.1016/j.bbamem.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 12.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, Bateman A. 2008. The Pfam protein families database. Nucleic Acids Res 36:D281–D288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mulder NJ, Apweiler R. 2008. The InterPro database and tools for protein domain analysis. Curr Protoc Bioinformatics Chapter 2:Unit 2.7. doi: 10.1002/0471250953.bi0207s21. [DOI] [PubMed] [Google Scholar]
- 14.Mouslim C, Cano DA, Casadesus J. 1998. The sfiX, rfe and metN genes of Salmonella typhimurium and their involvement in the HisC pleiotropic response. Mol Gen Genet 259:46–53. doi: 10.1007/s004380050787. [DOI] [PubMed] [Google Scholar]
- 15.Rida S, Caillet J, Alix JH. 1996. Amplification of a novel gene, sanA, abolishes a vancomycin-sensitive defect in Escherichia coli. J Bacteriol 178:94–102. doi: 10.1128/jb.178.1.94-102.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imlay JA. 2003. Pathways of oxidative damage. Annu Rev Microbiol 57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- 17.Imlay JA. 2013. The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Microbiol 11:443–454. doi: 10.1038/nrmicro3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corcoran A, Cotter TG. 2013. Redox regulation of protein kinases. FEBS J 280:1944–1965. doi: 10.1111/febs.12224. [DOI] [PubMed] [Google Scholar]
- 19.Arts IS, Gennaris A, Collet JF. 2015. Reducing systems protecting the bacterial cell envelope from oxidative damage. FEBS Lett 589:1559–1568. doi: 10.1016/j.febslet.2015.04.057. [DOI] [PubMed] [Google Scholar]
- 20.Saka K, Tadenuma M, Nakade S, Tanaka N, Sugawara H, Nishikawa K, Ichiyoshi N, Kitagawa M, Mori H, Ogasawara N, Nishimura A. 2005. A complete set of Escherichia coli open reading frames in mobile plasmids facilitating genetic studies. DNA Res 12:63–68. doi: 10.1093/dnares/12.1.63. [DOI] [PubMed] [Google Scholar]
- 21.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. 2008. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev 32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 22.Depuydt M, Leonard SE, Vertommen D, Denoncin K, Morsomme P, Wahni K, Messens J, Carroll KS, Collet JF. 2009. A periplasmic reducing system protects single cysteine residues from oxidation. Science 326:1109–1111. doi: 10.1126/science.1179557. [DOI] [PubMed] [Google Scholar]
- 23.Magnet S, Bellais S, Dubost L, Fourgeaud M, Mainardi JL, Petit-Frere S, Marie A, Mengin-Lecreulx D, Arthur M, Gutmann L. 2007. Identification of the l,d-transpeptidases responsible for attachment of the Braun lipoprotein to Escherichia coli peptidoglycan. J Bacteriol 189:3927–3931. doi: 10.1128/JB.00084-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magnet S, Dubost L, Marie A, Arthur M, Gutmann L. 2008. Identification of the l,d-transpeptidases for peptidoglycan cross-linking in Escherichia coli. J Bacteriol 190:4782–4785. doi: 10.1128/JB.00025-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shao F, Bader MW, Jakob U, Bardwell JC. 2000. DsbG, a protein disulfide isomerase with chaperone activity. J Biol Chem 275:13349–13352. doi: 10.1074/jbc.275.18.13349. [DOI] [PubMed] [Google Scholar]
- 26.Bessette PH, Cotto JJ, Gilbert HF, Georgiou G. 1999. In vivo and in vitro function of the Escherichia coli periplasmic cysteine oxidoreductase DsbG. J Biol Chem 274:7784–7792. doi: 10.1074/jbc.274.12.7784. [DOI] [PubMed] [Google Scholar]
- 27.Goemans C, Denoncin K, Collet JF. 2014. Folding mechanisms of periplasmic proteins. Biochim Biophys Acta 1843:1517–1528. doi: 10.1016/j.bbamcr.2013.10.014. [DOI] [PubMed] [Google Scholar]
- 28.Glauner B. 1988. Separation and quantification of muropeptides with high-performance liquid chromatography. Anal Biochem 172:451–464. doi: 10.1016/0003-2697(88)90468-X. [DOI] [PubMed] [Google Scholar]
- 29.Tomoyasu T, Mogk A, Langen H, Goloubinoff P, Bukau B. 2001. Genetic dissection of the roles of chaperones and proteases in protein folding and degradation in the Escherichia coli cytosol. Mol Microbiol 40:397–413. doi: 10.1046/j.1365-2958.2001.02383.x. [DOI] [PubMed] [Google Scholar]
- 30.Miot M, Betton JM. 2004. Protein quality control in the bacterial periplasm. Microb Cell Fact 3:4. doi: 10.1186/1475-2859-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sobiecka-Szkatula A, Polit A, Scire A, Gieldon A, Tanfani F, Szkarlat Z, Ciarkowski J, Zurawa-Janicka D, Skorko-Glonek J, Lipinska B. 2009. Temperature-induced conformational changes within the regulatory loops L1-L2-LA of the HtrA heat-shock protease from Escherichia coli. Biochim Biophys Acta 1794:1573–1582. doi: 10.1016/j.bbapap.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 32.Quan S, Koldewey P, Tapley T, Kirsch N, Ruane KM, Pfizenmaier J, Shi R, Hofmann S, Foit L, Ren G, Jakob U, Xu Z, Cygler M, Bardwell JC. 2011. Genetic selection designed to stabilize proteins uncovers a chaperone called Spy. Nat Struct Mol Biol 18:262–269. doi: 10.1038/nsmb.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neidhardt FC, VanBogelen RA, Vaughn V. 1984. The genetics and regulation of heat-shock proteins. Annu Rev Genet 18:295–329. doi: 10.1146/annurev.ge.18.120184.001455. [DOI] [PubMed] [Google Scholar]
- 34.Rutherford BJ, Dahl RH, Price RE, Szmidt HL, Benke PI, Mukhopadhyay A, Keasling JD. 2010. Functional genomic study of exogenous n-butanol stress in Escherichia coli. Appl Environ Microbiol 76:1935–1945. doi: 10.1128/AEM.02323-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srivastava SK, Lambadi PR, Ghosh T, Pathania R, Navani NK. 2014. Genetic regulation of spy gene expression in Escherichia coli in the presence of protein unfolding agent ethanol. Gene 548:142–148. doi: 10.1016/j.gene.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 36.Duguay AR, Silhavy TJ. 2004. Quality control in the bacterial periplasm. Biochim Biophys Acta 1694:121–134. doi: 10.1016/j.bbamcr.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 37.Raivio TL. 2014. Everything old is new again: an update on current research on the Cpx envelope stress response. Biochim Biophys Acta 1843:1529–1541. doi: 10.1016/j.bbamcr.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 38.Raivio TL, Silhavy TJ. 2001. Periplasmic stress and ECF sigma factors. Annu Rev Microbiol 55:591–624. doi: 10.1146/annurev.micro.55.1.591. [DOI] [PubMed] [Google Scholar]
- 39.Krojer T, Sawa J, Schafer E, Saibil HR, Ehrmann M, Clausen T. 2008. Structural basis for the regulated protease and chaperone function of DegP. Nature 453:885–890. doi: 10.1038/nature07004. [DOI] [PubMed] [Google Scholar]
- 40.Sklar JG, Wu T, Kahne D, Silhavy TJ. 2007. Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev 21:2473–2484. doi: 10.1101/gad.1581007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heras B, Edeling MA, Schirra HJ, Raina S, Martin JL. 2004. Crystal structures of the DsbG disulfide isomerase reveal an unstable disulfide. Proc Natl Acad Sci U S A 101:8876–8881. doi: 10.1073/pnas.0402769101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caparrós M, Quintela JC, de Pedro MA. 1994. Variability of peptidoglycan surface density in Escherichia coli. FEMS Microbiol Lett 121:71–76. [DOI] [PubMed] [Google Scholar]
- 43.Gardel C, Benson S, Hunt J, Michaelis S, Beckwith J. 1987. secD, a new gene involved in protein export in Escherichia coli. J Bacteriol 169:1286–1290. doi: 10.1128/jb.169.3.1286-1290.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nouwen N, Driessen AJ. 2005. Inactivation of protein translocation by cold-sensitive mutations in the yajC-secDF operon. J Bacteriol 187:6852–6855. doi: 10.1128/JB.187.19.6852-6855.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pogliano KJ, Beckwith J. 1993. The Cs sec mutants of Escherichia coli reflect the cold sensitivity of protein export itself. Genetics 133:763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. doi: 10.1016/0378-1119(95)00193-A. [DOI] [PubMed] [Google Scholar]
- 47.Alvarez L, Hernandez SB, de Pedro MA, Cava F. 2016. Ultra-sensitive, high-resolution liquid chromatography methods for the high-throughput quantitative analysis of bacterial cell wall chemistry and structure. Methods Mol Biol 1440:11–27. doi: 10.1007/978-1-4939-3676-2_2. [DOI] [PubMed] [Google Scholar]
- 48.Desmarais SM, De Pedro MA, Cava F, Huang KC. 2013. Peptidoglycan at its peaks: how chromatographic analyses can reveal bacterial cell wall structure and assembly. Mol Microbiol 89:1–13. doi: 10.1111/mmi.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.