

Abstract
Hypervalent iodine reagents constitute a powerful tool in modern synthetic organic chemistry, promoting several important reactions. One such reaction is the ring contraction of cycloalkenes and cycloalkanones promoted by iodine(III) compounds, such as iodobenzene diacetate, iodosylbenzene, iodotoluene difluoride, and [hydroxy(tosyloxy)-iodo]benzene (Koser´s reagent). This review covers all the literature related to the ring contraction of cyclic ketones and olefins promoted by iodine(III) species.
Keywords: Iodine(III), hypervalent iodine, ring contraction, oxidation, cycloalkenes, cycloalkanones
Introduction
Ring contraction reactions are an important method to increase molecular complexity in a single step, because, in several cases, the reorganization of the bonds occurs with a high level of selectivity, affording products not easily accessible by other approaches [1]. Ring contraction reactions can be effected by acids, by bases, by oxidizers or photochemically [1]. Among the oxidizers, one of the most used is thallium trinitrate (TTN) [2,3].
In the last years, hypervalent iodine reagents have become an essential tool in synthetic organic chemistry, due to the plethora of reactions that can be performed with them in excellent yield and selectivity. One such reaction is the oxidative rearrangement of cycloalkenes and cycloalkanones, which leads to a ring contraction. Although several reviews concerning hypervalent iodine chemistry have been published [4,5,6,7,8,9,10,11,12,13,14,15], none of them covered ring contraction reactions in a comprehensive manner. This review intends to cover the literature related to the ring contraction reaction of cyclic ketones and olefins promoted by hypervalent iodine. The definition of ring contraction through this article follows that previously mentioned by Redmore and Gutsche [1,16].
Ring Contractions of Cycloalkanones
Two articles concerning the ring contraction of steroidal ketones mediated by iodine(III) were released nearly simultaneously in 1984. In the first paper, Daum reported that the reaction of the androstan-3-one 1 with iodobenzene diacetate leads to a mixture of the ring contraction products 2 and 3 [17]. After recrystallization, 2 was obtained in 65% yield. A similar result was obtained when the reaction was performed with iodosobenzene, instead of iodobenzene diacetate. The configuration of the major product 2 has been explained by the formation of the adduct 7, which bears the iodine group at the equatorial position (Figure 1). The behavior of the 19-norandrostan-3-one 4 toward the oxidation of iodobenzene diacetate was different from 1, because 4 gave as the major product the hydroxyketal 5. In this case, the ring contraction product 6 was obtained in poor yield (Scheme 1). The author suggested that for the nor-androstanone 4 formation of the adduct 8 bearing the iodine(III) group in the axial position would occur, as shown in Figure 1. Thus, for substrates where there are no steric restrictions, such as 4, the addition would lead to an intermediate with the iodine(III) group in the axial position, as in 8. On the other hand, for cycloalkanones where an axial iodine(III) atom would lead to strong 1,3-diaxial interactions, such as 1, the iodine(III) occupies the equatorial position, as in 7.
Figure 1.

Structure of the Intermediates proposed by Daum.
Scheme 1.

In the second paper Moriarty and co-workers [18] described that the treatment of the 3-cholestanone 9 with trivalent iodine reagents, such as PhI(OAc)2, o-OIC6H4CO2H or PhIO2, led to an oxidative rearrangement, affording a 9:1 mixture of the ring contraction products 10 and 11, respectively (Scheme 2). The formation of the major product has been explained by the initial hyperiodination of the enolate 12, giving the adduct 13, which bears the iodine group at the axial position, which contrasts with the intermediate 7, proposed by Daum [17] (cf. Figure 1). Then, the adduct 13 would be converted into the corresponding twist-boat form 14, on which the rearrangement would take place giving the ring contraction product 10 (Scheme 3).
Scheme 2.

Scheme 3.

Under conditions similar to those used in the ring contraction of 9, the rearrangement of another cyclic ketone, namely 4-phenylcyclohexanone (15), has been performed by the same group in the synthesis of 1-piperidinobenzobicyclo[2.2.1]heptene [19]. In this example, however, the relative configuration of the ring contraction products 16 was not assigned (Scheme 4). The oxidative rearrangement of cyclohexanones can also be promoted by thallium(III) [20] and selenium(IV) [21], in good yield and diastereoselectivity.
Scheme 4.

The ring contraction reaction of cyclic ketones with iodine(III) can also be performed with substrates other than the abovementioned cyclohexanone derivatives. One of such examples is the treatment of the flavanones 17a-h with iodobenzene diacetate or with [hydroxy(tosylosy)iodo]benzene (Koser´s reagent) in trimethyl orthoformate (TMOF), as solvent, which gave the dihydrobenzofuran derivatives 18a-g, in 35 to 75% yield [22]. Although formed as a single diastereomer, the relative configuration of the ring contraction products was not assigned. Other products isolated in this study were the cis-3-methoxyflavanones 19 and the flavones 20. The formation of the rearrangement products 18a-g has been rationalized by the mechanism shown in Scheme 5, which is exemplified for 17a. A key feature of this mechanism is the electrophilic addition of the iodine(III) reagent in the enol ether moiety of 22 and the rearrangement of 24 in 25.
Scheme 5.

Table 1.
Reaction of Flavanones with Iodine(III)a.
| Entry | Substrate | Product (Yield)b |
| 1 | 17a (a: R=R1=H; Ar=Ph) | 18a (40%), 19a (27%), 20a (12%) |
| 2 | 17b (b: R=R1=H; Ar=p-ClC6H4) | 18b (60%), 20b (18%) |
| 3 | 17c (c: R=Cl; R1=H; Ar=Ph) | 18c (43%), 19c (36%), 20c (8%) |
| 4 | 17d (d: R=Cl; R1=H; Ar=p-ClC6H4) | 18d (35%), 19d (34%), 20d (16%) |
| 5 | 17e (e: R=Me; R1=H; Ar=Ph) | 18e (75%), 20e (16%) |
| 6 | 17f (f: R=Me; R1=H; Ar=p-ClC6H4) | 18f (75%), 20f (16%) |
| 7 | 17g (g: R=Cl; R1=Me; Ar=Ph) | 18g (47%), 20g (35%) |
| 8 | 17h (h: R=Cl; R1=H; Ar=p-OMeC6H4) | 20h (18%) |
a Conditions: (a) 1.1 eq. PhI(OAc)2, H2SO4 (cat.), TMOF, rt, overnight; or (b) 1.1 eq. PhI(OTs)OH, TMOF, rt, overnight. b Isolated yield.
Some years later, the reaction of the flavanones 17a and (-)-17a with iodobenzene diacetate was reinvestigated by Juhász et al. [23], which assigned a trans relationship for the substituent at the benzofuran ring. Based on the formation of this product, the relative configuration of the intermediates 22a and 24a was also proposed (Scheme 6).
Scheme 6.

The treatment of 2-arylidenecycloalkanones, such as 28-30, with iodobenzene diacetate led to an oxidative rearrangement, which culminated with the formation of the 2-arylidenecycloalkane carboxylates 31-33 in good yield [24]. It is worth mentioning that this approach can be used for the synthesis of five-, six-, and seven-membered ring derivatives, as shown in Scheme 7 and Scheme 8.
Scheme 7.

Scheme 8.

A two-step procedure to promote the ring contraction of 2-hydroxy-1,4-benzoquinones to 2-cyclo-pentene-1,4-diones has been developed by the group of Varvoglis and co-workers [25,26]. This approach is based on the thermolysis of a phenyliodonium species, which is obtained by reaction of a hydroxyquinone with iodobenzene diacetate. Representative examples of this transformation are shown in Scheme 9. Recently, the preparation of the intermediate phenyliodonium ylide 35 by rearrangement of the triptycene quinone 34 utilizing iodobenzene diacetate has been described [27]. The ring contraction step occurs through a sequence of events, initiated by a Wolff-type rearrangement of 35 to give a ketene. Reaction of the formed ketene with water gives a carboxylic acid that leads to 36 after a decarboxylation. The formation of the by-product 37 (Scheme 10) is evidence for the proposed mechanism. The treatment of a 2,5-dihydroxy-1,4-benzoquinone with iodobenzene diacetate gave a double iodonium zwitterion, which when submitted to the thermolysis conditions led to the ring contraction, as well as to a formal [3+2] cycloaddition reaction, giving a isoxazole derivative, albeit in low yield, as exemplified in Scheme 11 [26].
Scheme 9.

Scheme 10.

Scheme 11.

The ring contraction reaction is not the only reaction pathway that may take place in the oxidation of cyclic ketones with hypervalent iodine. A highly explored reaction is the α-oxidation of ketones, which occurs under different reaction conditions, including with o-iodosylbenzoic acid [28] (Table 2, entry 1) and with iodobenzene diacetate [29] (entry 2), both under basic conditions. It is interesting to note that the latter condition is similar to that employed in the ring contraction of cyclohexanone derivatives (see Scheme 2 and Scheme 4). Thus, the course of the oxidation of cyclic ketones with iodobenzene diacetate/KOH is determined by the structure of the substrate. Other important protocols developed for the α-oxidation are: i) the one-pot oxidation of alcohols to the corresponding carbonyl compound followed by α-tosyloxylation (entry 3) [30]; ii) the solvent-free reaction of ketones with PhI(OAc)2/p-TsOH (entry 4) [31]; and iii) the proline-catalyzed asymmetric α-oxidation of ketones (entry 5) [32].
Table 2.
Representative examples of the α-oxidation of cyclic ketones.
| Entry | Substrate | Conditions | Product (Yield) | Reference |
| 1 | 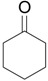 |
1.1 eq. o-H2OCC6H4IO, 3 eq. KOH, MeOH, rt, overnight |  |
Moriarty et al. [28] |
| 2 |  |
1.1 eq. PhI(OAc)2, 3 eq. KOH, MeOH, 0-5 °C for 1 h and 23-25 °C for 20 h | 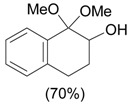 |
Moriarty et al. [29] |
| 3 | 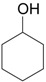 |
3.0 eq. PhIO, 2.5 eq. p-TsOH.H2O, CH3CN, 60 °C, 3 h | 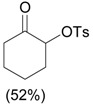 |
Ueno et al. [30] |
| 4 | |
PhI(OAc)2, p-TsOH grinding, 20 min |
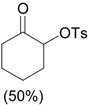 |
Yusubov and Wirth [31] |
| 5 | |
i) 0.33 eq. PhIO, L-proline (10-30 mol %), DMF, rt, 16-24 h; ii) NaBH4, MeOH, 0 °C |
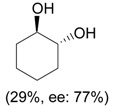 |
Engqvist et al. [32] |
Ring Contractions of Cycloalkenes
The oxidation of cycloalkenes with hypervalent iodine has been reported by several authors. The analysis of these papers reveals that products formed by an addition to the double bond is the most often observed reaction pathway. Selected examples of this transformation are shown in Table 3. However, treatment of cyclic olefins with iodine(III) reagents may also lead to ring contraction products, as discussed in the following paragraphs.
Table 3.
Representative examples of I(III)-promoted addition reactions of cyclohexene.
| Entry | Conditions | Product (Yield) | Reference |
| 1 | PhI(OTs)OH, rt, 1.5 h | 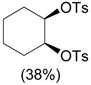 |
Koser et al. [33] |
| 2 |
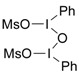 , ClCH2CH2Cl, 25 °C, 15-20 h , ClCH2CH2Cl, 25 °C, 15-20 h |
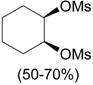 |
Zefirov et al. [34] |
| 3 |
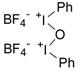 , AcOEt, LiClO4, rt, 2 h. , AcOEt, LiClO4, rt, 2 h. |
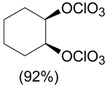 |
Zhdankin et al. [35] |
| 4 | CF3CH2I(OTs)OH, CH2Cl2, 0 °C, 3-4 h | 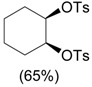 |
Zhdankin et al. [36] |
| 5 | PhI(O2PPh2)OH, I2, ClCH2CH2Cl, rt, 1 d | 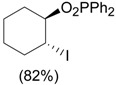 |
Togo et al. [37] |
Zefirov et al. [38] investigated the oxidation of cyclohexene (38) and 2,3-dihydropyran (40) with trivalent iodine under different conditions. Treatment of the olefin 38 with phenyl iodosulfate or with iodosobenzene led to the oxidative rearrangement product 39, in 20 and 60% yield, respectively. Furthermore, the complex iodosobenzene/BF3 and the sulfate bearing an iodine(III) atom 42 were utilized to carry out the transformation of the substrate 40 into the corresponding tetrahydrofuran 41 (Scheme 12).
Scheme 12.

In 1996, Moriarty et al. [39] described that the reaction of dihydropyran (40) with iodosobenzene in H2O or in MeOH, which gives the ring contraction product in moderate yield. Under similar conditions, no reaction between cyclohexene and PhIO has been observed. However, the addition of an acid catalyst (BF3.Et2O or H2SO4) led to the ring contraction product, albeit in low yield (Scheme 13).
Scheme 13.

The authors proposed that the reactive species in this reaction is PhI(OMe)2 or PhI(OH)2, which would be generated by the reaction of iodosobenzene with MeOH or H2O, respectively. Then, an electrophilic addition of iodine(III) to the double bond would give the carbocation 45, that would lead to the adduct 46 after addition of a molecule of solvent. The rearrangement would occur at this intermediate, producing the observed products, as exemplified for the cyclohexene in Scheme 14.
Scheme 14.

The ring contraction of cyclohexene can also be performed using the commercially available iodobenzene diacetate in the presence of an acid catalyst, giving 39, in 39-42% yield [40,41]. Under analogous conditions, the oxidation of 1-methylcyclohexene gave the cyclopentane derivative 51 in 22% yield. In both cases, the aldehyde was isolated as the corresponding 2,4-dinitrophenylhydrazone. In these reactions, the formation of products of addition of two molecules of solvent (48 and 52) and of allylic oxidation (49 and 53) was also observed (Scheme 15) [41].
Scheme 15.

A recent publication by Justik and Koser described the rearrangement of arylalkenes with [hydroxy(tosyloxy)iodo]benzene in methanol giving α-aryl ketones as products [42]. In the case of 1‑phenylcycloalkenes, the formation of two different rearrangement products was observed. The first is that originated by the migration of the phenyl group, as exemplified by the oxidation of 1-phenyl-cyclohexene (54). The second possibility would be the ring contraction reaction, which took place with 1-phenylcycloheptene (55) and with 1-phenylcyclooctene (56), although in these cases products of phenyl migration (59 and 61, respectively) were also isolated, as the major product (Scheme 16). The mechanism proposed by the authors shows the electrophilic addition of the [hydroxy(tosyloxy)iodo]benzene to the double bond, giving the adduct 63, after addition of a molecule of solvent to the carbocation 62. This adduct would then originate either the product of phenyl migration (Path a) or the ring contraction product (Path b), as exemplified for 1-phenylcycloheptene in Scheme 17.
Scheme 16.

Reagents and Conditions: 0.92 eq. PhI(OH)Ts; 95% MeOH, rt. Reaction times: 54: 20 min; 55: 1 h and; 56: 8h.
Scheme 17.

An efficient method to perform the rearrangement of cycloalkenes has been developed by Hara and co-workers [43,44]. This approach consists in the treatment of the olefin with iodotoluene difluoride in the presence of Et3N-5HF, leading to difluoride ring contraction products in poor to very good yield. This reaction occurs with high degree of diastereoselectivity, giving exclusively trans diastereomers (Table 4, entry 1). Moreover, such a protocol can be used for the ring contraction of cyclohexene (entries 1-3), cycloheptene (entry 4) and cyclooctene derivatives (entry 5). The formation of these compounds can be explained by a mechanism where the first step is the electrophilic addition of the hypervalent reagent, giving the fluoride adduct 70, which leads to the carbocation 72 after a ring inversion to reach the required anti-periplanarity for the rearrangement. Reaction of 72 with a fluoride anion gives the observed product 69 (Scheme 18). Contrasting with the above mentioned examples, the reaction of 4-substituted-cyclohexenes without substituents in the double bond, such as 73, with iodotoluene difluoride gives exclusively a cis-difluoro compound, such as 76, in good yield, as shown in Scheme 19 (compare to Table 4, entry 1) [45]. This different behavior has been explained considering the stability of the possible carbocation intermediates. The ring contraction of 68 would occur through the secondary carbocation 72, whereas the formation of the hypothetically product 75 would take place through the primary cation 74.
Table 4.
Ring Contraction of Cicloalkenes with 1.3 eq. p-Tol-IF2.a
| Entry | Cycloalkene | Product (Yield) |
| 1 | 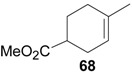 |
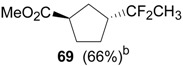 |
| 2 |  |
 |
| 3 | 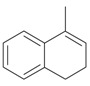 |
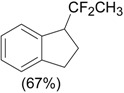 |
| 4 | 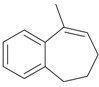 |
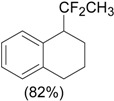 |
| 5 | 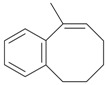 |
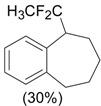 |
a Reagents and Conditions: 5 HF-Et3N, CH2Cl2, -20 °C, 1 h. b Reaction time: 2 h.
Scheme 18.

Scheme 19.

The ring contraction of carbohydrate derivatives has been used to obtain functionalized tetrahydrofurans by Kirsching and co-workers. [46,47,48] This transformation was carried out using PhI(Ots)OH in acetonitrile in the presence of molecular sieves and gave the corresponding ring contraction products from 16 to 35% yield (Scheme 20).
Scheme 20.

The mechanism proposed by the authors for the ring contraction of the carbohydrates derivatives is exemplified for 77 in Scheme 21.
Scheme 21.

The first step is the electrophilic addition of the iodine(III) reagent to the unsaturation, giving the oxonium ion 90. This intermediate would give the enone 80 through 91, by a reductive elimination. Alternatively, the oxonium 90 may give, after addition of water, the adduct 92, on which would occur the rearrangement leading to the tetrahydrofuran 82 or an intramolecular reductive displacement giving 83.
Conclusions
The oxidative rearrangement of cyclic alkenes and ketones promoted by iodine(III) reagents can be performed in an efficient manner under several different reaction conditions. However, considering the potential of hypervalent iodine reagents in synthetic organic chemistry, as well as the usefulness of ring contraction reactions to increase molecular complexity in a single step, we expect that applications of iodine(III)-mediated ring contractions will rapidly increase in the next years.
Acknowledgements
We are grateful for the financial support provided by FAPESP and TWAS.
References
- 1.Silva L. F., Jr. Tetrahedron. 2002;58:9137–9161. doi: 10.1016/S0040-4020(02)00990-0. [DOI] [Google Scholar]
- 2.Ferraz H. M. C., Silva L. F., Jr. Quim. Nova. 2000;23:216–224. [Google Scholar]
- 3.Ferraz H. M. C., Aguilar A. M., Silva L. F., Jr. Tetrahedron. 2003;59:5817. doi: 10.1016/S0040-4020(03)00974-8. [DOI] [Google Scholar]
- 4.Wirth T. Oxidations and rearrangements. In: Wirth T., editor. Hypervalent Iodine Chemistry: Modern Developments in Organic Synthesis. Springer-Verlag; Weinheim: 2003. pp. 185–208. [Google Scholar]
- 5.Dauban P., Dodd R. H. Synlett. 2003:1571–1586. doi: 10.1055/s-2003-41010. [DOI] [Google Scholar]
- 6.Stang P. J. J. Org. Chem. 2003;68:2997–3008. doi: 10.1021/jo030022e. [DOI] [PubMed] [Google Scholar]
- 7.Zhdankin V. V., Stang P. J. Chem. Rev. 2002;102:2523–2584. doi: 10.1021/cr010003+. [DOI] [PubMed] [Google Scholar]
- 8.Togo H., Sakuratani K. Synlett. 2002:1966–1975. doi: 10.1055/s-2002-35575. [DOI] [Google Scholar]
- 9.Moriarty R. M., Prakash O. Org. React. 1999;54:273. [Google Scholar]
- 10.Wirth T., Hirt U. H. Synthesis. 1999:1271–1287. doi: 10.1055/s-1999-3540. [DOI] [Google Scholar]
- 11.Varvoglis A. Tetrahedron. 1997;53:1179–1255. doi: 10.1016/S0040-4020(96)00970-2. [DOI] [Google Scholar]
- 12.Stang P. J., Zhdankin V. V. Chem. Rev. 1996;96:1123–1178. doi: 10.1021/cr940424+. [DOI] [PubMed] [Google Scholar]
- 13.Prakash O. Aldrichim. Acta. 1995;28:63–71. [Google Scholar]
- 14.Moriarty R. M. J. Org. Chem. 2005;70:2893–2903. doi: 10.1021/jo050117b. [DOI] [PubMed] [Google Scholar]
- 15.Wirth T. Angew. Chem. Int. Ed. 2005;44:3656. doi: 10.1002/anie.200500115. [DOI] [PubMed] [Google Scholar]
- 16.Redmore D., Gutsche C. D. Carbocyclic Ring Contraction Reactions. In: Hart H., Karabastos G. J., editors. Advances in Alicyclic Chemistry. Academic Press; New York, London: 1971. pp. 1–138. [Google Scholar]
- 17.Daum S. J. Tetrahedron Lett. 1984;25:4725–4728. [Google Scholar]
- 18.Moriarty R. M., Prakash I., Musallam H. A. Tetrahedron Lett. 1984;25:5867–5870. [Google Scholar]
- 19.Moriarty R. M., Enache L. A., Zhao L., Gilardi R., Mattson M. V., Prakash O. J. Med. Chem. 1998;41:468–477. doi: 10.1021/jm970059p. [DOI] [PubMed] [Google Scholar]
- 20.Ferraz H. M. C., Silva L. F., Jr. Tetrahedron Lett. 1997;38:1899–1902. doi: 10.1016/S0040-4039(97)00246-3. [DOI] [Google Scholar]
- 21.Giurg M., Mlochowski J. Synth. Commun. 1999;29:2281–2291. doi: 10.1080/00397919908086230. [DOI] [Google Scholar]
- 22.Prakash O., Tanwar M. P. Bull. Chem. Soc. Jpn. 1995;68:1168–1171. doi: 10.1246/bcsj.68.1168. [DOI] [Google Scholar]
- 23.Juhasz L., Szilagyi L., Antus S., Visy J., Zsila F., Simonyi M. Tetrahedron. 2002;58:4261–4265. doi: 10.1016/S0040-4020(02)00360-5. [DOI] [Google Scholar]
- 24.Varma R. S., Kumar D. Synthesis. 1999:1288–1290. doi: 10.1055/s-1999-3539. [DOI] [Google Scholar]
- 25.Hatzigrigoriou E., Spyroudis S., Varvoglis A. Liebigs Ann. Chem. 1989:167–170. doi: 10.1002/jlac.198919890132. [DOI] [Google Scholar]
- 26.Papoutsis I., Spyroudis S., Varvoglis A. Tetrahedron Lett. 1994;35:8449–8452. doi: 10.1016/S0040-4039(00)74430-3. [DOI] [Google Scholar]
- 27.Spyroudis S., Xanthopoulou N. J. Org. Chem. 2002;67:4612–4614. doi: 10.1021/jo020078t. [DOI] [PubMed] [Google Scholar]
- 28.Moriarty R. M., Hou K. C. Tetrahedron Lett. 1984;25:691–694. [Google Scholar]
- 29.Moriarty R. M., Engerer S. C., Prakash O., Prakash I., Gill U. S., Freeman W. A. J. Org. Chem. 1987;52:153–155. doi: 10.1021/jo00377a028. [DOI] [Google Scholar]
- 30.Ueno M., Nabana T., Togo H. J. Org. Chem. 2003;68:6424–6426. doi: 10.1021/jo030045t. [DOI] [PubMed] [Google Scholar]
- 31.Yusubov M. S., Wirth T. Org. Lett. 2005;7:519–521. doi: 10.1021/ol047363e. [DOI] [PubMed] [Google Scholar]
- 32.Engqvist M., Casas J., Sundén H., Ibrahem I., Córdova A. Tetrahedron Lett. 2005;46:2053–2057. doi: 10.1016/j.tetlet.2005.01.167. [DOI] [Google Scholar]
- 33.Koser G. F., Rebrovic L., Wettach R. H. J. Org. Chem. 1981;46:4324–4326. doi: 10.1021/jo00334a057. [DOI] [Google Scholar]
- 34.Zefirov N. S., Zhdankin V. V., Dankov Y. V., Kozmin A. S. Zh. Org. Khim. 1984;20:446–447. [Google Scholar]
- 35.Zhdankin V. V., Tykwinski R., Berglund B., Mullikin M., Caple R., Zefirov N. S., Kozmin A. S. J. Org. Chem. 1989;54:2609–2612. doi: 10.1021/jo00272a029. [DOI] [Google Scholar]
- 36.Zhdankin V. V., Kuehl C. J., Simonsen A. J. J. Org. Chem. 1996;61:8272–8276. doi: 10.1021/jo961336n. [DOI] [PubMed] [Google Scholar]
- 37.Muraki T., Yokoyama M., Togo H. J. Org. Chem. 2000;65:4679–4684. doi: 10.1021/jo000296r. [DOI] [PubMed] [Google Scholar]
- 38.Zefirov N. S., Caple R., Palyulin V. A., Berglund B., Tykvinskii R., Zhdankin V. V., Kozmin A. S. Bull. Acad. Sci. USSR Div. Chem. Sci. 1988;37:1289–1289. doi: 10.1007/BF00961969. [DOI] [Google Scholar]
- 39.Moriarty R. M., Prakash O., Duncan M. P., Vaid R. K., Rani N. J. Chem. Res. S. 1996:432–433. [Google Scholar]
- 40.Yusubov M. S., Zholobova G. A. Russ. J. Org. Chem. 2001;37:1179–1181. doi: 10.1023/A:1013156801567. [DOI] [Google Scholar]
- 41.Yusubov M. S., Zholobova G. A., Filimonova I. L., Chi K. W. Russ. Chem. Bull. 2004;53:1735–1742. doi: 10.1007/s11172-005-0027-8. [DOI] [Google Scholar]
- 42.Justik M. W., Koser G. F. Tetrahedron Lett. 2004;45:6159–6163. doi: 10.1016/j.tetlet.2004.06.029. [DOI] [Google Scholar]
- 43.Hara S., Nakahigashi J., Ishi-i K., Fukuhara T., Yoneda N. Tetrahedron Lett. 1998;39:2589–2592. doi: 10.1016/S0040-4039(98)00276-7. [DOI] [Google Scholar]
- 44.Sawaguchi M., Hara S., Yoneda N. J. Fluorine Chem. 2000;105:313–317. doi: 10.1016/S0022-1139(99)00276-6. [DOI] [Google Scholar]
- 45.Hara S., Nakahigashi J., Ishi-i K., Sawaguchi M., Sakai H., Furuhara T., Yoneda N. Synlett. 1998:495–496. [Google Scholar]
- 46.Kirschning A. Eur. J. Org. Chem. 1998:2267–2274. doi: 10.1002/(SICI)1099-0690(199811)1998:11<2267::AID-EJOC2267>3.0.CO;2-E. [DOI] [Google Scholar]
- 47.Harders J., Garming A., Jung A., Kaiser V., Monenschein H., Ries M., Rose L., Schoning K. U., Weber T., Kirschning A. Liebigs Ann.-Recueil. 1997:2125–2132. [Google Scholar]
- 48.Kirschning A. Liebigs Ann. 1995:2053–2056. doi: 10.1002/jlac.1995199511289. [DOI] [Google Scholar]