

Abstract
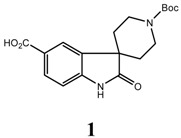
An efficient, scaleable synthesis approach towards the spirocyclic oxindole analogue 1’-(tert-butoxycarbonyl)-2-oxospiro[indoline-3,4’-piperidine]-5-carboxylic acid (1) is described. The key steps are dianion alkylation and cyclization of ethyl 2-oxindoline-5-carboxylate (4) and demethylation of the resulting spirocyclic oxindoleethyl 1’-methyl-2-oxospiro[indoline-3,4’-piperidine]-5-carboxylate (5). The target compound was obtained in an overall yield of 35 % over eight steps without resorting to chromatographic purification.
Keywords: Spirocyclic oxindole, dianion alkylation, N-demethylation
Introduction
Spiro compounds are becoming key building blocks for drug discovery as these templates have been shown to exhibit a variety of interesting biological activities, such as growth hormone secretagogues (Ibutamoren, MK-0677) [1], neurokinin antagonists [2], oxytocin antagonists [3], monoamine transporter inhibitors [4], bradykinin antagonists [5] and cell cycle inhibitors [6]. Due to their structure, they interact with a wide range of receptors and this activity has resulted in significant interest in developing efficient methods to prepare spiro compounds. We report herein the efficient synthesis of 1’-(tert-butoxycarbonyl)-2-oxospiro [indoline-3, 4’-piperidine]-5-carboxylic acid (1).
 |
Results and Discussion
The synthetic plan for the synthesis of the target molecule is outlined in Scheme 1. Our first objective was synthesis of the substituted oxindole 4. Of the reported synthetic routes to oxindoles [7], Lewis acid catalyzed cyclization of α-haloacetanilides [8] was proposed as the most efficient route to synthesize 4, but in practice this method gave a rather low yield (<10%), due to the strongly acidic conditions used in the transformation. Next, the synthesis of the target oxindole via the intermediacy of an azasulfonium salt [9] was investigated for its relatively mild conditions. Ethyl 4-aminobenzoate 2 was sequentially treated with tert-butyl hypochlorite, ethyl(methylthio) acetate and triethylamine, followed by dilute hydrochloric acid, to give substituted 3-methylthiooxindole 3 in 75% yield. Reductive desulfurization of 3 with Raney Ni afforded the target 4 in 85% yield.
Scheme 1.

(a) (i) t-BuOCl, ethyl(methylthio) acetate, CH2Cl2, -65 oC, (ii) Et3N, -65 oC to rt., (iii) HCl, ether, rt.; (b) Raney Ni, ethanol, reflux; (c) NaH, dichloroethamine hydrochloride, DMF, rt.; (d) (i) Trichloroethyl chloroformate, Et3N, 1,2-dichloroethane, (ii) Zinc, HOAc, (iii) (Boc)2O, dioxane; (e) NaOH, MeOH, H2O
Now we turned to the key step, the synthesis of the spirocyclic oxoindole 5. Goehring and others [5,10] have reported that treatment of a non substituted oxindole with excess sodium hexamethyldisilazide gave an oxoindole dianion [11], which when alkylated with N-methylbis(2-chloroethyl)amine afforded the corresponding spirocyclic oxindole. Unfortunately, in our case these conditions gave rather low yields. We found that the alkylation is difficult with various bases such as sodium hexamethyldisilazide, lithium hexamethyldisilazide, lithium diisopropylamide and potassium tert-butoxide in solvents such as tetrahydrofuran and dimethylformamide [12]. The low yields are due to the presence in 4 of the electron-withdrawing carboxylic ester group, which decreases the nucleophilicity of the enolate anion. Eventually we found that treatment of 4 with 4.5 equivalents of sodium hydride and dichloroethylmine hydrochloride in dimethylformamide for 5 hours gave the desired product 5 in 65% yield. With this intermediate in hand, we next embarked on the demethylation of 5 [13]. It is worth noting that, in our hands, attempts to alkylate the oxindole dianion with either N-Boc or N-benzyl dichloroethamine (to potentially shorten the synthesis) met with no success. However, treatment of 5 with 2,2,2-trichloroethyl chloroformate (TCE-Cl), followed by zinc reduction and subsequent Boc protection, gave a low yield (31%) of the desired protected amine 6.
After detailed investigation of the process, we found the following (Scheme 2): a) when 5 was treated with TCE-Cl, the mono-acylated intermediate 7 is first formed quantitatively, but it cannot be completely converted to 9, even when excess TCE-Cl is added. The acid, which was formed in situ during the formation of 7, would most likely result in the competition between the formation of hydrochloric salt 10, which would no longer react with TCE-Cl, and quaternary salt intermediate 8, which will lead to the desired di-acylated intermediate 9; b) when 7 was isolated and treated with TCE-Cl, the reaction is still reversible even under reflux conditions. This strongly indicates that the quaternary salt intermediate 8 undergoes two competitive reactions [13d], namely nucleophilic attack of the chloride ion on the O-alkyl portion, which has no net effect on 7, and nucleophilic attack of the chloride ion on the methyl group, leading to 9; c) eventually we found that upon treatment of 5 with 1-1.5 eq. triethylamine, followed by addition of TCE-Cl, the desired intermediate 9 was formed quantitatively and the reaction was complete in less than 10 minutes. These results suggest that triethylamine not only neutralizes the hydrochloric acid generated during the formation of 7, but also has significant influence on the breakup of quaternary salt 8. Presumably triethylamine is chelated with the chloride ion. The nucleophilic attack of the chelated chloride ion on the O-alkyl portion is then disfavored due to steric hindrance. To test the hypothesis, we investigated the dealkylation of N-ethylpiperidine, which was most often reported as the standard substrate for dealkylation [13b]. The report yield is less than 10% when TCE-Cl was used. As we expected, when one equivalent of triethylamine hydrochloride was added in the reaction mixture, the dealkylation was finished within 30 minutes with 95% yields. Finally, upon zinc reduction and subsequent Boc protection, the spirocyclic oxindole 6 was obtained in 92% from 5. Saponification of 6 gave 1 in 91% yield.
Scheme 2.

(a) Trichloroethyl chloroformate, 1,2-dichloroethane, rt.; (b) and (c) Trichloroethyl chloroformate, Et3N, 1,2-dichloroethane, rt.
Conclusions
We have described an efficient route to prepare the spirocyclic oxindole analogue 1’-(tert-butoxycarbonyl)-2-oxospiro[indoline-3,4’-piperidine]-5-carboxylic acid 1 in eight steps with an overall yield of 35%. This synthetic route is scaleable without chromatographic purification.
Experimental Section
General
All commercially available chemicals and reagents were used without further purification. The 1H- and 13C-NMR spectra were recorded on Bruker DPX-300 spectrometer. Chemical shifts (δ) are reported in ppm and are referenced to the deuterium solvent residual peak (for 1H-NMR, δ is 7.26 in CDCl3 and 2.50 in DMSO-d6, respectively; for 13C-NMR, δ is 77.2 in CDCl3 and 39.5 in DMSO-d6, respectively). Mass spectral analyses were performed by the Institute for Biomolecular Design of University of Alberta using an Applied Biosystems Voyager System 6064. TLC analysis was facilitated by the use of the following stains in addition to UV light with fluorescent-indicating plates (silica gel on glass, Silicycle): phosphomolybdic acid, anisaldehyde/EtOH, or KMnO4/H2O.
Ethyl 3-(methylthio)-2-oxindoline-5-carboxylate (3).
To a solution of ethyl 4-aminobenzoate 2 (30.0 g, 0.182 mol) in dichloromethane (600 mL) at 0 oC was added a solution of tert-butylhypochlorite (19.8 g, 0.182 mol) in dichloromethane (80 mL). The reaction mixture was stirred for 10 minutes and cooled to -65 °C. A solution of ethyl (methylthio) acetate (24.4 g, 0.182 mol) in dichloromethane (80 mL) was added dropwise and the reaction mixture was stirred for 1 hour, then a solution of triethylamine (18.4 g, 0.182 mol) in dichloromethane (80 mL) was added and the reaction mixture was warmed to room temperature. To this mixture was added water (300 mL) and the phases were separated. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated. The residue was dissolved in ether (600 mL) to which 2N HCl (80 mL) was added and the mixture was stirred for 18 hours. The solid formed was collected by filtration, washed with a small amount of hexane and dried in a vacuum oven to afford 3 (34.3 g, 75%) as an off white solid; 1H-NMR (CDCl3) δ 9.64 (br, 1H), 8.06 (s, 1H), 8.03 (d, 1H, J = 8.1 Hz), 6.98 (d, 1H, J = 8.1 Hz), 4.40 (dd, 2H, J = 7.1 Hz, 7.2 Hz), 2.03 (s, 3H), 1.41 (t, 3H, J = 7.1 Hz, 7.2 Hz); 13C-NMR (CDCl3) δ 176.4, 165.4, 147.0, 130.9, 127.1, 125.5, 123.4, 109.3, 60.5, 45.0, 14.2, 11.7; LRMS: MS (ES+) m/z = 252.3 (M+1)+
Ethyl 2-oxindoline-5-carboxylate (4)
To a solution of ethyl 3-(methylthio)-2-oxindoline-5-carboxylate 3 (60.0 g, 0.239 mol) in absolute ethanol (1200 mL) was added Raney Nickel 2800 (60 g). The reaction mixture was heated to reflux for 4 hours and quickly filtered while it was still hot. The filtrate was concentrated to half its volume and the product was crystallized. The solid was filtered and dried in a vacuum oven to afford 4 (41.6 g, 85 %) as a white solid; 1H-NMR (DMSO-d6) δ 10.72 (br, 1H), 7.83 (d, 1H, J = 8.1 Hz), 7.60 (s, 1H), 6.89 (d, 1H, J = 8.1 Hz), 4.25 (dd, 2H, J = 7.1 Hz, 7.2 Hz), 3.54 (s, 2H), 1.29 (t, 3H, J = 7.1 Hz, 7.2 Hz); 13C-NMR (DMSO-d6) δ 176.7, 165.7, 148.2, 129.8, 126.1, 125.1, 122.7, 108.8, 60.3, 35.5, 14.2; LRMS: MS (ES+) m/z = 206.2 (M+1)+
Ethyl 1’-methyl-2-oxospiro[indoline-3,4’-piperidine]-5-carboxylate (5)
To a solution of oxindole 4 (26.7 g, 0.130 mol) in dry dimethylformamide (900 mL) at 0 °C was added sodium hydride (14.1 g, 0.585 mol). The reaction mixture was stirred for 10 minutes. Dichloethylamine hydrochloride (25.0 g, 0.130 mol) was added in one portion. The reaction mixture was stirred for 10 minutes and warmed to room temperature for 5 hours. The mixture was cooled to 0°C and quenched with saturated ammonium chloride solution. The resulted mixture was diluted with water and extracted (4x) with ethyl acetate, dried over anhydrous sodium sulfate and concentrated. The product was crystallized to afford 5 (24.4 g, 65%) as a white solid; 1H-NMR (CDCl3) δ 8.18 (br, 1H), 8.04 (s, 1H), 7.98 (d, 1H, J = 8.1 Hz), 6.93 (d, 1H, J = 8.1Hz), 4.36 (dd, 2H, J = 7.1 Hz, 7.2 Hz), 3.07 – 2.93 (m, 2H), 2.84 – 2.72 (m, 2H), 2.50 (s, 3H), 2.13-2.02 (m, 2H), 2.02 – 1.93 (m, 2H), 1.39 (t, 3H, J = 7.1 Hz, 7.2 Hz); 13C-NMR (CDCl3) δ 182.1, 166.5, 144.2, 135.0, 130.7, 125.2, 124.9,, 109.2, 61.1, 50.5(x2), 46.4, 44.6, 33.1(x2), 14.6; LRMS: MS (ES+) m/z = 289.3 (M+1)+
1’-tert-Butyl 5-ethyl-2-oxospiro [indoline-3,4’-piperidine]-1’5-dicarboxylate (6)
To a solution of spirocyclic oxindole 5 (18.3 g, 0.0634 mol) in 1,2-dichloroethane (400 mL) was added triethylamine (9.6 g, 0.0951 mol). 2,2,2-Trichloroethylchloroformate (28.2 g, 0.133 mol) was then added dropwise. The reaction mixture was stirred for 15 minutes and was diluted with ether (800 mL). The mixture was washed with water and brine, dried over anhydrous sodium sulfate and concentrated. The residue was dissolve in acetic acid (300 mL). Zinc powder (9 g) was added in several portions and the reaction mixture was stirred for 2 hours. The solvent was evaporated under vacuum and water was added. The mixture was basified to pH = 10 by addition of ammonium hydroxide and then extracted with dichloromethane (3x). The organic layer was combined, dried over anhydrous sodium sulfate and concentrated. The residue was dissolve in dioxane (150 mL). To the solution, di-tert-butyl dicarbonate (13.8 g, 0.0634 mol) was added and the mixture was stirred for 1 hour. The solvent was evaporated and was recrystallized from 1:5 ethyl acetate/hexane to afford compound 6 (21.7 g, 92%) as a white solid; 1H-NMR (CDCl3) δ 8.61 (br, 1H), 8.01 (d, 1H, J = 8.1 Hz), 7.99 (s, 1H), 6.96 (d, 1H, J = 8.1 Hz), 4.37 (dd, 2H, J = 7.1 Hz, 7.2 Hz), 3.86 – 3.82 (m, 4H), 1.91 – 1.84 (m, 4H), 1.52 (s, 9H), 1.42 (t, 3H, J = 7.1 Hz, 7.2 Hz); 13C-NMR (CDCl3) δ 182.8, 166.5, 155.2, 144.6, 134.4, 130.9, 125.0, 124.8, 109.8, 80.1, 61.2, 46.7, 45.7 (x2), 32.6 (x2), 28.7(x3), 14.6; LRMS: MS (ES+) m/z = 375.4 (M+1)+
1’-(tert-Butoxycarbonyl)-2-oxospiro[indoline-3,4’-piperidine]-5-carboxylic acid (1)
To a solution of spirocyclic oxindole 6 (19.6 g, 0.0524 mol) in a mixture of methanol (450 mL) and water (80 mL) was added 2N sodium hydroxide (40 mL). The reaction mixture was heated to 55 °C and stirred for 8 hours. The methanol was evaporated under vacuum. The residue was cooled to 0 °C, acidified carefully to pH = 3 and extracted with ethyl acetate (3x). The organic layer was combined, washed with brine, dried over sodium sulfate and concentrated. The residue was recrystallized from ethyl acetate/hexane (2:1) to afford 1 (16.4 g, 91%) as an off white solid; 1H-NMR (DMSO-d6) δ 12.63 (br, 1H), 10.77 (s, 1H), 7.87 (s, 1H), 7.85 (d, 1H, J = 8.1 Hz), 6.93 (d, 1H, J = 8.1 Hz), 3.71 – 3.60 (m, 4H), 1.72 – 1.68 (m, 4H), 1.43 (s, 9H); 13C-NMR (DMSO-d6) δ 180.9, 167.3, 154.1, 145.5, 134.2, 130.4, 124.3, 124.1, 109.3, 78.8, 59.8, 44.6(x2), 31.9(x2), 28.1(x3); LRMS: MS (ES+) m/z = 347.4 (M+1)+
Footnotes
Sample Availability: Samples of compounds 1, 3, 4 and 5 are available from the authors.
References and Notes
- 1.(a) Patchett A. A., Nargund R. P., Tata J. R., Chen M.-H., Barakat K. J., Johnston D. B. R., Cheng K., Chan W. W.-S., Butler B., Hickey G. J., Jacks T., Schleim K., Pong S.-S., Chuang L.-Y. P., Chen H. Y., Frazier E., Leung K. H., Chiu S.-H. L., Smith R. G. Design and Biological Activities of L-163,191 (MK-0677): A Potent, Orally Active Growth Hormone Secretagogue. Proc. Natl. Acad. Sci. U.S.A. 1995;92:7001–7005. doi: 10.1073/pnas.92.15.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Barakat K. J., Cheng K., Chan W. W.-S., Butler B. S., Jacks T. M., Schleim K. D., Hora D. F., Jr., Hickey G. J., Smith R. G., Patchett A. A., Nargund R. P. Synthesis and biological activities of phenyl piperazine- based peptidomimetic growth hormone secretagogues. Bioorg. Med. Chem. Lett. 1998;8:1431–1436. doi: 10.1016/s0960-894x(98)00238-8. [DOI] [PubMed] [Google Scholar]
- 2.Eliott J. M., Cascieri M. A., Chicchi G., Davies S., Kellecher F. J., Kurtz M., Ladduwahetty T., Lewis R. T., Macleod A. M., Merchant K. J., Sadowski S., Stevenson G. I. Serine derived NK1 antagonists 1: The effect of modifications to the serine substituents. Bioorg. Med. Chem. Lett. 1998;8:1845–1850. doi: 10.1016/s0960-894x(98)00320-5. [DOI] [PubMed] [Google Scholar]
- 3.Evans B. E., Leighton J. L., Rittle K. E., Gilbert K. F., Lundell G. F., Gould N. P., Hobbs D. W., Dipardo R. M., Veber D. F., Pettibone D. J., Clineschmidt B. V., Anderson P. S., Freidinger R. M. Orally active, nonpeptide oxytocin antagonists. J. Med. Chem. 1992;35:3919–3927. doi: 10.1021/jm00099a020. [DOI] [PubMed] [Google Scholar]
- 4.Efange S. M. N., Kamath A. P., Khare A. B., Kung M.-P., Mach R. H., Parsons S. M. N-Hydroxyalkyl Derivatives of 3β-Phenyltropane and 1-Methylspiro[1H-indoline-3,4'-piperidine]: Vesamicol Analogues with Affinity for Monoamine Transporters. J Med. Chem. 1997;40:3905–3914. doi: 10.1021/jm970326r. [DOI] [PubMed] [Google Scholar]
- 5.Goehring R. R. Synthesis of a Spiro Oxindole Analogue as a Putative Replacement for Pro2-pro3-gly4-phe5 in Bradkinin Antagonists. Org. Prep. Proced. Int. 1995;27:691–694. [Google Scholar]
- 6.Edmondson S., Danishefsky S. J., Sepp-Lorenzino L., Rosen N. Total Synthesis of Spirotryprostatin A, Leading to the Discovery of Some Biologically Promising Analogues. J. Am. Chem. Soc. 1999;121:2147–2155. [Google Scholar]
- 7.For a review of oxindole synthesis, see Sundberg R. J. “The Chemistry of Indoles”. Academic Press; New York, N. Y.: 1970.
- 8.(a) Sumpter W. C. The chemistry of Oxindole. Chem. Rev. 1945;37:443–479. doi: 10.1021/cr60118a003. [DOI] [PubMed] [Google Scholar]; (b) Beckett A. H., Daisley R. W., Walker J. Substituted oxindole—I : The preparation and spectral characteristics of some simple oxindole derivatives. Tetrahedron. 1968;24:6093–6109. [Google Scholar]; (c) Walker J., Daisley R. W., Beckett A. H. Substituted oxindoles. III. Synthesis and pharmacology of some substituted oxindoles. J. Med. Chem. 1970;13:983–985. doi: 10.1021/jm00299a048. [DOI] [PubMed] [Google Scholar]
- 9.Gassman P. G., van Bergen T. J. Oxindoles. New, general method of synthesis. J. Am. Chem. Soc. 1974;96:5508–5512. [Google Scholar]
- 10. Freund R., Mederski W. W. K. R. A Convenient Synthetic Route to Spiro[indole-3,4′-piperidin]-2-ones. Helv. Chim. Acta. 2000;83:1247–1255.; (b) ref. [5]; Jackson A. H., Smith A. E. Electrophilic substitution in indoles—II : The formation of 3,3-spirocyclic indole derivatives from tryptamines and their rearrangement to β-carbolines. Tetrahedron. 1968;24:403–413.
- 11.For examples of C-3 alkylation of the oxindole dianion, see: Kende A. S., Hodges J. C. Regioselective C-3 alkylations of oxindole dianion. Synth. Commun. 1982;12:1–10. DeMarinis R. M., Gallagher G., Hall R. F., Franz R. G., Webster C., Huffman W. F., Schwarta M. S., Kaiser C., Ross S. T., Wilson J. W., Hieble P. Syntheses and in vitro evaluation of 4-(2-aminoethyl)-2(3H)-indolones and related compounds as peripheral prejunctional dopamine receptor agonists. J. Med. Chem. 1986;29:939–947. doi: 10.1021/jm00156a010. Carroll W. A., Grieco P. A. Biomimetic total synthesis of pseudotabersonine: a novel oxindole-based approach to construction of Aspidosperma alkaloids. J. Am. Chem. Soc. 1993;115:1164–1165. Karp G. M. Preparation and Reactions of Indolin-2(3H)-ones. A Review. Org. Prep. Proc. Int. 1993;25:481–514.
- 12.All reaction conditions are the same as the given procedure for compound 5. The N-methyl bis(2-chloroethyl)amine free base was generated in situ by neutralization of the dichloroethylmine hydrochloric salt with the base used.
- 13.For the dealkylation of tertiary amine, see: Katritzky A. R., Meth-Cohn O., Rees C. W. Comprehensive Organic Functional Group Transformations. Vol. 2. Pergamon; Oxford: 1995. pp. 302–304. Cooley J. H., Evain E. J. Amine Dealkylation with Acyl Chlorides. Synthesis. 1989:1–7. Olofson R. A., Martz J. T., Senet J.-P., Piteau M., Malfroot T. A new reagent for the selective, high-yield N-dealkylation of tertiary amines: improved syntheses of naltrexone and nalbuphine. J. Org. Chem. 1984;49:2081–2082. Howarth N. M. Manipulation of substituents at nitrogen in tropanes, homotropanes, and dehydro- derivatives. Tetrahedron. 1998;54:10899–10914. Olofson R. A., Schnur R. C., Bunes L., Pepe J. P. Selective N-dealkylation of tertiary amines with vinyl chloroformate: An improved synthesis of naloxone. Tetrahedron Lett. 1977;18:1567–1570. Montzka T. A., Matiskella J. D., Partyka R. A. 2,2,2-trichloroethyl chloroformate: A general reagent for demethylation of tertiary methylamines. Tetrahedron Lett. 1974;15:1325–1327.