

Introduction:
The parallel development of methods for heterologously expressing and mutating cloned ion channel subunits, and for voltage-clamp electrophysiological assessment of ion channel function has engendered multiple structure-function approaches that contributed to discovery of general anesthetic sites in pentameric ligand-gated ion channels. Here, we review structure-function experimental strategies combining electrophysiology with ion channel subunit chimeras, single-point mutations, and covalent modification of sulfhydryls and their application in identifying general anesthetic sites on nicotinic acetylcholine receptors (nAChRs) and γ-aminobutyric acid type A receptors (GABAARs). We review detailed methodological considerations for one method that has proven useful in identifying drug-receptor contacts on GABAA receptors: substituted cysteine modification and protection (SCAMP).
1). Challenges for Identifying General Anesthetic Binding Sites
Many anesthetic drugs, including alcohols and volatile alkanes and ethers are low affinity and probably act through multiple molecular targets. More potent anesthetics that display stereoselectivity and greater target specificity include certain barbiturates, etomidate, and alphaxalone. These potent anesthetics present fewer challenges in defining their mechanisms of action and represent bases for developing new drugs with improved clinical utility.
Many anesthetics inhibit the activity of excitatory pentameric ligand-gated ion channels (pLGICs). A well-studied case is inhibition of the muscle-type nAChR, although nAChR activity is also positively modulated by small alcohols [1]. Mechanisms of anesthetic inhibition in nAChRs include channel blockade, desensitization, and negative allosteric modulation [2, 3]. In contrast, most general anesthetics positively modulate inhibitory pLGICs such as GABAA receptors and glycine receptors [4]. Positive modulation at a subset of GABAA receptor isotypes is known to mediate major behavioral effects of many general anesthetics, including hypnosis, amnesia, and immobility [5]. In addition, high concentrations of some anesthetics directly agonize GABAA receptors and others also inhibit receptor activity. Thus, there are potentially many types of anesthetic binding sites mediating different anesthetic actions on each pLGIC. Identifying these sites is a first challenge, and a second is demonstrating which anesthetic actions, if any, each binding site mediates. It is also possible that one site can mediate multiple drug effects.
2). pLGIC Structural Models
Current structural models of heteromeric pLGICs are homology models, typically based on both cryo-electron microscopy and crystallography in channels from bacteria, nematodes, and mammals. The existence of many different subunit isotype genes indicates a very high number of potential structures, whereas a limited number of nAChRs and GABAA receptors are found in the central nervous system. The subunit arrangement of muscle nAChR is established as α(γ/ε)αδβ. The most common synaptic αβγ GABAA receptor structure is established as βαβαγ (Fig 1). All pLGIC subunits include an N-terminal extracellular domain (ECD) of about 200 amino acids, a transmembrane domain (TMD) consisting of 4 transmembrane helices (M1 to M4), a variable-size intracellular domain (ICD) between M3 and M4, and a short extracellular C-terminus. Sequence alignments in the TMD are well-established in M2 helices, where a universally conserved leucine is the 9th residue (L9´) after the positively charged residue defining the onset of the helix. However, sequence alignment of other transmembrane helices is more uncertain due to variation in the length of inter-helical linker sequences.
Figure 1: Pentameric Ligand-Gated Ion Channel Structure and Types of Anesthetic Binding Sites.
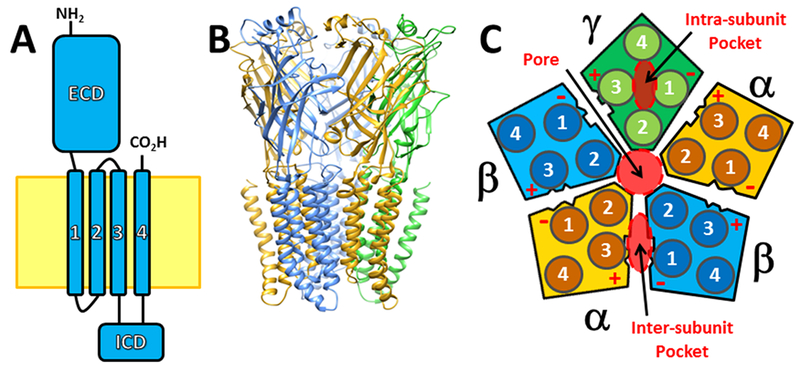
Panel A depicts the canonical subunit structure shared by all pLGICs, including a large N-terminal extracellular domain (ECD), a transmembrane domain consisting of four alpha-helices, and an intracellular domain (ICD) between M3 and M4. The cytoplasmic membrane is depicted as a yellow zone. Panel B shows a structural homology model of a synaptic GABAA receptor consisting of two α subunits (gold), two β subunits (blue), and one γ subunit (green). Peptide backbones are depicted as ribbons to highlight secondary structural folding. Subunit ICDs have been removed because their structure is not well-defined. Panel C is a diagram showing a cross-section of an αβγ GABAA receptor through the TMD. Each subunit is color-coded as in panel B, the arrangement of subunits is details, as are the positions of numbered alpha-helices in each subunit. Three types of anesthetic binding pockets in the TMD are highlighted with red overlays: the central transmembrane pore, intra-subunit pockets formed by subunit four-helix bundles, and inter-subunit pockets. Inter-subunit anesthetic binding pockets have also been imaged in the ECD. The faces of each subunit are also labeled “+” or “−” depending on whether they include M3 or M1 helical elements.
Structure-function studies have established that in muscle nAChRs orthosteric agonists (e.g. ACh and nicotine) bind to α+/δ− and α+/(γ/ε)− interfaces in the ECD, while in GABAARs, GABA binds to ECD β+/α− interfaces. Thus, nAChR α’s can be considered homologs of GABAAR β’s and in both pLGIC families these archetypal subunits can form functional homopentamers.
Several high-resolution structural studies have focused on anesthetic binding sites in crystallized pLGICs, all of which are homopentamers [6–10]. These confirm that small amphiphilic general anesthetics can fit into multiple types of intra-subunit or inter-subunit protein pockets formed in both ECDs and TMDs (Fig 1C).
3). Electrophysiological Approaches to Assessing pLGIC Function
3.1). Functional states of pLGICs.
Electrophysiological studies have demonstrated that most pLGICs undergo a similar set of functional transitions. Binding of orthosteric agonists to resting-state closed receptors stimulates rapid transition to activated or open ion-conductive states that can be observed in voltage-clamp experiments. Prolonged agonist application results in multiple phases of channel desensitization, leading to non-conductive states with high affinity for agonists. After discontinuing agonist application, pLGICs undergo deactivation as open channels return to the resting state. Recovery of desensitized receptors to the resting state may be via “silent” pathways or return to open states, and take up to tens of seconds, depending on the degree of receptor desensitization.
3.2). Cellular systems for heterologous expression and electrophysiology.
The most common cell systems used for heterologous expression and structure-function studies are Xenopus oocytes and cultured mammalian cell lines like HEK293. Xenopus oocytes are large cells (~ 1 mm diameter) that can express many receptors on the membrane surface and under ideal conditions, oocytes can provide stable currents for lengthy experiments lasting several hours. Oocytes are microinjected with messenger RNA or cDNA encoding wild-type or mutant receptor subunits. Oocytes can then be used for whole-oocyte two-electrode voltage-clamp studies or for patch-clamp studies in excised membrane patches [11]. Mammalian cells are transfected with cDNA plasmids that direct synthesis of receptor subunits that are assembled and transported to the cytoplasmic membrane and used for whole cell or patch current studies. Post-translational modification of receptors may vary between expression cell types or depending on cytoplasmic kinase activity. There are examples where either receptor assembly and/or functional properties differ when expressed in HEK293 cells vs. oocytes [12, 13].
3.3). Electrophysiological and solution exchange equipment.
Commercially available two-microelectrode voltage-clamp amplifiers can typically measure whole-oocyte voltage-clamped currents ranging from under 5 nA to 10 µA. Application of various solutions containing agonists and anesthetics or other chemicals is typically controlled by manual or electronically triggered valves from multiple reservoirs feeding into a manifold with outflow directed at the oocyte in a flow-chamber and then to a waste-collection system. Special care needs to be used with volatile anesthetics to prevent evaporation and adsorption into flow-system materials. When using very hydrophobic compounds such as potent anesthetics, reservoirs and tubing should be made of impermeable materials like glass, stainless steel, or PTFE. Depending on the rate of flow and the design of the reservoir around the oocyte, solution exchange can be as fast as 10 ms, but is typically around 0.1 s or longer. This is often too slow to observe the fastest rates of desensitization and deactivation observed in nAChRs and synaptic GABAA receptors. Another important consideration in studying hydrophobic anesthetics is that the large volume of intracellular lipid and protein in oocytes acts as a sink, slowing the rate of equilibration between drug in the oocyte membrane and the surrounding solution. Pre-equilibrating oocytes with anesthetics before agonist application can reduce errors due to this process. An associated challenge is prolonged washout times to fully remove hydrophobic drugs from oocytes.
Patch-clamp amplifiers can typically measure currents in the 5 pA to 10 µA range. When combined with “artificial synapse” techniques for rapid solution switching, solution exchange times under 1 ms can be achieved at excised membrane patches [14, 15]. Solution exchange times in whole HEK cells are typically 2 to 10 ms using rapid solution switching devices. Equilibration of hydrophobic drugs is fastest in excised patch experiments, and can be rate-limiting in measuring drug effects in voltage-clamped cells [16]. These approaches enable studies of state-dependent drug actions with analysis of desensitization and deactivation rates. Single-channel recordings can also be obtained using low-noise patch-clamp techniques.
4). Chimeric Subunits
Structure-function studies based on chimeric subunits represent an unbiased approach to identifying peptide sequences within receptor subunits that influence anesthetic sensitivity. Mihic et al [17] created a set of chimeric pLGIC subunits containing complementary portions of anesthetic-sensitive α1 glycine receptors and anesthetic-insensitive ρ GABAA receptors, and used electrophysiology to assess anesthetic modulation in each chimera at its EC20 agonist concentration (the agonist depended on which type of receptor donated the N-terminal domain). The results identified a subunit region including M2 and M3 helices that was both necessary and sufficient to confer positive modulation by volatile anesthetics and alcohols. Subsequent point mutations exchanging the non-homologous residues in M2 and M3 helices identified two amino acids that influenced anesthetic modulation in both parent receptors: the M2-15´ position, and an M3 residue 21 amino acids away (sometimes called 36´). Mutations at the homologs on GABAA receptor α and β subunits were subsequently shown to influence sensitivity to both volatile and intravenous anesthetics.
J.J. Lambert and colleagues found that GABAA receptors containing β2 or β3 subunits were sensitive to modulation and direct agonism by the intravenous drug etomidate, while those with β1 subunits were not [18]. Chimeras containing complementary portions of β1 and β2, co-expressed with α and γ subunits and studied electrophysiologically, showed that determinants of etomidate sensitivity were in the transmembrane domain [19]. Subsequent point mutation experiments showed that swapping β2 and β1 M2-15´residues determined drug sensitivity. At this position in β2, a N-to-S mutation suppressed etomidate sensitivity, while the complementary β1 S-to-N mutation increased sensitivity to etomidate modulation.
Interpretation of data from chimera experiments is critically dependent on the experimental framework. An important illustration is a study of β3 sequences involved in pentobarbital agonism [20]. Chimeric GABAA β3/ρ subunits were co-expressed with α1 subunits and pentobarbital vs. GABA agonism was assessed electrophysiologically in the resulting receptors. In one chimeric receptor where pentobarbital efficacy was reduced relative to that of GABA, noise and single-channel analyses indicated that the structural modification had increased GABA efficacy, without altering pentobarbital efficacy. Alternative analysis based on deactivation kinetics in the various chimeric receptors suggested that pentobarbital affinity determinants were downstream (toward the C-terminus) from the M2 helix. Another study of pentobarbital actions on GABAA receptor chimeras containing complementary sequences from γ and δ subunits [21] concluded that sequences upstream from M2 were critical. However, pentobarbital-induced currents were normalized to maximal GABA currents, and this study failed to account for evidence that GABA intrinsic efficacy in αβγ receptors is high, but is very low in αβδ receptors [22].
5). Hydrophobic Point Mutations in nAChRs
5.1). Introduction.
Single point mutations are the simplest hypothesis-based (biased) structure-function approach, because each site chosen for a mutation represents a hypothesis or set of hypotheses. Conceptually, one makes mutations and tests their effects on receptor function and sensitivity to drugs, leading to inferences about drug binding or modulation mechanisms.
5.2). Nicotinic ACh receptors.
Pioneering work by Henry Lester and colleagues focused on the ion conductive pore of muscle nAChRs, which was thought to be surrounded by M2 helices of the TMD [23]. They showed that M2-6´ S-to A mutations in mouse muscle nAChR alpha subunits reduced sensitivity to QX222, a charged quarternary ammonium analog of an amide local anesthetic thought to block nAChRs via open-channel block. A strength of this study was the use of single-channel kinetics, demonstrating reduced apparent QX222 residence time in its site, as evidence of reduced binding affinity. Voltage-dependent block was also demonstrated, supporting a transmembrane location of the site. Mutations at M2-10´ residues on nAChRs showed that increased hydrophobicity enhanced QX-222 affinity, suggesting that the charged end of the molecule interacted with 6´ residues, while the hydrophobic end interacted with 10´ residues.
Kinetic studies in mouse nAChRs also suggested that general anesthetics might act as open-channel blockers [24]. Following the work on QX222, Forman et al [25] tested M2-10´ mutations that enhanced hydrophobicity, and found that they dramatically increased mouse nAChR sensitivity to open state-dependent block by both alcohols and volatile anesthetics. Subsequent studies showed that mutations on all five M2-10´ residues affected sensitivity to anesthetic inhibition, suggesting a pore site [26]. Scanning mutagenesis studies defined a “hydrophobic patch” in the nAChR channel where anesthetics apparently interacted [27].
Unlike long-chain alcohols, ethanol enhances the apparent affinity of ACh for muscle and Torpedo nAChRs. Studies in nAChR mutants that impaired gating demonstrated that ethanol enhances ACh agonist efficacy rather than binding [28]. However, positive modulation by ethanol is unaffected by M2 mutations that affect alcohol inhibitory potency.
5.3). GABAA receptors.
In contrast to anesthetic inhibition of excitatory serotonin and nAChRs, many general anesthetics enhance activation of inhibitory glycine and GABAA receptors [4]. Moreover, high concentrations of general anesthetics directly activate (agonize) these receptors in the absence of GABA or other orthosteric agonists, which might indicate the presence of at least two classes of anesthetic sites and presented challenges in defining anesthetic potency for putative sites. A standard approach is to measure anesthetic enhancement of receptor activation by a low agonist concentration that alone generates a small fraction (5 to 20%) of maximal receptor-mediated responses.
Based on the findings of chimera studies [17], mutations on various GABAA receptor subunit M2-15´ and 36´ residues have been tested for effects on anesthetic modulation. These studies showed that βM286W mutations weaken modulation by propofol and related alkyl-phenols [29, 30] and that the size of the α1 M2-15´ (S270) sidechain determines volatile anesthetic sensitivity [31]. Tryptophan mutant sensitivity has been applied as a scanning method to discover anesthetic contacts [32, 33]. Similar approaches have also been used to discern selectivity for multiple GABAA receptor binding sites by various anesthetics [34].
5.4). MWC co-agonism as an interpretive framework for anesthetic actions on GABAA receptors.
The interpretive framework for mutant function studies is also critically important, because it is difficult, if not impossible, to rule out allosteric effects based on binding studies and functional analysis alone. This is particularly difficult when studying agonist sites [35]. Reframing anesthetic actions at GABAA receptors as allosteric co-agonism was based on pharmacological analysis using a formal Monod-Wyman-Changeux (MWC) two-state model [36–38]. These models provide a mechanistic framework for structure-function experiments that accounts for both direct anesthetic agonism and modulation of GABA-evoked responses based on drug interaction with a single class of allosteric agonist sites, providing simplified analysis in many cases. This approach has also proven insightful in explaining results that might otherwise lead to the conclusion that direct activation and modulation are mediated by independent sites. For example, based on azi-etomidate photolabeling results [39], we examined the effects of tryptophan mutations at both α1M236 and β2M286 in α1β2γ2L receptors [40]. Oocyte electrophysiology showed that α1M236Wβ2γ2L receptors were spontaneously active and that etomidate potently and efficaciously activated these receptors, whereas etomidate modulation of GABA responses was much weaker than that in wild-type receptors. MWC models predict that spontaneously active mutant channels should display increased sensitivity to agonists (both orthosteric and allosteric), and model-based analysis showed that all of the etomidate-dependent results were consistent with reduced drug efficacy. More importantly, this example illustrates how mutations that alter receptor gating or orthosteric agonist efficacy may produce the appearance of altered anesthetic sensitivity, even when underlying anesthetic site interactions are unaltered.
However, even though MWC mechanisms distinguish binding affinity to resting receptors from agonist efficacy as separate model parameters, we have consistently found that mutations alter apparent efficacy far more than binding affinity, even at residues thought to be important binding contact points. This is likely because agonist efficacy in MWC is defined as the ratio of binding dissociation constants in open vs. closed receptors, and molecules sufficiently efficacious to activate receptors bind much more tightly to open state receptors. At the same time, the very high anesthetic concentrations needed to occupy the majority of low-affinity resting state sites usually produce mixed functional effects that include positive modulation and inhibition. Some mutations that produce total insensitivity to anesthetics provide no data for differentiating binding vs. efficacy changes [41]. Thus, even using an analytical approach that differentiates binding and efficacy, identifying binding determinants is a challenge.
5.5). Comparing mutant sensitivity and anesthetic photolabeling.
Given the possibility that mutations allosterically alter apparent anesthetic sensitivity, an important question is how well hydrophobic mutagenesis and functional analysis performs in identifying drug-receptor contacts. We recently compared results of studies based on GABAA receptor photolabeling with derivatives of etomidate, propofol, mephobarbital, and alphaxalone to tryptophan mutagenesis experiments at two photolabeled amino acids [42]. With eight possible interactions (4 positive and 4 negative photolabeling results), Trp mutant studies agreed with photolabeling in 6 of 8 cases, with one false positive and one false negative. This correlation was too weak to support mutant function analysis as a reliable approach to discovering anesthetic contact residues in GABAA receptors.
6). Substituted Cysteine Accessibility Method: SCAM
6.1). Introduction.
Cysteine mutation-based approaches based on covalent sulfhydryl modification are potentially more sensitive to local steric rather than allosteric effects, because implicit in this method is a bi-molecular interaction between a chemical probe and the sulfhydryl group. These approaches include the Substituted Cysteine Accessibility Method (SCAM), disulfide mapping, and Substituted Cysteine Modification Protection (SCAMP). Below, we focus on how these methods have been applied to pLGIC receptor structure and mapping of anesthetic sites.
6.2). Nicotinic ACh receptors.
Akabas and Karlin were first to combine real-time agonist-dependent cysteine modification of pLGICs with electrophysiological analysis in nAChRs [43]. Their focus was cysteine accessibility, using a positively charged methane thiosulfonate-ethylammonium probe (MTS-EA), which they presumed was accessible only to the water-filled cation-conductive pore. Water is usually required for disulfide bond formation, which can also be influenced by the acidity of the targeted –SH group. At the time of these pioneering experiments, it was assumed that water was generally excluded from access to the M1, M3, and M4 helices that surrounded the M2s. Akabas and Karlin assumed that MTSEA modification in the nAChR pore would inhibit cation conduction. Thus, they compared maximal ACh-activated voltage-clamp currents mediated by cysteine-substituted receptors before and after MTSEA exposure. When inhibition was found, they inferred sidechain access from the water-filled ion pore. Other water-soluble reagents have also been used to probe water-accessible portions of pLGIC receptors. Using various modifying reagents at accessible substituted cysteines also enables studies of the effect of size, hydrophobicity, and charge on function.
6.3). GABAA receptors.
Akabas and colleagues extended the SCAM approach to M2 helices of GABAA receptors, and using EC50 GABA instead of high GABA when testing mutant receptors, found that modification both enhanced receptor activation and inhibited it at various positions [44]. The finding of enhanced GABA responses after modification meant that inferences regarding accessibility via the central ion pore were uncertain. Accessibility to water remained the sole inferential outcome of these studies. Importantly however, results in other TM helices showed that water could reach close to the presumed inner ends of TM helices, implying that these helices are flexible and that water intercalates between them, and not just in the ion-conductive pore [45, 46]. In short, SCAM revealed water-accessible pockets formed among the various transmembrane helices.
7). Disulfide Bond Formation Used to Probe Helix-Helix Interactions in GABAA Receptors.
Janson & Akabas [47] extended the use of cysteine substitutions in order to gain more information about the orientation of residues on adjacent transmembrane helices in the GABA α1 subunits. In this study, they inserted cysteines at different positions in both α1-M2 and α1-M3 helices, and used oocyte electrophysiology and chemical manipulations to either promote or reverse disulfide bond formation between the two helices. Their results helped constrain structural models and also suggested possible relative movements of M2 and M3 during channel gating.
Bali et al [48] utilized similar methods, but focused on creating disulfide linkages between two transmembrane helical faces across the β+/α− inter-subunit cleft. Their results study further constrained structural models and were consistent with azi-etomidate photolabeling data suggesting β+/α− inter-subunit etomidate binding sites based on photolabeling of residues on both β-M3 and α-M1 helices [39].
8). Substituted Cysteine Modification and Protection: SCAMP
8.1). Introduction.
Because covalent modification of a substituted cysteine requires bimolecular interactions between the probe reagent and the engineered sulfhydryl, this reaction can be hindered by non-covalent ligands that bind nearby. This inhibition of cysteine modification or “protection” has been developed as an approach to both complement and supplement other techniques, particularly photolabeling, in identifying anesthetic contacts on GABAA receptors. The first such study was that of Bali and Akabas [49], testing whether propofol contacted βN265 (M2-15´) or βM286 (36´) residues that had been identified in chimera and point mutation studies. To do this, they used oocyte electrophysiology to estimate the rates of modification at βN265C or βM286C by the sulfhydryl-reactive probe p-chloromercuribenzene sulfonate (pCMBS). They then tested whether addition of propofol slowed the rate of modification, and found protection at βM286C, but not βN265C.
8.2). SCAMP experimental design considerations.
For the SCAMP technique to enable strong inferences of steric interactions, a number of experimental conditions must be met [50]:
-
8.2.1
The background receptor must be insensitive to probe exposure. This may require removing native cysteines, usually by mutation to alanines or serines.
-
8.2.2
The cysteine-substituted mutant receptor should retain sensitivity to the anesthetic drug (usually measured as EC5 enhancement by a standard drug concentration), ideally similar to wild-type sensitivity. Loss of drug sensitivity might be caused by the cysteine mutation dramatically reducing drug binding, which could lead to false negative results. For example, we studied βN265C protection by etomidate, but also found that the mutation totally eliminated etomidate sensitivity [51]. Thus, the observed absence of etomidate protection could have been due to absence of drug binding, rather than binding at a site distant from βN265. It is possible that the lack of propofol protection at βN265C [49] was also due to low occupancy of these sites. In cases where drug sensitivity is moderately reduced, protection may still be tested, but using higher drug concentrations.
-
8.2.3
Modification of the mutant receptor must reliably produce quantifiable functional changes. Multiple probes may be teste to determine which produce the largest functional effects. However, very large probes may increase sensitivity to inhibition by ligands that bind farther from the study residue. Electrophysiological signals indicating GABAA receptor modification can include: a) spontaneous receptor activity, determined with picrotoxin inhibition; b) maximal receptor current; or c) EC5 GABA response, usually normalized to maximal response. In our experience, carefully designed SCAMP studies can lead to valid inferences in cases where receptor modification produces functional changes with magnitudes as small as two-fold.
-
8.2.4
Modification conditions must enable estimation of modification rates. Typically, sequential cycles of receptor modification are separated by repeated electrophysiological assessments of receptor function, with each cycle resulting in approximately 10% of the full modification effect (Fig 2). With many such cycles, full modification can be achieved and the electrophysiological results fitted to an exponential function to determine a rate parameter. Alternatively, three or four partial modification cycles can be used to establish an initial “apparent” modification rate based on a linear fit to the electrophysiological results. Normalization to each individual cell’s maximal current is essential when using “apparent” modification rate analysis.
-
8.2.5
The distribution of receptor states should be similar in both control modification experiments and in drug protection experiments. This aspect of study design can be challenging when using anesthetics that act as agonists at GABAA receptors. In our experience, the rate of pCMBS modification at cysteines in transmembrane helices of GABAA receptors is usually faster when pCMBS is co-applied with GABA than when applied alone [42, 52, 53]. If adding anesthetics activates receptors, then using a “closed state” modification control is inappropriate, because a fraction of receptors will be activated during protection experiments. To avoid this problem, we use control modification conditions with pCMBS + GABA. GABA-bound receptors also bind more avidly to anesthetics (allosteric agonists), resulting in higher anesthetic binding site occupancy. However, GABA may become a partial agonist at with introduction of some cysteine mutations, introducing the possibility that the fractions of closed vs. open vs. desensitized receptors differ in control experiments and anesthetic protection experiments. In such cases, addition of another positive modulator that binds to distinct sites may help establish similar distributions of receptor states in control modification and protection experiments [52]. For example, pharmacological and photolabeling evidence indicates that alphaxalone and some barbiturate sites are distinct from those for propofol or etomidate.
-
8.2.6
Drug site occupancy should correlate with the degree of observed protection. Demonstrating concentration-dependent protection, particularly when anesthetic functional effects parallels protection, supports the presumed local steric hindrance mechanism [52]. By extension, SCAMP will be more sensitive when high anesthetic concentrations are used, leading to higher binding site occupancy. As noted above, this is particularly important when studying mutations that are relatively insensitive to modulation by one or more study drugs. Including GABA usually enhances mutant receptor affinity for anesthetics, and increases drug site occupancy (see above).
-
8.2.7
In protection experiments, potent anesthetics should be pre-applied before adding modifiers, to assure anesthetic site occupancy before modifiers reach their target. This approach is particularly important when studying very hydrophobic anesthetics that slowly equilibrate in oocyte membranes due to intracellular binding to protein and lipid. Higher sulfhydryl probe concentrations will more rapidly reach the target sulfhydryl, so selecting conditions where lower probe concentrations are used (i.e. apply GABA with the modifier) will also reduce this potential artifact.
-
8.2.8
Even with all these conditions met, it is important to perform additional studies to help rule out allosteric drug inhibition of modification at each substituted cysteine. Allosteric inhibition of modification at a particular cysteine should correlate with receptor functional state, rather than anesthetic site occupancy. Thus, all anesthetics that modulate the mutant receptor should inhibit modification of that cysteine. Our approach has been to compare multiple anesthetics that all produce similar functional effects in GABAA receptors, but that are also known to bind to distinct sets of sites, based on pharmacological and photolabeling evidence. Sets of such drugs perform as negative controls for each other, and lend support to a steric hindrance mechanism in most cases where SCAMP experiments show inhibition.
Figure 2: Substituted Cysteine Modification Protection (SCAMP) Experimental Approach.
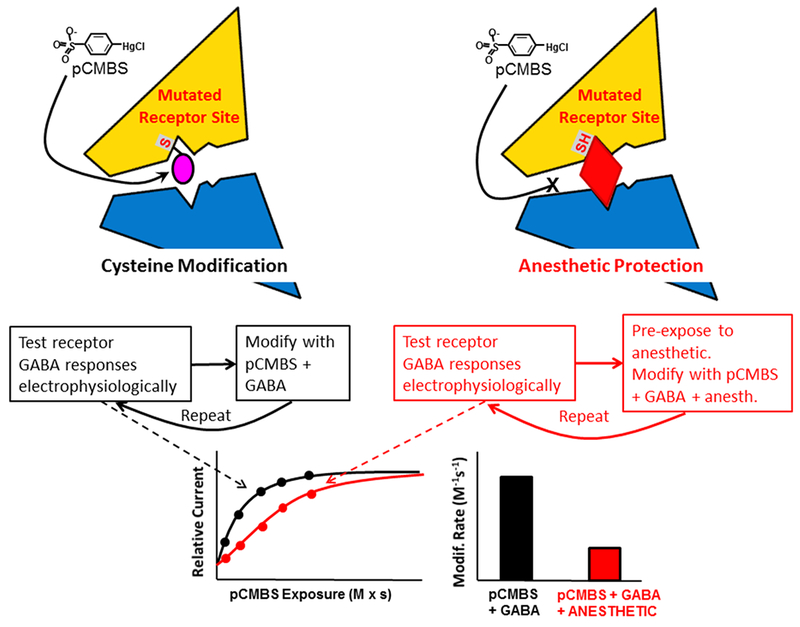
The top and middle left panels depict a mutant receptor with a cysteine-substitution in an inter-subunit anesthetic binding pocket as it is covalently modified by para-chloromercuribenzene sulfonate (pCMBS). Repeated cycles of partial modification produce functional changes that are measured electrophysiologically. The top and middle right panels depict similar experiments on anesthetic-bound receptors, with inhibition of the bimolecular reaction between pCMBS and the engineered sulfhydryl group on the receptor. The bottom left panel shows how electrophysiological measurements for both control modification (black dots) and protection (red dots) conditions are plotted against cumulative pCMBS exposure and fitted with exponential functions (black and red lines) to determine modification rates. The bottom right panel shows a summary of the analyzed rates indicating that bound anesthetic “protects” this cysteine from modification.
8.3). SCAMP Limitations.
SCAMP doesn’t enable every hypothesized residue to be tested against every drug of interest. Some cysteine mutations prevent functional expression of receptors (e.g. β3Y284C) and some (e.g. α1I239C and α1Q242C) show no functional effects when exposed to sulfhydryl modifiers [53, 54]. Several cases where cysteine mutants are totally insensitive to drugs of interest have also been reported. In the case of β2N265, we used an indirect SCAMP approach, based on protection of α1M236C by etomidate, to show that β2N265M, which also eliminates etomidate sensitivity, dramatically reduces apparent etomidate affinity for its site in both closed and GABA-activated states. Indirect SCAMP experiments may also be used to distinguish between allosteric and steric effects of other mutations. Despite negative control experiments, we have also encountered cases where anesthetics only partially inhibit sulfhydryl modification. These may represent cases where substituted cysteines are located near the periphery of anesthetic sites, so that steric hindrance is incomplete. This possibility may be tested with different size modifiers that should show size-dependent interactions with bound anesthetic.
8.4). Comparing SCAMP and anesthetic photolabeling.
We have also formally compared SCAMP results against photolabeling at two positions with four different drugs. The two approaches show 100% agreement for the eight residue-drug pairs we examined, a correlation that is statistically significant and strengthened when other photolabeled residues studied with SCAMP are added. However, in one case, β3H267, SCAMP disagrees with photolabeling, reminding us that both approaches are based on structural variants of either the receptor or the ligand, and therefore potentially inaccurate [55]. The preponderance of evidence supports SCAMP as a technique that is useful both for confirming photolabeling results and for testing potential drug contacts that have not been photolabeled. Indeed, SCAMP experiments have substantially expanded the list of anesthetic contact residues in GABAA receptors beyond those based on photolabeling alone.
Acknowledgments
Funding Sources: This work was supported by the National Institutes of Health (R01-GM089745 and P01-GM058448).
Abbreviations:
- SCAM
Substituted Cysteine Accessibility Method.
- SCAMP
Substituted Cysteine Modification-Protection.
- GABAA
gamma-aminobutyric acid type A.
References
- [1].Miller KW, Firestone LL, Forman SA, General anesthetic and specific effects of ethanol on acetylcholine receptors, Ann.N.Y.Acad.Sci 492 (1987) 71–87. [DOI] [PubMed] [Google Scholar]
- [2].Dilger JP, Brett RS, Lesko LA, Effects of isoflurane on acetylcholine receptor channels. 1. Single-channel currents, Mol Pharmacol 41(1) (1992) 127–33. [PubMed] [Google Scholar]
- [3].Dilger JP, Brett RS, Mody HI, The effects of isoflurane on acetylcholine receptor channels.: 2. Currents elicited by rapid perfusion of acetylcholine, Mol Pharmacol 44(5) (1993) 1056–63. [PubMed] [Google Scholar]
- [4].Harrison NL, Kugler JL, Jones MV, Greenblatt EP, Pritchett DB, Positive modulation of human gamma-aminobutyric acid type A and glycine receptors by the inhalation anesthetic isoflurane, Mol Pharmacol 44(3) (1993) 628–32. [PubMed] [Google Scholar]
- [5].Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F, Zaugg M, Vogt KE, Ledermann B, Antkowiak B, Rudolph U, General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta3 subunit, FASEB J 17(2) (2003) 250–2. [DOI] [PubMed] [Google Scholar]
- [6].Nury H, Van Renterghem C, Weng Y, Tran A, Baaden M, Dufresne V, Changeux JP, Sonner JM, Delarue M, Corringer PJ, X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel, Nature 469(7330) (2011) 428–31. [DOI] [PubMed] [Google Scholar]
- [7].Pan J, Chen Q, Willenbring D, Mowrey D, Kong XP, Cohen A, Divito CB, Xu Y, Tang P, Structure of the pentameric ligand-gated ion channel GLIC bound with anesthetic ketamine, Structure 20(9) (2012) 1463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Spurny R, Billen B, Howard RJ, Brams M, Debaveye S, Price KL, Weston DA, Strelkov SV, Tytgat J, Bertrand S, Bertrand D, Lummis SC, Ulens C, Multisite binding of a general anesthetic to the prokaryotic pentameric Erwinia chrysanthemi ligand-gated ion channel (ELIC), J Biol Chem 288(12) (2013) 8355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Laurent B, Murail S, Shahsavar A, Sauguet L, Delarue M, Baaden M, Sites of Anesthetic Inhibitory Action on a Cationic Ligand-Gated Ion Channel, Structure 24(4) (2016) 595–605. [DOI] [PubMed] [Google Scholar]
- [10].Fourati Z, Ruza RR, Laverty D, Drege E, Delarue-Cochin S, Joseph D, Koehl P, Smart T, Delarue M, Barbiturates Bind in the GLIC Ion Channel Pore and Cause Inhibition by Stabilizing a Closed State, J Biol Chem 292(5) (2017) 1550–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Stuhmer W, Electrophysiological recording from Xenopus oocytes, Methods Enzymol 207 (1992) 319–39. [DOI] [PubMed] [Google Scholar]
- [12].Shu HJ, Bracamontes J, Taylor A, Wu K, Eaton MM, Akk G, Manion B, Evers AS, Krishnan K, Covey DF, Zorumski CF, Steinbach JH, Mennerick S, Characteristics of concatemeric GABA(A) receptors containing alpha4/delta subunits expressed in Xenopus oocytes, Br J Pharmacol 165(7) (2011) 2228–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lewis TM, Harkness PC, Sivilotti LG, Colquhoun D, Millar NS, The ion channel properties of a rat recombinant neuronal nicotinic receptor are dependent on the host cell type., J.Physiol 505 (1997) 299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Brett RS, Dilger JP, Adams PR, Lancaster B, A method for the rapid exchange of solutions bathing excised membrane patches, Biophys.J 50(5) (1986) 987–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Forman SA, A hydrophobic photolabel inhibits nicotinic acetylcholine receptors via open-channel block following a slow step., Biochemistry 38 (1999) 14559–14564. [DOI] [PubMed] [Google Scholar]
- [16].Akk G, Shu HJ, Wang C, Steinbach JH, Zorumski CF, Covey DF, Mennerick S, Neurosteroid access to the GABAA receptor, J Neurosci 25(50) (2005) 11605–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD, Finn SE, Mascia MP, Valenzuela CF, Hanson KK, Greenblatt EP, Harris RA, Harrison NL, Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors, Nature 389 (1997) 385–389. [DOI] [PubMed] [Google Scholar]
- [18].Hill-Venning C, Belelli D, Peters JA, Lambert JJ, Subunit-dependent interaction of the general anaesthetic etomidate with the gamma-aminobutyric acid type A receptor, Br J Pharmacol 120 (1997) 749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Belelli D, Lambert JJ, Peters JA, Wafford K, Whiting PJ, The interaction of the general anesthetic etomidate with the gamma-aminobutyric acid type A receptor is influenced by a single amino acid, Proc Natl Acad Sci U S A 94(20) (1997) 11031–11036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Serafini R, Bracamontes J, Steinbach JH, Structural domains of the human GABAA receptor beta3 subunit involved in the actions of pentobarbital, Journal of Physiology 524 Pt 3 (2000) 649–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Feng HJ, Macdonald RL, Barbiturates require the N terminus and first transmembrane domain of the delta subunit for enhancement of alpha1beta3delta GABAA receptor currents, J Biol Chem 285(31) (2010) 23614–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Feng HJ, Jounaidi Y, Haburcak M, Yang X, Forman SA, Etomidate Produces Similar Allosteric Modulation in alpha-1/beta-3/delta and alpha-1/beta-3/gamma2L GABA-A Receptors. , Br J Pharmacol 171(3) (2014) 789–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Leonard RJ, Labarca CG, Charnet P, Davidson N, Lester HA, Evidence that the M2 membrane-spanning region lines the ion channel pore of the nicotinic receptor, Science 242(4885) (1988) 1578–81. [DOI] [PubMed] [Google Scholar]
- [24].Dilger JP, Vidal AM, Mody HI, Liu Y, Evidence for direct actions of general anesthetics on an ion channel protein. A new look at a unified mechanism of action, Anesthesiology 81(2) (1994) 431–42. [DOI] [PubMed] [Google Scholar]
- [25].Forman SA, Miller KW, Yellen G, A discrete site for general anesthetics on a postsynaptic receptor, Mol.Pharm 48(4) (1995) 574–581. [PubMed] [Google Scholar]
- [26].Forman SA, Homologous mutations on different subunits cause unequal but additive effects on n-alcohol block in the nicotinic receptor pore., Biophys.J 72 (1997) 2170–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhou QL, Zhou Q, Forman SA, The n-alcohol site in the nicotinic receptor pore is a hydrophobic patch, Biochemistry 39(48) (2000) 14920–6. [DOI] [PubMed] [Google Scholar]
- [28].Forman SA, Zhou Q, Novel modulation of a nicotinic receptor channel mutant reveals that the open state is stabilized by ethanol, Mol.Pharm 55(1) (1999) 102–108. [DOI] [PubMed] [Google Scholar]
- [29].Krasowski MD, Koltchine VV, Rick CE, Ye Q, Finn SE, Harrison NL, Propofol and other intravenous anesthetics have sites of action on the gamma-aminobutyric acid type A receptor distinct from that for isoflurane, Mol Pharmacol 53 (1998) 530–538. [DOI] [PubMed] [Google Scholar]
- [30].Krasowski MD, Nishikawa K, Nikolaeva N, Lin A, Harrison NL, Methionine 286 in transmembrane domain 3 of the GABAA receptor beta subunit controls a binding cavity for propofol and other alkylphenol general anesthetics, Neuropharmacology 41(8) (2001) 952–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Koltchine VV, Finn SE, Jenkins A, Nikolaeva N, Lin A, Harrison NL, Agonist gating and isoflurane potentiation in the human gamma- aminobutyric acid type A receptor determined by the volume of a second transmembrane domain residue, Mol.Pharmacol 56 (1999) 1087–1093. [DOI] [PubMed] [Google Scholar]
- [32].Jenkins A, Andreasen A, Trudell JR, Harrison NL, Tryptophan scanning mutagenesis in TM4 of the GABA(A) receptor alpha1 subunit: implications for modulation by inhaled anesthetics and ion channel structure, Neuropharmacology 43(4) (2002) 669–78. [DOI] [PubMed] [Google Scholar]
- [33].Ueno S, Lin A, Nikolaeva N, Trudell JR, Mihic SJ, Harris RA, Harrison NL, Tryptophan scanning mutagenesis in TM2 of the GABA(A) receptor alpha subunit: effects on channel gating and regulation by ethanol, Br J Pharmacol 131(2) (2000) 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Maldifassi MC, Baur R, Sigel E, Functional sites involved in modulation of the GABA receptor channel by the intravenous anesthetics propofol, etomidate and pentobarbital, Neuropharmacology 105 (2016) 207–214. [DOI] [PubMed] [Google Scholar]
- [35].Colquhoun D, Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors, Br J Pharmacol 125(5) (1998) 924–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rüsch D, Zhong H, Forman SA, Gating allosterism at a single class of etomidate sites on alpha1beta2gamma2L GABA-A receptors accounts for both direct activation and agonist modulation., J Biol Chem 279(20) (2004) 20982–92. [DOI] [PubMed] [Google Scholar]
- [37].Rüsch D, Neumann E, Wulf H, Forman SA, An Allosteric Coagonist Model for Propofol Effects on alpha1beta2gamma2L gamma-Aminobutyric Acid Type A Receptors, Anesthesiology 116(1) (2012) 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ziemba AM, Forman SA, Correction for Inhibition Leads to an Allosteric Co-Agonist Model for Pentobarbital Modulation and Activation of alpha1beta3gamma2L GABAA Receptors, PloS one 11(4) (2016) e0154031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Li GD, Chiara DC, Sawyer GW, Husain SS, Olsen RW, Cohen JB, Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog, J Neurosci 26(45) (2006) 11599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Stewart DS, Desai R, Cheng Q, Liu A, Forman SA, Tryptophan mutations at azi-etomidate photo-incorporation sites on α1 or β2 subunits enhance GABAA receptor gating and reduce etomidate modulation Mol Pharmacol 74(6) (2008) 1687–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Desai R, Rüsch D, Forman SA, Gamma-amino butyric acid type A receptor mutations at beta2N265 alter etomidate efficacy while preserving basal and agonist-dependent activity, Anesthesiology 111(4) (2009) 774–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nourmahnad A, Stern AT, Hotta M, Stewart DS, Ziemba AM, Szabo A, Forman SA, Tryptophan and Cysteine Mutations in M1 Helices of α1β3γ2L γ-Aminobutyric Acid Type A Receptors Indicate Distinct Intersubunit Sites for Four Intravenous Anesthetics and One Orphan Site, Anesthesiology 125(6) (2016) 1144–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Akabas MH, Kaufmann C, Archdeacon P, Karlin A, Identification of acetylcholine receptor channel-lining residues in the entire M2 segment of the alpha subunit, Neuron 13(4) (1994) 919–27. [DOI] [PubMed] [Google Scholar]
- [44].Xu M, Akabas MH, Identification of channel-lining residues in the M2 membrane-spanning segment of the GABA(A) receptor alpha1 subunit, J Gen Physiol 107(2) (1996) 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Williams DB, Akabas MH, Gamma-aminobutyric acid increases the water accessibility of M3 Membrane-spanning segment residues in GABA-A receptors., Biophysical Journal 77 (1999) 2563–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Goren EN, Reeves DC, Akabas MH, Loose protein packing around the extracellular half of the GABA(A) receptor beta1 subunit M2 channel-lining segment, J Biol Chem 279(12) (2004) 11198–205. [DOI] [PubMed] [Google Scholar]
- [47].Jansen M, Akabas MH, State-dependent cross-linking of the M2 and M3 segments: Functional basis for the alignment of GABAA and acetylcholine receptor M3 segments, J Neurosci 26(17) (2006) 4492–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bali M, Jansen M, Akabas MH, GABA-induced intersubunit conformational movement in the GABAA receptor alpha1M1-beta2M3 transmembrane subunit interface: Experimental basis for homology modeling of an intravenous anesthetic binding site, J Neurosci 29(10) (2009) 3083–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bali M, Akabas MH, Defining the propofol binding site location on the GABAA receptor, Mol Pharmacol 65(1) (2004) 68–76. [DOI] [PubMed] [Google Scholar]
- [50].Forman SA, Miller KW, Mapping General Anesthetic Sites in Heteromeric gamma-Aminobutyric Acid Type A Receptors Reveals a Potential For Targeting Receptor Subtypes, Anesth Analg 123(5) (2016) 1263–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Stewart DS, Pierce DW, Hotta M, Stern AT, Forman SA, Mutations at Beta N265 in Gamma-Aminobutyric Acid Type A Receptors Alter Both Binding and Efficacy of Potent Anesthetics, PloS one October 27;9(10):e111470 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Stewart DS, Hotta M, Desai R, Forman SA, State-Dependent Etomidate Occupancy of Its Allosteric Agonist Sites Measured in a Cysteine-Substituted GABAA Receptor, Mol Pharmacol 83(6) (2013) 1200–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Stewart DS, Hotta M, Li GD, Desai R, Chiara DC, Olsen RW, Forman SA, Cysteine Substitutions Define Etomidate Binding and Gating Linkages in the alpha-M1 Domain of gamma-Aminobutyric Acid Type A (GABAA) Receptors, J Biol Chem 288(42) (2013) 30373–30386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Borghese CM, Hicks JA, Lapid DJ, Trudell JR, Harris RA, GABA(A) receptor transmembrane amino acids are critical for alcohol action: disulfide cross-linking and alkyl methanethiosulfonate labeling reveal relative location of binding sites, J Neurochem 128(3) (2014) 363–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Stern AT, Forman SA, A Cysteine Substitution Probes beta3H267 Interactions with Propofol and Other Potent Anesthetics in alpha1beta3gamma2L gamma-Aminobutyric Acid Type A Receptors, Anesthesiology 124(1) (2016) 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]