

Summary
Alopecia with mental retardation syndrome (APMR) is a rare autosomal recessive disorder characterized by total or partial absence of hair from the scalp and other parts of the body and associated with mental retardation. Previously, we have reported the mapping of two alopecia and mental retardation genes (APMR1 and APMR2) on human chromosome 3. In the present study, after excluding both of these loci through linkage analysis, a whole genome scan was performed by genotyping 396 polymorphic microsatellite markers located on 22 autosomes and the X and Y chromosomes. A disease locus was mapped to a 10.9 cM region, flanked by markers D18S866 and D18S811, on chromosome 18q11.2–q12.2. A maximum two-point LOD score of 3.03 at θ = 0.0 was obtained with marker D18S1102. Multipoint linkage analysis resulted in maximum LOD scores of 4.03 with several markers in the candidate region. According to the Rutgers combined linkage-physical map of the human genome (build 36) this region covers 12.17 Mb. DNA sequence analysis of nine candidate genes including DSC3, DSC1, DSG1, DSG4, DSG3, ZNF397, ZNF271, ZNF24 and ZNF396 did not reveal any sequence variants in the affected individuals of the family presented here.
Keywords: Alopecia with mental retardation syndrome (APMR3), 18q11.2–q12.2, Candidate genes
Introduction
Alopecia or hair loss in humans occurs either as an isolated abnormality or in association with defects in ectodermal and other structures. To date, several genetic defects for isolated forms of alopecia have been mapped. These include hypotrichosis simplex (MIM 605389) at 6p21.3, Marie Unna hereditary hypotrichosis (MIM 146550) at 8p21, congenital atrichia (MIM 203655) at 8p21, localized hereditary hypotrichosis (LAH, MIM 607903) at 18q12.1, and autosomal recessive hereditary hypotrichosis (AH, MIM 609167) at 3q26.33. Four of these conditions (hypotrichosis simplex, congenital atrichia, LAH and AH) have been reported to result from mutations in the corneodesmosin (CDSN, MIM 602593), hairless (HR, MIM 225060), desmoglein 4 (DSG4, MIM 607892) and lipase H (LIPH, MIM 607365) genes, respectively (Levy-Nissenbaum et al. 2003; Ahmad et al. 1998; John et al. 2005, 2006b; Kljuic et al. 2003; Rafique et al. 2003; Kazantseva et al. 2006).
Syndromic forms of alopecia show association of loss of hair with a variety of clinical conditions including lack or dysgenesis of teeth, nails and sweat glands. Alopecia with mental retardation syndrome [APMR, MIM 203650] is a rare autosomal recessive form of alopecia (Benke & Hajianpour, 1985; Baraitser et al. 1983; John et al. 2006a; Perniola et al. 1980; Shokeir, 1977; Vogt et al. 1988; Wali et al. 2006). Affected individuals with APMR syndrome show loss of hair on the scalp, absence of eyebrows, eyelashes, axillary and pubic hair, and mild to severe mental retardation. Recently, we have reported the mapping of two different loci for alopecia and mental retardation syndrome on chromosome 3. John et al. (2006a) mapped the APMR1 locus to human chromo-some 3q26.33–q27.3 in a family with complete hair loss and severe mental retardation (IQ from 25–30). The second locus for alopecia and mental retardation (APMR2) was mapped to chromosome 3q26.2–q26.31 (Wali et al. 2006) in a family with total alopecia and mild to moderate mental retardation (IQ from 53–61).
In the present study, we report the mapping of a third locus for alopecia and mental retardation syndrome (APMR3) in a large family from a remote region of Pakistan. The gene region has been mapped to chromosome 18q11.2–q12.2, and is flanked by markers D18S866 and D18S811. Analysis of nine candidate genes has failed to reveal any pathogenic mutation in the affected individuals of the family.
Materials and Methods
Human Subjects
Before the onset of the study, approval was obtained from the Quaid-i-Azam University Institutional Review Board (IRB). Informed consent was obtained from all family members who participated in the study. A consanguineous family segregating an autosomal recessive form of alopecia and mental retardation syndrome was studied, in which two males and two females were affected. The pedigree (Figure 1) provided convincing evidence of an autosomal recessive mode of inheritance. All patients showed typical features of hereditary APMR syndrome. All of the affected individuals underwent examination at the Children’s Hospital of Lahore, Pakistan. IQ tests were performed by the Wechsler method of calculating IQ values.
Figure 1.
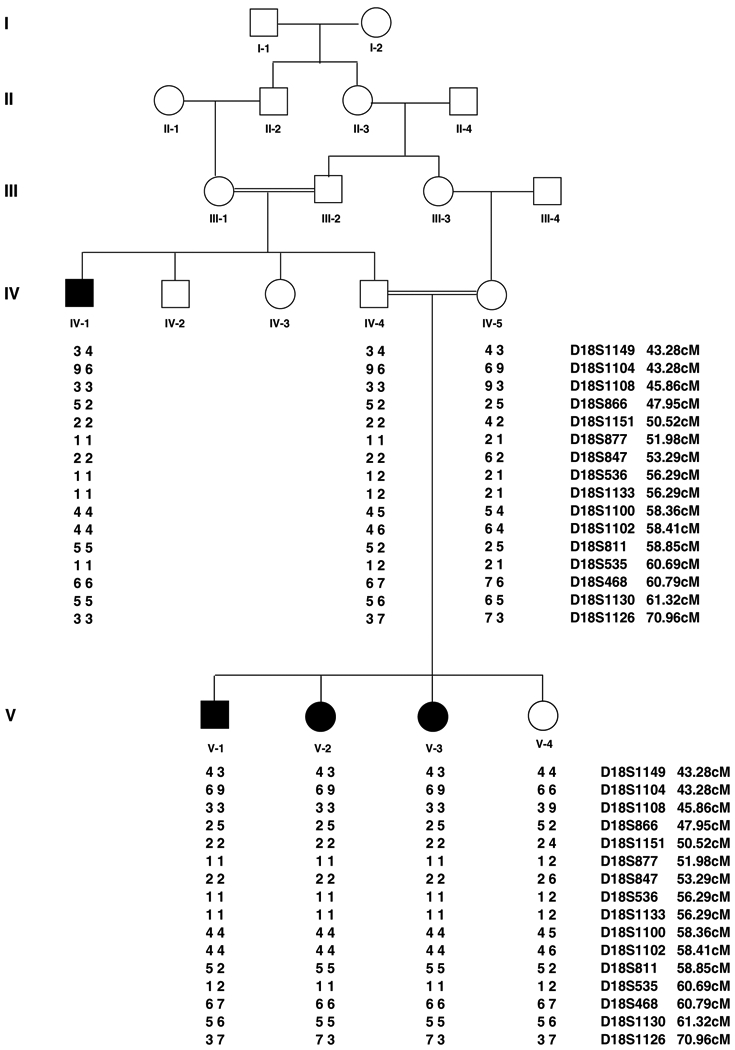
Pedigree structure and haplotype analysis of a five generation family which segregates APMR3. Affected males and females are indicated by filled squares and circles, respectively. Markers are ordered from centromeric (top) to telomeric (bottom) and the disease associated haplotypes are shown beneath each symbol. Haplotype showing a region of homozygosity flanked by markers D18S866 and D18S811. Haplotypes were generated using SIMWALK2.
Extraction of Genomic DNA and Genotyping
Venous blood samples were collected from a total of seven individuals, including four individuals affected with APMR syndrome. Genomic DNA was extracted from whole blood following a standard protocol (Grimberg et al. 1989), quantified by spectrophotomet ric readings at optical density 260nm, and diluted to 40ng/μl for polymerase chain reaction (PCR) amplification. PCR amplification of microsatellite markers was performed according to standard procedures in a total volume of 25 μl, containing 40 ng genomic DNA, 20 pmol of each primer, 200 μM of each deoxynucleo-side triphosphate (dNTP), 1 U of Taq DNA polymerase and 2.5 μl reaction buffer (MBI Fermentas York, UK). The thermal cycling conditions used were 95°C for 1 min, followed by 35 cycles of 95°C for 1 min, 57°C for 1 min, 72°C for 1 min, and a final extention at 72°C for 10 minutes. PCR products were resolved on an 8% nondenaturing polyacrylmide gel and genotypes were assigned by visual inspection.
The family was tested first for linkage to previously identified APMR loci, by using microsatellite markers which are tightly linked to alopecia and mental retardation syndrome (APMR1) at 3q26.33 (D3S2314, D3S3578, D3S3583, D3S3592, D3S1530, D3S1617, D3S1602, D3S1262) and (APMR2) at 3q26.2 (D3S3656, D3S2433, D3S3053, D3S1556, D3S2425, D3S2421, D3S2309).
After exclusion of known loci a whole genome scan was performed using 396 short tandem repeat (STR) microsatellite markers from the Linkage Mapping Set (Invitrogen Co, San Diego, California, U.S.A.). These markers were spaced approximately 10 cM apart and are located on the 22 autosomes and the X and Y chromosomes.
Linkage Analysis
The National Center for Biotechnology Information (NCBI) Human Genome Build 36 sequence-based physical map was used to determine the order of the genome scan and fine mapping markers (International Human Genome Sequence Consortium, 2001). The Rutgers combined linkage-physical map of the human genome (Kong et al. 2004) was utilized for genetic map distances in linkage analysis for the fine mapping and genome scan markers. PEDCHECK (O’Connell & Weeks, 1998) was used to identify Mendelian inconsistencies. Haplotypes were constructed using SIMWALK2 (Sobel & Lange, 1996; Weeks et al. 1995). Two-point linkage analysis was carried out using the MLINK program of the FASTLINK computer package for the genome scan and fine mapping marker loci (Cottingham et al. 1993). Multipoint linkage analysis was performed using ALLEGRO (Gudbjartsson et al. 2005). An autosomal recessive mode of inheritance with complete penetrance and a disease allele frequency of 0.001 was used for the analysis. Equal allele frequencies were used for the analysis in two-point and multipoint analysis for fine mapping markers, because it was not possible to estimate allele frequencies from the founders since the markers were only genotyped in this family.
Sequencing of Candidate Genes
To screen for mutations in the nine candidate genes, including DSC3 (MIM 600271), DSC1 (MIM 125643), DSG1 (MIM 125670), DSG4 (MIM 607892), DSG3 (MIM 169615), ZNF397 (MIM 609601), ZNF271 (MIM 604754), ZNF24 (MIM 194534) and ZNF396 (MIM 609600), exons and splice junctions were PCR amplified from the genomic DNA of two affected and one normal individual, then purified by the Rapid PCR amplification system (Marligen Bio-sciences, Ijamsville, MD, USA). Sequencing was performed using a Big Dye Terminator v3.1 cycle sequencing kit, together with an ABI-Prism 310 Automated DNA Sequencer (Applera Corporation, Cleveland, OH, USA).
Results
Clinical Findings
All the four affected individuals in the family were born with total alopecia. At birth, hair was completely absent from the scalp. The affected individuals were devoid of eyebrows, eyelashes, axillary and pubic hair. However, these individuals showed normal teeth, nails, sweating and hearing. In addition all the affected individuals in the APMR family were severely mentally retarded (IQ from 25–30) and were described as ‘difficult to handle’ (Figure 2). Head circumferences of the affected individuals (51 ±/−1 cm) were comparable to those of normal individuals. No other clinical signs, including seizures, were detected in any of the affected individuals. Heterozygous carriers had normal hair, and were clinically indistinguishable from genotypically normal individuals.
Figure 2.

Clinical features of the APMR3 phenotype. An affected individual (V-3) showing complete absence of hair on scalp and missing eyebrows and eyelashes.
Localization of APMR3 to 18q
To identify the underlying APMR locus in our family we performed haplotype analysis for the microsatellite markers linked to the previously reported APMR1 (John et al. 2006a) and APMR2 (Wali et al. 2006) loci. After exclusion of these two loci from linkage, a genome scan was carried out using the DNA samples of four affected individuals (IV–1, V–1, V–2, V–3). In the course of screening 396 markers we found preliminary evidence for linkage with marker D18S1133 (56.29 cM) at 18q12.3. Upon genotyping all family members this marker was found to be informative; the four affected members of the family (IV–1, V–1, V–2, V–3) were homozygous and three unaffected members (IV–4, IV–5, V–4) were heterozygous.
In order to fine map the locus, 23 additional markers were selected from the Rutgers combined linkage-physical map of the human genome (Kong et al. 2004). Thirteen markers (D18S1149, D18S1104, D18S1108, D18S866, D18S1151, D18S877, D18S965, D18S847, D18S36, D18S457, D18S456, D18S1124, D18S536) were proximal to marker D18S1133, while ten markers (D18S1162, D18S1100, D18S1102, D18S811, D5S535, D18S1096, D18S468, D18S1130, D18S1145, D18S1126) were distal to D18S1133. Eight of the markers (D18S965, D18S36, D18S457, D18S456, D18S1124, D18S1162, D18S1096, D18S1145) were un-informative in the present family, and therefore not included in the analysis. Analysis of the marker geno-types within this region with PEDCHECK (O’Connell & Weeks, 1998) did not elucidate any genotyping errors. After genotyping these markers the data was analyzed using two-point and multipoint linkage analyses. The maximum two-point LOD score of 3.03 (θ = 0) was obtained with marker D18S1102 (Table 1). Another marker, D18S1100, which is proximal to D18S1102, generated a LOD score of 2.94. The maximum multi-point LOD score of 4.03 was obtained with several markers, including D18S536, D18S1133, D18S1100 and D18S1102, which supports linkage to 18q11.2–q12.2. The one unit support interval for the APMR3 locus is flanked by the markers D18S866 (47.95 cM) and D18S811 (58.85 cM). This defines a genetic interval of 10.9 cM on the Rutgers combined linkage-physical map of the human genome (Kong et al. 2004), comprising 12.17 Mb according to build 36 of the sequence-based physical map (International Human Genome Sequence Consortium, 2001).
Table 1.
Two-point LOD score results between the APMR3 locus and chromosome 18 markers. Also displayed are the genetic and sequence-based physical map distances. Markers displayed in bold flank the region of homozygosity
| Marker | cMa | Physical locationb | LOD score at recombination fraction θ = | |||||
|---|---|---|---|---|---|---|---|---|
| 0.00 | 0.01 | 0.03 | 0.05 | 0.10 | 0.30 | |||
| D18S1149 | 43.28 | 17,315,222 | −2.26 | −0.69 | −0.27 | −0.11 | 0.03 | 0.00 |
| D18S1104 | 43.28 | 17,413,680 | −2.35 | −0.76 | −0.32 | −0.14 | 0.03 | 0.03 |
| D18S1108 | 45.86 | 20,351,017 | 1.66 | 1.65 | 1.61 | 1.57 | 1.43 | 0.68 |
| D18S866 | 47.95 | 21,624,595 | −2.57 | −0.99 | −0.54 | −0.35 | −0.14 | −0.00 |
| D18S1151 | 50.52 | 23,435,752 | 1.50 | 1.47 | 1.41 | 1.35 | 1.20 | 0.53 |
| D18S877 | 51.98 | 24,979,188 | 1.23 | 1.20 | 1.14 | 1.09 | 0.95 | 0.41 |
| D18S847 | 53.29 | 25,957,311 | 1.60 | 1.58 | 1.53 | 1.47 | 1.32 | 0.60 |
| DSC3 | N/A | 26,850,908 | ||||||
| DSC2 | N/A | 26,918,190 | ||||||
| DSC1 | N/A | 26,980,015 | ||||||
| DSG1 | N/A | 27,171,251 | ||||||
| DSG4 | N/A | 27,229,307 | ||||||
| DSG3 | N/A | 27,296,115 | ||||||
| DSG2 | N/A | 27,356,528 | ||||||
| D18S536 | 56.29 | 29,842,284 | 2.35 | 2.30 | 2.18 | 2.07 | 1.77 | 0.63 |
| D18S1133 | 56.29 | 30,352,603 | 2.35 | 2.30 | 2.18 | 2.07 | 1.77 | 0.63 |
| ZNF397 | N/A | 31,083,686 | ||||||
| ZNF271 | N/A | 31,133,185 | ||||||
| ZNF24 | N/A | 31,174,180 | ||||||
| ZNF396 | N/A | 31,205,979 | ||||||
| D18S1100 | 58.36 | 33,176,142 | 2.94 | 2.87 | 2.73 | 2.60 | 2.25 | 0.84 |
| D18S1102 | 58.41 | 33,177,191 | 3.03 | 2.96 | 2.82 | 2.68 | 2.33 | 0.88 |
| D18S811 | 58.85 | 33,797,091 | -Infinity | 0.87 | 1.22 | 1.32 | 1.29 | 0.51 |
| D18S535 | 60.69 | 36,402,787 | -Infinity | 0.30 | 0.67 | 0.79 | 0.82 | 0.32 |
| D18S468 | 60.79 | 36,483,496 | -Infinity | 1.04 | 1.39 | 1.48 | 1.44 | 0.59 |
| D18S1130 | 61.32 | 37,027,637 | -Infinity | 0.97 | 1.31 | 1.41 | 1.38 | 0.55 |
| D18S1126 | 70.96 | 45,610,471 | −2.46 | −0.32 | 0.07 | 0.22 | 0.35 | 0.20 |
Average-sex distance in cM according to Rutgers combined linkage-physical human genome map (Kong et al. 2004).
The physical position is based on build 36 of the human genome (International Human Genome Sequence Consortium, 2001).
Haplotypes were constructed using SIMWALK2 (Sobel & Lange, 1996; Weeks et al. 1995) to determine the linkage interval. The proximal boundary of the interval was defined by a recombination event between markers D18S866 and D18S1151 as observed in the affected individuals (V–1, V–2, V–3). Another recombination event was observed between markers D18S1102 and D18S811 in individual (V–1), which defined the distal boundary of this interval. The region of homozygosity was the same as the one-unit support interval and flanked by markers D18S866 (centromeric) and D18S811 (telomeric).
Out of seventeen genes located within the APMR3 interval of 12.17 Mb, nine genes (DSC3 (MIM 600271), DSC1 (MIM 125643), DSG1 (MIM 125670), DSG3 (MIM 169615), DSG4 (MIM 607892), ZNF397 (MIM 609601), ZNF271 (MIM 604754), ZNF24 (MIM 194534), ZNF396 (MIM 609600)) were screened in two affecteds and one normal individual from the family, and were found to be negative for functional sequence variants.
Discussion
The linkage data presented here map the third novel alopecia and mental retardation syndrome (APMR3) locus to chromosome 18q11.2–q12.2 in a five generation consanguineous Pakistani family. According to Rutgers combined linkage-physical map of the human genome the APMR3 interval spans 10.9 cM, and comprises 12.17 Mb according to build 36 of the sequence-based physical map. Previously we reported the localization of two alopecia and mental retardation syndrome loci, APMR1 and APMR2 on chromosome 3q26.33–q27.3 (John et al. 2006a) and 3q26.2–q26.31 (Wali et al. 2006), respectively. The APMR1 linkage interval overlaps with the autosomal recessive hair loss (AH) locus (Aslam et al. 2004). Recently, Kazantseva et al. (2006) have reported the identification of a deletion mutation in the lipase H (LIPH) gene, located in the AH candidate region on chromosome 3q27. Clinical features of the affecteds individuals with APMR1, APMR2 and APMR3 include total alopecia and mental retardation. However, affected individuals linked to APMR1 and APMR3 are severely mentally retarded (IQ from 25–30), while individuals linked to APMR2 are mildly to moderately retarded (IQ from 53–61).
Through a UCSC Genome Browser database search (Karolchik et al. 2003) we have identified several known and predicted genes and expressed sequence tags (ESTs) in the linkage interval between markers D18S866 and D18S811. Among others, this interval contains four desmogleins, three desmocollins and four zinc finger protein encoding genes. Desmogleins are expressed in the inner epithelial layers of the hair follicles, where their functions appear to be crucial during differentiation of the hair follicle layers (Kljuic et al. 2003). Mutations in one member of the desmoglein family of genes, DSG4, have recently been shown to be responsible for localized autosomal recessive hypotrichosis (LAH, Kljuic et al. 2003; Rafiq et al. 2004; John et al. 2006b). These patients suffer from hypotrichosis limited to the scalp, chest, arms and legs (Kljuic et al. 2003; Rafique et al., 2003). Their eyebrows and eyelashes are sparse, with normal beard hair in the male affected individuals (Rafique et al. 2003). Kljuic et al. (2003) demonstrated that human LAH is allelic with the lanceloate hair (lah) mouse. Mutations in mouse Dsg3 have been shown to result in a balding phenotype, characterized by cyclical hair loss (Koch et al. 1997; Pulkkinen et al. 2002).
In the present study, nine genes including three desmoglein genes (DSG1, DSG3, DSG4) and two desmocollin genes (DSC1, DSC3), and four other genes encoding zinc finger proteins (ZNF397, ZNF271, ZNF24, ZNF396) which function as transcriptional repressors, were sequenced in two affecteds and one normal individual from the APMR3 family, but no sequence variants were identified. In addition to these nine genes the linkage interval of APMR3 contains eight other well-characterized genes. Two of these genes, desmocollin 2 (DSC2, MIM 125645) and desmoglein 2 (DSG2, MIM 125671), are components of inter-cellular desmosome junctions and were previously reported to be involved in arrhythmogenic right ventricular dysplasia (Syrris et al. 2006) and arrhythmogenic right ventricular dysplasia/cardiomyopathy (Awad et al. 2006), respectively. Therefore, these two genes were not screened for mutations in the present study. The APMR3 linkage interval contains six other genes, nucleolar protein 4 (NOL4, MIM 603577), dystrobrerin alpha (DTNA, MIM 601239), microtubule-associated protein RP/EB family member 2 (MAPRE2, MIM 605789), polypeptide N-acetylgalactosaminyltransferase 1 (GALNT1, MIM 602273), solute carrier family 39 member 6 (SLC39A6, MIM 608731), formin homology 2 domain containing 3 (FHOD3, MIM 609691). However functions assigned to these genes make them unsuitable to be considered as candidates for the APMR3 phenotype.
In conclusion, we report the localization of a novel locus for alopecia with mental retardation syndrome (APMR3) in a 10.9 cM region on chromosome 18q11.2–q12.2. The mapping of the APMR3 locus further confirms the heterogeneity underlying autosomal recessive forms of the alopecia with mental retardation syndrome.
Acknowledgments
We would like to thank the patients and other members of the family for participation in this study. The work presented here was funded by the Higher Education Commission (HEC), Islamabad, Pakistan. Abdul Wali was supported by HEC Indigenous PhD fellowship program.
References
- Ahmad W, Faiyaz ul Haque M, Brancolin V, Tsou HC, ul Haque S, Lam H, Aita VM, Owen J, de Blaquiere M, Frank J, Cserhalmi-Friedman PB, Leask A, McGrath JA, Peacocke M, Ahmad M, Ott J, Chris-tiano AM (1998) Alopecia universalis associated with a mutation in the human hairless gene. Science 279, 720–724. [DOI] [PubMed] [Google Scholar]
- Aslam M, Chahrour MH, Razzaq A, Haque S, Yan K, Leal SM, Ahmad W (2004) A novel locus for auto-somal recessive form of hypotrichosis maps to chromosome 3q26.33–q27.3. J Med Genet 41, 849–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad MM, Dalal D, Cho E, Amat-Alarcon N, James C, Tichnell C, Tucker A, Russell SD, Bluemke DA, Dietz HC, Calkins H, Judge DP (2006) DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Hum Genet 79, 136–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraitser M, Carter CO, Brett EM (1983) A new alopecia/mental retardation syndrome. J Med Genet 20, 64–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benke PJ, Hajianpour MJ (1985) Alopecia universalis-mental retardation is an autosomal recessive syndrome disorder. Am J Hum Genet 37, 44. [Google Scholar]
- Cottingham RW jr., Jonasson K, Frigge ML, Kong A (1993) Faster sequential genetic linkage computations. Am J Hum Genet 53, 252–263. [PMC free article] [PubMed] [Google Scholar]
- Grimberg J, Nawoschik S, Belluscio L, McKee R, Turck A, Eisenberg A (1989) A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res 17, 8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudbjartsson DF, Thorvaldsson T, Kong A, Gunnarsson G, Ingolfsdottir A (2005) Allegro version 2. Nat Genet 37, 1015–1016. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequence Consortium. (2001) Initial sequence and analysis of the human genome. Nature 409, 860–921. [DOI] [PubMed] [Google Scholar]
- John P, Ali G, Chishti MS, Naqvi SM, Leal SM, Ahmad W (2006a) Localization of a novel locus for alopecia with mental retardation syndrome to chromosome 3q26.33–q27.3. Hum Genet 118, 665–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John P, Aslam M, Rafiq MA, Amin-ud-din M, Haque S, Ahmad W (2005) Atrichia with papular lesions in two Pakistani consanguineous families resulting from mutations in the human hairless gene. Arch Dermatol Res 297, 226–230. [DOI] [PubMed] [Google Scholar]
- John P, Tariq M, Arshad Rafiq. M., Amin-ud-din M, Muhammad D, Waheed I, Ansar M, Ahmad W (2006b) Recurrent intragenic deletion mutation in desmoglein 4 gene underlies autosomal recessive hypotrichosis in two Pakistani families of Balochi and Sindhi origins. Arch Dermatol Res 298, 135–137. [DOI] [PubMed] [Google Scholar]
- Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, Roskin KM, Schwart M, Sugnet CW, Thomas DJ, Weber RJ, Haussler D, Kent WJ (2003) The UCSC Genome Browser Database. Nucleic Acids Res 31, 51–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantseva A, Goltsov A, Zinchenko R, Grigorenko AP, Abrukova AV, Moliaka YK, Kirillov AG, Guo Z, Lyle S, Ginter EK, Rogaev EI (2006). Human hair growth deficiency is linked to a genetic defect in the phospholipase gene LIPH. Science 314, 982–985. [DOI] [PubMed] [Google Scholar]
- Kljuic A, Bazzi H, Sundberg JP, Martinez-Mir A, O’Shaughnessy R, Mahoney MG, Levy M, Montagutelli X, Ahmad W, Aita VM, Gordon D, Uitto J, Whiting D, Ott J, Fischer S, Gilliam TC, Jahoda CA, Morris RJ, Panteleyev AA, Nguyen VT, Christiano AM (2003) Desmoglein-4 in hair follicle differentiation and epidermal adhesion: evidence from inherited hypotrichosis and aquired pempsigus vulgaris. Cell 113, 249–260. [DOI] [PubMed] [Google Scholar]
- Koch PJ, Mahoney MG, Ishikawa H, Pulkkinen L, Uitto J, Shultz L, Murphy HF, Whitaker-Menezes D, Stanley JR (1997). Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a pheno-type similar to pemphigus vulgaris. J Cell Biol 137, 1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, Murphy K, Raj T, He C, White PS, Matise TC (2004) A combined linkage-physical map of the human genome. Am J Hum Genet 75, 1143–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy-Nissenbau E, Betz RC, Frydman M, Simon M, Lahat H, Goldman B, Bygam A, Pierick M, Hillmer AM, Jonca N, Toribio J, Kruse R, Dewald G, Cichon S, Kubisch C, Guerrin M, Serre G, Nothen NN (2003) Hypotrichosis simplex of the scalp is associated with nonsense mutations in CDSN encoding corneodesmosin. Nat Genet 34, 151–153. [DOI] [PubMed] [Google Scholar]
- O’Connell JR, Weeks DE (1998) PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 63, 259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perniola T, Krajewska G, Carnevale F, Lospalluti M (1980) Congenital alopecia, psychomotor retardation, convulsions in two sibs of a consanguineous marriage. J Inherit Metab Dis 3, 49–53. [DOI] [PubMed] [Google Scholar]
- Pulkkinen L, Choi YW, Simpson A, Montagutelli X, Sundberg J, Uitto J, Mahoney MG (2002). Loss of cell adhesion in Dsg3bal-Pas mice with homozygous deletion mutation (2079del14) in the desmoglein 3 gene. J Invest Dermatol 119, 1237–1243. [DOI] [PubMed] [Google Scholar]
- Rafiq MA, Ansar M, Mahmood M, Haque S, Faiyazul-Haque M, Leal SM, Ahmad W (2004). A Recurrent Intragenic Deletion Mutation in DSG4 Gene in Three Pakistani Families with Autosomal Recessive Hypotrichosis. J Invest Dermatol 123, 247–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafique MA, Ansar M, Jamal SM, Malik S, Sohail M, Faiyaz-Ul-Haque M, Haque S, Leal SM, Ahmad W (2003) A locus for hereditary hypotrichosis localized to human chromosome 18q21.1. Eur J Hum Genet 11, 623–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokeir MHK (1977) Universal permanent alopecia, psychomotor epilepsy, pyorrhea and mental subnormaliy. Clin Genet 11, 13–17. [DOI] [PubMed] [Google Scholar]
- Sobel E, Lange K (1996) Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet 58, 1323–1337. [PMC free article] [PubMed] [Google Scholar]
- Syrris P, Ward D, Evans A, Asimaki A, Gandjbakhch E, Sen-Chowdhry S, McKenna WJ (2006) Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet 79, 978–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt BR, Traupe H, Ham H (1988) Congenital atrichia with nail dystrophy, abnormal facies, and retarded psychomotor development in two siblings: a new autosomal recessive syndrome. Pediatr Dermatol 5, 236–242. [DOI] [PubMed] [Google Scholar]
- Wali A, John P, Gul A, Lee K, Chishti MS, Ali G, Hassan MJ, Leal SM, Ahmad W (2006) A novel locus for alopecia with mental retardation syndrome (APMR2) maps to chromosome 3q26.2–q26.31. Clin Genet 70, 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks DE, Sobel E, O’Connell JR, Lange K (1995) Computer programs for multilocus haplotyping of general pedigrees. Am J Hum Genet 56, 1506–1507. [PMC free article] [PubMed] [Google Scholar]