

Abstract
HIV infection is characterized by persistent immune activation, even in the context of suppressive antiretroviral therapy. This persistent activation, which appears to be fueled by microbial translocation from the gut resulting from HIV-related damage, is associated with deficits in immune function that in turn contribute to persistent activation. The presence of latent HIV reservoirs in lymphoid tissues also provokes immune activation in the context of immune suppression, resulting in expansion of the viral reservoir and potential viral replication, even with suppressive antiretroviral therapy. Therapeutic strategies are being devised to reduce persistent immune activation and limit the size of the HIV reservoir. This article summarizes a presentation by Daniel C. Douek, MD, PhD, at the IAS–USA continuing education program held in San Francisco, California, in March 2013.
Keywords: HIV, immune activation, persistence, gut, microbial translocation, reservoir, raltegravir intensification, lymphoid tissue
Viral infection is accompanied by an innate immune response during the acute phase of infection. For most types of viral infection, immune activation is reduced as the immune response acts to reduce viral load. In HIV infection, however, immune activation persists despite the initial decline in viral load. Persistent activation is observed in numerous components of the innate immune system, including cells (eg, activated phenotypes of macrophages and dendritic cells), cytokines and chemokines (tumor necrosis factor, interleukin [IL]-1, IL-6, IL-8, IL-15, and IL-10), acute phase proteins (serum amyloid A, C-reactive protein), elements of the coagulation cascade (D-dimers, tissue factor), elements of fibrosis (matrix metalloproteinase activation, collagen deposition), and microbial sensors (lipopolysaccharide binding protein, soluble CD14). In the adaptive immune system, there is increased turnover and exhaustion of T cells, low thymic output, and establishment of a viral reservoir. Similarly, there is increased turnover of B cells, an altered phenotypic profile, and hyperimmunoglobulinemia.
With regard to the causes of chronic immune activation in HIV infection, it does not appear that HIV alone can account for the persistence of immune activation. In HIV infection, viral load is a poor predictor of disease progression, whereas measures of immune activation, including the frequency of activated T cells, are independent predictors of progression. Elite controllers—individuals who, in the absence of therapy, spontaneously control viral replication to undetectable levels—exhibit high levels of activated CD38+ CD8+ T cells on progression.1 Further, when viral load is suppressed with antiretroviral therapy, immune activation persists and is predictive of disease progression. Potential contributors to chronic immune activation include increased antigen load, bacterial overgrowth, the presence of herpes viruses, and translocation of proinflammatory mediators across the gut mucosa.
Consequences of HIV Infection in the Gastrointestinal Tract
A healthy gut is characterized by tight epithelial junctions and a layer of mucus (Figure 1). Antimicrobial peptides and large quantities of antibodies are secreted into the lumen. The majority of CD4+ T cells in the body is contained in the gut. Cross-communication occurs between microbes and epithelial cells or immune cells as part of a complex system that protects what is essentially the greatest surface area of tissue in contact with the outside environment. In HIV infection, there is a massive loss of CD4+ T cells in the gut during acute infection, with enteropathy caused by enterocyte apoptosis and a 2- to 10-fold increased permeability (leakiness) of the gut. Gut permeability allows translocation of microbial products into the systemic circulation, causing systemic immune activation.
Figure 1.
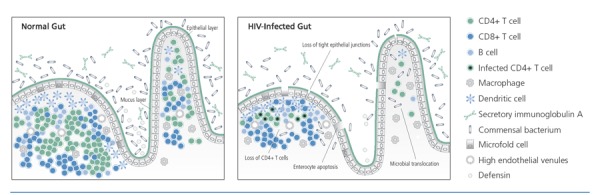
The effects of HIV infection in the gastrointestinal tract. Adapted from Mowat,12 and Brenchley and Douek.13
The emerging picture of the role of microbial translocation from the gut in fueling persistent immune activation is of a cyclic process in which more activated T cells are produced, providing more target cells for HIV, and resulting in immune deficiency via increased infection of the T cells, low thymic output, lymphoid tissue fibrosis, and T and B cell dysfunction (Figure 2). Increased immune deficiency results in persistent microbial translocation and poor pathogen control, allowing other viruses (eg, cytomegalovirus) and bacteria to replicate and further priming immune activation. Ongoing immune activation results in systemic inflammation, tissue damage (including fibrosis of lymphoid tissue) in the heart, lungs, liver, and kidneys, and coagulopathy, all of which are associated with non–HIV-related morbidity and mortality. By controlling HIV replication, antiretroviral therapy subdues some of these processes and allows recovery of some T cell populations. However, it does not stop immune activation, resultant inflammation, or tissue damage completely.
Figure 2.
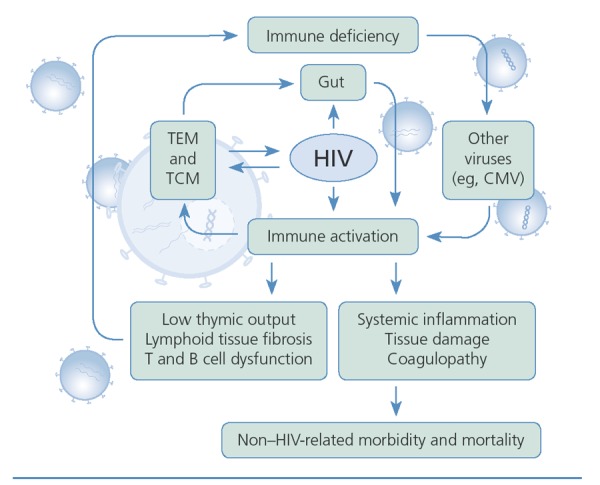
The role translocatof microbialion from the gut in fueling immune activation. CMV indicates cytomegalovirus; TCM, central memory T cells; TEM, effector memory T cells. Adapted from Klatt et al.14
As shown by Hunt and colleagues,1,2 T cell activation does decline during antiretroviral therapy but remains at higher than normal levels even after many years of viral suppression. The higher the level of immune activation (indicated by the percentage of activated CD38+ CD8+ T cells persisting during antiretroviral therapy), the lower the recovery of CD4+ T cell levels. Overall, these investigators have shown that numerous markers of inflammation and gut barrier dysfunction are associated with an increased risk of mortality among HIV-infected patients, independent of CD4+ cell count and viral load (Figure 3).3 For example, elevated blood levels of intestinal fatty acid binding protein (iFABP), a marker of damage in the gut epithelial lining, are associated with an 8-fold increased risk of mortality among HIV-infected patients.
Figure 3.
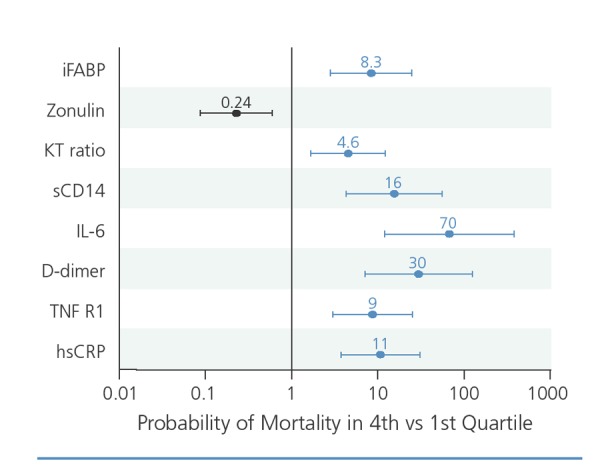
Markers of inflammation and gastrointestinal dysfunction that predict mortality in HIV infection. hsCRP indicates high-sensitivity C-reactive protein; iFABP, intestinal fatty acid binding protein; IL, interleukin; KT, kynurenine-to-tryptophan; sCD14, soluble CD14; TNF R1, tumor necrosis factor receptor 1. Adapted with permission from Hunt et al.3
Immune Activation and HIV Persistence
In addition to the other deleterious effects noted, persistent inflammation appears to be associated with the persistence of HIV. Ongoing HIV replication in host reservoirs also appears to contribute to persistent inflammation. Research is under way to determine whether novel investigational approaches can reduce HIV reservoir size, through, for example, the use of antiinflammatory drugs or by enhancement of HIV-specific immunity.
An association between HIV persistence and persistent immune activation in patients with undetectable plasma HIV RNA levels has been shown in studies of raltegravir intensification. In one study, antiretroviral therapy with raltegravir intensification resulted in a substantially greater reduction in immune activation (higher percentage of CD38+ memory CD8+ T cells) than standard antiretroviral therapy.4 Another study showed that raltegravir intensification resulted in a substantial reduction in the viral reservoir of latently infected memory CD4+ T cells (reduction in infectious units per million cells, or IUPM) and in CD8+ T cell activation in patients with viral suppression.5 However, yet another study showed that the addition of raltegravir to an antiretroviral regimen did not further reduce low-level plasma viremia and found no association between persistence of plasma HIV RNA levels and T cell activation.6
One explanation for the seemingly divergent findings with raltegravir intensification may be that its effects are more prominent in gut tissue than in peripheral blood. A number of studies have shown a strong association between cell-based measures of viral persistence and T cell activation in gastrointestinal tissue.7 As shown in Figure 4, raltegravir intensification reduced immune activation more in gut tissues than in peripheral blood and reduced viral persistence (measured as HIV RNA level) in the terminal ileum.8
Figure 4.
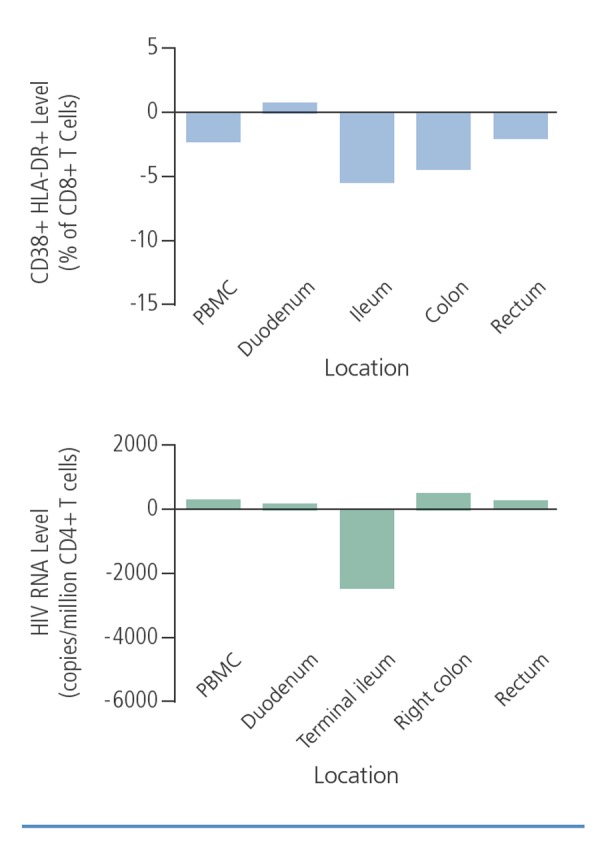
The effects of raltegravir intensification on immune activation (top) and HIV RNA levels (bottom) in peripheral blood mononuclear cells (PBMCs) and gut tissue sites. Adapted from Yukl et al.8
Ongoing HIV Replication During Antiretroviral Therapy?
Although complete inhibition of viral replication is an unlikely curative strategy for HIV infection, all functional cure strategies are based on first having achieved complete suppression of virus. There are substantial data that indicate HIV replication is not ongoing during suppressive antiretroviral therapy, but there are also data that suggest ongoing replication occurs and is associated with immune activation. In determining whether ongoing HIV replication takes place, the source of the sample and the assay used to measure viral reservoir are crucial. Blood tests may indicate the absence of ongoing viral replication, whereas gut or other lymphoid tissue assays that are sensitive enough to detect to 1 viral copy per million cells may demonstrate ongoing replication. Lymphoid tissue is likely the major HIV reservoir in patients on antiretroviral therapy. The gut is actually the largest lymphoid tissue in the body, but widespread destruction of CD4+ T cells in the gut make the lymph nodes or spleen the more likely primary viral reservoir during chronic infection. It is in these tissues that evidence of viral reservoir expansion and resultant ongoing immune activation can be found.
Results of the recently reported VISCONTI (Viro-Immunological Sustained Control after Treatment Interruption) study9 indicate an association between ongoing viral replication and immune activation. In this study, 14 patients started taking antiretroviral therapy soon after HIV infection and continued it for many years. When antiretroviral therapy was stopped, the patients did not exhibit viral rebound. Like elite controllers, these patients had very low levels of cell-associated HIV DNA. However, unlike elite controllers, who may exhibit immune activation and disease progression, patients in the VISCONTI study had very low levels of T cell activation. A potential implication of these findings is that very early treatment limits the size of the latent viral reservoir, which thus limits the magnitude of immune activation from the outset, potentially providing a lower set point for ongoing inflammation.
Relationship Between HIV-Specific Immunity and HIV Persistence
Ongoing immune activation adversely affects the HIV-specific T cell response, as well as overall CD4+ T cell reconstitution. Thus, ongoing immune activation may interfere with reconstitution of the HIV-specific T cell response. An example of the potential relationship between HIV-specific immunity in the gut and viral persistence is provided in a study by Hatano and colleagues.10 The study showed that in patients taking antiretroviral therapy, a stronger HIV-specific T cell response in the gut mucosa was associated with lower levels of proviral DNA in peripheral blood mononuclear cells (PBMCs).
Model for Immune Activation and HIV Persistence
If the mechanisms for the association between immune activation and HIV persistence can be identified, it may be possible to direct therapeutic strategies at these mechanisms. Figure 5 shows a potential model for more fully explaining HIV pathogenesis in a way that incorporates persistent immune activation and the role of the viral reservoir in fueling immune activation. Persistent immune activation causes many problems in the immune system, including low thymic output, lymphoid tissue fibrosis, poor immune reconstitution and renewal of CD4+ T cells, and dysfunction of T and B cells. It also continues to cause mucosal damage at the gut surface. These problems can result in immune suppression, with subsequent poor control of pathogens (eg, Streptococcus, Staphylococcus, herpes viruses), even in patients with low or undetectable HIV RNA levels.
Figure 5.
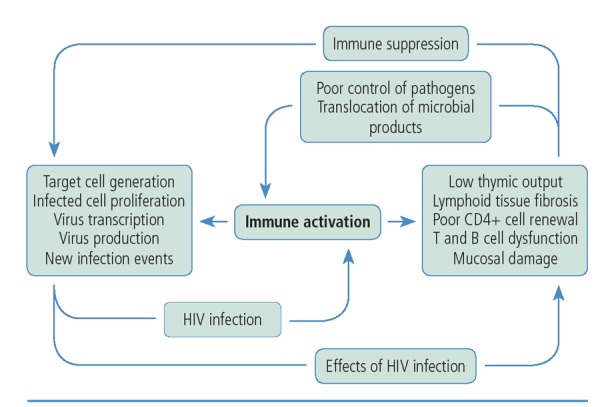
The central role of persistent immune activation in HIV infection. Adapted from Klatt et al.14
Immune activation affects the reservoir of HIV, even in patients taking antiretroviral therapy. It causes the generation of activated T cells, the target cells for the virus. Cells that are already infected with HIV will proliferate, producing more HIV-infected cells and expanding the viral reservoir. Viral transcription may begin, and some viral replication may occur. There is increasing evidence of the risk of new infection events, even in patients taking fully suppressive antiretroviral therapy. This increase in HIV replication results in further immune activation and, because the patient remains in an immune-suppressed state, poor immune control of newly produced virus. Antiretroviral therapy can profoundly reduce viral replication but does not eliminate residual immune activation nor stop the replication of virus-containing cells or the expansion of the viral reservoir that can result from persistent immune activation.
Although this picture of pathogenesis may appear daunting with regard to achieving a cure for HIV infection, it actually suggests many points in the processes underlying persistent immune activation that can serve as therapeutic targets. Among the therapeutic interventions in development are chemokine receptor inhibitors, antiinfective therapies, drugs that may reduce microbial translocation, drugs that may enhance T cell renewal, antifibrotic drugs, anti-aging strategies, antiinflammatory agents, and anticoagulants (Table). Combination approaches will likely be necessary to reduce persistent immune activation and counteract its effects in the body, given the multifactorial pathogenesis of HIV infection.
Table.
Therapeutic Interventions in Development to Reduce Persistent Immune Activation
| Intervention | Examples |
|---|---|
| Antiinfective therapy | Cytomegalovirus, Epstein-Barr virus, herpes simplex virus, HCV/HBV |
| Antiaging | Caloric restriction, sirtuin activators, vitamin D, omega-3 fatty acids, rapamycin, diet, exercise |
| Enhance T-cell renewal | Growth hormone, interleukin 7 |
| Chemokine receptor inhibitors | Maraviroc, cenicriviroc |
| Microbial translocation | Sevelamer, colostrum, rifaximin |
| Antifibrotic drugs or agents | Pirfenidone, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, keratinocyte growth factor |
| Anticoagulants | Low-dose warfarin, dabigatran, aspirin, clopidogrel |
| Antiinflammatory drugs | Chloroquine, hydroxychloroquine |
| Minocycline | |
| NSAIDs: COX-2i, aspirin | |
| Statins | |
| Methotrexate | |
| Thalidomide, lenalidomide, pentoxyfyline (weak TNF inhibitors) | |
| Biologics | TNF inhibitors, interleukin 6 inhibitors, anti–alpha-interferon antibodies, anti-PD1 antibodies |
COX-2i indicates cyclooxygenase-2 inhibitor; HCV, hepatitis C virus; NSAID, nonsteroidal antiinflammatory drug; PD1, programmed death 1; TNF, tumor necrosis factor.
Conclusion: In the Context of a Cure
Numerous mechanisms contribute to HIV persistence, many of which are currently being addressed therapeutically. The unifying theme in these efforts is to reduce the size of the HIV reservoir. Strategies to achieve this include reducing persistent inflammation and increasing immune function, potentially including the strategies of early antiretroviral therapy and antiretroviral therapy intensification. It is also possible that gene therapy could be used to reduce the viral reservoir. It has already been shown that stem-cell transplants can lead to a reduction in the size of the viral reservoir (ie, in the Berlin patient11), serving as proof of principle that such genetic approaches may be viable. Drugs with biologic activity against latent virus exist and are being assessed, and vaccines may be developed to enhance host clearance mechanisms (ie, by boosting the damaged HIV-specific immune response). As noted, a combination of approaches will be necessary to address the many aspects of HIV infection and persistent immune activation.
References
- 1.Hunt PW, Brenchley J, Sinclair E, et al. Relationship between T cell activation and CD4+ T cell count in HIV-seropositive individuals with undetectable plasma HIV RNA levels in the absence of therapy. J Infect Dis. 2008;197:126-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hunt PW, Martin JN, Sinclair E, et al. T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus-infected patients with sustained viral suppression during antiretroviral therapy. J Infect Dis. 2003;187(10): 1534-1543. [DOI] [PubMed] [Google Scholar]
- 3.Hunt P, Rodriguez B, Shive C, et al. Gut epithelial barrier dysfunction, inflammation, and coagulation predict higher mortality during treated HIV/AIDS [Abstract 278]. 19th Conference on Retroviruses and Opportunistic Infections (CROI) March 5-8, 2012; Seattle, Washington. [Google Scholar]
- 4.Llibre JM, Buzon MJ, Massanella M, et al. Treatment intensification with raltegravir in subjects with sustained HIV-1 viraemia suppression: a randomized 48-week study. Antivir Ther. 2012;17(2):355-364. [DOI] [PubMed] [Google Scholar]
- 5.Vallejo A, Gutierrez C, Hernandez-Novoa B, et al. The effect of intensification with raltegravir on the HIV-1 reservoir of latently infected memory CD4 T cells in suppressed patients. AIDS. 2012;26(15):1885-1894. [DOI] [PubMed] [Google Scholar]
- 6.Ghandi RT, Zheng L, Bosch RJ, et al. The effect of raltegravir intensification on low-level residual viremia in HIV-infected patients on antiretroviral therapy: a randomized controlled trial. PLoS Clin Trials. 2010;7(8):pii:e1000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheth PM, Yi TJ, Kovacs C, et al. Mucosal correlates of isolated HIV semen shedding during effective antiretroviral therapy. Mucosal Immunol. 2012;5(3):248-257. [DOI] [PubMed] [Google Scholar]
- 8.Yukl SA, Shergill AK, McQuaid K, et al. Effect of raltegravir-containing intensification on HIV burden and T-cell activation in multiple gut sites of HIV-positive adults on suppressive antiretroviral therapy. AIDS. 2010;24(16):2451-2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saez-Cirion A, Bacchus C, Hocqueloux L, et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 2013;9(3):e1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hatano H, Hayes TL, Dahl V, et al. A randomized, controlled trial of raltegravir intensification in antiretroviral-treated, HIV-infected patients with a suboptimal CD4+ T cell response. J Infect Dis. 2011;203(7):960-968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hutter G, Nowak D, Mossner M, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360(7):692-698. [DOI] [PubMed] [Google Scholar]
- 12.Mowat AM., Anatomical basis of tolerance and immunity to intestinal antigens. Nat Rev Immunol. 2003;3(4):331-341. [DOI] [PubMed] [Google Scholar]
- 13.Brenchley JM, Douek DC. HIV infection and the gastrointestinal immune system. Mucosal Immunol. 2008;1(1): 23-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klatt NR, Chomont N, Douek DC, Deeks SG. Immune activation and HIV persistence: implications for curative approaches to HIV infection. Immunol Rev. 2013;254(1):326-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional Suggested Reading
- Armah KA, McGinnis K, Baker J, et al. HIV status, burden of comorbid disease, and biomarkers of inflammation, altered coagulation, and monocyte activation. Clin Infect Dis. 2012;55(1):126-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks SG, Autran B, Berkhout B, et al. Towards an HIV cure: a global scientific strategy. Nat Rev Immunol. 2012;12(8):607-614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler NG, Douek DC. Microbial translocation in HIV infection: causes, consequences and treatment opportunities. Nat Rev Microbiol. 2012;10(9):655-666. [DOI] [PubMed] [Google Scholar]