

Abstract
The ultimate goal of hepatitis C virus (HCV) treatment is the eradication of the virus. Ongoing research continues to add to knowledge of the HCV life cycle, revealing new potential viral and host targets for the development of therapy. Understanding of HCV was initially hampered by the inability to achieve viral replication in cell culture. Advances such as the HCV replicon and complete cell culture systems, however, have permitted rapid growth in knowledge and accelerated testing of candidate antiviral agents. Among potential targets are viral entry factors, including scavenger receptor type B1 (SR-B1) and CD81, as well as neutralizing antibodies against the viral glycoproteins. Popular targets related to translation and replication are the NS3/4A protease (inhibited by telaprevir and boceprevir) and the NS5B polymerase, as well as the NS2/3 autoprotease, the NS3 helicase, and nonenzymatic targets such as NS4B and NS5A proteins. Host targets are also available, including microRNAs and cyclophilins. This article summarizes a presentation by Charles M. Rice, PhD, at the IAS–USA live continuing medical education course, Management of Hepatitis C Virus in the New Era: Small Molecules Bring Big Changes, held in New York City in April 2011.
History: Roadblocks to Hepatitis C Virus Research
The development of antiviral agents effective against hepatitis C virus (HCV) was hindered for many years by the inability to achieve viral replication in laboratory cell cultures and by the absence of a small-animal model for infection. These roadblocks slowed both the understanding of the HCV life cycle and the investigation of potential antiviral drugs.
HCV was discovered in 1989 as the major causative agent of non-A, non-B hepatitis.1 This positive-strand RNA virus has a genome approximately 9.6 kilobases (kb) in length. It encodes a single long polyprotein that is processed into 10 individual proteins by viral enzymes (NS2/3 and NS3/4A) and cellular enzymes (signal peptidase and signal peptide peptidase) (Figure 1).2-4 The NS3/4A serine protease, which cleaves the viral polyprotein at 4 sites, is the target of the only direct-acting HCV antivirals that have been approved by the US Food and Drug Administration (FDA) to date—telaprevir and boceprevir.
Figure 1.
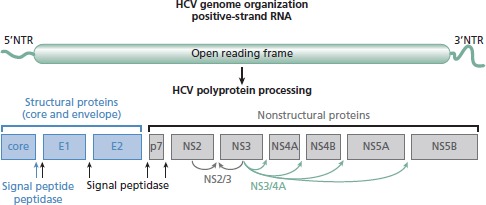
Delineation of hepatitis C virus (HCV) genome organization and polyprotein processing. Schematic shows structural proteins, nonstructural proteins, and enzymatic activities required for cleaving the polyprotein. Adapted from Bartenschlager et al,2 Grakoui et al,3 and Hijikata et al.4
It was almost a decade after the discovery of HCV that the first infectious complementary DNA (cDNA) clone was constructed. Full-length viral RNA transcribed from the cDNA in vitro was capable of establishing infection in chimpanzees but, somewhat paradoxically, could not replicate in cell culture. A major breakthrough in the development of cell-based systems was the construction of the HCV replicon in 1999.5 In the initial version, the genome of a genotype-1b HCV isolate was modified to contain only the key viral RNA replication genes and a neomycin resistance marker for drug selection. Transfection of this “subgenome” into a human hepatoma cell line, Huh-7, resulted in a low frequency of neomycin-resistant cell colonies containing persistently replicating HCV RNA. The replicon provided the first tractable system for the study of HCV RNA replication in the laboratory and the first opportunity to investigate candidate inhibitors in cells.
Improvements to the replicon system followed its initial description. It was recognized that adaptive mutations, which emerged spontaneously in the subgenomic replicons during their growth in cells, could dramatically increase RNA replication levels and transduction efficiency. Furthermore, using interferon alfa to cure cells that had previously harbored the viral RNA resulted in new hepatoma lines that were much more permissive to HCV replication. Despite these improvements, however, the system suffered a major drawback: replicons encoding the full-length HCV genome failed to produce infectious virus in cell culture.
It was not until 2005 that this hurdle was overcome and the first complete HCV cell culture system was developed.6-8 The breakthrough came when an HCV genotype 2a clone, derived by Takaji Wakita from a rare case of acute fulminant HCV infection (Japanese fulminant hepatitis 1 [ JFH-1] strain), was shown to initiate efficient replication in hepatoma cells without the need for adaptive mutations. Remarkably, electroporation of JFH-1 RNA into cells also led to the release of filterable infectious particles, meeting the classic criterion for a virus. These virus particles were capable of infecting naive hepatoma cells, chimpanzees, and a mouse model harboring human hepatocytes. Both animal models produced virus particles that were still infectious in cell culture. Thus, almost 20 years after the discovery of the virus, all steps in the HCV life cycle could finally be recapitulated in the laboratory.
What We Have Learned: Viral Entry and Spread
HCV is an enveloped virus that displays 2 glycoproteins on its surface: E1 (up to 6 glycosylation sites), the exact role of which is unknown, and E2 (11 glycosylation sites), which appears to be responsible for receptor binding. The proteins are embedded in a lipid bilayer surrounding a nucleocapsid composed of core protein and the genomic RNA. Surprisingly, the virus is often associated with serum lipoproteins (ie, very-low-density and low-density lipoproteins), which are thought to play crucial roles in entry, virus assembly, and possibly in immune evasion. The association of lipoproteins and virus results in material that is heterogeneous in terms of buoyant density; particles with the lowest density often exhibit the highest infectivity. This heterogeneity has made it difficult to gain precise structural information on the HCV particle.
The list of molecules that are active in HCV lipoviral particle entry into liver cells continues to grow. At present, glu-cosaminoglycans (GAGs) and low-density lipoprotein receptors (LDLRs) on the hepatocyte surface are implicated as putative initial attachment factors. Entry factors scavenger receptor type B1 (SR-B1), CD81, and tight-junction proteins CLDN-1 (claudin-1) and OCLN (occludin) are essential for uptake. The restriction of HCV to human and chimpanzee hepatocytes appears to be related to the characteristics of the CD81 and OCLN orthologs.
The current model for initial infection posits that the virus enters the liver through the bloodstream, passing through the endothelium and the space of Disse. The particle then interacts with GAG, LDLR, SR-B1, and CD81 on the basolateral surface of the hepatocyte, followed by movement to tight junctions formed by CLDN-1 and OCLN (Figure 2). The mechanisms by which HCV utilizes all 4 entry factors are not entirely clear, as so far only SR-B1 and CD81 have been shown to interact directly with the E2 glycoprotein. It is also not completely understood to what degree spread through the liver occurs via virus released into circulation versus by cell-to-cell transmission between closely connected hepatocytes.
Figure 2.
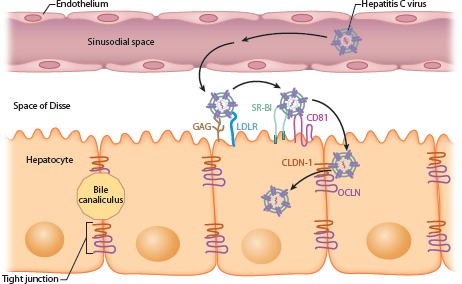
Current model of hepatitis C virus entry into the hepatocyte. GAG indicates glu-cosaminoglycan; LDLR, low-density lipoprotein receptor; SR-B1, scavenger receptor type B1; CLDN-1, claudin-1; OCLN, occludin. Adapted from an animated slide from Shihyun You, PhD, and Charles Rice, PhD.
The processes of viral entry and spread are potential drug targets, although uptake inhibitors might best be used in combination with other antiviral agents inhibiting replication. Such combinations might protect cured or uninfected cells and could delay spread of resistant variants. Furthermore, inhibition of entry might prevent reinfection of liver allografts in HCV-infected patients receiving transplants. Potential treatments targeting entry include antibodies or small molecules against the entry factors and virus-neutralizing antibodies.
What We Have Learned: HCV Translation and Replication
Following translation and processing of the 10 viral proteins, all of the gene products remain associated with intracellular membranes (Figure 3).9 This is a feature common to all positive-strand RNA viruses and is not yet well understood. The HCV nonstructural proteins, which include NS3, NS4A, NS4B, NS5A, and NS5B, comprise the RNA replication machinery. The membrane-associated replicase works by copying incoming genome RNA into a negative-strand intermediate, which is then used to generate additional positive-strand RNAs for subsequent rounds of translation and packaging into virus particles. The replication factories use microtubules to move around the cell as they function, coalescing into vesicular structures termed the membranous web.
Figure 3.
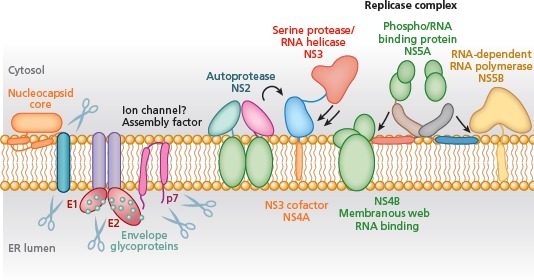
Hepatitis C virus proteins and functions. ER indicates endoplasmic reticulum. Scissors indicate cleavage by host enzymes, arrows represent processing by viral proteases. Adapted from Moradpour et al.9
The most popular targets for the development of HCV antiviral drugs have been 2 enzymatic components of the replicase: the NS3/4A serine protease and the NS5B RNA-dependent RNA polymerase. These are attractive in part because both are enzymes required for HCV replication. In addition, their functions are amenable to the development of biochemical assays for inhibitor screening, an important consideration especially in the era before cell-based systems. Other enzymatic targets have been less well explored, including the NS2/3 autoprotease, which mediates a single cleavage in the polyprotein, and an RNA helicase encoded in the C-terminal two-thirds of NS3, which appears to unwind RNA structures during replication.
A number of nonenzymatic targets have also received considerable attention. The NS4B protein may scaffold the RNA replication machinery and is sufficient to form the membranous web. It is the target of several compounds recently identified in novel biochemical assays. The NS5A protein is involved in both RNA replication and infectious virus assembly. Despite these important roles, the protein was initially an unpopular target because it has no known enzymatic activity. Recently, however, a compound acting on NS5A, BMS-790052, has emerged as one of the most potent and broadly active inhibitors of HCV replication observed to date. In a study by Gao and colleagues, the drug exhibited 50% effective concentration (EC50) values of 9 pM to 146 pM against replicons and infectious HCV in culture. Early-phase clinical trials revealed promising pharmacokinetics and a rapid decrease in HCV RNA levels.10
Host factors involved in translation and replication present an intriguing alternative strategy for drug design. MicroRNAs are small noncoding RNAs that fine-tune cellular gene expression, usually by sequestering an mRNA or inducing template turnover in a highly selective manner. In the liver, microRNA-122 (miR-122) is responsible for regulating the expression of approximately 200 cellular genes, including those involved in cholesterol biosynthesis and lipid metabolism. Interestingly, miR-122 was found to bind to 2 short recognition sequences near the 5ʹ end of the HCV genome. Rather than downregulating HCV gene expression, however, miR-122 binding augments viral RNA replication. Recently, Lanford and colleagues have shown that a stabilized “locked nucleic acid” antagonist of miR-122 can be efficiently delivered to the livers of HCV-infected chimpanzees. Delivery of the antagonist reduced HCV viremia by several orders of magnitude, with the effect persisting for several weeks after treatment was stopped.11
HCV Assembly and Release
Thus far, there are relatively few data on how virus assembly and release might be inhibited. In addition to the viral structural proteins (core, E1, and E2), the nonstructural proteins p7, NS2, NS3, and NS5A are required for production of infectious virus. This hints at coordination between the machinery involved in making new copies of the genome RNA and the machinery packaging it into particles.
As mentioned above, HCV particles are typically associated with serum lipoproteins in circulation. In fact, HCV uses the lipoprotein production pathway to assemble viral particles and to move them out of the infected cell. Thus, interfering with lipoprotein components or disrupting the pathways involved in lipoprotein secretion are potential areas for intervention in assembly and release, as well as entry.
Host Targets for Antiviral Drug Development
HCV is dependent on host cells to provide numerous factors for its replication, and there is ongoing discussion about whether viral targets or host targets are optimal for antiviral drug development. Strategies for identifying potential host targets include screening small interfering RNA (siRNA) or short hairpin RNA (shRNA) libraries to identify human genes required for HCV replication. Another strategy is to pursue already-known targets, such as SR-B1 (eg, with anti-SR-B1 antibody), CD81 (eg, with anti-CD81 antibody), lipoproteins (eg, with anti-apolipoprotein E antibody), or microRNAs (eg, with locked nucleic acid compounds). Furthermore, existing pharmacologically active compounds, for example, those potent against other pathogens, can be screened using cell-based systems for inhibiting HCV.
One of the most interesting outcomes of a screen of preexisting drugs was the finding that cyclophilins, which are targeted by cyclosporin A, are required for HCV replication. Cyclophilin A is a cellular peptidyl-prolyl isomerase, which appears to act on the HCV NS5A protein, as well as possibly on NS2 and NS5B. It has been shown that nonimmunosuppressive analogues of cyclosporin A, such as the compound Debio 025, can bind cyclophilin A and inhibit HCV replication. Debio 025 was found to markedly reduce HCV viremia in clinical trials when used alone or in combination with pegylated interferon alfa.
Although host factors may be feasible antiviral targets, the debate continues on the benefits of this approach. On the plus side, host proteins present a wide array of strategies; viral targets are limited to 10 proteins and 1 RNA molecule. Drug development time may be reduced by evaluating compounds already known to target host processes relevant to HCV. In addition, host proteins are well conserved. This is in contrast to the huge genetic variability of the virus, which can lead to rapid emergence of resistance. The genetic heterogeneity of HCV can be appreciated when it is considered that approximately 1012 viral particles are produced each day in a chronically infected individual. As with HIV and other RNA viruses, replication of HCV is error prone, easily resulting in patients who harbor all single- and double-substitution resistance-associated mutations prior to starting any drug treatment. Agents that act on host targets may also be more potent across the different viral genotypes.
Importantly, inhibiting a host factor is inherently less virus-specific than targeting an HCV protein. Disrupting important roles of a cellular protein could lead to increased side effects. Host factor polymorphisms may also affect drug activity. Ideally, therefore, a combination of host and viral inhibitors will provide a variety of drug regimens appropriate for different patients.
Summary
HCV is a cytoplasmic RNA virus that does not integrate into the host cell DNA, making eradication a seemingly achievable goal. Understanding of the HCV life cycle and potential drug targets has increased considerably over the last decades, and there is now a large number of candidate antivirals in the development pipeline. Very recently, the first direct-acting antiviral drugs were approved in combination with peginterferon alfa and ribavirin. For the near future, a major goal is to develop interferon–free therapies that can consistently clear the virus. As more agents effective in inhibiting the HCV life cycle are identified, the chances of achieving cure in most, if not all, HCV-infected patients are likely to increase dramatically.
References
- 1.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359-362. [DOI] [PubMed] [Google Scholar]
- 2.Bartenschlager R, Ahlborn-Laake L, Mous J, Jacobsen H. Nonstructural protein 3 of the hepatitis C virus encodes a serine-type proteinase required for cleavage at the NS3/4 and NS4/5 junctions. J Virol. 1993;67:3835-3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grakoui A, Wychowski C, Lin C, Fein-stone SM, Rice CM. Expression and identification of hepatitis C virus polyprotein cleavage products. J Virol. 1993;67:1385-1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hijikata M, Mizushima H, Tanji Y, et al. Proteolytic processing and membrane association of putative nonstructural proteins of hepatitis C virus. Proc Natl Acad Sci USA. 1993;90:10773-10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lohmann V, Körner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110-113. [DOI] [PubMed] [Google Scholar]
- 6.Wakita T, Pietschmann T, Kato T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhong J, Gastaminza P, Cheng G, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci USA. 2005;102:9294-9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindenbach BD, Evans MJ, Syder AJ, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623-626. [DOI] [PubMed] [Google Scholar]
- 9.Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5:453-463. [DOI] [PubMed] [Google Scholar]
- 10.Gao M, Nettles RE, Belema M, et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature. 2010;465:96-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lanford RE, Hildebrandt-Eriksen ES, Petri A, et al. Therapeutic silencing of micro-RNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327: 198-201. [DOI] [PMC free article] [PubMed] [Google Scholar]