

Abstract
With the overall objective of assessing the potential of utilizing plasma protein binding interactions in combination with the prodrug approach for improving the pharmacokinetics of drug substances, a series of model carbonate ester prodrugs of phenol, encompassing derivatives with fatty acid-like structures, were characterized in vitro. Stability of the derivatives was studied in aqueous solution, human serum albumin solution, human plasma, and rat liver homogenate at 37oC. Stability of the derivatives in aqueous solution varied widely, with half-lives ranging from 31 to 1.7 × 104 min at pH 7.4 and 37oC. The carbonate esters were subject to catalysis by plasma esterases except for the t-butyl and acetic acid derivatives, which were stabilized in both human plasma and human serum albumin solutions relative to buffer. In most cases, however, hydrolysis was accelerated in the presence of human serum albumin indicating that the derivatives interacted with the protein, a finding which was confirmed using the p-nitrophenyl acetate kinetic assay. Different human serum albumin binding properties of the phenol model prodrugs with fatty acid-like structure and neutral carbonate esters were observed. In the context of utilizing plasma protein binding in combination with the prodrug approach for optimizing drug pharmacokinetics, the esterase-like properties of human serum albumin towards the carbonate esters potentially allowing the protein to act as a catalyst of parent compound regenerations is interesting.
Keywords: Bioreversible derivatives, carbonate ester, esterase-like properties, human serum albumin, prodrug, plasma protein binding
Introduction
Poor pharmacokinetics, together with toxicological issues, are major causes of drug failure during early development. High affinity leads generated in drug discovery seldom possess the physico-chemical properties required for transport to the site of action in the body. Although considerable measures are taken to improve the drug-like properties of the hits generated in drug screening programs, e.g. [1,2,3,4,5], simultaneous optimization of pharmacodynamic and pharmacokinetic properties may not always be achievable. Bioreversible derivatization [6,7,8,9,10], i.e. the prodrug approach, allowing transient modification of pharmacokinetic properties may constitute a suitable means for improving drug performance of active agents characterized by possessing favorable receptor profiles but poor transport properties.
Plasma protein binding is a major factor determining the fate of drug substances upon entry into the body [11,12,13,14]. Human serum albumin (HSA) interacts with a wide range of endo- and exogenous substances, primarily hydrophobic organic anionic compounds including fatty acids. HSA is quantitatively the most important protein as regards drug-plasma protein binding [15,16]. Exploitation of reversible drug-HSA binding in order to obtain improved drug pharmacokinetics has only been utilized in a few cases. The protracted effect of insulin fatty acid analogues is, at least in part, due to reversible binding to albumin [17,18,19]. Also acylation of glucagon-like peptide-1 derivatives with fatty acids led to protracted action facilitated by albumin binding [20]. Further, sustained release of 5-fluorouracil prodrugs with affinity for HSA was found to be effective against sarcoma in a mouse model [21,22,23]. In addition to the well-known ligand binding characteristics, HSA has been associated with esterase-like properties [9,24,25,26] which may be of potential interest in the prodrug setting in relation to regeneration of parent compound.
To assess the potential of utilizing plasma protein binding interactions in combination with the prodrug approach for improving pharmacokinetics, we have studied a series of bioreversible derivatives of the model drug phenol. It is known that several phenolic drugs are susceptible to extensive first-pass metabolism [27]. Therefore, it appeared of interest to investigate whether HSA binding may aid in decreasing the extent of first-pass metabolism of drug candidates containing a phenolic hydroxyl group. Thus, the overall objective of this study was to investigate the potential feasibility of optimizing the pharmacokinetic profile of phenol, i.e. minimizing hepatic first-pass metabolism of phenol, by design of bioreversible derivatives exhibiting a high affinity for HSA. In the current work, we present the in vitro characterization of a series of carbonate esters of phenol encompassing derivatives with fatty acid-like structures (Table 1) with respect to stability in phosphate buffer (pH 7.40), diluted plasma and rat liver homogenate as well as initial studies on the interaction between the carbonate esters and HSA and the implications hereof on carbonate ester reconversion rates. The synthesis of the carbonate ester derivatives is reported in the accompanying work together with detailed studies on the susceptibility of the carbonate esters towards hydrolysis as a function of pH [28]. In the following, bioreversible derivative is used as the preferred term, since phenol is not a drug and consequently the carbonate esters subject to study not prodrug derivatives.
Table 1.
Chemical structures and numbering of the carbonate ester derivatives subject to investigation. The carboxylic acid ester phenyl acetate was also included in the study.
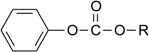 |
| Compound | R | Name |
|---|---|---|
| 1 | -C2H5 | Ethyl phenyl carbonate |
| 2 | -C(CH3)3 | t-Butyl phenyl carbonate |
| 3 | -C6H5 | Diphenyl carbonate |
| 4 | -CH2COOH | 2-(Phenoxycarbonyloxy)-acetic acid |
| 5 | -(CH2)5COOH | 6-(Phenoxycarbonyloxy)-hexanoic acid |
| 6 | -(CH2)7COOH | 8-(Phenoxycarbonyloxy)-octanoic acid |
| 7 | -(CH2)11COOH | 12-(Phenoxycarbonyloxy)-dodecanoic acid |
| 8 | -(CH2)15COOH | 16-(Phenoxycarbonyloxy)-hexadecanoic acid |
| 9 | - | Phenyl acetate |
Results and Discussion
Phenol was selected as a model for drug substances containing a phenolic hydroxyl group because its metabolism has been studied extensively [29,30,31,32] and phenol only possess a very weak affinity for HSA [33,34,35]. Phenol is subject to hepatic metabolism [29,30,31,32], the main metabolites being phenyl glucuronide and phenyl sulphate. Furthermore, it has previously been used as a model compound in prodrug related studies aiming at the circumvention of first-pass metabolism [36,37,38,39].
Hydrolysis in biological media
Rates of hydrolysis of the carbonate esters and phenyl acetate were determined at 37oC in 0.067 M phosphate buffer (pH 7.40) and 80% (v/v) human plasma. The hydrolysis rates of the carbonate esters in aqueous buffer (pH 7.40) were found to follow pseudo-first-order kinetics, however, at widely different rates, with half-lives ranging from 31 min to 280 h. The results obtained are in agreement with those previously obtained under slightly different conditions [28]. The stability of the carbonate esters in plasma was monitored for up to 8 h and the degradation reactions were also found to follow pseudo-first-order kinetics (Table 2). Hydrolysis of most of the esters was catalyzed plasma enzymes, however, to varying extents. The neutral esters 1 and 3 are hydrolysed rapidly with t½ < 2 min, unlike the sterically hindered t-butyl derivative 2 (Table 2). The plasma catalyzed hydrolysis for phenyl acetate (9) was too fast to be monitored. In fact, 2 is more stable in 80% plasma than in buffer at pH 7.4. This behavior was also observed for the charged acetic acid derivative 4. Plasma protein binding of these esters may be the cause of the observed stabilizing effect. Stabilization of bioreversible derivatives in vitro in human plasma relative to aqueous buffer due to plasma protein binding has been found previously for a glutaric acid derivative of phenol [37], various alkyl and aromatic esters of timolol [40,41], a t-butyl ester of L-dopa [42], a basic carbamate of 4-hydroxyanisole [43], and haloperidol decanoate [44]. With the exception of t-butyl phenyl carbonate (2), reconversion to phenol proceeds more slowly for the carbonate esters 4-8 negatively charged at physiological pH than for the neutral derivatives. However, for the esters with fatty acid-like structure no correlation between the number of methylene groups in the promoiety or lipophilicity [45] and the hydrolysis rates in 80% human plasma is apparent (Table 2). Generally, esters having a negative charge possess a higher resistance towards plasma catalyzed hydrolysis by being poor substrates for plasma esterases [46,47,48]. Examples include hemiesters of phenols [37] and metronidazole [49,50], corticosteroids [47] and benzoylglycolic acid [48].
Table 2.
Rate data for the decomposition of various carbonate esters and phenyl acetate in 67 mM buffer solution, 4% HSA solution, 80% human plasma, and 20% rat liver homogenate at 37oC a).
| Compound | Half-life ± SD (min) | log P d) | ||||
|---|---|---|---|---|---|---|
| Buffer, pH 7.40 | 4.3% HSA | 4.3% HSAc) | 80% Human plasma | 20% rat liver homogenate | ||
| 1 | 7.9 (± 1.0) × 103 | 77 ± 3.2 | 82 ± 2.5 | 1.13 ± 0.10 | b) | 2.17 |
| 2 | 31 ± 0.6 | 268 ± 14 | 248 ± 24 | 82 ± 7.4 | 0.10 ± 0.02 | 3.02 |
| 3 | 4.6 (± 0.03) × 102 | 14 ± 2.3 | 16.9 ± 1.3 | 0.18 ± 0.002 | b) | 3.21 |
| 4 | 31 ± 0.2 | 148 ± 1.3 | 147 ± 5.0 | 74 ± 1.0 | 0.90 ± 0.16f) | 1.72 |
| 5 | 1.7 (± 0.02) × 104 | 421 ± 20 | 422 ± 4.7 | 38 ± 2.8 | b) | 2.56 |
| 6 | 1.7 (± 0.15) × 104 | 72 ± 1.6 | 70 ± 1.3 | 7.5 ± 1.1 | b) | 3.39 |
| 7 | 1.4 (± 0.07) × 104 | 1.0 (± 0.2) × 104 | 7.0 (± 2.2) × 103 | 661 ± 86 | b) | 4.26 |
| 8 | n.d.g) | 142 ± 3.9 | 146 ± 2.2 | 40 ± 2.0 | 0.12 ± 0.03 | 4.67 |
| 9 | 3.0 (± 0.36) × 103 | 11.9 ± 0.3 | 12.6 ± 0.3 | b) | b) | 1.49e) |
-
a)Experiments performed in triplicate.
-
b)Degraded within 15 s.
-
c)1 × 10-4 M physostigmine added to HSA solution.
-
d)Logarithm to the octanol-water partition coefficient (log P) taken from [45].
-
e)From [51].
-
f)Mixed kinetics observed. Half-life calculated from t½ = ln2/(Vmax/Km). Vmax and Km obtained by nonlinear regression analysis using an integrated form of the Michaelis-Menten equation [52].
-
g)n.d., not determined due to solubility limitations.
Hydrolysis in HSA solution
For the negatively charged esters, binding to plasma proteins may also be a contributing factor to the observed high resistance towards plasma esterase catalyzed hydrolysis. This aspect has to our knowledge not been investigated. However, based on the binding specificity of HSA (e.g. references: [15,53,54,55,56]) it may be reasonable to suggest that negatively charged derivatives are bound to a higher degree than their neutral counterparts and, thus, rendering the negatively charged esters less accessible for the hydrolytic enzymes. It is well recognized that fatty acids are extensively bound to HSA [15,53,56], the affinity increasing with the chain length of the acid. Targeting to HSA has previously been shown to be successful for various delivery purposes in the case of proteins [17,18,19,20] and small organic molecules [57,58,59]. The bioreversible derivatives 4-8 may show some resemblance to fatty acids having a carboxylic acid group and a lipophilic part including an alkyl chain of varying length. Therefore, their stability in aqueous buffer was investigated in the presence of a physiological concentration of HSA (662 μM) to shed light on the effects of protein interactions on the stability of the derivatives. The course of degradation was adequately described by pseudo-first-order kinetics. For the derivative 7 being very stable in the HSA solution, first-order kinetics was assumed and for 8, the half-live was determined from the rate of phenol formation. Enhanced lability of the derivatives 1, 3, 5, 6 and 8 in the presence of HSA as compared to 0.067 M phosphate buffer (pH 7.40) was observed, indicating that interaction with HSA takes place. In contrast, derivatives 2 and 4 were more stable in the HSA containing solution than in buffer suggesting that protein binding is these cases causes increased stability in plasma of the latter derivatives. To this end stabilization towards hydrolysis due to HSA binding has been observed for ester derivatives of salicylic acid and diflunisal [60]. Interestingly, the stability of 7 was essentially unaffected by the presence of HSA.
HSA has been found to possess esterase-like properties [24,25,26,61,62,63]. Best characterized is probably the catalysis of p-nitrophenyl acetate decomposition [26,63,64] which will discussed further below. Compounds studied in a prodrug context include nicotinic acid esters [65,66], acetylsalicylic acid [61], and carbamates of phenol [36,38]. Furthermore, hydrolysis of long chain aryl esters has been found to be catalyzed by HSA [62]. Caution should however be exercised when assigning the catalytic effect to HSA. Commercially available HSA preparations have been found to contain cholinesterase as an impurity, leading to an erroneous assignment of HSA as a catalyst [67,68] as has probably been the case for nicotinic acid esters [65,66]. Therefore, the hydrolysis of the esters in HSA containing solution was also investigated in the presence of 10-4 M of physostigmine, a pseudocholinesterase inhibitor. As seen from Table 2, the hydrolysis rates for the carbonate esters in HSA were not affected by the presence of physostigmine, suggesting that cholinesterases are not present in the HSA preparation used or that the carbonate esters are poor substrates for the enzyme. The half-life of phenyl acetate (9) was slightly increased in the presence of the inhibitor (*P > 0.05). Detailed studies are required to assign the hydrolysis of the esters to particular sites on HSA and to entirely exclude hydrolysis occurring due to the presence of other contaminant enzymes. However, to the best of our knowledge this is the first report of the HSA catalysis of carbonate ester degradation. The reconversion of ester prodrug derivatives to parent compound in blood is usually mediated by plasma enzymes or by chemical hydrolysis, however, based on the obtained data it may be suggested that specific prodrugs using HSA as a facilitator of parent drug liberation may be designed.
Hydrolysis in rat liver homogenate
Degradation of the esters was also studied in 20% (w/w) rat liver homogenate at pH 7.4 and 37oC. For most of the derivatives degradation was too fast to be followed (Table 2). Mixed zero- and first-order kinetics was observed for the acetic acid derivative 4. The rapid hydrolysis of all the esters suggests that none of the promoieties are capable of protecting phenol from hepatic metabolism upon entry into the hepatocyte. In order to protect phenol from hepatic first-pass metabolism the model prodrugs should be able to prevent the derivatives from being taken up by the liver (due to extensive HSA binding) otherwise they will most likely be subject to sequential metabolism [69], i.e. hydrolysis followed by conjugation in the present case.
Affinity of carbonate esters for human serum albumin as studied by a spectrophotometric assay
The altered stability of the carbonate esters in HSA solution as compared to phosphate buffer solution strongly indicated that the carbonate esters interact with the plasma protein. HSA possesses two major drug binding sites termed site I and site II, respectively, according to the Sudlow nomenclature [15,53,70,71]. It should be appreciated that site I and II are binding areas or regions rather than binding sites [55,72,73] as more than one ligand may be accommodated at a time without considerable interference. Site I ligands are typically heterocyclic anions with the charge placed in the central part of the molecule [71]. However, a large degree of variability in the chemical structure is found among these ligands [55]. Site II ligands are generally aromatic hydrophobic anions with the charge situated in one end of the molecule away from the apolar region [71]. However, site II ligands may also be neutral. The site II binding area is characterized as a hydrophobic pocket with estimated dimensions of 8 × 16 Å [74]. Irikura et al. estimated a length of 21-25 Å for the long dimension [75]. Thus, certain size limitations apply for the ligand to be accommodated by HSA. For instance, long-chain fatty acids cannot bind to site II in contrast to medium-chain fatty acids with 6 to 10 carbon atoms [63,74]. A priori, based on their structure, elongated shape and negative charge positioned at the end of the molecule, the carbonate esters 4-8 with fatty acid-like structure were expected to interact with the HSA binding site II or the long-chain fatty acid binding sites.
The rapid hydrolysis of most of the carbonate esters in the presence of HSA (Table 2) precludes the use of equilibrium methods, and other slow methods, for characterization of the interaction. In order to get some initial insights on the interaction of the bioreversible derivatives of phenol with HSA, a rapid spectrophotometric kinetic assay exploiting the esterase-like properties of HSA was applied [26,63,76,77]. The ability of HSA to facilitate the hydrolysis of p-nitrophenyl acetate is primarily associated with the Sudlow site II [64,78]. The amino acid residues primarily involved are Tyr-411 and Arg-410 [64,78], however, secondary less reactive sites have been recognized [26,77,79,80]. The ester p-nitrophenyl acetate reacts rapidly with HSA leading to the formation of p-nitrophenolate and HSA acetylated (Ac-HSA) at the amino acid residue Tyr-411. The addition of ligands (L) may inhibit the acetylation reaction. In the most simple cases, this occurs in a competitive manner through the formation of an unreactive ligand-HSA complex (L·HSA) [26,63,76,77,79,80,81]. However, it has been found that binding to other sites on albumin, including the long-chain fatty acid binding sites, may also affect the rate of acetylation. The reaction scheme proposed by Means and co-workers [26,63] for the p-nitrophenyl acetate – HSA affinity assay is depicted in Scheme 1.
Scheme 1.

Reaction scheme for the inhibition of the acetylation of human serum albumin (HSA) by p-nitrophenyl acetate (pNphOAc) by ligand (L) addition [26,63].
In accordance with previous investigations, the degradation of p-nitrophenyl acetate was found to follow pseudo-first-order kinetics when HSA was present in excess of p-nitrophenyl acetate. This was the case both in the absence and presence of added ligands. Using a HSA concentration of 7 x 10-5 M, a half-life of ~10 s for the degradation of p-nitrophenyl acetate was observed in the absence of ligand in the 0.067 M phosphate buffer at pH 7.40 and 37oC. The t½ of p-nitrophenyl acetate in the phosphate buffer at pH 7.40 without HSA was 355 min. At the selected conditions, the reaction was of first-order with respect to p-nitrophenyl acetate and proportional to the concentration of free (unoccupied) HSA when taking into account the formation of p-nitrophenolate due to reaction with secondary HSA sites and chemical hydrolysis [26]:
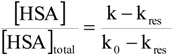 |
(1) |
where [HSA] and [HSA]total are the concentration of primary free binding sites on HSA in the presence and absence of added ligand, respectively. k and k0 are the pseudo-first-order rate constants in the presence and absence of added ligand, respectively, and kres is the apparent rate constant due to other reactions of p-nitrophenyl acetate with HSA leading to the formation of p-nitrophenolate. The magnitude of kres was determined from experiments conducted with ibuprofen added in large excess of HSA (10-20 equivalents) and the rate was found to correspond to 6% of the uninhibited reaction. All of the carbonate esters investigated inhibited the reaction between p-nitrophenyl acetate and HSA. The neutral carbonate esters (ethyl phenyl, t-butyl phenyl and diphenyl, 1-3), did so, however, to a much lesser extent than the carbonate esters with fatty-acid like structure, as can be seen from Figure 1. According to convention the ratio of k/k0 was plotted as a function of the ligand/HSA ratio rather than the right hand side of Equation 1. The ability of the parent compound phenol and phenyl acetate (9) to inhibit the actylation reaction was also investigated and results qualitatively similar to that of the carbonate esters 1-3 were obtained (data not shown). Figure 1B shows that 2-(phenoxycarbonyloxy)-acetic acid (4) inhibited the reaction more strongly than the longer hexanoic acid and octanoic acid derivatives. Also, it is apparent that the inhibition profiles of the derivatives are different from those of the site II ligands ibuprofen and octanoic acid, the differences being most prominent for 6, 7 and 8. For comparison, the inhibitory effect of the site II ligands ibuprofen and octanoic acid was investigated as well. In agreement with previous reports, the affinity for HSA of ibuprofen was higher than that of octanoic acid [79]. Ibuprofen and octanoic acid proved to be more efficient inhibitors of the acetylation reaction than any of the carbonate esters (Figure 1). Plotting of the obtained rate data according to Koh and Means [63] indicated that only 2-(phenoxycarbonyloxy)-acetic acid (4), in addition to octanoic acid and ibuprofen, had Sudlow site II as the primary and/or only binding site. Apparent dissociation constants were estimated using linear regression analysis, the values being 2.6 × 10-5 M, 5.4 ×10-6 M and 5.7 ×10-6 M for 2-(phenoxycarbonyloxy)-acetic acid (4), octanoic acid and ibuprofen, respectively. It may tentatively be suggested that the longer carbonate ester derivatives (5-8) may have other binding sites than site II, possible the long-chain fatty acid binding sites, as their primary area of interaction. The spectrophotometric affinity assay did not allow quantitative assessment of the binding parameters of the carbonate esters, except for derivative 4. However, the assay revealed that the mode and degree of interaction with HSA is different for derivatives with fatty acid-like structure than the neutral carbonate esters. From the rate data and the stability studies in 80% plasma and HSA containing solution it may also be stipulated that the charged derivatives interact strongly with HSA.
Figure 1.


Inhibition of the reaction of p-nitrophenyl acetate with human serum albumin (HSA) by the addition of ligand in 0.067 M sodium phosphate buffer pH 7.40 and 37oC. The degree of inhibition, expressed as the ratio of the observed first-order rate constants in the presence of ligand, k, and absence of ligand, k0, as a function of the ligand to HSA concentration ratio. (A). Ligands: 1 (●), 2 (○), 3 (▲), ibuprofen (△), and octanoic acid (▼). (B) Ligands: 4 (●), 5 (○), 6 (×), ibuprofen (△), and octanoic acid (▼). (C) Ligands: 7 (●), 8 (○), ibuprofen (△), and octanoic acid (▼).
Conclusions
The in vitro fate of eight carbonate esters of phenol, some with fatty acid-like structure, upon incubation in diluted human plasma and a phosphate buffer solution with a near physiological human serum albumin (HSA) concentration, was studied. The degradation of the carbonate esters was catalyzed by plasma as compared to their stability in buffer at pH 7.4, except for the relatively unstable t-butyl and acetic acid derivatives 2 and 4, which were stabilized by the presence of plasma proteins. The presence of HSA in the incubation medium was found to significantly influence the degradation rates of the model prodrug carbonate esters. Again, for the t-butyl and acetic acid derivatives a stabilizing effect was observed, however, for five of the model prodrugs a catalytic effect of HSA was observed for the first time. These results, indicating that the carbonate esters interacted with HSA were confirmed by the p-nitrophenyl acetate kinetic affinity assay. The inhibition patterns obtained from the kinetic assay suggest that bioreversible derivatives with fatty acid-like structure, negatively charged at physiological pH, interacted more strongly with HSA than the neutral counterparts although quantitive estimates of affinity were not obtained.
As relates to the potential of exploiting plasma protein binding together with the prodrug approach for improving drug pharmacokinetics, the possibility of using HSA as a catalyst of parent compound regeneration is highly interesting and warrants further investigation. The model prodrug derivatives of phenol subject to the conducted in vitro characterization appear suitable for further investigations of this concept. Especially derivatives 4, 7 and 8 may be suited for further testing e.g. in hepatocyte suspensions or single pass rat liver perfusion studies due to their in vitro affinity and stability properties. Protein binding has been suggested to be a potentially limiting step in liver uptake [82]. Optimal bioreversible derivatization for preventing phenol from being first-pass metabolized in the liver most likely requires extensive HSA binding in order to minimize prodrug uptake into hepatocytes. Current studies in our lab are directed at developing methods suitable for quantitative evaluation of the binding between bioreversible derivatives and HSA.
Experimental
Chemicals
Eserine hemisulfate salt (physostigmine), ethyl phenyl carbonate, and human serum albumin (≥ 96% albumin, essentially fatty acid free, A-1887, lot 90K7604), and ibuprofen were obtained from Sigma Chemical Co. (St. Louis, MO, USA). Phenyl acetate was obtained from Acros Organics (Geel, Belgium). Diphenyl carbonate, phenol, p-nitrophenyl acetate, octanoic acid, and t-butyl phenyl carbonate were obtained from Aldrich-Chemie (Steinheim, Germany). 2-(Phenoxycarbonyloxy)-acetic acid, 6-(phenoxycarbonyloxy)-hexanoic acid, 8-(phenoxycarbonyloxy)-octanoic acid, 12-(phenoxy-carbonyloxy)-dodecanoic acid, and 16-(phenoxycarbonyloxy)-hexadecanoic acid were synthesized and characterized as described elsewhere [28]. All other chemicals and solvents were of analytical grade or better. Purified water from a Milli-Q deionization unit (Millipore, Bedford, MA, USA) was used throughout.
Apparatus
An Aquarius CE7200 UV-spectrophotometer (Cecil Instruments, Cambridge, England) equipped with a thermostatted cell compartment, using 10 mm quartz cuvettes, was used for measurement of the p-nitrophenyl acetate decomposition. Data was collected and transferred to Microsoft Excel using HyperAccess Ver. 8.4 software (Hilgraeve, Monroe, MI, USA). HPLC was performed using a Merck-Hitachi L-6200 pump, a L-4000 UV-detector, and a D-2000 Chromato-Integrator (Merck-Hitachi, Tokyo, Japan) equipped with a Rheodyne 7125 injection valve with a 20 μL loop. A ChromSpher C18 (150 × 4.6 mm; 5 μm particles) column (Chrompack Varian, The Netherlands) was used and the mobile phases consisted of acetonitrile - 0.1 % v/v phosphoric acid in suitable proportions (10 to 70 % v/v of acetonitrile). The flow rate was set at 1 mL/min and the effluent was monitored at 200 nm.
Kinetic measurements
Stability of the phenol derivatives was studied in 0.067 M sodium phosphate buffer solution at 37 ± 0.2oC. The buffer solutions were kept in a water bath at constant temperature, and at appropriate time intervals samples were withdrawn and chromatographed immediately. The experiments were initiated by addition of a stock solution in acetonitrile to the buffer solution providing an organic solvent concentration of 1% or less and an initial ester concentration of approximately 1 x 10-4 M. The pseudo-first-order rate constants were determined by linear plots of the logarithm of intact derivative versus time or by using the initial rate method [83]. Carbonate ester degradation was also studied in 4.3% (w/v) HSA in 0.067 M phosphate buffer (pH 7.40), human plasma diluted to 80% (v/v) with 0.067 M phosphate buffer (pH 7.40), and 20% (w/w) rat liver homogenate at 37oC. The rat liver homogenate was prepared by addition of a calculated amount of 0.067 M phosphate buffer (pH 7.40) to the livers followed by homogenization at 4oC. Samples were withdrawn at appropriate intervals and deproteinized by addition of methanol, acetonitrile or 5% (v/v) perchloric acid. Upon centrifugation the supernatant was analyzed by HPLC.
p-Nitrophenyl acetate – human serum albumin affinity assay
The formation of p-nitrophenolate due to the reaction between p-nitrophenyl acetate and HSA in the presence or absence of ligand (carbonate esters, phenyl acetate, ibuprofen, or octanoic acid) added was monitored as a function of time at 401 nm by UV-Vis spectrophotometry. The cause of reaction was followed in thermostatted quarts cuvettes at 37 ± 0.2oC filled with 0.067 M sodium phosphate buffer (pH 7.40) containing 1% v/v acetonitrile. The reaction solution contained 7 x 10-5 M HSA and the investigated ligand in the concentration range 0 – 3.5 x 10-4 M which was preheated for 10 – 15 min prior to the start of experiments. Reaction was initiated by addition of 10 μL of p-nitrophenyl acetate providing a final concentration of 5 x 10-6 M followed by rapid mixing. The degradation of p-nitrophenyl acetate followed pseudo-first-order kinetics and the rate constants were obtained from the slopes of linear plots of ln (A∞ – At) versus time t, where A∞ and At are the absorbance measured at time infinity and t, respectively [83].
References
- 1.Lipinski C. A., Lombardo F., Dominy B. W., Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Del. Rev. 1997;23:3–25. doi: 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- 2.Avdeef A. Physicochemical profiling (Solubility, permeability and charge state) Curr. Topics Med. Chem. 2001;1:277–351. doi: 10.2174/1568026013395100. [DOI] [PubMed] [Google Scholar]
- 3.Muegge I. Selection criteria for drug-like compounds. Med. Res. Rev. 2003;23:302–321. doi: 10.1002/med.10041. [DOI] [PubMed] [Google Scholar]
- 4.van de Waterbeemd H., Smith D. A., Beumont K., Walker D. K. Property-based design: optimization of drug absorption and pharmacokinetics. J. Med. Chem. 2001;44:1313–1333. doi: 10.1021/jm000407e. [DOI] [PubMed] [Google Scholar]
- 5.Walters W. P., Murcko A., Murcko M. A. Recognizing molecules with drug-like properties. Curr. Opin. Chem. Biol. 1999;3:384–387. doi: 10.1016/S1367-5931(99)80058-1. [DOI] [PubMed] [Google Scholar]
- 6.Bundgaard H. Design of prodrugs: Bioreversible derivatives for various functional groups and chemical entities. In: Bundgaard H., editor. Design of prodrugs. Elsevier; Amsterdam: 1985. pp. 1–92. [Google Scholar]
- 7.Larsen C. S., Østergaard J. Design and application of prodrugs. In: Krogsgaard-Larsen P., Liljefors T., Madsen U., editors. Textbook of drug design and discovery. 3 ed. Taylor & Francis; London: 2002. pp. 410–458. [Google Scholar]
- 8.Testa B., Mayer J. M. Concepts in prodrug design to overcome pharmacokinetic problems. In: Testa B., van de Waterbeemd H., Folkers G., Guy R., editors. Pharmacokinetic optimization in drug research. Verlag Helvetica Chimica Acta/Wiley-VCH; Zürich: 2001. pp. 85–95. [Google Scholar]
- 9.Testa B., Mayer J. M. Hydrolysis in drug and prodrug metabolism. Chemistry, biochemistry, and enzymology. Verlag Helvetica Chimica Acta; Zürich: 2003. [Google Scholar]
- 10.Stella V. J., Borchardt R. T., Hageman M. J., Oliyai R., Maag H., Tilley J. W., editors. Prodrugs: challenges and rewards. Part 1. Springer-AAPS Press; New York: 2007. [Google Scholar]
- 11.Jusko W. J., Gretch M. Plasma and tissue protein binding of drugs in pharmacokinetics. Drug Metab. Rev. 1976;5:43–140. doi: 10.3109/03602537608995839. [DOI] [PubMed] [Google Scholar]
- 12.Kwong T. C. Free drug measurements: methodology and clinical significance. Clin. Chim. Acta. 1985;151:193–216. doi: 10.1016/0009-8981(85)90082-8. [DOI] [PubMed] [Google Scholar]
- 13.Vallner J. J. Binding of drugs by albumin and plasma protein. J. Pharm. Sci. 1977;66:447–465. doi: 10.1002/jps.2600660402. [DOI] [PubMed] [Google Scholar]
- 14.Meyer M. C., Guttmann D. E. The binding of drugs by plasma proteins. J. Pharm. Sci. 1968;57:895–918. doi: 10.1002/jps.2600570601. [DOI] [PubMed] [Google Scholar]
- 15.Peters T., Jr. All about albumin. Biochemistry, genetics, and medical applications. Academic Press; San Diego: 1996. [Google Scholar]
- 16.Kragh-Hansen U., Chuang V. T. G., Otagiri M. Practical aspects of the ligand-binding and enzymatic properties of human serum albumin. Biol. Pharm. Bull. 2002;25:695–704. doi: 10.1248/bpb.25.695. [DOI] [PubMed] [Google Scholar]
- 17.Kurtzhals P., Havelund S., Jonassen I., Kiehr B., Larsen U. D., Ribel U., Markussen J. Albumin binding of insulins acylated with fatty acids: characterization of the ligand-protein interaction and correlation between binding affinity and timing of the insulin effect in vivo. Biochem. J. 1995;312:725–731. doi: 10.1042/bj3120725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurtzhals P., Havelund S., Jonassen I., Markussen J. Effect of fatty acids and selected drugs on the albumin binding of a long-acting, acylated insulin analogue. J. Pharm. Sci. 1997;86:1365–1368. doi: 10.1021/js9701768. [DOI] [PubMed] [Google Scholar]
- 19.Markussen J., Havelund S., Kurtzhals P., Andersen A. S., Halstrøm J., Hasselager E., Larsen U. D., Ribel U., Schäffer L., Vad K., Jonassen I. Soluble, fatty acid acylated insulins bind to albumin and show protracted action in pigs. Diabetologia. 1996;39:281–288. doi: 10.1007/BF00418343. [DOI] [PubMed] [Google Scholar]
- 20.Knudsen L. B., Nielsen P. F., Huusfeldt P. O., Johansen N. L., Madsen K., Pedersen F. Z., Thøgersen H., Wilken M., Agersø H. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J. Med. Chem. 2000;43:1664–1669. doi: 10.1021/jm9909645. [DOI] [PubMed] [Google Scholar]
- 21.Oku N., Yamashita S., Sakuragi n., Doi K., Okada S., Shimidzu K., Sumi M., Nadai T., Kusumoto S., Suda Y. Therapeutic efficacy of 5-fluorouracil prodrugs using endogenous serum proteins as drug carriers: A new strategy in drug delivery system. Biol. Pharm. Bull. 1995;18:181–184. doi: 10.1248/bpb.18.181. [DOI] [PubMed] [Google Scholar]
- 22.Suda Y., Shimidzu K., Sumi M., Oku N., Kusumoto S., Nadai T., Yamashita S. The synthesis and in vitro and in vivo stability of 5-fluorouracil prodrugs which possess serum albumin binding potency. Biol. Pharm. Bull. 1993;16:876–878. doi: 10.1248/bpb.16.876. [DOI] [PubMed] [Google Scholar]
- 23.Yamashita S., Suda Y., Masada M., Nadai T., Sumi M. 5-Fluorouracil derivatives with serum protein binding potencies. Chem. Pharm. Bull. 1989;37:2861–2863. doi: 10.1248/cpb.37.2861. [DOI] [PubMed] [Google Scholar]
- 24.Tove S. B. The esterolytic activity of serum albumin. Biochim. Biophys. Acta. 1962;57:230–235. doi: 10.1016/0006-3002(62)91115-0. [DOI] [PubMed] [Google Scholar]
- 25.Taylor R. P. Enzyme-like activities associated with albumin. In: Rosenoer V. M., Oratz M., Rotschild M. A., editors. Albumin structure, function and uses. 1 ed. Pergamon Press; Oxford: 1977. pp. 183–201. [Google Scholar]
- 26.Means G. E., Bender M. L. Acetylation of human serum albumin by p-nitrophenyl acetate. Biochemistry. 1975;14:4989–4994. doi: 10.1021/bi00693a031. [DOI] [PubMed] [Google Scholar]
- 27.Pond S. M., Tozer T. N. First-pass elimination. Basic concepts and clinical consequenses. Clin. Pharmacokinet. 1984;9:1–25. doi: 10.2165/00003088-198409010-00001. [DOI] [PubMed] [Google Scholar]
- 28.Østergaard J., Larsen C. Bioreversible derivatives of phenol. II. Reactivity of carbonate esters with fatty-acid like structure towards hydrolysis in aqueous solutions. Molecules. 2007;12:2396–2412. doi: 10.3390/12102396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ballinger L. N., Cross S. E., Roberts M. S. Availability and mean transit times of phenol and its metabolites in the isolated perfused rat liver: Normal and retrograde studies using tracer concentrations of phenol. J. Pharm. Pharmacol. 1995;47:949–956. doi: 10.1111/j.2042-7158.1995.tb03276.x. [DOI] [PubMed] [Google Scholar]
- 30.Scott D. O., Lunte C. E. In vivo microdialysis sampling in the bile, blood, and liver of rats to study the disposition of phenol. Pharm. Res. 1993;10:335–342. doi: 10.1023/A:1018971818689. [DOI] [PubMed] [Google Scholar]
- 31.Cassidy M. K., Houston J. B. In vivo assessment of extrahepatic conjugative metabolism in first pass effects using the model compound phenol. J. Pharm. Pharmacol. 1980;32:57–59. doi: 10.1111/j.2042-7158.1980.tb12846.x. [DOI] [PubMed] [Google Scholar]
- 32.Cassidy M. K., Houston J. B. In vivo capacity of hepatic and extrahepatic enzymes to conjugate phenol. Drug Metab. Disp. 1984;12:619–624. [PubMed] [Google Scholar]
- 33.stergaard J., Schou C., Larsen C., Heegaard N. H. H. Evaluation of capillary electrophoresis frontal analysis for the study of low molecular weight drug-human serum albumin interactions. Electrophoresis. 2002;23:2842–2853. doi: 10.1002/1522-2683(200209)23:17<2842::AID-ELPS2842>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 34.Judis J. Binding of selected phenol derivatives to human serum proteins. J. Pharm. Sci. 1982;71:1145–1147. doi: 10.1002/jps.2600711017. [DOI] [PubMed] [Google Scholar]
- 35.Ogata N., Shibata T. Binding of alkyl- and alkoxy-substituted simple phenolic compounds to human serum proteins. Res. Commun. Chem. Pathol. Pharmacol. 2000;107:167–173. [PubMed] [Google Scholar]
- 36.Hansen J., Mørk N., Bundgaard H. Phenyl carbamates of amino acids as prodrug forms for protecting phenols against first-pass metabolism. Int. J. Pharm. 1992;81:253–261. [Google Scholar]
- 37.Fredholt K., Mørk N., Begtrup M. Hemiesters of aliphatic dicarboxylic acids as cyclization-activated prodrug forms for protecting phenols against first-pass metabolism. Int. J. Pharm. 1995;123:209–216. [Google Scholar]
- 38.Thomsen K. F., Bundgaard H. Cyclization-activated phenyl carbamate prodrug forms for protecting phenols against first-pass metabolism. Int. J. Pharm. 1993;91:39–49. [Google Scholar]
- 39.Thomsen K. F., Strøm F., Sforzini B. V., Begtrup M., Mørk N. Evaluation of phenyl carbamates of ethyl diamines as cyclization-activated prodrug forms for protecting phenols against first-pass metabolism. Int. J. Pharm. 1994;112:143–152. [Google Scholar]
- 40.Bundgaard H., Buur A., Chang S.-C., Lee V. H. L. Timolol prodrugs: synthesis, stability and lipophilicity of various alkyl, cycloalkyl and aromatic esters of timolol. Int. J. Pharm. 1988;46:77–88. [Google Scholar]
- 41.Bundgaard H., Buur A., Chang S.-C., Lee V. H. L. Prodrugs of timolol for improved ocular delivery: synthesis, hydrolysis kinetics and lipophilicity of various timolol esters. Int. J. Pharm. 1986;33:15–26. [Google Scholar]
- 42.Brunner-Guenat M., Carrupt P.-A., Lisa G., Testa B., Rose S., Thomas K., Jenner P., Ventura P. Esters of L-dopa: Structure-hydrolysis relationships and ability to induce circling behaviour in an experimental model of hemiparkinsonism. J. Pharm. Pharmacol. 1995;47:861–869. doi: 10.1111/j.2042-7158.1995.tb05755.x. [DOI] [PubMed] [Google Scholar]
- 43.Saari W. S., Schwering J. E., Lyle P. A., Smith S. J., Engelhardt E. L. Cyclization-activated prodrugs. Basic carbamates of 4-hydroxyanisole. J. Med. Chem. 1990;33:97–101. doi: 10.1021/jm00163a016. [DOI] [PubMed] [Google Scholar]
- 44.Nambu K., Miyazaki H., Nakanishi Y., Oh-E Y., Matsunaga Y., Hashimoto M. Enzymatic hydrolysis of haloperidol decanoate and its inhibition by proteins. Biochem. Pharmacol. 1987;36:1715–1722. doi: 10.1016/0006-2952(87)90058-x. [DOI] [PubMed] [Google Scholar]
- 45.stergaard J., Hansen S. H., Larsen C., Schou C., Heegaard N. H. H. Determination of octanol-water partition coefficients for carbonate esters and other small organic molecules by microemulsion electrokinetic chromatography. Electrophoresis. 2003;24:1038–1046. doi: 10.1002/elps.200390120. [DOI] [PubMed] [Google Scholar]
- 46.Krisch K. Carboxylic ester hydrolases. In: Boyer P. D., editor. The enzymes. 3 ed. Academic Press; New York: 1971. pp. 43–69. [Google Scholar]
- 47.Anderson B. D., Conradi R. A., Spilman C. H., Forbes A. D. Strategies in the design of solution-stable, water-soluble prodrugs III: Influence of the pro-moiety on the bioconversion of 21-esters of corticosteroids. J. Pharm. Sci. 1985;74:382–387. doi: 10.1002/jps.2600740404. [DOI] [PubMed] [Google Scholar]
- 48.Nielsen N. M., Bundgaard H. Prodrug as drug delivery systems. 68. Chemical and plasma-catalyzed hydrolysis of various esters of benzoic acid: a reference system for designing prodrug esters of carboxylic acid agents. Int. J. Pharm. 1987;39:75–85. [Google Scholar]
- 49.Larsen C., Kurtzhals P., Johansen M. Kinetics of regeneration of metronidazole from hemiesters of maleic acid, succinic acid and glutaric acid in aqueous buffer, human plasma and pig liver homogenate. Int. J. Pharm. 1988;41:121–129. [PubMed] [Google Scholar]
- 50.Johansen M., Larsen C. Stability and kinetics of hydrolysis of metronidazole monosuccinate in aqueous solution and in plasma. Int. J. Pharm. 1984;21:201–209. [Google Scholar]
- 51.Hansch C., Leo A., Hoekman D. Exploring QSAR. Hydrophobic, electronic, and steric constants. 1 ed. Vol. 2 ACS; Washington: 1995. [Google Scholar]
- 52.Nielsen N. M., Bundgaard H. Glycolamide esters as biolabile prodrugs of carboxylic acid agents: Synthesis, stability, bioconversion, and physicochemical properties. J. Pharm. Sci. 1988;77:285–298. doi: 10.1002/jps.2600770402. [DOI] [PubMed] [Google Scholar]
- 53.Carter D. C., Ho J. X. Structure of serum albumin. Adv. Protein Chem. 1994;45:153–203. doi: 10.1016/S0065-3233(08)60640-3. [DOI] [PubMed] [Google Scholar]
- 54.Kragh-Hansen U. Molecular aspects of ligand binding to serum albumin. Pharmacol. Rev. 1981;33:17–53. [PubMed] [Google Scholar]
- 55.Kragh-Hansen U. Structure and ligand binding properties of human serum albumin. Dan. Med. Bull. 1990;37:57–84. [PubMed] [Google Scholar]
- 56.Vorum H. Reversible ligand binding to human serum albumin. Theoretical and clinical aspects. Dan. Med. Bull. 1999;46:379–399. [PubMed] [Google Scholar]
- 57.Charbon V., Latour I., Lambert D. M., Buc-Calderon P., Neuvens L., De Keyser J.-L., Gallez B. Targeting of drug to the hepatocytes by fatty Acids. Influence of the carrier (albumin or galactosylated albumin) on the fate of the fatty acids and their analogs. Pharm. Res. 1996;13:27–31. doi: 10.1023/a:1016012913664. [DOI] [PubMed] [Google Scholar]
- 58.Gallez B., Debuyst R., Demeure R., Dejehet F., Grandin C., Van Beers B., Taper H., Pringot J., Dumont P. Evaluation of a Nitroxyl Fatty Acid as Liver Contrast Agent for Magnetic Resonance Imaging. Magn. Reson. Med. 1993;30:592–599. doi: 10.1002/mrm.1910300510. [DOI] [PubMed] [Google Scholar]
- 59.Lambert D. M. Rationale and applications of lipids as prodrug carriers. Eur. J. Pharm. Sci. 2000;11(Suppl. 2):S15–S27. doi: 10.1016/S0928-0987(00)00161-5. [DOI] [PubMed] [Google Scholar]
- 60.Hung D. Y., Mellick G. D., Prankerd R. J., Roberts M. S. Synthesis, identification, characterization, stability, solubility, and protein binding of ester derivatives of salicylic acid and diflunisal. Int. J. Pharm. 1997;153:25–39. [Google Scholar]
- 61.Aarons L., Clifton P., Fleming G., Rowland M. Aspirin binding and the effect of albumin on spontaneous and enzyme-catalysed hydrolysis. J. Pharm. Pharmacol. 1980;32:537–543. doi: 10.1111/j.2042-7158.1980.tb12991.x. [DOI] [PubMed] [Google Scholar]
- 62.Wolfbeis O. S., Gürakar A. The effect of fatty acid chain length on the rate of arylester hydrolysis by various albumins. Clin. Chim. Acta. 1987;164:329–337. doi: 10.1016/0009-8981(87)90308-1. [DOI] [PubMed] [Google Scholar]
- 63.Koh S.-W. M., Means G. E. Characterization of a small apolar anion binding site of human Serum albumin. Arch. Biochem. Biophys. 1979;192:73–79. doi: 10.1016/0003-9861(79)90072-9. [DOI] [PubMed] [Google Scholar]
- 64.Sakurai Y., Ma S.-F., Watanabe H., Yamaotsu N., Hirono S., Kurono Y., Kragh-Hansen U., Otagiri M. Esterase-like activity of serum albumin: characterization of its structural chemistry using p-nitrophenyl esters as substrates. Pharm. Res. 2004;21:285–292. doi: 10.1023/B:PHAM.0000016241.84630.06. [DOI] [PubMed] [Google Scholar]
- 65.Salvi A., Carrupt P.-A., Mayer J. M., Testa B. Esterase-like activity of human serum albumin toward prodrug esters of nicotinic acid. Drug Metabol. Dispos. 1997;25:395–398. [PubMed] [Google Scholar]
- 66.Steiner A., Mayer J. M., Testa B. Nicotinate esters: Their binding to and hydrolysis by human serum albumin. J. Pharm. Pharmacol. 1992;44:745–749. doi: 10.1111/j.2042-7158.1992.tb05512.x. [DOI] [PubMed] [Google Scholar]
- 67.Whelpton R., Hurst P. R. The binding of physostigmine to human serum albumin. J. Pharm. Pharmacol. 1990;42:804–805. doi: 10.1111/j.2042-7158.1990.tb07027.x. [DOI] [PubMed] [Google Scholar]
- 68.Chapuis N., Brühlmann C., Reist M., Carrupt P.-A., Mayer J. M., Testa B. The esterase-like activity of serum albumin may be due to cholinesterase contamination. Pharm. Res. 2001;18:1435–1439. doi: 10.1023/a:1012204906502. [DOI] [PubMed] [Google Scholar]
- 69.Stella V. J., Charman W. N., Naringrekar V. H. Prodrugs. Do they have advantages in clinical practice? Drugs. 1985;29:455–473. doi: 10.2165/00003495-198529050-00002. [DOI] [PubMed] [Google Scholar]
- 70.Sudlow G., Birkett D. J., Wade D. N. The characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol. 1975;11:824–832. [PubMed] [Google Scholar]
- 71.Sudlow G., Birkett D. J., Wade D. N. Further characterization of specific drug binding sites on human serum albumin. Mol. Pharmacol. 1976;12:1052–1061. [PubMed] [Google Scholar]
- 72.Fehske K. J., Müller W. E., Wollert U. The location of drug binding sites in human serum albmin. Biochem. Pharmacol. 1981;30:687–692. doi: 10.1016/0006-2952(81)90151-9. [DOI] [PubMed] [Google Scholar]
- 73.Kragh-Hansen U. Evidence for a large and flexible region of human serum albumin possessing high affinity binding sites for salicylate, warfarin, and other ligands. Mol. Pharmacol. 1988;34:160–171. [PubMed] [Google Scholar]
- 74.Wanwimolruk S., Birkett D. J., Brooks P. M. Structural requirements for drug binding to site II on human serum albumin. Mol. Pharmacol. 1983;24:458–463. [PubMed] [Google Scholar]
- 75.Irikura M., Takadate A., Goya S., Otagiri M. 7-Alkylaminocoumarin-4-acetic acids as fluorescent probe for studies of drug-binding sites on human serum albumin. Chem. Pharm. Bull. 1991;39:724–728. doi: 10.1248/cpb.39.724. [DOI] [PubMed] [Google Scholar]
- 76.Sollenne N. P., Means G. E. Characterization of a specific drug binding site of human serum albumin. Mol. Pharmacol. 1979;15:754–757. [PubMed] [Google Scholar]
- 77.Kurono Y., Ohta N., Yotsuynagi T., Ikeda K. Effect of drug binding on the esterase-like activity of human serum albumin. III. Evaluation of reactivities of the two active sites by using clofibric acid as an inhibitor. Chem. Pharm. Bull. 1981;29:2345–2350. doi: 10.1248/cpb.29.2345. [DOI] [PubMed] [Google Scholar]
- 78.Watanabe H., Tanase S., Nakajou K., Maruyama T., Kragh-Hansen U., Otagiri M. Role of Arg-410 and Tyr-411 in human serum albumin for ligand binding and esterase-like activity. Biochem. J. 2000;349:813–819. doi: 10.1042/bj3490813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kurono Y., Ozeki Y., Yamada H., Takeuchi T., Ikeda K. Effects of drug binding on the esterase-like activity of human serum albumin. VII. Subdivision of R-type drugs inhibiting the activity towards p-nitrophenyl acetate. Chem. Pharm. Bull. 1987;35:734–739. doi: 10.1248/cpb.35.734. [DOI] [PubMed] [Google Scholar]
- 80.Ozeki Y., Kurono Y., Yotsuynagi T., Ikeda K. Effects of drug binding on the esterase activity of human serum albumin: Inhibition modes and binding sites of anionic drugs. Chem. Pharm. Bull. 1980;28:535–540. doi: 10.1248/cpb.28.535. [DOI] [PubMed] [Google Scholar]
- 81.Kurono Y., Ikeda K. Effect of drug binding on the esterase-like activity of human serum albumin. IV. Application of an analog computer to determination of the multiple dissociation constants. Chem. Pharm. Bull. 1981;29:2993–3002. doi: 10.1248/cpb.29.2993. [DOI] [PubMed] [Google Scholar]
- 82.Weisiger R. A. Dissociation from albumin: A potentially rate-limiting step in the clearance of substances by the liver. Proc. Natl. Acad. Sci. USA. 1985;82:1563–1567. doi: 10.1073/pnas.82.5.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Connors K. A. Chemical kinetics. The study of reaction rates in solution. VCH Publishers; New York: 1990. [Google Scholar]