

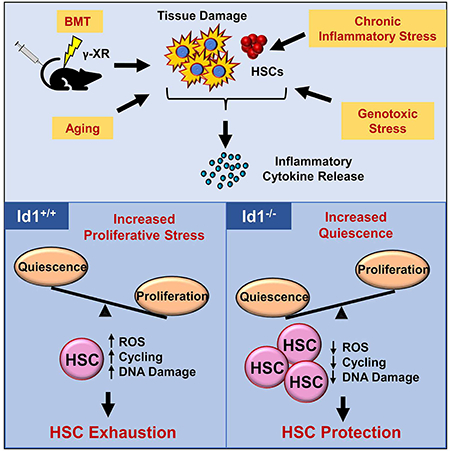
SUMMARY
Defining mechanisms that maintain tissue stem cells during homeostasis, stress and aging, is important for improving tissue regeneration and repair, and enhancing cancer therapies. Here we show Id1 is induced in hematopoietic stem cells (HSCs) by cytokines that promote HSC proliferation and differentiation, suggesting that it functions in stress hematopoiesis. Genetic ablation of Id1 increases HSC self-renewal in serial bone marrow transplantation (BMT) assays, correlating with decreases in HSC proliferation, mitochondrial biogenesis, and ROS production. Id1−/− HSCs have a quiescent molecular signature and harbor less DNA damage than control HSCs. Cytokines produced in the hematopoietic microenvironment after γ-irradiation induce Id1 expression. Id1−/− HSCs display a blunted proliferative response to such cytokines and other inducers of chronic proliferation including genotoxic and inflammatory stress, and aging, protecting them from chronic stress and exhaustion. Thus, targeting Id1 may be therapeutically useful for improving HSC survival and function during BMT, chronic stress, and aging.
Keywords: Stem cells, hematopoiesis, self-renewal, quiescence, Id proteins, proliferative stress, bone marrow transplantation, aging
eTOC:
Singh et al. show that Id1 is an important mediator of stress hematopoiesis. They found that Id1 deletion protects HSCs from exhaustion in multiple paradigms of chronic and physiologicallyrelevant stress and promotes their quiescence, suggesting that targeting Id1 may improve HSC survival and function during chronic stress and aging.
INTRODUCTION
Hematopoiesis is an excellent vertebrate developmental system to study adult stem cell biology, and to uncover mechanism(s) that regulate tissue stem cell quiescence, proliferation, self-renewal, cell fate and differentiation (Orkin and Zon, 2008; Seita and Weissman, 2010). Most hematopoietic stem cells (HSCs) are quiescent under homeostasis, and cycle rarely to selfrenew or differentiate into multipotent progenitors (MPPs), and more committed progenitors, with limited self-renewal potential (Nakamura-Ishizu et al., 2014). Hematopoiesis is tightly regulated by both intrinsic and extrinsic mechanism(s), which balance quiescence, self-renewal and differentiation to maintain normal multi-lineage reconstitution (Mendelson and Frenette, 2014; Morrison and Scadden, 2014). While our knowledge of gene function and their mechanism(s) of action has greatly expanded, the precise pathways that maintain HSC quiescence, and prevent HSC exhaustion and subsequent bone marrow failure, remain to be fully elucidated.
Inhibitor of DNA binding (Id1-4) proteins are helix-loop-helix (HLH) transcription factors that lack a basic region found in other family members, which is required for DNA binding (Ling et al., 2014; Perk et al., 2005). Id proteins bind to ubiquitously expressed basic(b)HLH E proteins and disrupt their ability to bind DNA, thus inhibiting their transcriptional activity. E proteins are required for proper differentiation and cell cycle arrest of adult muscle, neural, endothelial, and hematopoietic lineage cells (Kee, 2009; Wang and Baker, 2015). As key regulators of E proteins, Id proteins have been implicated in neural, epithelial, and hematopoietic stem and progenitor cell proliferation and self-renewal (Lasorella et al., 2014). Id proteins are often deregulated in human cancers, where they contribute to tumor growth, invasiveness, metastasis, and self-renewal of cancer stem cells (CSCs) (Lasorella et al., 2014; Perk et al., 2005). Thus, a complete understanding of the function of Id genes in normal adult stem cells and CSCs could lead to the development of novel therapies.
Mice that lack Id1 (Id1−/−) develop normally and show no overt phenotypes; however, these mice have dysregulated hematopoiesis which includes decreased bone marrow cellularity, increased hematopoietic progenitor cycling, decreased B cell numbers, and increased myeloid cell output in the bone marrow (Jankovic et al., 2007; Perry et al., 2007; Suh et al., 2009). These phenotypes are due, in part, to cell non-autonomous effects of Id1 in the hematopoietic microenvironment (HME), since Id1−/− bone marrow cells (BMCs) show normal development when transplanted into γ-irradiated (γIR) Id1+/+ recipient mice (Suh et al., 2009). The number of HSCs are roughly the same in Id1−/− and Id1+/+ under homeostasis and Id1 is expressed at low levels in HSCs, suggesing that Id1 may not be required to maintain HSCs during steady-state hematopoiesis. However, Id1 is induced in HSPCs by growth factors that promote myeloid proliferation and differentiation including IL-3, and enforced expression of Id1 in HSPCs promotes myeloid proliferation, implicating Id1 as a potential modulator of HSC function, including proliferation, self-renewal and differentiation under conditions of hematopoietic stress (Cochrane et al., 2009; Leeanansaksiri et al., 2005; Suh et al., 2008). Therefore, we examined the intrinsic role of Id1 in HSC stress using serial bone marrow transplantation (BMT) assays.
We found that Id1−/− HSCs show enhanced self-renewal potential, and are maintained during serial BMT. Id1−/− HSCs show reduced cycling and proliferation and increased quiescence after BMT. Id1−/− HSC quiescence is associated with reduced levels of γH2AX phosphorylation, reduced mitochondrial biogenesis and stress, and lower ROS levels. Id1−/− HSCs are protected from cytokine-induced proliferative stress in vitro. HSCs express low levels of Id1 under homeostasis; however, Id1 is induced in HSCs after BMT, in part, by proinflammatory cytokines present in the HME after γIR. Id1−/− HSCs are protected from exhaustion by other conditions that model chronic physiological stress including toll-like receptor (TLR) signaling and aging.
RESULTS
Hematopoietic stem cells that lack Id1 have enhanced self-renewal potential.
Since Id1 is induced in HSPCs by cytokines in vitro, and overexpression of Id1 promotes HSPC proliferation (Cochrane et al., 2009; Suh et al., 2008), we hypothesized that Id1 may have an important function in stress hematopoiesis. First, we backcrossed conventional Id1 knockout mice (Id1−/−) maintained on a mixed B6;129 background with C57BL/6 mice for ten generations, to study mice on a pure genetic background. We found that the cell-non autonomous hematopoietic phenotype previously reported for Id1−/− mice on the mixed background to be less severe on the pure C57BL/6 background (Suh et al., 2009). Specifically, loss of Id1 in the C57BL/6 background did not result in differences in myeloid and lymphoid cell development in peripheral blood cells (PBCs) or BMCs (Figures S1A-B). In addition, the previously observed reduction in BM cellularity was not as pronounced (Figure S1C), the increase in lineage-negative Sca-1+c-Kit+(LSK) and HSPC populations was less severe (Figure S1D-E), and no effect on HSC numbers was observed (Figure S1E, and summarized Figure1F). We performed competitive serial repopulation assays to evaluate the function of Id1−/− BMCs, and found that mice transplanted with Id1+/+ BMCs did not survive beyond the fourth serial BMT due to HSC exhaustion, while donor Id1−/− BMCs survived a fourth, fifth and sixth BMT, and succumbed to exhaustion after the seventh BMT (Figure 1A). This observation was confirmed using noncompetitive serial BMTs, in which Id1+/+ BMCs failed to support hematopoiesis after the third BMT, while Id1−/− donor derived BMCs survived through tertiary transplantation. The Id1−/− BMCs failed to promote the survival of quaternary BMT recipient mice (Figure S2A). Collectively, these data suggest that Id1−/− HSCs have enhanced self-renewal potential.
Figure 1. Ablation of Id1 enhances self-renewal of HSCs.

(A) Kaplan-Meier survival curves of mice competitively transplanted with Id1+/+ and Id1−/− BMCs. (B) Percentage of donor-derived (CD45.2+) BMCs in serially transplanted recipients. (C) Quantitation of of lineage repopulation in BM and spleen cells from primary BMT recipient mice. (D) Frequency of donor-derived Id1−/− and Id1+/+ HSCs in serial BMT recipient mice. using the gating strategy in Figure S2B. (E) Total number of donor-derived Id1−/− and Id1+/+ HSCs in serial BMT recipient mice. Representative FACS analysis of donor HSCs in BMCs from secondary BMT recipient mice (left). See also Figure S1/S2.
To understand how Id1−/− HSCs are maintained during serial BMT, we examined donor cell engraftment by flow cytometry. No statistically significant differences in the percentage of donor reconstitution in primary recipients transplanted with Id1+/+ or Id1−/− BMCs was observed, suggesting that Id1−/− and Id1+/+ BMCs contain roughly equivalent numbers of HSCs (Figure 1B). No differences in lineage development were found, other than the expected decreased percentage of mature B cells, previously reported for Id1−/− mice (Figure 1C) (Cochrane et al., 2011). However, donor reconstitution was significantly decreased in second and third serial recipients that received Id1+/+ BMCs, and the fourth serial transplant recipients did not survive. Strikingly, Id1−/− donor BMC engraftment was sustained through 7 serial transplants, suggesting that Id1−/− HSCs have enhanced self-renewal potential (Figure 1B). As predicted, the total number of donor-derived HSCs was roughly equivalent in primary Id1+/+ and Id1−/− BMT recipients 14 weeks after BMT (Figure 1D-E and Figure S2B). However, donor Id1−/− HSCs were significantly increased in the 2nd and 3rd BMT recipient mice compared to Id1+/+ HSCs, and gradually declined in serially transplanted mice, but were sustained through six serial BMTs. Recipients of the fourth Id1+/+ BMT failed to reconstitute due to exhaustion of donor HSCs. Additional immunophenotypic analysis using antibodies that recognize CD34, confirmed that Id1+/+ HSCs were significantly decreased in the 2nd and 3rd serial BMT recipients, while Id1−/− HSCs were maintained through six serial BMTs (Figure 1E). No difference in the homing of Id1−/− and Id1+/+ Lin−, LSK, or HSCs was observed, suggesting alternative mechanism(s) for the increased self-renewal of Id1−/− HSCs (Figure S2C). Thus, Id1−/− HSCs have enhanced self-renewal potential and are maintained during serial BMT with normal lineage developmental potential and homing function.
Loss of Id1 in HSCs could affect the development of progenitors downstream of HSCs; therefore, we analyzed HSPC populations in secondary BMT recipients. As expected, we observed an increase in the number of donor HSCs and ST-HSCs in mice transplanted with Id1−/− BMC; however, there were no detectable differences in MPPs (Figure S2D). Thus, while the total number of Id1−/− HSCs were higher in secondary BMT recipient mice compared to Id1+/+ recipients, the number of committed MPPs was the same, suggesting a critical function for Id1 in HSCs under conditions of BMT related stress.
Hematopoietic stem cells that lack Id1 show increased quiescence after bone marrow transplantation
Ionizing radiation (γ-IR) is used to condition patients that receive BMT; however, γ-IR induces a proinflammatory cytokine storm and increases the production of reactive oxygen species (ROS), promoting DNA damage in hematopoietic and HME cells (Kim et al., 2014). Quiescent donor HSCs are induced to proliferate and differentiate in primary γ-IR recipients, and serial BMT eventually leads to bone marrow failure and aplasia due to HSC exhaustion. Since Id1 promotes HSPC proliferation (Leeanansaksiri et al., 2005), and replication stress can drive the functional decline of HSCs (Beerman et al., 2013; Flach et al., 2014; Walter et al., 2015), we examined the proliferative status of Id1−/− HSCs in primary BMT recipient mice. Id1−/− HSCs, but not Id1−/− STHSCs and Id1−/− MPPs, showed a significant increase in G0, and a reduction in G1 and S/G2/M, compared to Id1+/+ HSCs 12–14 weeks after BMT, indicating that fewer Id1−/− HSCs are in cycle compared to Id1+/+ HSCs (Figure 2A and Figure S3A). We compared HSC cycling 4 and 8 weeks after BMT, and found that few donor Id1+/+ and Id1−/− HSCs were in G0 4 weeks after BMT, and most (>90%) were in G1 and S/G2/M (Figure S3B). There was a decrease in the number of Id1−/− HSCs in G1 compared to Id1+/+ BMCs, and a trend toward an increase in the number of Id1−/− HSCs in G0. In comparison, 8 weeks after BMT, there are increased numbers of Id1−/− and Id1+/+ HSCs in G0; however, there is a significant decrease in the number of Id1−/− HSCs in S/G2/M and an increase in G1 compared to Id1+/+ HSCs (Figure S3B). Taken together, Id1−/− HSCs show decreased cycling that begins 4–8 weeks after BMT, and is maintained for 3–4 months after BMT, suggesting that Id1−/− HSCs show prolonged resistance to proliferative stress after BMT.
Figure 2. Increased quiescence of Id1−/− HSCs after bone marrow transplantation.

(A-B) Quantitation of Id1−/− and Id1+/+ HSC cycling in primary recipient mice by Ki-67/FxCycle analysis. (n=5 mice/group). (C) Quantitation of γH2AX phosphorylation expression levels of Id1−/− and Id1+/+ HSC in primary BMT recipient mice by flow cytometry ( n=5 mice/group). (D) Immunoflourescence analysis of γ-H2AX foci in Id1−/− and Id1+/+ HSCs FACS sorted from primary BMT recipient mice by confocal microscopy. Mann-Witney test p<0.00086, and z-score −3.33. (E) Kaplan-Meier survival curves of chimeric Id1+/+ and Id1−/− recipient mice treated with 5-FU. Mantel-Cox test P=0.034 (n=10 mice/group). (F) Quanitification of HSCs in chimeric Id1+/+ and Id1−/− BMT recipient mice treated with 5-FU (n=5 mice/group). (G) Quantification of HSC proliferation in Id1−/− and Id1+/+ chimeric mic after 5-FU-induced proliferative stress by BrdU incorporation assay (n=5 mice/group). See also Figure S3/S4.
Since DNA replicative stress is accompanied with γH2AX phosphorylation at stalled replication forks (Branzei and Foiani, 2010; Flach et al., 2014; Walter et al., 2015), we measured the phosphorylation of histone H2AX S139 phosphoyrlation (γH2AX) in Id1−/− HSCs 12–14 wks after BMT. Flow cytometry analysis of Id1−/− HSCs showed a significant reduction in γH2AX levels compared with Id1+/+ HSCs, suggesting that Id1−/− HSCs accumulated less DNA damage after BMT in primary recipient mice (Figure 2C). Id1−/− HSCs showed reduction in γH2AX 4 weeks after BMT, indicating that this occurs relatively early after BMT (Figure S3C). Photomicrographs of γH2AX stained HSCs show reduced γH2AX phosphorylation in Id1−/− HSCs compared to Id1+/+ HSCs (Figure 2D). To validate the specificity of γH2AX staining, we analyzed levels of γH2AX phosphorylation in host versus donor Lin− cells in primary recipient mice. As expected, the level of γH2AX staining was higher in host cells, which received a lethal dose of γIR compared to the non irradiated donor cells (Figure S4A). Examination of another marker of DNA damge, 53BP1, showed increased 53BP1 flourescence in Id1−/− HSCs compared to Id1+/+ in HSCs 12 weeks after BMT (Figure S4B). Recent evidence suggests that 53BP1 binding is increased on unreplicated DNA, while newly replicated DNA shows reduced 53BP1 binding (Pellegrino et al., 2017). Thus, the increased 53BP1 floursecence that we observe in Id1−/− HSC directly correlates with reduced replicative stress and increased quiescence.
Furthermore, we provide evidence that the reduced cell cycling and DNA damage in Id1−/− HSCs does not lead to increased cell death, as no difference in the number of apoptotic and dead cells was observed in Id1−/− and Id1+/+ transplanted HSPCs (Figure S4C).
To rule out that Id1 has direct role in DNA damage repair, Id1+/+ and Id1−/− purified BMCs (Lin− or LSK) were cultured for 16 hours in serum free media with mSCF and hTPO, exposed to 6 Gy γ-IR, then analyzed for γH2AX staining after 1, 4.5 and 24 hours (Figure S4D). No differences in the percentages of γH2AX+ cells were detected during the 24-hour period, indicating that Id1+/+ and Id1−/− Lin− and LSK cells have similar repair kinetics after γ-IR, and suggest no direct role for Id1 in DNA damage repair. Thus, the reduced levels of γH2AX phosphorylation in Id1−/− HSCs compared to Id1+/+ after serial BMT is not a direct effect of Id1 in the DNA damage response. Rather, the data suggest that the levels of γH2AX phosphorylation reflect reduced replication stress and increased quiescence of Id1−/− HSC during serial BMT. Thus, loss of Id1 reduces replicative stress, thereby helping to preserve the number and function of Id1−/− HSCs during serial BMT.
Since Id1−/− HSCs show reduced cycling after BMT, we asked if Id1−/− HSCs might proliferate less in other in vivo models of chronic proliferative stress. Therefore, we examined serial administration of a sublethal dose of 5-fluorouracil (5-FU) to chimeric Id1−/− and Id1+/+ mice, which induces chronic proliferative stress, stem cell exhaustion and bone marrow failure (Harrison and Lerner, 1991). We observed a significant delay in 5-FU induced hematopoietic failure in Id1−/− recipients, where 50% of the Id1−/− mice survived a third administration of 5-FU, while most Id1+/+ recipient mice did not survive beyond the second 5-FU treatment (Figure 2E). HSPC analysis of BMCs from chimeric mice treated with 5-FU (2x) confirm that BMCs from mice transplanted with Id1−/− BMCs contain roughly twice as many donor HSCs as mice transplanted with Id1+/+ BMCs (Figure 2F). In addition, the Id1−/− HSCs show a significant reduction in proliferation compared to Id1+/+ cells, as measured by BrdU incorporation assays (Figure 2G). Finally, BMCs from 5-FU-treated Id1−/− mice show increased donor repopulation in competitively transplanted mice 12 weeks after BMT (Figure S4E), and give rise to increased numbers of immunophenotypic HSCs compared to Id1+/+(Figure S4F). This suggests that there are increased numbers of of functional HSCs after 5-FU treatment in chimeric Id1−/− mice. These results are consistent with the interpretation that Id1−/− HSCs remain quiescent after 5-FU induced proliferative stress compared to Id1+/+ HSCs, and thus, are able to provide hematopoietic protection from serial 5-FU-induced proliferative stress.
Id1−/− HSCs show reduced mitochondrial and cellular stress after bone marrow transplantation
HSCs induced to proliferate in vitro and in vivo show increased mitochondrial biogenesis, increased mitochondrial stress, and increased reactive oxygen species (ROS) generation, which damage DNA and can lead to functional exhaustion of HSCs (Flach et al., 2014; Walter et al., 2015). Therefore, we examined the metabolic activity of Id1−/− and Id1+/+ HSCs in secondary BMT recipient mice, since these mice show a significant difference in the number of HSCs. Id1−/− HSCs, but not ST-HSCs or MPPs, showed reduce mitochondrial biogenesis compared to Id1+/+ HSCs after BMT (Figure 3A). Furthermore, Id1−/− HSCs showed reduced mitochondrial stress compared to Id1+/+ HSCs by JC-1 fluorescent staining, which measures mitochondrial membrane depolarization (Figure 3B). Recent studies indicate that HSCs with low mitochondrial membrane potential are enriched for HSCs with increased competitive repopulation ability (Sukumar et al., 2016; Vannini et al., 2016). We observed a significant decrease in ROS production in Id1−/− HSCs compared to Id1+/+ HSCs (Figure 3C), which agrees with reduced mitochondrial metabolic activity of Id1−/− HSCs (Figure 3B). Finally, Id1−/− HSCs show significantly higher levels of reduced thiols, known to correlate with reduced glutathione levels, as indicated by ThiolTracker Violet staining (Figure 3D). Collectively, these data show that Id1−/− HSCs have reduced mitochondrial biogenesis and metabolic activity, less ROS production, and reduced signs of oxidative stress. Together, these results are consistent with the proposed model that Id1−/− HSCs are more quiescent and resistant to BMT-induced hematopoietic stress.
Figure 3. Reduced mitochondrial stress and ROS production in Id1−/− HSCs after bone marrow transplantation.

(A) Quantification of mitochondrial biogenesis in Id1−/− and Id1+/+ HSPCs from secondary BMT recipients using MitoTracker Green and flow cytometry(n=5 mice/group). (B) Quantification of mitochondrial stress in Id1−/− and Id1+/+ HSCs from secondary BMT recipients using JC-1 to measure mitochondrial membrane depolarization. (Percentage JC-1 ratios = % FITC/(% FITC + % PE) x100) (n=5 mice/group). (C) ROS expression levels in Id1−/− and Id1+/+ HSCs from secondary BMT recipients using CellRox Deep Red (n=5 mice/group). (D) Quantification of intracellular reduced thiols in Id1−/− and Id1+/+ HSCs from secondary BMT recipients using ThiolTraker Violet (n=5 mice/group). See also Figure S5.
Since Id1 is expressed at low levels in HSCs, and is induced under conditions of replicative stress, we hypothesized that absence of Id1 would show little effect on HSCs during steady-state hematopoiesis. Therefore, we examined mitochondrial biogenesis/stress and ROS production in Id1+/+ and Id1−/− HSCs under homeostasis. No difference in mitochondrial biogenesis, and mitochondrial stress, as measured by Tetramethyrhodamine methyl ester (TMRM) staining was observed in Id1−/− and Id1+/+ HSCs in young (2 month) and middle aged (1 year) mice (Figure S5B-C). In addition, no difference in ROS production was observed in Id1−/− and Id1+/+ HSCs in young mice; however, we observed reduced levels of ROS in middle aged Id1−/− HSCs compared to Id1+/+ HSCs (Figure S5A). Thus, no differences in mitochondrial biogenesis/stress or production of ROS is observed in young and middle aged Id1−/− and Id1+/+ HSCs under homeostasis, except that Id1−/− HSCs from middle aged mice show reduced ROS production compared to Id1+/+ HSCs.
Id1 is induced in HSCs after bone marrow transplantation and by proinflammatory cytokines
Id1 mRNA is expressed at low levels in LSK cells, with elevated levels observed in committed myeloid progenitors (CMPs, GMPs, and neutrophils) (Leeanansaksiri et al., 2005; Perry et al., 2007), suggesting that steady-state levels of Id1 are low in HSPCs, and increase during normal myeloid differentiation. Myeloid growth factors, including interleukin-3 (IL-3), have been shown to induce expression of Id1 mRNA in LSK, and overexpression of Id1 in normal HSPCs promotes proliferation (Cochrane et al., 2009; Suh et al., 2008). Thus, we reasoned that Id1 may be induced in HSCs by the cytokine storm produced in the HME after γIR, which promotes HSC proliferation and hematopoietic reconstitution (Kim et al., 2014; Schaue et al., 2012). To measure induction of Id1 expression in HSCs, we used Id1-GFP reporter knock-in mice (Id1GFP/+), which accurately reports Id1 expression in normal BMCs (Perry et al., 2007). We confirmed that HSCs, ST-HSCs, and MPPs expressed low levels of Id1(GFP), and that Id1(GFP) expression increases in GMPs, MEPs and neutrophils (Figure 4A). BMCs from Id1GFP/+ mice were cultured in serum free medium containing cytokines or known proinflammatory agents for 36 hours, and then examined for Id1(GFP) expression in HSCs by flow cytometry. SCF did not induce Id1(GFP ) expression in HSCs, while SCF with IFNγ, TPO, IL-6, or LPS induced high levels of Id1(GFP) expression. SCF plus IL-3, GM-CSF, FLT3L, pI:pC, IL-1β, IFNα, or TNFα induced comparatively lower levels of GFP expression in HSCs (Figure 4B). To confirm that GFP expression accurately reflects Id1 protein levels, we purified GFPHi and GFPLo/Med cells cultured in SCF/TPO/IFNγ for 36 hours by FACS, and show that Id1 protein levels directly correlate with the level of GFP expression in these cells (Figure S6A). Taken together, HSCs express low levels of Id1 under steady-state conditions, and Id1 expression can be increased in these cells by cytokines and inflammatory agents in vitro.
Figure 4. Id1 expression is upregulated in hematopoietic stem and progenitor cells by cytokines and proinflammatory agents.

(A) Expression of Id1(GFP) in HSPCs in Id1GFP/+ mice (n=5 mice/group). (B) Cytokine/growth factor induced Id1(GFP) expression in HSCs from Lin- Id1GFP/+ reporter BMC cultures. Dashed line indicates percent Id1(GFP) expression for cells cultured in SCF alone. (C) Cytokine/growth factor induced proliferation of Id1(GFP+) and Id1(GFP−) HSCs from Lin- Id1GFP/+ reporter BMC cultures. (D) γ-irradiation, cytokines and proinflammatory mediator induction of Id1(GFP) expression in HSCs from Id1GFP/+ reporter BMCs in vivo. Dashed lined indicates control value for mice treated with saline (n=5 mice/group). (E) Effect of pan-JAK and pan-STAT inhibitors on the induction of Id1(GFP) expression in HSCs following BMT of Id1GFP/+ reporter BMCs. (n=5 mice/group). See also Figure S6.
We have shown that overexpression of Id1 transgene in HSPCs promotes progenitor cell proliferation in vitro and in vivo (Suh et al., 2008). To determine if growth-factor-induced Id1 expression correlates with increased proliferation, we measured BrdU incorporation in Id1(GFP+) HSCs compared to Id1(GFP−) HSCs. Lin- BMCs from Id1GFP/+ reporter mice were cultured in SCF, SCF/TPO or SCF/IFNγ for 48 hours, and then BrdU was added for the last 3 hours of incubation. GFP+ HSCs show increased BrdU incorporation in cultures containing SCF/TPO and SCF/IFNγ compared to cultures containing SCF alone. In comparison, GFP- showed significantly less BrdU incorporation (Figure 4C), indicating that cytokine induced Id1(GFP) expression correlates with increased proliferation of HSCs.
We hypothesize that radiation-induced damage to the HME can increase Id1 expression in HSCs; therefore, we treated Id1GFP/+ reporter mice with increasing doses of γ-IR and examined GFP expression in HSCs after 48 and 72 hrs. The expression of Id1(GFP) in HSCs was upregulated with increasing doses of γIR (4, 6, or 8 Gy) from an average of 7% to 25% 48 hours post γ-IR, and up to 45% 72 hours after treatment (Figure S6B). Further, we examined if the administration of individual cytokines could increase the expression of Id1(GFP) in HSCs in vivo. To rule out any effect of the Id1−/− HME, we generated chimeric mice by transplanting BMCs from Id1GFP/+ reporter mice (CD45.2+) into Id1+/+ recipient mice (CD45.1+). Six Gy γIR induced high levels of Id1(GFP) in donor HSCs. Administration of IFNγ, pI:pC and, to a lesser extent, IL-6 also induced Id1(GFP) expression in HSCs compared to control mice (Figure 4D). Thus, Id1 expression is induced in HSCs after γIR, and by cytokines produced in the HME after γIR.
Cytokines produced as part of the cytokine storm in the HME after γIR may function alone, or in combination, to induce Id1 expression in HSCs. Because of the complexity of this milieu, it would be difficult to predict which individual cytokines to knock down to reduce Id1 induction in HSCs in vivo. We previously found that JAK2 inhibitors block IL-3-induced Id1 expression in the MPP cell line, EML, suggesting that Id1 is downstream of JAK-2 activation (data not shown). Furthermore, we show here that IFNγ, IL-6, TPO, GM-CSF and other cytokines, which signal through the JAK/STAT pathway also induce GFP (Id1). Therefore, we reasoned that treating mice with pan-JAK (momelotinib) or pan-STAT (SH-4–54) inhibitors would inhibit the signaling of multiple cytokines, and would thus inhibit induction of Id1 expression in HSCs in vivo. To test this, mice were pretreated with momelotinib and SH-4–54, exposed γIR, and transplanted with Lin− Id1GFP/+ BMCs, whereby a subsequent analysis of GFP (Id1) expression in HSCs was performed (Figure 4E). Momelotinib and SH-4–54 significantly inhibited the induction of GFP (Id1) expression in donor LSK and HSCs 48 hours after BMT. This demonstrates that Id1 is induced in HSCs after BMT in γIR recipient mice, in part, via cytokines that signal through the JAK/STAT pathway.
Id1−/− HSCs are resistant to cytokine-induced proliferative stress in vitro
Since Id1 drives HSPC cycling, and Id1 induction is downstream of cytokines and inflammatory signals, we speculated that Id1−/− HSCs may be protected from cytokine induced proliferation in vitro. To test this, we cultured Id1−/− and Id1+/+ LSK cells in cytokine conditions known to promote HSC expansion/proliferation in vitro, and determined how many HSCs persist in culture over time (Dahlberg et al., 2011; Walasek et al., 2012). While the total number of HSCs declined in all cultures after 6 days as expected, there were significantly more Id1−/− HSCs surviving in culture compared to Id1+/+ HSCs (Figure 5A and Figure S6C). To determine how Id1−/− HSCs are maintained in these cultures, we examined the cycling status of HSCs, and found that half as many Id1−/− HSCs were in cycle (6%) compared to Id1+/+ HSCs (12%) (Figure 5B). Furthermore, we found that Id1−/− HSCs are proliferating less after 6 days in culture compared to Id1+/+ HSCs in BrdU-incorporation assays (Figure 5C). Finally, Id1−/− LSK cells cultured for 6 days in vitro showed increased donor repopulation in competitive repopulation assays compared to Id1+/+ LSK cultured cells, indicating that increased numbers of Id1−/− HSCs are maintained in LSK cultures after 6 days (Figure S6D). These results are consistent with the interpretation that Id1−/− HSCs proliferate and differentiate less in response to cytokines in vitro, which may be a part of the mechanism by which the Id1−/− HSCs maintain their presence and function during serial BMT.
Figure 5. Increased survival and quiescence of Id1−/− HSCs in vitro.

(A) Total number of HSCs present in Id1+/+ and Id1−/− LSK cell cultures after 6 days in vitro culture. (B) HSC cycling in LSK cell cultures after 6 days determined by Ki-67/FxCycle staining, and (C) proliferation by BrdU incorporation. (D) HSC single cell division assay. (E) Total number of HSCs in Id1−/− and Id1−/− chimeric mice treated with LPS (0.043 mg/kg) every other day for 30 days (n=5 mice/group). Dashed lines indicate values from untreated control mice. See Figure S6C.
To confirm that Id1−/− HSCs remain quiescent in these cultures, we measured the proliferation of single Id1−/− and Id1+/+ HSCs sorted into Terasaki plates. Greater than 80% of single Id1−/− and Id1+/+ HSCs did not divide after 24 hours in the two different cytokine conditions tested (Figure 5D). After 48 hours in culture, an average of 50% of the HSCs remained undivided in cultures containing SCF/TPO, and fewer than 30% remained undivided in SCF/TPO/IL-3 cultures (data not shown), since addition of IL-3 is a stronger proliferative signal, with no observed difference between Id1+/+ and Id1−/− HSCs. However, 60 hours after the initiation of culture, significantly more Id1−/− HSCs remained undivided in cultures containing SCF/TPO (Figure 5D) and SCF/TPO/IL-3 (data not shown) compared to Id1+/+ HSCs, suggesting that Id1−/− HSCs proliferate less in response to cytokines in vitro. These data suggest that Id1−/− HSCs show increased quiescence and are less responsive to proliferative stimuli, which results in increased surivival in vitro.
Id1−/− HSCs are resistant to exhaustion caused by chronic toll-like receptor signaling
Since Id1−/− HSCs were protected from the chronic proliferative stress of serial BMT and 5-FU treatment, we asked if Id1−/− HSCs might be protected from chronic inflammatory proliferative stress in vivo, including prolonged exposure to toll-like receptor (TLR) ligands, lipopolysaccharides (LPS). Repeated exposure to LPS mimics chronic inflammation and results in increased HSC cycling, increased myeloid bias of HSCs, and ultimately, HSC exhaustion, resembling an aging phenotype in mice (Esplin et al., 2011). We treated Id1−/− or Id1+/+ chimeric mice with low dose LPS every other day for 30 days, and found that the absolute number of Id1+/+ HSCs in LPS-treated mice was significantly reduced compared to untreated mice, confirming that chronic LPS treatment leads to HSC exhaustion. In comparison, the number of Id1−/− HSCs in LPS-treated mice was significantly increased compared to Id1+/+ HSCs in LPStreated mice (Figure 5E). Thus, Id1−/− HSCs are resistant to chronic LPS-induced HSC exhaustion.
Reduced aging of Id1−/− HSCs
Aging of the hematopoietic system is associated with increased numbers of myeloid biased CD150-high HSC (CD150Hi HSC) and decreased numbers of lymphoid biased HSCs, CD150low (CD150Lo HSC), decreased repopulation ability of HSCs, and increased DNA damage in HSCs (Beerman et al., 2010a; Beerman et al., 2010b; Geiger et al., 2013; Wahlestedt et al., 2015). Serial BMT, chronic genotoxic (5-FU), and inflammatory stress (LPS) resemble premature aging of the hematopoietic system; therefore, we speculated that HSCs in Id1−/− mice might show reduced signs of aging (Beerman et al., 2013; Dykstra et al., 2011). To measure this, we bred Id1fl/fl mice (Figure S7A) to male Tie2-Cre transgenic mice, which intrinsically deletes Id1 in HSCs and endothelial cells, but not other cells in the HME, and then compared HSCs in aged (24 month) and young (2 month) Tie2-Cre+;Id1fl/fl (Tie-2;Id1−/−) and Tie2-Cre+;Id1+/+ (Tie2;Id1+/+) mice. Aged Tie2;Id1−/− BMCs show a significant decrease in CD150Hi HSCs (myeloid biased), and an increase in the percentage of CD150Lo HSCs (lymphoid biased) compared to aged Tie2;Id1+/+ BMCs, which is consistent with an immunophenotype young HSCs (Figure 6A). Total numbers of CD150Hi HSCs were also decreased in aged Tie2;Id1−/− mice compared to Tie2;Id1+/+ mice (Figure S7B). The expression of CD41 (increased on aging HSCs) was decreased on aged CD150Hi and CD150Lo Tie2;Id1−/− HSCs compared to aged Tie2;Id1+/+ HSCs, which is consistent with a youthful immunophenotype of Tie2;Id1−/− HSCs (Figure 6B) (Gekas and Graf, 2013). Finally, no difference in myeloid and lymphoid development was observed in PBCs from young Tie2;Id1−/− and Tie2;Id1+/+ mice, while there was an increase in T cells in PBCs from 15 mos Tie2;Id1−/− compared to Tie2;Id1+/+ mice, which is consistent with a youthful phenotype of Tie2;Id1−/− HSCs (Figure S7C-D).
Figure 6. Reduced aging of Id1−/− HSCs in vivo.

(A) CD150Hi and CD150Lo HSCs in aged (2 year) Id1+/+ and Id1−/− HSCs from Tie2;Id1−/− and Tie2;Id1+/+ mice. HSC Ratio calculated as [(% CD150Hi/ % CD150Hi + %CD150Lo ) x 100)] (n=4 mice/group). (B) Expression of CD41 on aged CD150Hi and CD150Lo Id1−/− and Id1+/+ HSCs. (C) Kaplan-Meier survival curves of secondary BMT recipients of 100 FACS sorted aged Id1−/− and Id1+/+ HSCs (Mantel-Cox Test P <0.05), and donor cell repopulation in PBCs from primary recipient mice after 4 months (n=5 mice/group) (D) Cell cycling of aged and young (2 month) HSCs (n=4 mice/group). (E) Percentage of CD150Hi and CD150Lo HSCs from aged and young mice that express phosphorylated γH2AX (n=4 mice/group). (F) Percentage CellRox Deep Red+ HSCs, ST-HSCs and MPPs from aged and young mice (n=4 mice/group). See also Figure S7.
To determine if the aged Id1−/− HSCs also have improved functional capacity, we transplanted equal numbers of aged Tie2;Id1−/− or Tie2;Id1+/+ HSCs into 10 Gy γIR treated recipient mice, and examined HSC function in serial BMT assays. Although no differences were observed in the survival of primary BMT recipients, aged Tie2;Id1−/− HSCs have increased ability to repopulate secondary BMT recipient mice compared to Tie2;Id1+/+ HSCs, demonstrating that aged Tie2;Id1−/− HSCs BMCs have improved HSC function (Figure 6C). We determined the lineage repopulation potential in primary recipient mice, and found that aged Tie2;Id1−/− BMCs showed a significant increase in T cell repopulation 4 months after transplantation, but not in B220+ B cells (Figure 1B), though Id1−/− BMCs have reduced B cell potential in BMT assays (Cochrane et al., 2011). No difference in myeloid repopulation was observed. Thus, aged Tie2;Id1−/− HSCs show increased lymphoid-biased HSCs, no myeloid bias, and greatly improved HSC function compared to the aged Tie2;Id1+/+ BMCs, suggesting that Id1−/− HSCs show functional hallmarks of youthful HSCs.
To examine how Id1−/− HSCs are protected from functional decline during aging, we compared cell cycling in young and old HSCs by Ki-67 staining. We found no differences in the cycling status of young Tie2;Id1−/− and Tie2;Id1+/+ HSCs, with few HSCs in cell cycle, as expected (Figure 6D). While the number of HSCs in cycle (S/G2/M) were the same in aged Tie2;Id1−/− and Tie2;Id1+/+ HSCs, we found that the number of aged Tie2;Id1−/− HSCs in G0 was increased, while HSCs in G1 were decreased compared to the aged Tie2;Id1+/+ HSCs (Figure 6D). These data suggest that there are increased numbers of quiescent HSCs in aged Tie2;Id1−/− mice.
Since aged Tie2;Id1−/− HSCs showed increased quiescence, we asked if these HSCs might also show reduced γH2AX phophorylation. As expected, young Tie2;Id1+/+ HSCs showed low levels of γH2AX staining, which was not affected by the loss of Id1 expression; however, the level of γH2AX staining in aged Id1−/− CD150Hi and CD150Lo HSCs was significantly lower compared to Id1+/+ HSCs (Figure 6E). Thus, increased quiescence of aged Tie2;Id1−/− HSCs is correlated with reduced phosphorylation of γH2AX. We also evaluated ROS levels in the HSPC of aged Tie2;Id1−/− and Tie2;Id1+/+ since ROS levels increase in aged HSCs, and can affect their function during aging (Ito et al., 2006). We found that ROS levels were significantly lower in aged Tie2;Id1−/− HSCs compared with Tie2;Id1+/+ HSCs (Figure 6F). We showed above that ROS levels are low and similar in young Id1+/+ and Id1−/− HSCs, and are increased in Id1+/+ HSCs in middle aged (1 year) mice (Figure S5A), and that ROS levels are reduced in middle aged Id1−/− HSCs compared to Id1+/+ HSCs (Figure S5A). Thus, we confirm that ROS levels increase in HSCs as they age, and that Id1−/− HSCs are protected from increased ROS levels during aging. Further analysis of conventional mice demonstrate that aged Id1−/− HSCs show reduced mitochondrial stress (TMRM+) and mitochondrial biogenesis (Mitotraker green+) compared to Id1+/+ HSCs, which can account, in part, for the observed decrease in ROS expression in aged mice (Figure S5B-C). Collectively, these data suggest that during aging, loss of Id1 protects HSCs from increased proliferation, which results in reduced mitochondrial biogenesis/stress and ROS-induced damage, rendering Id1−/− HSCs more resistant to functional decline.
Quiescent molecular signature of Id1−/− HSC after BMT
To explore the molecular basis by which Id1 loss promotes HSC quiescence under stress we compared the transcriptome of Id1−/− and Id1+/+ HSCs isolated from primary BMT recipient mice by RNA-seq. We found 179 upregulated and 1476 downregulated genes in Id1−/− HSCs compared to Id1+/+ HSCs using a fold-change cut-off of 2 (Table S1). Ingenuity pathway analysis (IPA) of differentially expressed genes revealed that the Top Biological Functions affected in Id1−/− HSCs included, cell stress and injury, protein and gene expression, cell proliferation and growth, and cell signaling, which were all decreased (Figure 7A). The top canonical pathways and networks affected in Id1−/− compared to Id1+/+ HSCs included, Cellular Stress and Injury Pathways (EIF2 Signaling, mTOR signaling, Regulation of eIF4 and p70S6K, AMPK, ATM signaling, NRF2Mediated Oxidative Stress Response), Cell Cycle Pathways, and Cell Signaling Pathways, which were all decreased in Id1−/− HSCs (Figure S7E and Table S2 and S3). Collectively, the IPA analysis shows that Id1−/− HSCs have gene expression profiles consistent with reduced response to stress, reduced cycling and proliferation, and reduced ribosomal biogenesis and protein synthesis, indicating that Id1−/− HSCs have a quiescent molecular signature compared to Id1+/+ HSCs. GSEA analysis of differentially expressed genes confirmed the IPA analysis, and showed significantly reduced expression of genes involved in cell cycle and cell division (Cdk2, Cdk6, Wee1, Cyclin D2/D3, PCNA and others) (Figures 7B and S7F), mitochondrial biogenesis and oxidative phosphorylation (Complex IV-cytochrome C oxidase: Cox8A/6A1/6B1, Complex I: NdufA8/V1/V4 and Complex V-ATP Sythase: ATP5F1/ATP5J/ATP5G1) (Figures 7C and S7G), and ribosome biogenesis (Rp12/5/32, Rps6/8/19 and others) (Figures 7D and S7H). Thus, Id1−/− HSCs show a quiescent molecular signature including reduced cycling, decreased mitochondrial biogenesis and oxidative phosphorylation, and decreased ribosomal biogenesis which is consistent with the hypothesis that Id1−/− HSCs remain quiescent during conditions that promote chronic proliferative stress.
Figure 7.

Quiescent Molecular Signature of Id1−/− HSCs after bone marrow transplantation.
(A) IPA analysis of differentially expressed genes revealed the top molecular functions affected by loss of Id1 in HSCs. All pathways were significantly downregulated in Id1−/− HSCs. GSEA analysis of differentially expressed genes shows (B) a reduction in GO Cell Division genes in Id1−/− HSC compared to Id1+/+ HSCs, (C) a reduction in GO Mitochondrial Organization genes that regulate mitochondrial biogenesis and oxidative phosphorylation in Id1−/− HSCs compared to Id1+/+ HSCs, and (D) a reduction in GO Ribosome genes that regulate ribosomal function in Id1−/− HSCs compared to Id1+/+ HSCs. Heat maps on the right side of enrichment plots show expression of some of the gene hits in the enrichment plots that are up or down downregulated in Id1−/− HSCs following BMT (blue/red indicates low/high level of gene expression). (E) Expression of p16, p21 and p27 in Id1−/− and Id1+/+ HSCs in Lin- expansion assays after 6 days determined by flow cytometry. (F) Expression of p16, p21 and p27 in BMCs were harvested from mice competitively transplanted with Id1−/− and Id1+/+ HSCs after 14 wks (n=5/mice group). (G) Proliferation of Id1−/− and Id1+/+ HSCs in Lin- cell expansion assays after knockdown of p16 and E2A expression by BrdU incorporation. See also Figure S7.
Id1 increases HSC proliferation by antagonizing E protein function.
E-proteins inhibit cellular proliferation by upregulating cyclin dependent kinase inhibitors (CDKI’s) (p21, p27, p57, and p16), and Id1 inhibits E-protein function, which results in increased cell proliferation (Lasorella et al., 2014; Roschger and Cabrele, 2017). E2A−/− HSCs show decreased expression of p21 and p27, increased HSC cycling, and decreased self-renewal in serial BMT experiments (Semerad et al., 2009; Yang et al., 2011). Furthermore, HSCs that lack p21 show decreased self-renewal in serial BMT experiments (Cheng et al., 2000). These data suggest that E proteins restrain HSC proliferation, in part, by increasing CDKI gene expression. Id1−/− HSCs show decreased proliferation and enhanced self-renewal after BMT; therefore, we speculate that Id1 increases HSC proliferation by antagonizing E protein function (Id1-E2A-CDKI pathway). To test this hypothesis, we evaulated the expression of p21, p27 and p16 in HSCs cultured in vitro, and in HSCs after 14 weeks competitive BMT in vivo. Id1−/− HSCs showed increased expression of p27 and p16 after 6 days in expansion cultures, and increased expression of p21 and p27 after 14 weeks BMT compared to Id1+/+ HSCs (Figure 7E-F). Thus, increased CDKI expression in Id1−/− HSCs correlates with decreased proliferation of HSCs. Furthermore, knockdown of E2A and p16 using shRNA expressing lentiviral vectors significantly increased the proliferation of Id1−/− LSK CD48- HSCs in expansion cultures compared to control shRNA treated cells (Figure 7G). Quantitative RT-PCR confirmed shRNAmediated knockdown of E2A and p16 gene expression in the Id1−/− HSC expansion assay in vitro (Figure S7I). Taken together, these experiments suggest that Id1 increases HSC proliferation by restraining E protein function, and by reducing p16 expression.
DISCUSSION
Id proteins are expressed at low levels in most tissues and differentiated cell types, but can be induced by a wide array of extracellular signals in response to stress or injury resulting in increased proliferation, growth, tissue repair and regeneration; this suggests that Id genes may be part of the functional response to hematopoietic stress. Since Id1 expression is induced in HSPC by growth factors that promote proliferation and differentiation, we examined if Id1 might function in conditions of hematopoietic stress including BMT. We show here that Id1−/− HSCs have enhanced self-renewal potential and are protected from exhaustion during serial BMT. Specifically, Id1−/− HSCs show reduced cell cycling and proliferation, and reduced DNA damage after BMT. These are indicative of reduced proliferative stress, and could protect Id1−/− HSCs from exhaustion. Id1−/− HSCs show a reduction in mitochondrial biogenesis and metabolic activity, reduced intracellular ROS levels, and increased levels of reduced glutathione, which suggests that Id1−/− HSCs are more quiescent after BMT in vivo. We provide evidence that HSCs express low levels of Id1 under steady state hematopoiesis. Moreover, cytokines and other proinflammatory stimuli present in the HME after γ-IR can induce Id1 expression and proliferation in HSCs, suggesting that HSC exhaustion is mediated, in part, by the proinflammatory HME after BMT. Consistent with these observations, we found that administration of pan-JAK and STAT inhibitors, which prevent signaling of many proinflammatory cytokines, partially block Id1 induction in HSCs after BMT. Furthermore, Id1−/− HSCs demonstrate reduced ability to proliferate and differentiate in response to cytokines in vitro and after transplantation in vivo, while enforced expression of Id1 promotes HSC proliferation (Suh et al., 2008), suggesting that Id1 promotes HSC proliferation in response to extracellular signals produced under conditions of hematopoietic stress. BMT stress resembles other chronic stress including chronic exposure of BMCs to IFNs, IL-1, or LPS in vivo, which induce stem cell proliferation and exhaustion (Baldridge et al., 2010; Esplin et al., 2011; Pietras et al., 2016; Trumpp et al., 2010). We found that Id1−/− HSCs are protected from exhaustion in other models of chronic stress including chronic genotoxic stress (5-FU treatment), chronic inflammatory stress (LPS treatment), and aging, suggesting that reducing Id1 levels during chronic stress could have therapeutic potential by preserving HSC pools.
Quiescent HSCs with limited divisional history have the greatest repopulating ability, while HSCs that have divided show reduced repopulating ability (Qiu et al., 2014; Sawen et al., 2016). Thus, HSCs that exit dormancy, divide and differentiate, and progressively lose repopulation potential and eventually exhaust. These observations are consistent with the protective function of Id1 in stress hematopoiesis, whereby Id1−/− HSCs show reduced proliferation and increased quiescence in vitro and after BMT in vivo and, thus, are protected from cell division, differentiation and exhaustion. Under conditions of acute stress, some HSCs are induced to express Id1, and are pushed into division to differentiate and enter the active pool of MPPs to provide myeloid cell reconstitution and protection after the stress or injury. After which, hematopoiesis returns to steady-state conditions with minimal loss of HSC function. However, under conditions of chronic stress, Id1−/− HSCs have an apparent advantage over Id1+/+ HSCs and are protected from exhaustion by maintaining quiescence. Interestingly, it has been shown that HSCs maintain a high proliferation rate even 4 months after BMT compared to non-transplanted mice using assays to track the division history of HSCs after BMT, which suggests that the γIR (conditioned) HME may remain damaged long after and BMT resulting in chronic proliferative stress to the transplanted HSCs (Rodrigues-Moreira et al., 2017; Sawen et al., 2016; Schaue et al., 2015). Thus, it would be predicted that inhibiting Id1 expression in HSCs during BMT, especially in BMTs with poor graft function, or those with concurrent infections, and other situations of chronic stress including aging, might protect the HSC pool from depletion.
Since it is difficult to predict which proinflammatory cytokines produced in the HME after γIR induce Id1 and promote HSC proliferation; we used small molecule inhibitors of common cytokine signal transduction pathways that target multiple cytokines. We found that inhibitors of the JAK and STAT pathways partially reverse the induction of Id1 in HSCs after BMT, confirming that Id1 is downstream of the JAK/STAT pathway in vivo, suggesting that other signal transduction pathways might contribute to the induction of Id1 to promote HSC proliferation after BMT. For example, we found that LPS induces Id1 in HSCs, and that Id1−/− HSCs are protected from the chronic inflammatory stress induced by repeated LPS treatment, suggesting a potential role for other TLRs and their ligands in HSC exhaustion during BMT. Thus, we speculate that inhibiting TLRs that recognize pathogen associated molecular patterns (PAMPs) other than LPS, and TLRs that recognize damage-associated molecular patterns (DAMPS), or alarmins, produced after bone marrow conditioning (γIR) and tissue damage might also inhibit Id1 expression, and protect HSCs from exhaustion during BMT. In support of this hypothesis, BMCs that lack TLR4 or TLR9 show significantly enhanced reconstitution after BMT compared to WT mice (Ichii et al., 2010), suggesting a potential role for these receptors and their downstream signaling pathways in protecting HSCs from chronic stress of BMT. Similarly, since TNF and IL-1 induce Id1 expression in HSCs, inhibitors of TNFRs, IL-1Rs and possibly IL-8R signaling might also block induction of Id1 in HSCs. Interestingly, BMCs that lack TNFR-p55 and TNFR-p75 show greatly enhanced competitive serial repopulation ability compared to WT mice during BMT (Markus et al., 2009), suggesting that inhibiting TNFR signaling might protect HSCs during chronic stress. Thus, it will be important in future studies to examine additional small molecule inhibitors of TLRs, TNFRs and other pathways to determine if they prevent Id1 induction in HSCs, and if they preserve or rescue HSCs from exhaustion during BMT or from other chronic proliferative stress (Matatall et al., 2016; Zhang et al., 2016).
Current methods to expand HSCs in vitro for BMT and cell therapy, including gene editing, have been challenging due to the differentiation of the initial stem cell population (Walasek et al., 2012). Recent evidence suggests that HSCs with extensive proliferative history in vivo have poor hematopoietic repopulation ability, suggesting that methods to expand HSCs may result in HSC populations with poor function. Therefore, it will be necessary to develop methods that promote HSC survival and expansion without loss of function in vitro. Our results show that HSCs that lack Id1 are preserved in cytokine expansion cultures in vitro. Thus, for purposes of gene editing or transgene expression, inhibiting Id1 function in current expansion media could result in a significant increase in functional HSCs.
In summary, chronic stress from BMT, chronic inflammatory diseases and infections, hematopoietic malignancies, bone marrow failure syndromes, and aging promote HSC exhaustion. The data shown here provide evidence that Id1 is induced in quiescent HSCs by the proinflammatory HME after BMT, which promotes proliferation, differentiation and hematopoietic reconstitution. However, under conditions of chronic stress, Id1−/− HSCs have an advantage and are protected from proliferative stress by remaining quiescent without losing engraftment and repopulation ability. Thus, moderating levels of Id1 in HSCs could be therapeutically beneficial under conditions of chronic stress including BMT, bone marrow failure, and chronic inflammatory conditions.
STAR METHODS
KEY RESORUCES TABLE
CONTACT FOR REAGENTS AND SOURCE SHARING
Further information and requests for resources and reagents should be directed to, and will be fulfilled by the Lead Contact, J.R.K. (kellerjo@mail.nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Id1−/− mice on C57BL/6 background were derived from Id1−/− mixed background mice (B6;129 generously provided by Robert Benezra) after 10 (or greater) generations of backcross with C57BL/6 mice (Charles River). Id1fl/fl mice were generated by inserting LoxP sequences around exon 2 and 3 by the CCR Gene Targeting Facility, NCI Frederick, MD, USA (summarized in Figure S7A). Id1fl/fl mice were bred with male Tie2-Cre+ mice (Koni et al., 2001) to generate Tie2-Cre+;Id1fl/fl (Tie2;Id1−/−)and Tie2-Cre+;Id1+/+ (Tie2;Id1+/+) mice. C57BL/6 Id1-EGFP knock-in reporter mice (Id1GFP/+) were generously provided by Xiao-Hong Sun, Oklahoma Medical Research Foundation, Oklahoma, US. C57BL/6 Ly5.2 (CD45.1) mice were obtained from Charles River. All mice were maintained on C57BL/6 genetic background. Recipient mice used in BMT experiments were 8–12 week old females. Experiments involving the use of mice were approved by the NCI at Frederick Animal Care and Use Committee in accordance with the eighth edition “Guide for the Care and Use of Laboratory Animals.”
Chronic LPS treatment
1 × 106 total BMCs from Id1+/+ and Id1−/− mice were transplanted into 10 Gy irradiated CD45.1 recipients. Twenty weeks after BMT, transplanted mice were treated with LPS (Sigma) 0.043 mg/kg every alternate day for 30 days. On the 31st day, mice were euthanized and HSPCs were analyzed as described above.
METHOD DETAILS
Bone marrow transplantation
For competitive serial BM transplantation assays, BMCs (5×105) from Id1+/+ and Id1−/− mice (CD45.2) were mixed with 5×105 C57BL/6 (CD45.1) BMCs and transplanted into γ-irradiated (10 Gy, 137Cs source) congenic CD45.1 mice by tail vein injection (iv). Recipient mice were pretreated one week before and two weeks after transplantation with antibiotic-containing water (pH 2.5–3.0, 0.5 mg/mL amoxicillin, 0.17 mg/mL enrofloxacin). Donor reconstitution was determined in peripheral blood cells (PBCs) 4 and 8 weeks after BMT, and after 12–14 weeks in PBCs and bone marrow. For serial transplantation experiments, 1 × 106 total BMCs from pooled recipients were transplanted into 10 Gy irradiated secondary recipient, and similarly for the 3rd, 4th, 5th, 6th, and 7th BMT. Donor reconstitution was determined by flow cytometry in PBCs 4 and 8 weeks after BMT, and in the bone marrow and/or peripheral blood after 12 weeks for all BMT recipient mice. HSCs were purified from aged Tie2;Id1−/− or Tie2;Id1+/+ mice mice by FACS. HSCs (100) CD45.2+ were co-transplanted with 1 × 105 CD45.1+ radioprotective BMCs into primary γ-IR recipient mice. BMCs from primary recipient mice were pooled, and then transplanted (1 ×106 cells) into secondary recipient mice and mice were monitored for survival (n=5 mice/group). PBCs from primary recipient mice were analyzed for donor lineage reconstitution after 4 months by flow cytometry.
Lineage and hematopoietic stem and progenitor cell analysis
Single-cell suspensions were prepared from Id1+/+ or Id1−/− BMCs or from BMCs and PBCs of animals transplanted with Id1+/+ or Id1−/− bone marrow cells (BMCs). BMCs were harvested from femurs and tibias by flushing with PBS (1% BSA) using 25 gauge needle and syringe. The light density BMC fraction was isolated using lymphocyte separation media (LSM) (MO Biomedicals, LLC., Solon, OH). PBCs collected from mandibular bleed, were incubated with ACK lysis buffer (Lonza) to lyse red cells followed by a wash in staining buffer (1% BSA in PBS). PBCs and BMCs were incubated with the following fluorochrome conjugated monoclonal antibodies for lineage analysis: CD45.2 (104), Mac-1 (M1/70), Gr-1 (RB6–8C5), B220 (RA3–6B2), CD4 (GK1.5), CD8 (53–6.7), CD71 (R17217) and Ter119 (Ter119). For HSPC analysis LSM purified BMCs were treated with Fc receptor blocking antibodies (anti-mouse CD16/32), then incubated with biotinylated lineage markers (Mac-1, Gr-1, B220, TER119, CD4, CD8, and IL-7Rα), and fluorochrome conjugated streptavidin, followed by the addition of directly flourochrome labeled antibodies (as indicated): c-Kit (ACK2), Sca-1(D7), CD150 (mShad150), CD48 (HM48–1), CD34 (RAM34), Flk2 (A2F10), anti-FcγRII/III (FcR) (93). Lin-negative (Lin−) c-Kit+ and Sca-1+ (LSK) cells were analyzed according to the following marker expression: HSC: LSK Flk2CD150+ CD48−, short-term HSC (ST-HSCs): LSK Flk2−CD150−CD48−, and multipotent progenitors (MPP): LSK Flk2−CD150−CD48+ and LSK Flk2−CD150+CD48+. All cells were incubated in staining buffer for 45 min on ice with intermittent mixing, and then washed in buffer prior to analysis. All the antibodies used were purchased from BD Biosciences (San Jose, CA) or eBiosciences (San Diego, CA). For annexin V staining, BMCs stained for HSPC analysis were further incubated with annexin V-FITC (BD Biosciences) and analyzed immediately by flow cytometry. For cell cycle/quiescence and DNA damage assays, BMCs stained for HSPC analysis were fixed and permeabilized with Cytofix/Cytoperm buffer (BD Biosciences) followed by intracellular staining overnight with Ki-67-FITC (B56) (BD Biosciences) and γH2AX-PE (20E3) (phosphor Ser139) (Cell Signaling, Danvers, MA) antibody. Cells were incubated with FxCycle Violet dye (ThermoFisher, Waltham, MA) 2–4 hrs before acquisition. BD LSRII SORP and BD Fortessa were used for FACS acquisition and Aria II for cell sorting. Data analysis was done using FlowJo V9 software (Tree Star Inc., Ashland OR). For analysis of cyclin dependent kinase inhibitor expression (CDKI), BMCs stained for HSPC analysis were fixed and permeabilized with BD cytofix/perm buffer, and then blocked with respective purified isotype control antibody for 40 min on ice and then stained with p16 (Santacruz Biotech, sc-166760 PE), p21 (Santacruz Biotech, sc-6246 Alexa fluor 488) and p27 (Santacruz Biotech, sc-1641PE) for 30 min at room temperature and analyzed by flow cytometry.
Immunoflourescence Analysis of HSCs
BMCs were harvested from primary BMT recipient mice 12 weeks after competitive repopulation of Id1+/+ and Id1−/− BMCs. BMCs were further separated using LSM density gradient and stained for HSC cell surface markers. Donor-derived CD45.2+ LSK CD150+48- HSCs were isolated from Id1+/+ and Id1−/− transplanted mice by FACS. Approximately 1000 sorted HSCs were incubated for 1 hr. at 37°C in 200ul of Stemspan medium, on retronectin (20ug/ml) coated 8-well chamber slides (Thermo Fischer Scientific). The cells were fixed with 4% paraformaldehyde for 15 min at RT, washed with PBS (3X) and then permeabilized by 0.25% Triton X-100 for 10 min at RT. The cells were then blocked with 10% goat and donkey serum overnight at 4°C. Afterwhich, the cells were incubated with 1:1000 diluted anti-γH2AX (S139) (20E3) (Rabbit, #9718, Cell Signaling Technology), 4°C overnight or anti-53BP1 (Rabbit, Abcam), 4°C overnight. After washing four times with PBST (PBS + 0.1% tween 20) the cells were incubated with 1:2000 diluted Alexa Fluor594 -conjugated goat anti-rabbit IgG(H + L) antibody (Jackson ImmunoResearch laboratories) at RT for 2 hrs. The cells were again washed four times with PBST and counterstained and mounted with Prolong Gold antifade reagent with DAPI (Life Technologies). Cells were imaged on Leica SP8 microscope equipped with Spinning Disk Confocal CSU-W1 (Yokogava) and Zyla4.2 sCMOS camera (Andor), using x63 oil immersion lens. Z-stacks with 0.3um step was acquired over the 12 μm range, so that full Z range was scanned for all cells. Images were processed and analyzed in ImageJ. The antibody signal was normalized to DAPI signal to account for different amounts of DNA in cells. Total fluorescence across the Z-stack was measured for antibody and DAPI, and arbitray units (A.U.) represents the ratio of H2AX and 53BP1 flourescence to DNA in each cell.
Homing Assay
LSM separated BMCs were incubated with biotinylated lineage markers as described above. Then labeled cells were incubated with sheep anti-rat IgG-conjugated magnetic beads (20:1 beads/cell) (Invitrogen, Carlsbad, CA), and Lin-positive (Lin+) cells were removed using a magnetic bead separater. Lin- cells were stained with 5 μM CFSE (Invitrogen) in pre-warmed PBS at 37°C for 20 minutes, washed, and transplanted into 8 Gy irradiated recipients (CD45.1). Recipients were euthanized 2.5 days after BMT and analyzed by flow cytometry for HSPC engraftment.
Serial 5-FU assay
Chimeric mice were generated by transplanting Id1−/− and Id1+/+ BMCs (1×106) (i.v.) into 10 Gy irradiated recipient mice. Ten weeks after BMT, recipient mice were treated with 5-fluorouracil (5-FU) (135mg/kg), 4 times at 7 day intervals and monitored for survival. Alternatively, chimeric mice were treated with two does of 5-FU (150 mg/kg) separated by 2 weeks. Then 2 weeks after the last injection BMCs were harvested for HSPC immunophenotype analysis (n=5 mice/group), and BMCs (1 ×106) were transplanted with competitor BMCs (1 × 106) into irradiated (10 Gy) recipient mice to evaluate HSC function. Finally, chimeric Id1+/+ and Id1−/− BMT recipients were treated with 5-FU (135 mg/kg mouse) twice, two weeks apart, and mice were administered BrdU continuously for 3 days before harvesting BMCs 10 days after the last 5-FU treatment to determine the percentage of BrdU+ HSCs by flow cytometry using the gating strategy shown in Figure S2B.
Mitochondrial activity and ROS measurement
Mononuclear cells were isolated by LSM density gradient separation from Id1+/+ and Id1−/− mice, and from secondary competitive BMT recipient mice after 12 weeks. BMCs (5×106 cells/mL) were incubated in StemSpan media (Stem Cell Technologies, Vancouver, CA) at 37°C with either (a) MitoTracker Green (Thermo Fisher Scientific) 50nM for 15 mins, (b) JC-1 (Thermo Fisher Scientific) 2uM for 15 mins, (c) CellRox Deep Red (Thermo Fisher Scientific) 500nM for 30 mins, (d) Thiol tracker violet (Thermo Fisher Scientific) 10μM for 15 mins, or (e) TMRM (Thermo Fisher Scientific) 100nM for 30mins, as per the manufacturer’s instructions. Cells were then washed with PBS and stained for HSPC markers, as described above, and analyzed immediately.
In vitro Id1-GFP reporter cell assays
BMCs were harvested from Id1GFP/+ mice, and LSM separated BMCs, and HSCs, ST-HSCs, MPPsures, common myeloid progenitors (CMP), Lin- c-Kit+ (LK) CD34+ FcR−, granulocyte/macrophage progenitors (GMP), LK CD34+FcR+, megakaryocyte erythroid progenitors (MEP) LK CD34−FcR−, and neutrophils (Mac-1+Gr-1+) were analyzed for GFP expression by flow cytometry. Alternatively, LSM cells were cultured in StemSpan SFEM (Stem Cell Technologies, Vancouver, Canada) serum free medium containing mSCF (100 ng/mL), with or without the additional following cytokines and proinflammatory agents as indicated in the results section: pI:pC 1μg/mL (InvivoGen, San Diego, CA), LPS 1 μg/mL (Sigma, St. Louis, MO), IL-1β 25 ng/mL, IFNγ (type II) 1000 units/mL, IFNα (type I) 1000 units/mL, TNFα 20ng/mL, mIL-3 20 ng/mL, hTPO 100ng/mL, hFLT3L-100 ng/mL, GM-CSF 50 ng/mL, mIL-6 50 ng/mL. All cytokines were from Peprotech, Rocky Hill, NJ. Cells were removed from triplicate cultures after 36 hrs and HSPCs were analyzed for GFP expression by flow cytometry. For BrdU incorporation assays, BMCs were harvested from Id1GFP/+ reporter mice and Lin- cells were isolated as described above. Lin- cells were cultured for 48 hours in StemSpan along with SCF (100 ng/mL) alone or in combination with TPO (100 ng/mL) and IFNγ (1000 units/mL). The cells were pulsed with 10 μM BrdU (Sigma) for the last 3 hours of the culture, stained with HSPC markers, fixed and permeabilized by Cytofix/Cytoperm buffer (BD Biosciences), treated with DNase and stained with anti-BrdU as per the manufacturer’s instructions. Cells were washed with Perm/wash buffer (BD Biosciences) twice and then analyzed by flow cytometry.
In vivo Id1-GFP assays
Irradiation induction of Id1 expression.
Id1GFP/+ reporter mice and C57BL/6 controls were exposed to either 0 Gy, 4 Gy, 6 Gy or 8 Gy γIR, and BMCs were harvested 48 and 72 hours after irradiation. BMCs were harvested from treated mice and HSPCs were analyzed for the expression of GFP by flow cytometry as described above.
Cytokine/Growth Factor Induction of Id1 expression.
BMCs were harvested from Id1GFP/+ reporter mice (CD45.2+) and were transplanted (1×106 cells) into 10 Gy irradiated CD45.1 recipients (chimeric mice). Twenty weeks after BMT chimeric mice (5 mice/group) were administered, SCF (0.4mg/kg SC), IFNγ (0.43mg/kg IV), mIL6 (0.4mg/kg IP), hTPO (0.4mg/kg SC) (all from Peprotech) or pI:pC (5mg/kg IP) (InvivoGen), or a 6 Gy γIR dose. Recipients and non-transplanted controls were sacrificed 48 hours post-treatment, and BMCs were harvested and HSPCs were analyzed for the expression of GFP as describe above.
JAK/STAT inhibitor effect on Id1 expression.
C57BL/6 (CD45.1) recipient mice were treated daily for 4 days with either Momelotinib (Selleckchem, Houston, TX) dissolved in 1-methyl-2pyrrolidinone to 120mg/ml and diluted with 0.1M Capsitol to 4mg/ml SC at 22.4 mg/kg, or SH4–54 (Selleckchem) dissolved in 50% polyethylene glycol and 50% PBS IP at 10mg/kg. On day 2, mice were irradiated (8 Gy) and transplanted with 5 × 105 lineage-negative BMCs from Id1GFP/+ reporter mice (CD45.2). Recipients were euthanized on day 4 and BMCs were harvested and HSPCs were analyzed for GFP expression as described above.
In vitro culture of hematopoietic stem and progenitor cells
BMCs were harvested from Id1+/+ and Id1−/− mice and stained with biotinylated antibodies that recognize lineage markers,and fluorochrome-conjugated streptavidin, and antibodies that recognize c-Kit and Sca-1. LSK cells were purified by FACS and seeded into StemSpan medium containing mSCF (100 ng/mL), hTPO (100 ng/mL), mFGF1 (10ng/mL), hIGF2 (20 ng/mL), and Angiopoietin 2 (50ng/mL) at 3 × 104 cells/ml. Cells were removed from culture after 6 days and analyzed for HSPCs cell cycle using Ki-67/FxCycle, and proliferation using BrdU by flow cytometry as described above. The harvested cells were also analysed for CDKI expression. Briefly, the cells were stained with HSPC surface markers, fixed and permeabilized with BD cytofix/perm buffer, and then stained with p16, p21 and p27 antibodies as described above.
Single cell assay.
BMCs were labled with antibodies that recognize HSPC as described above, and HSCs (LSK Flk2−CD150+CD48−) were sorted one cell per well directly into Terasaki plates (Greiner Bio-One) containing StemSpan serum free media supplemented with mSCF plus hTPO, plus mIL-3 (30ng/mL) in vitro. Cells were counted at 24, 48 and 60 hours after plating.
Lentiviral mediated shRNA knockdown
Mouse p16(CDKN2A) (SHCLNG-NM_009877), E2a (TCF3) (SHCLNG-NM_011548) shRNA target sequences were purchased from Sigma-Aldrich. Infectious Lentivirus was generated by transfecting the shRNA constructs and packaging plasmids (PMD2G and pCMV8.74) into 293T/17 cells using LipoD293 (SignaGen, Rockville, MD, USA). Virus containing supernatants were collected 48 hours post transfection. To calculate viral titers, NIH3T3 cells were infected with serial dilutions of viral supernatants. For knockdown, lineage depleted cells from mouse bone marrow were transduced with shRNA lentivirus by spinoculation. Lineage depleted cells (Depleted for Mac-1, Gr-1, B220, Ter119, CD4, CD8 and Il7R) were isolated from Id1−/− mice using immune-magnetic bead separation and cultured in Stemspan medium containing mSCF (100 ng/mL), hTPO (100 ng/mL), mFGF1 (10ng/mL), hIGF2 (20 ng/mL), and Angiopoietin 2 (50ng/mL) at 5 × 105 cells/0.5mL for 12 hours. Twelve hours after culture, the cells were subjected to first round of shRNA mediated lentiviral transduction lentivirus, where lineage depleted cells were spun at 2000 x g for 90 minutes at 37°C. The cells were then washed and reseeded with fresh complete medium. After 24 hours, a second round of transduction was performed as described above, and the cells were cultured in puromycin (2 μg/mL). Twenty-four hour after the second lentiviral mediated transduction, the cells were pulsed with 10uM BrdU for 45 min., and then harvested for qRT-PCR and flow cytometry.
RNA-seq analysis
RNA was purified from HSCs isolated from Id1−/− and Id1+/+ mice by flow cytometry as described above, and from HSCs isolated from primary recipient mice 4 weeks after transplantation of Lin- Id1−/− and Id1+/+ BMCs. The SMARTer Ultra Low Input RNA Kit (Thermo Fisher) was used to generate high-quality cDNA. The cDNA was made into a sequencing ready library using the Nextera® XT DNA Sample Preparation Kit (Illumina). The final purified product is then quantitated by qPCR before cluster generation and sequencing on the Illumina HiSeq 2500 sequencer. The HiSeq Real Time Analysis software (RTA 1.18) was used for processing image files, the Illumina CASAVA_v1.8.4 was used for demultiplex and converting binary base calls and qualities to fastq format. The sequencing reads were trimmed for adapters and low quality bases using Trimmomatic (version 0.30). The trimmed reads were aligned to mouse mm9 reference genome (NCBIM37 /UCSC mm9) and Ensembl annotation version 67 using TopHat_v2.0.8 software. Quantification was carried out with RSEM using the transcriptome bam file created by STAR. Differentially expressed genes were obtained by comparing (Id1−/− HSCs-stressed - Id1−/− HSCs-normal) and (Id1+/+ HSCs-stressed - Id1+/+ HSCsnormal). Differentially expressed genes were then analyzed and selected by Limma pipeline with adjusted p value < 0.05, and fold change ± 2 (Table S1).
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise indicated, data in all figures are expressed as the mean ± SD and are representative of at least 2 trials. Statistical differences between groups was determined by Student’s unpaired t test using Prism 5 software (GraphPad, San Diego, CA). Statistical differences between survival curves were determined using the og-rank (Mant-Cox) test. For all graphs, data are presented as mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001.
Supplementary Material
Table S1. Related to Figure 7, Differentially expressed genes in Id1−/− and Id1+/+ HSCs from primary BMT Recipients. RNA was obtained from Id1−/− and Id1+/+ HSCs isolated from primary BMT recipient mice 4 weeks after transplantation of Lin− BMCs, and analyzed by RNA-seq for significant differential gene expression.
Table S2. Related to Figure 7, Decreased Molecular Functions in Id1−/− HSC from Primary BMT Recipient Mice. IPA analysis of differentially expressed genes in Table S1 revealed the top molecular functions affected by loss Id1 in HSCs. All functions were significantly decreased in Id1−/− HSCs.
Table S3. Related to Figure 7. Top Networks affected in Id1−/− HSC from Primary BMT Recipient Mice. IPA analysis of differentially expressed genes in Table S1 revealed the top networks affected by loss of Id1 in HSCs.
Table S4. Related to Figure 7. Oligonucleotides for PCR Analysis.
Highlights.
Id1−/− HSCs have enhanced self-renewal potential and are maintained during BMT
Id1−/− HSCs show reduced oxidative stress and increased quiescence after BMT
Id1−/− HSCs are protected from inflammatory cytokine-induced proliferative stress
Id1−/− HSCs are protected from exhaustion by chronic inflammatory stress and aging
ACKNOWLEDGEMENTS
We wish to thank FNLCR LASP Animal Research and Technical Support Staff, the NCIFrederick Flow Cytometry Core. Dr. Philip Oberdoeffer for his thoughtful review of this manuscript. This project has been funded in part with Federal funds from the Frederick National Laboratory for Cancer Research, NIH, under Contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsements by the US Government.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-seq data in this paper is GEO: GSE114533.
SUPPLEMENTAL INFORMATION
Supplemental information includes seven figures and three tables.
REFERENCES
- Baldridge MT, King KY, Boles NC, Weksberg DC, and Goodell MA (2010). Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 465, 793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerman I, Bhattacharya D, Zandi S, Sigvardsson M, Weissman IL, Bryder D, and Rossi DJ (2010a). Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci U S A 107, 5465–5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerman I, Bock C, Garrison BS, Smith ZD, Gu H, Meissner A, and Rossi DJ (2013). Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 12, 413–425. [DOI] [PubMed] [Google Scholar]
- Beerman I, Maloney WJ, Weissmann IL, and Rossi DJ (2010b). Stem cells and the aging hematopoietic system. Current opinion in immunology 22, 500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, and Foiani M (2010). Maintaining genome stability at the replication fork. Nature reviews Molecular cell biology 11, 208–219. [DOI] [PubMed] [Google Scholar]
- Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, and Scadden DT (2000). Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 287, 1804–1808. [DOI] [PubMed] [Google Scholar]
- Cochrane SW, Zhao Y, Perry SS, Urbaniak T, and Sun XH (2011). Id1 has a physiological role in regulating early B lymphopoiesis. Cellular & molecular immunology 8, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochrane SW, Zhao Y, Welner RS, and Sun XH (2009). Balance between Id and E proteins regulates myeloid-versus-lymphoid lineage decisions. Blood 113, 1016–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlberg A, Delaney C, and Bernstein ID (2011). Ex vivo expansion of human hematopoietic stem and progenitor cells. Blood 117, 6083–6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykstra B, Olthof S, Schreuder J, Ritsema M, and de Haan G (2011). Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J Exp Med 208, 2691–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esplin BL, Shimazu T, Welner RS, Garrett KP, Nie L, Zhang Q, Humphrey MB, Yang Q, Borghesi LA, and Kincade PW (2011). Chronic exposure to a TLR ligand injures hematopoietic stem cells. J Immunol 186, 5367–5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM, Reynaud D, Alvarez S, Diolaiti ME, Ugarte F, Forsberg EC, et al. (2014). Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 512, 198–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger H, de Haan G, and Florian MC (2013). The ageing haematopoietic stem cell compartment. Nature reviews Immunology 13, 376–389. [DOI] [PubMed] [Google Scholar]
- Gekas C, and Graf T (2013). CD41 expression marks myeloid-biased adult hematopoietic stem cells and increases with age. Blood 121, 4463–4472. [DOI] [PubMed] [Google Scholar]
- Harrison DE, and Lerner CP (1991). Most primitive hematopoietic stem cells are stimulated to cycle rapidly after treatment with 5-fluorouracil. Blood 78, 1237–1240. [PubMed] [Google Scholar]
- Ichii M, Shimazu T, Welner RS, Garrett KP, Zhang Q, Esplin BL, and Kincade PW (2010). Functional diversity of stem and progenitor cells with B-lymphopoietic potential. Immunol Rev 237, 10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, Ohmura M, Naka K, Hosokawa K, Ikeda Y, et al. (2006). Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nature medicine 12, 446–451. [DOI] [PubMed] [Google Scholar]
- Jankovic V, Ciarrocchi A, Boccuni P, DeBlasio T, Benezra R, and Nimer SD (2007). Id1 restrains myeloid commitment, maintaining the self-renewal capacity of hematopoietic stem cells. Proc Natl Acad Sci U S A 104, 1260–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee BL (2009). E and ID proteins branch out. Nature reviews Immunology 9, 175–184. [DOI] [PubMed] [Google Scholar]
- Kim JH, Jenrow KA, and Brown SL (2014). Mechanisms of radiation-induced normal tissue toxicity and implications for future clinical trials. Radiation oncology journal 32, 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, and Flavell RA (2001). Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med 193, 741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasorella A, Benezra R, and Iavarone A (2014). The ID proteins: master regulators of cancer stem cells and tumour aggressiveness. Nature reviews Cancer 14, 77–91. [DOI] [PubMed] [Google Scholar]
- Leeanansaksiri W, Wang H, Gooya JM, Renn K, Abshari M, Tsai S, and Keller JR (2005). IL-3 induces inhibitor of DNA-binding protein-1 in hemopoietic progenitor cells and promotes myeloid cell development. J Immunol 174, 7014–7021. [DOI] [PubMed] [Google Scholar]
- Ling F, Kang B, and Sun XH (2014). Id proteins: small molecules, mighty regulators. Current topics in developmental biology 110, 189–216. [DOI] [PubMed] [Google Scholar]
- Markus T, Cronberg T, Cilio C, Pronk C, Wieloch T, and Ley D (2009). Tumor necrosis factor receptor-1 is essential for LPS-induced sensitization and tolerance to oxygen-glucose deprivation in murine neonatal organotypic hippocampal slices. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 29, 73–86. [DOI] [PubMed] [Google Scholar]
- Matatall KA, Jeong M, Chen S, Sun D, Chen F, Mo Q, Kimmel M, and King KY (2016). Chronic Infection Depletes Hematopoietic Stem Cells through Stress-Induced Terminal Differentiation. Cell reports 17, 2584–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelson A, and Frenette PS (2014). Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nature medicine 20, 833–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ, and Scadden DT (2014). The bone marrow niche for haematopoietic stem cells. Nature 505, 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura-Ishizu A, Takizawa H, and Suda T (2014). The analysis, roles and regulation of quiescence in hematopoietic stem cells. Development 141, 4656–4666. [DOI] [PubMed] [Google Scholar]
- Orkin SH, and Zon LI (2008). Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132, 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino S, Michelena J, Teloni F, Imhof R, and Altmeyer M (2017). Replication-Coupled Dilution of H4K20me2 Guides 53BP1 to Pre-replicative Chromatin. Cell reports 19, 1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perk J, Iavarone A, and Benezra R (2005). Id family of helix-loop-helix proteins in cancer. Nature reviews Cancer 5, 603–614. [DOI] [PubMed] [Google Scholar]
- Perry SS, Zhao Y, Nie L, Cochrane SW, Huang Z, and Sun XH (2007). Id1, but not Id3, directs long-term repopulating hematopoietic stem-cell maintenance. Blood 110, 2351–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, Lakshminarasimhan R, Chin CP, Techner JM, Will B, et al. (2016). Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of selfrenewal. Nature cell biology 18, 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Papatsenko D, Niu X, Schaniel C, and Moore K (2014). Divisional history and hematopoietic stem cell function during homeostasis. Stem cell reports 2, 473–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues-Moreira S, Moreno SG, Ghinatti G, Lewandowski D, Hoffschir F, Ferri F, Gallouet AS, Gay D, Motohashi H, Yamamoto M, et al. (2017). Low-Dose Irradiation Promotes Persistent Oxidative Stress and Decreases Self-Renewal in Hematopoietic Stem Cells. Cell reports 20, 3199–3211. [DOI] [PubMed] [Google Scholar]
- Roschger C, and Cabrele C (2017). The Id-protein family in developmental and cancer-associated pathways. Cell Commun Signal 15, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawen P, Lang S, Mandal P, Rossi DJ, Soneji S, and Bryder D (2016). Mitotic History Reveals Distinct Stem Cell Populations and Their Contributions to Hematopoiesis. Cell reports 14, 2809–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaue D, Kachikwu EL, and McBride WH (2012). Cytokines in radiobiological responses: a review. Radiation research 178, 505–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaue D, Micewicz ED, Ratikan JA, Xie MW, Cheng G, and McBride WH (2015). Radiation and inflammation. Seminars in radiation oncology 25, 4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seita J, and Weissman IL (2010). Hematopoietic stem cell: self-renewal versus differentiation. Wiley interdisciplinary reviews Systems biology and medicine 2, 640–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semerad CL, Mercer EM, Inlay MA, Weissman IL, and Murre C (2009). E2A proteins maintain the hematopoietic stem cell pool and promote the maturation of myelolymphoid and myeloerythroid progenitors. Proc Natl Acad Sci U S A 106, 1930–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh HC, Ji M, Gooya J, Lee M, Klarmann KD, and Keller JR (2009). Cell-nonautonomous function of Id1 in the hematopoietic progenitor cell niche. Blood 114, 1186–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh HC, Leeanansaksiri W, Ji M, Klarmann KD, Renn K, Gooya J, Smith D, McNiece I, Lugthart S, Valk PJ, et al. (2008). Id1 immortalizes hematopoietic progenitors in vitro and promotes a myeloproliferative disease in vivo. Oncogene 27, 5612–5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M, Liu J, Mehta GU, Patel SJ, Roychoudhuri R, Crompton JG, Klebanoff CA, Ji Y, Li P, Yu Z, et al. (2016). Mitochondrial Membrane Potential Identifies Cells with Enhanced Stemness for Cellular Therapy. Cell metabolism 23, 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpp A, Essers M, and Wilson A (2010). Awakening dormant haematopoietic stem cells. Nature reviews Immunology 10, 201–209. [DOI] [PubMed] [Google Scholar]
- Vannini N, Girotra M, Naveiras O, Nikitin G, Campos V, Giger S, Roch A, Auwerx J, and Lutolf MP (2016). Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nature communications 7, 13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlestedt M, Pronk CJ, and Bryder D (2015). Concise review: hematopoietic stem cell aging and the prospects for rejuvenation. Stem cells translational medicine 4, 186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walasek MA, van Os R, and de Haan G (2012). Hematopoietic stem cell expansion: challenges and opportunities. Annals of the New York Academy of Sciences 1266, 138–150. [DOI] [PubMed] [Google Scholar]
- Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, Moehrle B, Brocks D, Bayindir I, Kaschutnig P, et al. (2015). Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 520, 549–552. [DOI] [PubMed] [Google Scholar]
- Wang LH, and Baker NE (2015). E Proteins and ID Proteins: Helix-Loop-Helix Partners in Development and Disease. Developmental cell 35, 269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Esplin B, and Borghesi L (2011). E47 regulates hematopoietic stem cell proliferation and energetics but not myeloid lineage restriction. Blood 117, 3529–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Rodriguez S, Wang L, Wang S, Serezani H, Kapur R, Cardoso AA, and Carlesso N (2016). Sepsis Induces Hematopoietic Stem Cell Exhaustion and Myelosuppression through Distinct Contributions of TRIF and MYD88. Stem cell reports 6, 940–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Related to Figure 7, Differentially expressed genes in Id1−/− and Id1+/+ HSCs from primary BMT Recipients. RNA was obtained from Id1−/− and Id1+/+ HSCs isolated from primary BMT recipient mice 4 weeks after transplantation of Lin− BMCs, and analyzed by RNA-seq for significant differential gene expression.
Table S2. Related to Figure 7, Decreased Molecular Functions in Id1−/− HSC from Primary BMT Recipient Mice. IPA analysis of differentially expressed genes in Table S1 revealed the top molecular functions affected by loss Id1 in HSCs. All functions were significantly decreased in Id1−/− HSCs.
Table S3. Related to Figure 7. Top Networks affected in Id1−/− HSC from Primary BMT Recipient Mice. IPA analysis of differentially expressed genes in Table S1 revealed the top networks affected by loss of Id1 in HSCs.
Table S4. Related to Figure 7. Oligonucleotides for PCR Analysis.