

Abstract
Synthesis and characterization of bile acid-porphyrin conjugates (BAPs) are reported. Binding of saccharides with BAPs in aqueous methanol was studied by monitoring changes in the visible absorption spectral of the porphyrin-moieties. Although these studies clearly showed absorbance changes, suggesting quite high if non-selective binding, the mass spectral studies do not unambiguously support these results.
Keywords: Bile acids, porphyrin, saccharide, visible spectroscopy, 1H-NMR, ESI MS.
Introduction
Bile acids possess a large number of applications in pharmaceutical and supramolecular chemistry, as recently reviewed [1]. For example, bile acids are potential carriers for liver specific drugs and they can be used as cholesterol level lowering agents [2]. Bile acids and their derivatives have also been used for treatment of bile acid deficiency and liver diseases [3]. Furthermore, Matile et al. have reported that some oligomeric bile acid derivatives can act as carriers − folding like umbrellas around hydrophilic guests [4]. More recently, Janout et al. have reported bile acid derived molecular umbrella-assisted transport of an oligonucleotide across cholesterol-rich phospholipid bilayers [5]. Porphyrin conjugates are potential supramolecular hosts as revealed by their use in saccharide sensing [6,7,8,9], anion binding [10,11,12], and nucleotide recognition in protic media [13,14]. We have recently reported the synthesis and characterization, as well as nucleotide binding studies, of tetrameric cholic acid-porphyrin conjugates 1-4 (Figure 1) [15].
Figure 1.

Structures of 1-16.
The design of these conjugates stems from the idea that four bile acid moieties (recognition arms of the conjugate) are attached to one tetratolylporphyrin unit (signaling group of the conjugate) via an alkylammonium amido linkage [15]. In addition to hydrogen bonding, these structures can also provide binding mechanisms via π-stacking and coulombic interactions. All these interaction mechanisms increase the functionality and capability of these conjugates to interact with different biologically active substrates. As an example of the medicinal significance of porphyrin derivatives, their use as sensitizers in photodynamic therapy can be mentioned [16].
The principal aim of this work was to study the suitability of bile acid-porphyrin conjugates as saccharide receptors. However, owing to some contradictory results, as described below, this problem remains unsolved and requires further studies, probably with other experimental techniques.
Results and Discussion
Synthesis and characterization
Conjugates 5-16 (Figure 1) were prepared according to the previously reported procedure for the synthesis of 1-4 (Scheme 1) [15]. In designing these structures we were especially interested in varying the number and position of hydroxyls in monohydroxy (lithocholic), dihydroxy (deoxycholic and chenodeoxycholic), and trihydroxy (cholic) bile acids, thus forming a smoothly changing structural continuum in this series of receptors.
Scheme 1.
Synthesis of 5.

Reagents and conditions: (a) (i) N,N’-di-iso-propylcarbodiimide, N-hydroxysuccinimide, CHCl3, r.t. 2 h; (ii) 2-N,N-dimethylaminoethylamine, pyridine, CHCl3, r.t, 1 day; (b) H2TPP(p-BrCH2)4, CHCl3, reflux, 1 day.
The 1H-NMR spectral characterization of 5-16 was based on our results on bile acids and their derivatives [1], as well as on cholic acid-porphyrin conjugates 1-4 reported before [15]. As in the case of cholic acid derivatives 1-4 [15], the C18 and 19 angular methyls of the steroidal moieties in 5-16 are strongly shielded, due to the anisotropic ring current effect of the BAP porphyrin core. However, the separation of the angular methyl signals into two clearly resolved resonances with 1:3 intensity ratio, as seen in the case of compound 1, was not that clear in 5-16. For example, in 5 the shift difference between the C18 and 19 methyls was only 0.04 ppm, while in 1 it was 0.2 ppm [15]. In compound 6 four resonances in a 1:1:1:2 intensity ratio exist in the most shielded spectral range (from 0.3 to -0.8 ppm). This means that different BAPs possess different conformational characteristics, depending on how many hydroxyls are present in the steroidal moiety and whether they are linked with a m- or p-tolyl-derived porphyrin core. Many BAPs displayed complex 1H- and 13C-NMR spectra in CD3OD at 30 °C, owing to a conformational equilibrium of the steroidal moieties. However, the 1H-NMR spectra simplified remarkably when measured in DMSO-d6 at elevated temperatures. As an example the 1H-NMR spectra of 8 in DMSO-d6 at 30 °C and at 130 °C are shown in Figure 2. A subsequent remeasurement of 8 at 30 °C afforded the same spectrum which was observed before heating, thus revealing the thermal stability of the receptor.
Figure 2.

1H-NMR spectra of 8 in DMSO-d6 measured at 30 °C and 130 °C.
Binding studies
Binding studies with BAPs 1-12 were conducted using three common saccharides, viz. d(+)-glucose, d(+)-sucrose, and maltotriose, and also for sialic acid (Figure 3) with 1-4, by detecting the changes in the visible absorption (419-421 nm) of the porphyrin-moiety of the BAP.
Figure 3.

Structures of the saccharides used.
These experiments have been performed in a 2:1 (v:v) water-methanol solvent mixture because in a pure aqueous medium the aggregation tendency of this kind of conjugates is obvious [16]. In spite of the addition of methanol, we were unable to study the binding in lithocholyl derivatives 13-16 owing to their poor solubility in this solvent system, therefore we are only reporting the synthesis and spectral characterization of the latter compounds. Before UV/Vis spectral measurements, the BAP and saccharide solutions were adjusted to pH 7.4 with dilute hydrochloric acid and sodium hydroxide solutions. It was previously shown that chloride and other inorganic small anions do not cause any changes in the visible absorption of BAPs 1-4 [15]. Titration curves (absorbance as a function of [S]/[BAP]) showed that all saccharides tested had clear influences on the absorbance levels of 1-12. Titrations were performed by adding a solution containing both BAP (2.3 μM) and saccharide (92 μM) into the BAP solution (2.3 μM), covering the range of 0-20 equivalents of saccharide. Measurements were carried out at the wavelength λmax = 419-421 nm depending on the BAPs’ Soret maximum. Binding constants (Ka) can be calculated from the absorbance changes of the porphyrins by equation 1 using the Soret band maximum (ΔA) and assuming 1:1 stoichiometry [17].
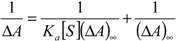 |
(1) |
where [S] is the saccharide concentration and (ΔA)∞ is the extrapolated absorbance change where saccharide concentration goes to infinity. In analyzing the titration curves, their Job’s plots did not show any definite stoichiometries for BAP and saccharide interactions. Attempts to fit experimental data to a 1:1 model gave very high apparent binding constants (Figure 4).
Figure 4.

Titration curve of 1 with maltotriose. Dots denote measured data points and the solid line is a calculated curve based on a 1:1 binding model. Insert: Job’s plot of the titration of [maltotriose]/[1].
Figure 4 shows a typical titration curve of absorbance as a function of [maltotriose]/[1], and its corresponding Job’s plot. The calculated line follows the measured points well at lower saccharide concentrations but deviates significantly from them when the absorbance starts to rise at higher concentrations. Reasons for that phenomenon might be the self-aggregation of saccharide molecules at high saccharide concentrations or conformational changes of BAP. Titration of 1 with glucose in DMSO was performed to check whether an aprotic solvent has some effect to the titration curve, but a similar increase of absorbance was detected also in that case.
ESI mass spectrometric measurements
Because the results of the visible absorption spectral studies were somewhat confusing, ESI-TOF mass spectrometric measurements were also carried out. Equal volumes of equimolar (50 μM) solutions of the BAP and each individual saccharide, viz. d(+)-glucose, d(+)-sucrose, and maltotriose, were prepared. The same solvent mixture (H2O-MeOH 2:1 v:v) as in the UV/Vis experiments was used in order to enable comparison between the results. In general, the intensities of the ions of the BAP-saccharide adducts were weak in comparison with the spectral base peak. Comparison between mass spectra measured for pure receptor 1 and 1 with glucose or sucrose, revealed no explicit differences. With maltotriose additional weak signals were detected in the spectrum measured for the mixture of 1 and the saccharide, compared to the spectrum measured for the pure 1. These additional signals were identified to correspond to the multiply charged ions [1 + maltotriose]4+, [1 + maltotriose + Br-]3+, and [1 + maltotriose + 2Br-]2+. An additional experiment performed for a 1:1:1:1 mixture of 1 and all three saccharides supported the above observations. No ions resulting from adduct formation between glucose or sucrose with 1 could be identified. However, weak multiply charged ions corresponding to maltotriose adducts of 1 were detected. An example is shown in Figure 5.
Figure 5.

Partial ESI-TOF mass spectra for a) 1:1:1:1 mixture of receptor 1 and three saccharides [d(+)-glucose, d(+)-sucrose, maltotriose] and b) pure receptor 1 measured in H2O-MeOH (2:1 v:v) solution.
Thus, these mass spectral measurements did not unambiguously support the above mentioned high binding hypothesis drawn from the visible spectral studies. Although ESI MS results did not clearly reveal high binding, 1H-NMR spectral studies of 5 with glucose have been performed to clarify the possible interaction site of the saccharide with bile acid-porphyrin conjugate (Figure 6). In DMSO-d6 glucose shows a spectrum where all protons are well resolved. For a 1:1 [glucose]:[5] mixture in DMSO-d6 all glucose hydroxyl signals merge into one signal owing to a fast exchange in NMR time scale. Because no significant ASIS (aromatic solvent induced shift)-effect due to the aromatic porphyrin-moiety is observed in the remaining resolved glucose protons, this merging is most likely due to traces of acidic impurities and cannot be construed as proof of any host-guest interaction.
Figure 6.

1H-NMR spectra of d(+)-glucose (bottom), 5 (middle), and 5 + d(+)-glucose (top) in DMSO-d6.
Conclusions
Novel bile acid-porphyrin conjugates (BAPs) have been synthesized and characterized. Binding of four common representatives of mono-, di-, and trisaccharides (S), viz. d(+)-glucose, sialic acid, d(+)-sucrose and maltotriose in these conjugates has been studied by detecting changes in the visible absorption spectra of the porphyrin moieties as a function of [S]/[BAP]. The Job’s plots of these titrations did not reveal any definite stoichiometry for the saccharide binding. Also, the apparent binding constants calculated by assuming 1:1 stoichiometry, although being quite high, did not show any significant selectivity. In contrast with the hypothesis based on the visible absorption spectral studies, ESI-MS investigations did not unambiguously support the notion of high binding between the saccharides and BAPs. Additional work is needed to clarify the basis of this discrepancy between the results obtained with different spectral techniques. Syntheses of related bile acid derivatives with small aromatic cores are in progress. More comprehensive MS studies (not only at a 1:1 molar ratio) could also be useful. Nevertheless, and in spite of their somewhat unclear saccharide binding properties, these BAPs might be useful sensitizers in photodynamic therapy and in distinguishing between cancercerous and normal cells. These studies are also in progress.
Experimental
General
NMR experiments were run on a Bruker Avance DRX 500 FT NMR spectrometer equipped with a z-gradient accessory and a 5 mm diameter inverse detection probehead working at 500.13 MHz for proton spectra. Unless otherwise stated spectra were recorded at 30 °C in CD3OD. 1H-NMR chemical shifts are referenced to the solvent signals of CHD2OD [δ(1H) = 3.31] and DMSO-d5 [δ(1H) = 2.50 ppm] from internal TMS. All NMR acquisition and processing parameters are available from E.K. on request. UV/Vis measurements were performed with a Perkin Elmer Lambda 25 UV/VIS spectrometer using a 1.0 cm glass cuvette. Mass spectrometric measurements were performed using a Micromass LCT time of flight (TOF) mass spectrometer with electrospray ionization (ESI). Elemental analysis was performed using a VarioEL III elemental analyzer. All reagents and solvents (purity ≥ 96% or better) were purchased from Sigma-Aldrich or Fluka and used without further purification. Receptors 5-16 were synthesized by a previously reported procedure [15]. For the analytical and spectral data of the compounds 5,10,15,20-tetrakis[dimethyl (3α,7α,12α-trihydroxy-5β-cholan-24-amidoethyl)-ammoniomethylphen-4-yl] porphyrin tetrabromide (1), 5,10,15,20-tetrakis[dimethyl (3α,7α,12α-trihydroxy-5β-cholan-24-amidoethyl)ammoniomethylphen-3-yl] porphyrin tetrabromide (2), 5,10,15, 20-tetrakis[dimethyl (3α,7α,12α-trihydroxy-5β-cholan-24-amidopropyl)ammoniomethylphen-4-yl] porphyrin tetrabromide (3), and 5,10,15,20-tetrakis-[dimethyl (3α,7α,12α-trihydroxy-5β-cholan-24-amidopropyl)ammoniomethylphen-3-yl] porphyrin tetrabromide (4) see refs. [15,18].
Analytical and spectral data for 5-16.
5,10,15,20-Tetrakis[dimethyl (3α,12α-dihydroxy-5β-cholan-24-amidoethyl)ammoniomethylphen-4-yl] porphyrin tetrabromide (5): Yield 77 %; m.p. > 300 oC (decomp.); 1H-NMR: δ = 0.16−0.22 (18H), 0.23−2.40 (130H), 3.43 (24H, d), 3.70−3.90 (14H), 3.86 (4H, m), 3.92 (4H, m), 4.80 (8H, s), 5.00 (8H, s), 8.10 (8H, d), 8.39 (8H, d), 8.95 (8H, br); MS: m/z = 629 [M]4+, 866 [M+Br]3+, 1338 [M+2Br]2+; M.W. (C160H234N12O12Br4) = 2837.34; Elemental analysis: calcd. (%) for C160H234N12O12Br4 + CHCl3: C, 65.40; H, 8.01; N, 5.68. Found C, 65.46; H, 8.10; N, 5.52.
5,10,15,20-Tetrakis[dimethyl(3α,12α-dihydroxy-5β-cholan-24-amidoethyl)ammoniomethylphen-3-yl] porphyrin tetrabromide (6): Yield 87 %; m.p. > 300 oC (decomp.); 1H-NMR: δ = -0.84 (2H, s), -0.15 (4H, s), -0.10 (4H, s), 0.20−2.30 (136H), 3.36 (24H, d), 3.40−3.60 (8H), 3.65 (8H, m), 3.81 (8H, m), 4.80 (8H, s), 4.96 (8H, s), 8.01 (4H, d), 8.12 (4H, d), 8.45 (8H, m), 8.89 (8H, br); MS: m/z = 629 [M]4+, 866 [M+Br]3+, 1338 [M+2Br]2+; M.W. (C160H234N12O12Br4) = 2837.34; Elemental analysis: calcd (%) for C160H234N12O12Br4: C, 67.73; H, 8.31; N, 5.92. Found C, 67.40; H, 8.54; N, 5.37.
5,10,15,20-Tetrakis[dimethyl (3α,12α-dihydroxy-5β-cholan-24-amidopropyl)ammoniomethylphen-4-yl]porphyrin tetrabromide (7): Yield 83 %; m.p. > 300 oC (decomp.); 1H-NMR: δ = -1.00 − -0.70 (4H), -0.66 (6H, s), 0.00−2.40 (142H), 3.39 (24H, d), 3.35 (12H, m), 3.50−3.70 (14H), 4.80 (8H, s), 4.85 (8H, s), 8.04 (8H, d), 8.39 (8H, d), 8.94 (8H, br); MS: m/z = 643 [M]4+, 885 [M+Br]3+, 1367 [M+2Br]2+; M.W. (C164H242N12O12Br4) = 2893.45; Elemental analysis: calcd (%) for C164H242N12O12Br4 + 1.5 CHCl3: C, 64.70; H, 7.99; N, 5.47. Found C, 64.56; H, 8.12; N, 5.35.
5,10,15,20-Tetrakis[dimethyl (3α,12α-dihydroxy-5β-cholan-24-amidopropyl)ammoniomethylphen-3-yl] porphyrin tetrabromide (8): Yield 95%; m.p. > 300 oC (decomp.); 1H-NMR: δ = -0.33 (2H, s), 0.00 (3H, s), 0.06 (2H, s), 0.17 (2H, s), 0.30 (4H, s), 0.33−2.30 (150H), 3.31 (24H, s), 3.45 (5H, m), 3.59 (10H, m), 4.80 (8H, s), 4.92 (8H, s), 8.02 (4H, d), 8.09 (4H, d), 8.43 (8H, m), 8.92 (8H, br); MS: m/z = 643 [M]4+, 884 [M+Br]3+, 1367 [M+2Br]2+; M.W. (C164H242N12O12Br4) = 2893.45; Elemental analysis: calcd (%) for C164H242N12O12Br4 + CHCl3: C, 65.78; H, 8.13; N, 5.58. Found C, 65.51; H, 8.15; N, 5.31.
5,10,15,20-Tetrakis[dimethyl (3α,7α-dihydroxy-5β-cholan-24-amidoethyl)ammoniomethylphen-4-yl] porphyrin tetrabromide (9): Yield 70 %; m.p. > 300 oC (decomp.); 1H-NMR: δ = 0.04−0.22 (18H), 0.30−2.40 (132H), 3.44 (24H, d), 3.66 (12H, m), 3.85 (4H, m), 3.94 (4H, m), 4.80 (8H, s), 5.01 (8H, s), 8.11 (8H, d), 8.39 (8H, d), 8.94 (8H, br); MS: m/z = 629 [M]4+, 865 [M+Br]3+, 1337 [M+2Br]2+; M.W. (C160H234N12O12Br4) = 2837.34; Elemental analysis: calcd (%) for C160H234N12O12Br4 + 1.5 CHCl3: C, 64.31; H, 7.85; N, 5.57. Found C, 64.54; H, 7.95; N, 5.76.
5,10,15,20-Tetrakis[dimethyl (3α,7α-dihydroxy-5β-cholan-24-amidoethyl)ammoniomethylphen-3-yl] porphyrin tetrabromide (10): Yield 81 %; m.p. > 300 oC (decomp.); 1H-NMR: δ = -0.76 (2H, s), -0.31 (2H, s), -0.01 (2H, s), 0.03 (4H, s), 0.20 (6H, s), 0.30−2.20 (136H), 3.34 (24H, d), 3.50−3.80 (18H), 4.80 (8H, s), 4.91 (8H, d), 7.99 (4H, d), 8.09 (4H, d), 8.42 (8H, m), 8.90 (8H, br); MS: m/z = 629 [M]4+, 866 [M+Br]3+, 1338 [M+2Br]2+; M.W. (C160H234N12O12Br4) = 2837.34; Elemental analysis: calcd (%) for C160H234N12O12Br4 + CHCl3: C, 65.40; H, 8.01; N, 5.68. Found C, 65.73; H, 8.12; N, 5.65.
5,10,15,20-Tetrakis[dimethyl (3α,7α-dihydroxy-5β-cholan-24-amidopropyl)ammoniomethylphen-4-yl] porphyrin tetrabromide (11): Yield 94 %; m.p. > 300 oC (decomp.); 1H-NMR: δ = -1.04 (2H, s), -0.93 (1H, s), -0.80 − -0.60 (8H), 0.10−0.23 (8H), 0.32 (9H, s), 0.33−2.40 (135H), 3.39 (24H, d), 3.50−3.70 (15H), 4.80 (8H, s), 4.96 (8H, d), 8.04 (8H, d), 8.39 (8H, d), 8.93 (8H, br); MS: m/z = 643 [M]4+, 884 [M+Br]3+, 1366 [M+2Br]2+. M.W. (C164H242N12O12Br4) = 2893.45; Elemental analysis: calcd (%) for C164H242N12O12Br4 + 0.5 CHCl3: C, 66.91; H, 8.28; N, 5.69. Found C, 66.43; H, 8.29; N, 5.54.
5,10,15,20-Tetrakis[dimethyl (3α,7α-dihydroxy-5β-cholan-24-amidopropyl)ammoniomethylphen-3-yl] porphyrin tetrabromide (12): Yield 92 %; m.p. > 300 oC (decomp.); 1H-NMR: δ = -0.31 (2H, s), 0.00 (6H, s), 0.10−0.23 (158H), 3.30 (24H, d), 3.50−3.70 (12H), 4.80 (8H, s), 4.89 (8H, d), 8.01 (4H, d), 8.09 (4H, d), 8.43 (8H, m), 8.92 (8H, br); MS: m/z = 643 [M]4+, 884 [M+Br]3+, 1366 [M+2Br]2+; M.W. (C164H242N12O12Br4) = 2893.45; Elemental analysis: calcd (%) for C164H242N12O12Br4 + CHCl3: C, 65.78; H, 8.13; N, 5.58. Found C, 65.48; H, 8.15; N, 5.23.
5,10,15,20-Tetrakis[dimethyl (3α-hydroxy-5β-cholan-24-amidoethyl)ammoniomethylphen-4-yl] porphyrin tetrabromide (13): Yield 71 %; m.p. > 300 oC (decomp.); 1H-NMR: δ = 0.11−0.23 (17H), 0.30−2.40 (141H), 3.44 (24H, d), 3.66 (8H, m), 3.86 (4H, m), 3.94 (4H, m), 4.80 (4H, s), 5.01 (8H, d), 8.11 (8H, d), 8.39 (8H, d), 8.92 (8H, br); MS: m/z = 613 [M]4+, 844 [M+Br]3+, 1306 [M+2Br]2+; M.W. (C160H234N12O8Br4) = 2773.34; Elemental analysis: calcd (%) for C160H234N12O8Br4 + 0.5 CHCl3: C, 68.05; H, 8.34; N, 5.93. Found C, 67.98; H, 8.42; N, 5.86.
5,10,15,20-Tetrakis[dimethyl (3α-hydroxy-5β-cholan-24-amidoethyl)ammoniomethylphen-3-yl] porphyrin tetrabromide (14): Yield 73 %; m.p. > 300 oC (decomp.); 1H-NMR: δ = -0.76 (1H, s), -0.18 (2H, s), -0.04 (3H, s), 0.06 (3H, s), 0.20−2.30 (141H), 3.36 (24H, d), 3.40−3.60 (8H), 3.66 (8H, m), 3.79 (8H, m), 4.80 (4H, s), 4.96 (8H, d), 7.99 (4H, d), 8.11 (4H, d), 8.38 (4H, m), 8.46 (4H, m), 8.90 (8H, br); MS: m/z = 613 [M]4+, 845 [M+Br]3+, 1306 [M+2Br]2+; M.W. (C160H234N12O8Br4) = 2773.34; Elemental analysis: calcd (%) for C160H234N12O8Br4 + CHCl3: C, 66.85; H, 8.19; N, 5.81. Found C, 66.63; H, 8.16; N, 5.60.
5,10,15,20-Tetrakis[dimethyl (3α-hydroxy-5β-cholan-24-amidopropyl)ammoniomethylphen-4-yl] porphyrin tetrabromide (15): Yield 79 %; m.p. > 300 oC (decomp.); 1H-NMR: δ = -0.83 (3H, m), -0.61 (6H, s), 0.00−2.40 (161H), 3.38 (24H, d), 3.54 (12H, m), 4.80 (4H, s), 4.95 (8H, d), 8.04 (8H, d), 8.38 (8H, d), 8.93 (8H, br); MS: m/z = 627 [M]4+, 863 [M+Br]3+, 1334 [M+2Br]2+; M.W. (C164H242N12O8Br4) = 2829.45; Elemental analysis: calcd (%) for C164H242N12O8Br4 + 0.5 CHCl3: C, 68.39; H, 8.46; N, 5.82. Found C, 68.51; H, 8.60; N, 5.73.
5,10,15,20-Tetrakis[dimethyl (3α-hydroxy-5β-cholan-24-amidopropyl)ammoniomethylphen-3-yl] porphyrin tetrabromide (16): Yield 91 %; m.p. > 300 oC (decomp.); 1H-NMR: δ = -0.50−2.40 (168H), 3.31 (24H, d), 3.40−3.70 (14H), 4.80 (4H, s), 4.91 (8H, m), 8.00 (4H, d), 8.08 (4H, d), 8.41 (8H, m), 8.93 (8H, br); MS: m/z = 627 [M]4+, 863 [M+Br]3+, 1334 [M+2Br]2+; M.W. (C164H242N12O8Br4) = 2829.45; Elemental analysis: calcd. (%) for C164H242N12O8Br4 + 0.5 CHCl3: C, 68.39; H, 8.46; N, 5.82. Found C, 68.50; H, 8.62; N, 5.61.
Acknowledgements
The authors acknowledge Special Laboratory Technicians Mirja Lahtiperä and Reijo Kauppinen for their help in running the ESI TOF mass and NMR spectra. E.S. wishes to thank the Academy of Finland for financial support (project no. 7105950). This work was funded by a grant from the Grant Agency of the Czech Republic (Grant No. 203/06/1038) and supported in part by project LC06077 awarded by the Ministry of Education of the Czech Republic MSM.
Footnotes
Sample Availability: Samples of the compounds 1-16 are available from authors.
References and Notes
- 1.Virtanen E., Kolehmainen E. Use of bile acids in pharmacological and supramolecular applications. Eur. J. Org. Chem. 2004:3385–3399. and references cited therein. [Google Scholar]
- 2.Enhsen A., Kramer W., Wess G. Bile acids in drug discovery. Drug Discov. Today. 1998;3:409–418. doi: 10.1016/S1359-6446(96)10046-5. [DOI] [Google Scholar]
- 3.Hofmann A.F. Bile acids as drugs: principles, mechanisms of action and formulation. Ital. J. Gastroenterol. 1995;27:106–113. [PubMed] [Google Scholar]
- 4.Matile S., Som A., Sordé N. Recent synthetic ion channels and pores. Tetrahedron. 2004;60:6405–6435. doi: 10.1016/j.tet.2004.05.052. [DOI] [Google Scholar]
- 5.Janout V., Jing B., Regen S.L. Molecular Umbrella-Assisted Transport of an Oligonucleotide across Cholesterol-Rich Phospholipid Bilayers. J. Am. Chem. Soc. 2006;127:15862–15870. doi: 10.1021/ja053930x. [DOI] [PubMed] [Google Scholar]
- 6.a) Dukh M., Šaman D., Lang K., Pouzar V., Černý I., Drašar P., Král V. Steroid–porphyrin conjugate for saccharide sensing in protic media. Org. Biomol. Chem. 2003;1:3458–3463. doi: 10.1039/b302947f. [DOI] [PubMed] [Google Scholar]; b) Charvatová J., Rusin O., Král V., Volka K., Matejka P. Novel porphyrin based receptors for saccharide recognition in water. Sens. Actuators, B: Chem. 2001;76:366–372. doi: 10.1016/S0925-4005(01)00635-9. [DOI] [Google Scholar]
- 7.a) Hirata O., Kubo Y., Takeuchi M., Shinkai S. Mono- and oligosaccharide sensing by phenylboronic acid-appended 5,15-bis(diarylethynyl)porphyrin complexes. Tetrahedron. 2004;60:11211–11218. doi: 10.1016/j.tet.2004.08.061. [DOI] [Google Scholar]; b) Hirata O., Yamamoto M., Sugiyasu K., Kubo Y., Ikeda M., Takeuchi M., Shinkai S. Allosteric saccharide sensing by a phenylboronic-acids-appended 5,15-bis(triaryl-ethynyl)porphyrin. J. Supramol. Chem. 2003;2:133–142. [Google Scholar]
- 8.Mizutani T., Kurahashi T., Murakami T., Matsumi N., Ogoshi H. Molecular recognition of carbohydrates by zinc porphyrins: Lewis acid/Lewis base combinations as a dominant factor for their selectivity. J. Am. Chem. Soc. 1997;119:8991–9001. doi: 10.1021/ja9713183. [DOI] [Google Scholar]
- 9.Nakata E., Nagase T., Shinkai S., Hamachi I. Coupling a natural receptor protein with an artificial receptor to afford a semisynthetic fluorescent biosensor. J. Am. Chem. Soc. 2004;126:490–495. doi: 10.1021/ja035631i. [DOI] [PubMed] [Google Scholar]
- 10.Kim Y-H., Hong J-I. Carbamate-appended Zn-porphyrin: a neutral receptor for anions. Tetrahedron Lett. 2000;41:4419–4423. doi: 10.1016/S0040-4039(00)00671-7. [DOI] [Google Scholar]
- 11.Lee C., Lee D.H., Hong J-I. Colorimetric anion sensing by porphyrin-based anion receptors. Tetrahderon Lett. 2001;42:8665–8668. doi: 10.1016/S0040-4039(01)01876-7. [DOI] [Google Scholar]
- 12.Starnes S.D., Arungundram S., Saunders C.H. Anion sensors based on β,β’-disubstituted porphyrin derivatives. Tetrahedron Lett. 2002;43:7785–7788. doi: 10.1016/S0040-4039(02)01828-2. [DOI] [Google Scholar]
- 13.Kejík Z., Záruba K., Michalík D., Šebek J., Dian J., Pataridis S., Volka K., Král V. Optical sensing system for ATP using porphyrin–alkaloid conjugates. Chem. Commun. 2006:1533–1535. doi: 10.1039/b518239e. [DOI] [PubMed] [Google Scholar]
- 14.Král V., Shishkanová T.V., Sessler J.L., Brown C.T. Cytosine-substituted metalloporphyrins: receptors for recognition of nucleotides in ion-selective electrodes. Org. Biomol. Chem. 2004;2:1169–1175. doi: 10.1039/b400880d. [DOI] [PubMed] [Google Scholar]
- 15.Kolehmainen E., Koivukorpi J., Sievänen E., Král V. Novel porphyrin-cholic acid conjugates as receptors for biologically important anions. Supramol. Chem. 2005;17:437–441. doi: 10.1080/10610270500127048. [DOI] [Google Scholar]
- 16.Král V., Vasek P., Cigler P., Králová J., Poucková P. Preparation of porphyrin derivatives with polymethine substitution for photodynamic therapy. 20022272. Czech Patent. 2004
- 17.Kubát P., Lang K., Král V., Anzenbacher P., Jr. Preprogramming of porphyrin-nucleic acid assemblies via variation of the alkyl/aryl substituents of phosphonium tetratolylporphyrins. J. Phys. Chem. B. 2002;106:6784–6792. doi: 10.1021/jp014405x. [DOI] [Google Scholar]
- 18.Corrigendum for ref 15: Kolehmainen E., Koivukorpi J., Sievänen E., Král V. Novel porphyrin-cholic acid conjugates as receptors for biologically important anions. Supramol. Chem. 2005;17:437–441. doi: 10.1080/10610270500127048. Supramol. Chem. in press.