

Abstract
Microwave-assisted hydrodistillation was used to isolate an essential oil from the leaves of Cinnamomum iners Reinw. ex Bl., and the results compared with those obtained by conventional hydrodistillation. The composition of the oil from both methods was found to be similar, and (-)-linalool was found as the main component (30-50 %). The antioxidant activity of the essential oil obtained by both methods was evaluated using DPPH, ABTS, FRAP and lipid peroxidation methods, all of which indicated the same but insignificant activity.
Keywords: Microwave, (-)-Linalool, Essential oil, Cinnamomum iners
Introduction
Microwave heating has an incontestable place in analytical and organic laboratory practices as a very effective and non-polluting method of activation. There are numerous examples of applications of this technology in sample digestion, organic synthesis, analytical chemistry and the food industry [1,2,3,4,5,6,7,8]. Microwave energy, with a frequency of 2.45 GHz, is well known to have a significant effect on the rates of a variety of processes. The number of reported applications, especially in the food industry, is increasing rapidly [4,5,6,8]. The main reason for this increased interest lies in the much shorter operation times achievable. Microwave–assisted extraction of natural compounds is also an alternative to conventional techniques. Essential oils are among the products which have been extracted efficiently from a variety of matrices by this method, and many microwave-assisted essential oil extractions from several plants and subsequent product analyses have been reported [9,10,11,12,13,14,15]. However, no such report was found for Cinnamomum iners, a plant in the Cinnamomum genus. This is a large genus, belonging to the family Lauraceae, many species of which yield an essential oil upon distillation [16]. The composition of the oil, and its value and uses to which it is put depend very much on the species that is distilled, as well as the part of the plant which is utilized. The Cinnamomum genus comprises several hundred species which occur naturally in Asia. C. iners is one such species widespread throughout Southest Asia. Known as ‘Ob Chuei’ in Thai, this plant grows mostly in the northern part of Thailand. Its leaves are traditionally used as an analgesic and antipyretic [17].
A number of papers are available reporting both bioassays and component analysis of Cinnamomum in naturally occurring samples. Antifungal properties of essential oils from eleven species of Cinnamomum were reported, viz C. camphora, C. iners, C. microphyllum, C. mollisimum, C. porectum, C. pubescens, C. rhyncophyllum, C. scortechinii, C. sintoc, C. suvabenium and C. zeylanicum [18]. Amylase inhibitor properties were reported for C. cassia Blume, C. zeylanicum Nees, C. obtusifolium Nees, C. sieboldii Meisn, C. burmanni Blume and C. iners Reinw. ex Blume [19]. The chemical components of the leaf, bark and wood oils of seven Malaysian Cinnamomum species were analyzed using GC-MS and selective proton NMR [20]. Our literature search revealed one study of C. iners Reinw. oil from India reporting 1,8-cineole as a major component (40 %), but this oil was obtained from stem bark [21]. The objective of the work described in this communication was to investigate the components and antioxidant activity of the essential oil from the leaves of C. iners obtained by microwave-assisted hydrodistillation, as compared with the normal hydrodistillation.
Results and Discussion
The extraction time of 30 min with microwave-assisted hydrodistillation using 800 W of microwave power provided the same yield of oil (0.12 %) as that obtained after a 5-hour normal hydrodistillation. The compositions of the oils from both methods were analyzed using GC/MS. Both oils were rich in linalool (ca. 35-50 %). Identification of all components was performed by a comparison of their mass spectra with literature data (NIST and WILEY) and by comparison of their retention indices (RI) with those in the literature [22,23]. The gas chromatograms of the essential oils from both methods are presented in Figure 1 and Figure 2, and Table 1 lists the identified components of the oils. The most prominent components found (conventional/microwaved) were linalool (35.56/50.48 %), caryophyllene oxide (6.61/0.86 %), cadinol (6.65/3.37 %), α-cadinol (5.86/3.44 %), delta-cadinene (4.36/3.54 %), viridiflorol (3.87/3.06 %), unidentified (3.58/2.98 %), and caryophyllene (2.83/1.54 %). It can be seen that most of these components and a few more were obtained in smaller amounts in the microwaved oil, compared with the conventional oil. Furthermore, a few other minor components present in the normal oil were missing altogether in the microwaved oil. On the other hand, there were some minor constituents which appeared only in the microwaved oil.
Figure 1.

Gas chromatogram of the hydrodistillate essential oil of C. iners leaves. See Experimental for GC conditions and Table 1 for peak identification.
Figure 2.

Gas chromatogram of the microwave-assisted hydrodistillate essential oil of C. iners leaves. See Experimental for GC conditions and Table 1 for peak identification.
Table 1.
Volatile components in leaves of C. iners.
| Peak No. | Kovát Retention Index | Compounds | % Area | |
|---|---|---|---|---|
| Hydrodistillation | Microwave-assisted | |||
| 1 | - | cis-linalool oxide | 0.22 | - |
| 2 | - | trans-linaool oxide | 0.53 | - |
| 3 | 1103 | linalool | 36.90 | 55.27 |
| 4 | 1176 | borneol L | 0.52 | 1.02 |
| 5 | 1186 | terpinen-4-ol | 0.19 | 0.14 |
| 6 | 1198 | alpha-terpineol | 1.50 | 0.91 |
| 7 | 1232 | 2,6-octadien-1-ol | - | 0.12 |
| 8 | 1258 | geraniol | 0.37 | 0.25 |
| 9 | 1261 | propanoic acid | - | 0.17 |
| 10 | 1291 | (-)-bornyl acetate | 0.60 | 1.08 |
| 11 | 1343 | cyclohexene | - | 0.17 |
| 12 | 1362 | 3-allyl-6-methoxyphenol | - | 1.29 |
| 13 | 1382 | alpha-copaene | 1.32 | 1.19 |
| 14 | 1396 | beta-elemene | 2.32 | 1.82 |
| 15 | 1410 | dodecanal | 0.82 | 1.51 |
| 16 | 1420 | cis-alpha-bergamotene | 0.46 | 0.30 |
| 17 | 1427 | caryophyllene | 2.94 | 1.69 |
| 18 | 1446 | (+)-aromadendrene | 0.45 | - |
| 19 | 1448 | 2-propen-1-ol | - | 0.22 |
| 20 | 1460 | alpha-humulene | 1.63 | 0.97 |
| 21 | 1465 | aromadendrene | - | 0.12 |
| 22 | 1466 | alloaromadendrene | 0.52 | - |
| 23 | 1481 | alpha-amorphene | 0.61 | - |
| 24 | 1485 | germacrene-D | 1.38 | 1.87 |
| 25 | 1490 | beta-selinene | 0.34 | - |
| 26 | 1503 | alpha-muurolene | 0.75 | 0.60 |
| 27 | 1511 | beta-bisabolene | 0.58 | 0.53 |
| 28 | 1519 | alpha-cubebene | 1.22 | 3.30 |
| 29 | 1527 | delta-cadinene | 4.53 | 3.88 |
| 30 | 1564 | epiglobulol | 0.38 | - |
| 31 | 1566 | nerolidol | 0.53 | - |
| 32 | 1571 | palustrol | 0.67 | - |
| 33 | 1577 | unidentified | - | 1.26 |
| 34 | 1581 | spathulenol | 2.04 | 0.46 |
| 35 | 1585 | caryophyllene oxide | 6.85 | 0.95 |
| 36 | 1594 | viridiflorol | 4.01 | 3.36 |
| 37 | 1608 | unidentified | 3.63 | 3.26 |
| 38 | 1617 | tetradecanal | 1.38 | 2.09 |
| 39 | 1639 | calarene | 0.43 | - |
| 40 | 1643 | naphthalene, 1,2,3,4,4a,7-hexahydro | 2.18 | 0.93 |
| 41 | 1656 | isospathulenol | 0.50 | - |
| 42 | 1662 | alpha-cadinol | 6.10 | 3.78 |
| 43 | 1667 | alpha-copaene | 1.71 | 0.72 |
| 44 | 1679 | cadinol | 7.03 | 3.70 |
| 45 | 1712 | alpha-longipinene | 0.58 | 0.19 |
| 46 | 1742 | unidentified | 0.48 | 0.38 |
| 47 | 1774 | benzyl benzoate | 0.80 | 0.51 |
A possible reason for these minor differences may be the different heat sources used in the two methods. It is known that a microwaved solution is sometimes superheated, with the temperature of the solution being as much as 20 degress higher than normal. This occurs also when tap water is used instead of distilled water [24], as was the case in this present work. Thus, those oil components that are less heat stable, e.g. caryophyllene oxide, might have undergone some decomposition to other compounds not present in the conventional oil.
The identification of the major component, linalool, was also performed by separating the pure linalool from the crude oil by PLC. The optical rotation of the isolated linalool was measured, whereby the levorotatory (-) form of linalool was identified, [α]D24.5 = -22.2 (4.5, ethanol). Confirmation was obtained from spectroscopic data. The 1H-NMR spectrum indicated the presence of three CH3 singlets at δ 1.20, 1.53 and 1.61, which was confirmed by DEPT technique, whereby three CH3, three CH2 and two CH were identified. The protons located on adjacent carbons were assigned by performing a standard COSY experiment. The 1H-NMR spectrum was also identical to that given in the literature [25]. The 13C-NMR spectrum showed 10 signals, of which two at δ 73.57 and 131.90 were assigned to the quaternary carbons. The IR spectrum exhibited a strong broad hydroxy group absorption band (3,396 cm-1).
The antioxidant activities of the oils were evaluated using DPPH, ABTS, FRAP, and lipid peroxidation methods and the results are shown in Table 2. It can be seen that both oils had more or less of the same activity. However, it is obvious that these values for the activity all point to the same conclusion that the activity appears insignificant.
Table 2.
Antioxidant activity of C. iners volatile oil.
| DPPH method IC50 (μg/mL) |
ABTS method TEAC (mM/mg) |
||
| Hydrodistillation | Microwave-assisted | Hydrodistillation | Microwave-assisted |
| 218.88 | 216.66 | 0.176 | 0.175 |
| FRAP method EC1 (μM) |
Lipid peroxidation method AA (%) |
||
| Hydrodistillation | Microwave-assisted | Hydrodistillation | Microwave-assisted |
| 387.77 | 387.41 | 13.22 | 13.28 |
Conclusions
The use of microwave-assisted hydrodistillation to isolate an essential oil from the leaves of Cinnamomum iners Reinw. ex Bl. provided another example of the advantages of utilizing microwave irradiation as a heat source in isolating essential oils, viz. the much shorter time used in the isolation process to give the same yield of oil. In the case of the essential oil from the leaves of C. iners, it was found to be a linalool-rich oil, containing 30-50% linalool. However, the use of the microwave as heat source for distillation might be the cause for a slight difference of the composition of the oil from that of the conventionally obtained oil. The anti-oxidant activity of the oil from both methods was found to be the same but insignificant.
Experimental Section
Materials
Fresh leaves of C. iners were collected in Nonghan district, Sansai, Chiang Mai, Thailand on 19 December 2003. The plant material was identified by J. Maxwell from the Department of Biology at Chiang Mai University. Voucher specimen was deposited at the Department of Biology Herbarium, Chiang Mai University with Accession No. 22738.
Isolation and Analysis of the Essential Oil
Fresh leaves (700 g) were homogenized and hydrodistilled for 5 h using a Clevenger-type apparatus to yield about 0.12% of yellowish-colored oil with a strong odor. Fresh leaves (200 g) were also homogenized and hydrodistilled at 800 W for 30 min using an adapted microwave distillation apparatus which consists of a microwave oven connected to a Clevenger-type apparatus as illustrated in Figure 3, to yield 0.12 % of yellowish-colored oil with a strong odor.
Figure 3.

Adapted microwave distillation apparatus.
The maximum output power of this adapted microwave apparatus was 1,000 W with 2,450 MHz of microwave irradiation frequency. The reactor was a 500 mL short-necked flask. Analytical instruments with the following specifications were used: GC/MS was an Agilent 6890/HP 5975 with a 25m x 0.25 mm capillary column, (HP-5MS/CB5, film thickness 0.25 mm); temperature program: 3 min isothermal at 100 oC (no peaks before 100 oC after first injection), then at 4 oC/min to 280 oC (5 min isothermal); carrier gas He. The ionization voltage was 70 eV. Programmed-temperature Kováts retention indices (RI) were obtained by GC/MS analysis of an aliquot of the essential oil spiked with an n-alkane mixture containing each homologue from n-C8 to n-C30. IR spectra were run on a Perkin-Elmer FTIR spectrometer. 1H-NMR, 1H-COSY, 13C-NMR and DEPT were recorded on a Varian 300 spectrometer, the spectra being measured in CDCl3 at 25 oC at 300 MHz (100 MHz for 13C-NMR) with TMS as internal reference. Identification of the compounds was based on a comparison of their mass spectra with database (Wiley & NIST) and spectroscopic data. The optical rotation was measured on a Bellingham + Stanley (Model D) polarimeter. The isolation of linalool from the essential oil was carried out by PLC, with silica gel as adsorbent, and dichloromethane as eluent. The linalool was obtained in 37.5 and 55.7 % yields from the oil obtained from normal and microwave hydrodistillation, respectively. The structural confirmation of the isolated linalool was done spectroscopically by optical rotation, IR, MS and NMR: [α]D24.5 = -22.2 (4.5, ethanol); IR (neat) cm-1: 3369, 2969, 2926, 2854, 1451, 1375, 1113, 995, 920; MS m/z (% relative intensity): 71 (100), 93 (83), 43 (51), 55 (49), 80 (35), 121 (30); 1H-NMR: 1.20 (3H, s, CH3), 1.4-1.5 (2H, m, CH2), 1.53 (3H, s, CH3), 1.61 (3H, s, CH3), 1.95 (2H, q, CH2, J = 0.02 Hz), 2.08 (1H, broad s, OH), 4.97 (1H, d, CH, J = 0.032 Hz), 5.04 (1H, t, CH, J = 0.02 Hz), 5.13 (1H, d, CH, J = 0.054 Hz), 5.84 (1H, dd, CH, J = 0.035, 0.016 Hz); 13C-NMR: 17.82 (CH3), 22.97 (CH2), 25.84 (CH3), 27.89 (CH3), 42.28 (CH2), 73.57 (C), 111.83 (CH2), 124.58 (CH), 131.90 (C), 145.24 (CH).
Antioxidant activity: DPPH method [26]
The hydrodistillate was dissolved in methanol to give a solution of 100 ppm concentration. This sample was further diluted to obtain seven different concentrations (two-fold dilutions). Each concentration was tested in triplicate. A portion of sample solution (1 mL) was mixed with an equal volume of 0.2 mM DPPH (1,1-diphenyl-2-picrylhydrazyl) in absolute methanol and allowed to stand at room temperature for 30 min. The absorbance (A) was then measured at 514.5 nm (Hitachi U-2001 UV-spectrophotometer). Vitamin E was tested in the same system as a positive control. The results were expressed as percentage inhibition. The percent inhibition was calculated from the equation: % inhibition = [(A control – A sample )/ A control]×100. IC50 value (inhibition concentration of sample required to scavenge DPPH radical by 50 %) was obtained by linear regression analysis of the dose-response curve plot (% inhibition versus concentration).
ABTS Method [27]
The antioxidant activity of the essential oils was measured by the ABTS (2,2'-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) radical cation (ABTS·+) decolourisation assay involving preformed ABTS radical cation (ref). ABTS was dissolved in water to a 7 mM concentration. ABTS radical cation was produced by reacting ABTS stock solution with 2.45 mM potassium persulfate in water and allowing the mixture to stand in the dark at low temperature for 16-18 h before use. The solution was diluted in ethanol (1:20 v/v) to give an absorbance at 750 nm of 0.800±0.200. The stock solutions of volatile oil and BHT (1 mg/ml) were prepared in ethanol.
Ferric Reducing Antioxidant Power (FRAP) [28]
The FRAP assay was carried out by the method of Benzie and Strain [26] with some modifications. FRAP reagent containing 10 mmol/L TPTZ in 40 mmol/L HCl (5 mL) plus 20 mmol/L FeCl3·6H2O (5 mL) and 0.3 mol/L acetate buffer (50 mL, pH 3.6) was used. The FRAP reagent (180 μL) was mixed with sample or standard (20 μL) and the absorbance at 595 nm was measured at 15 min, using a microplate reader. Ethanol was used for control. Ethanolic solutions of FeSO4·7H2O of known Fe(II) concentration, ranging from 5-50 μmol/L (final concentration), was used for the preparation of the calibration curve. The parameter Equivalent Concentration (EC1) was defined as the concentration of antioxidant having a ferric-TPTZ reducing ability equivalent to that of 1 mmol/L FeSO4·7H2O. EC1 was calculated as the concentration of antioxidant giving an increase in the FRAP assay equivalent to the theoretical absorbance value of a 1 mmol/l concentration of Fe (II) solution, determined using the corresponding regression equation.
Lipid Peroxidation (β-Carotene Bleaching Method) [29]
Lipid Peroxidation assay was modifed by the method of Miller [30]. A solution of β-carotene was prepared by dissolving β-carotene (2 mg) in chloroform (2 mL) and mixing this with linoleic acid (50 mg) and Tween 20 (50 mg). This solution (2 mL) was transferred into a 100 mL round bottom flask. After the chloroform was removed under vacuum, aerated distilled water (50 mL) was added to the flask with vigorous shaking. The same procedure was repeated without β-carotene as negative control. Aliquots (180 µL) of this emulsion were transferred into different plates containing the essential oil (20 µL). As soon as the emulsion was added to each plate, the zero time absorbance was measured at 490 nm, using a microplate reader. The plates were placed at 45 °C in an incubator and the absorbance was measured after 150 minutes. A blank, devoid of β-carotene, was prepared for background subtraction. The same procedure was repeated with the volatile oil (1 mg/mL), as positive control. Antioxidant activity was calculated using the following equation:
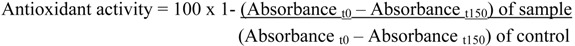 |
Acknowledgments
The financial support from Thailand Research Foundation (grant code: RMU5080006) to W. P. is gratefully acknowledged. We would like to thank Mr Suttiruk Plonjarean from the Department of Agronomy, Faculty of Agricultural Production, Maejo University for his help in the collection of plant material.
Footnotes
Sample Availability: Samples of (–)-linalool are available from authors.
References and Notes
- 1.Phutdhawong W., Buddhasukh D., Pyne S., Rujiwatra A., Pakawatchai C. Microwave-assisted facile synthesis and crystal structure of cis-9,10,11,15-tetrahydro-9,10[3',4']-furanoanthracene-12,14-dione. Synth. Comm. 2006;36:881–883. doi: 10.1080/00397910500466025. [DOI] [Google Scholar]
- 2.Phutdhawong W., Buddhasukh D. Facile microwave-assisted synthesis of 9,10-dihydro-9,10-ethanoanthracene-11-carboxylic acid methyl ester. Molecules. 2005;10:1409–1412. doi: 10.3390/10111409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bougrin K., Loupy A., Soufiaoui M. Microwave-assisted solvent-free heterocyclic synthesis. J. Photochem. Photobiol. C: Photochem. Rev. 2005;6:139–167. doi: 10.1016/j.jphotochemrev.2005.07.001. [DOI] [Google Scholar]
- 4.Venkatesh M. S., Raghavan G. S. V. An Overview of Microwave Processing and Dielectric Properties of Agri-food Materials. Biosyst. Eng. 2004;88:1–18. doi: 10.1016/j.biosystemseng.2004.01.007. [DOI] [Google Scholar]
- 5.Wang L., Sun D.-W. Recent developments in numerical modelling of heating and cooling processes in the food industry-a review. Trends Food Sci. Tech. 2003;14:408–423. doi: 10.1016/S0924-2244(03)00151-1. [DOI] [Google Scholar]
- 6.Li B., Sun D.-W. Novel methods for rapid freezing and thawing of foods - a review. J. Food Eng. 2002;54:175–182. [Google Scholar]
- 7.Lidstrom P., Tierney J., Wathey B., Westman J. Microwave assisted organic synthesis-a review. Tetrahedron. 2001;57:9225–9283. doi: 10.1016/S0040-4020(01)00906-1. [DOI] [Google Scholar]
- 8.Zhang M., Tang J., Mujumdar A. S., Wang S. Trends in microwave-related drying of fruits and vegetables. Trends Food Sci. Tech. 2006;17:524–534. doi: 10.1016/j.tifs.2006.04.011. [DOI] [Google Scholar]
- 9.Lucchesi M. E., Smadja J., Bradshaw S., Louw W., Chemat F. Solvent free microwave extraction of Elletaria cardamomum L.: A multivariate study of a new technique for the extraction of essential oil. J. Food Eng. 2007;79:1079–1086. doi: 10.1016/j.jfoodeng.2006.03.029. [DOI] [Google Scholar]
- 10.Ferhat M. A., Meklati B. Y., Smadja J., Chemat F. An improved microwave Clevenger apparatus for distillation of essential oils from orange peel. J. Chromatogr. A. 2006;1112:121–126. doi: 10.1016/j.chroma.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 11.Deng C., Xu X., Yao N., Li N., Zhang X. Rapid determination of essential oil compounds in Artemisia Selengensis Turcz by gas chromatography-mass spectrometry with microwave distillation and simultaneous solid-phase microextraction. Anal. Chim. Acta. 2006;556:289–294. doi: 10.1016/j.aca.2005.09.038. [DOI] [Google Scholar]
- 12.Wang Z., Ding L., Li T., Zhou X., Wang L., Zhang H., Liu L., Li Y., Liu Z., Wang H., Zeng H., He H. Improved solvent-free microwave extraction of essential oil from dried Cuminum cyminum L. and Zanthoxylum bungeanum Maxim. J. Chromatogr. A. 2006;1102:11–17. doi: 10.1016/j.chroma.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 13.Chemat F., Lucchesi M. E., Smadja J., Favretto L., Colnaghi G., Visinoni F. Microwave accelerated steam distillation of essential oil from lavender: A rapid, clean and environmentally friendly approach. Anal. Chim. Acta. 2006;555:157–160. doi: 10.1016/j.aca.2005.08.071. [DOI] [Google Scholar]
- 14.Tigrine-Kordjani N., Meklati B. Y., Chemat F. Microwave 'dry' distillation as a useful tool for extraction of edible essential oils. Int. J. Aromather. 2006;16:141–147. doi: 10.1016/j.ijat.2006.09.007. [DOI] [Google Scholar]
- 15.Lucchesi M. E., Chemat F., Smadja J. Solvent-free microwave extraction of essential oil from aromatic herbs: comparison with conventional hydro-distillation. J. Chromatogr A. 2004;1043:323–327. doi: 10.1016/j.chroma.2004.05.083. [DOI] [PubMed] [Google Scholar]
- 16.Wannissorn B., Jarikasem S., Siriwangchai T., Thubthimthed S. Antibacterial properties of essential oils from Thai medicinal plants. Fitoterapia. 2005;76:233–236. doi: 10.1016/j.fitote.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 17.Pormjit S. Pharmacognosy. R. D. P. Press; BKK: 1989. pp. 86–87. [Google Scholar]
- 18.Mastura M., Azah M. A. N., Khozirah S., Mawardi R., Manaf A. A. Anticandidal and antidermatophytic activity of Cinnamomum species essential oils. Cytobios. 1999;98:17–23. [PubMed] [Google Scholar]
- 19.Iida N., Ishii R., Hakamata J., Myamoto S., Oozeki H. Amylase inhibitors for food and pharmaceutical. No. JP 09040572. Jpn. Kokai Tokkyo Koho. Patent. 1997
- 20.Bin Jantan I., Goh S. H. Essential oils of Cinnamomum species from Peninsular Malaysia. J. Essent. Oil Res. 1992;4:161–171. doi: 10.1080/10412905.1992.9698038. [DOI] [Google Scholar]
- 21.Baruah A., Nath S. C., Hazarika A. K. Stem bark oil of Cinnamomum iners Reinw. Indian Perfumer. 2001;45:261–263. [Google Scholar]
- 22.Adams R. P. Identification of Essential Oil Components by GC/MS. Allured Publishing Corporation; Carol Stream, ILL, USA: 1995. [Google Scholar]
- 23.Davies N. W. Gas chromatographic retention indices of monoterpenes and sesquiterpenes on methyl silicon and Carbowax 20M phases. J. Chromatogr. A. 1990;503:1–24. doi: 10.1016/S0021-9673(01)81487-4. [DOI] [Google Scholar]
- 24.Pelle L., Jason T., Bernard W., Jacob W. Microwave Assisted Organic Synthesis_a Review. Tetrahedron. 2001;57:9225–9283. doi: 10.1016/S0040-4020(01)00906-1. [DOI] [Google Scholar]
- 25.Bryliakov K. P., Talsi E. P., Stas’ko S. N., Kholdeeva O. A., Popov S. A., Tkachev A. V. Stereoselective oxidation of linalool with tert-butyl hydroperoxide, catalyzed by a vanadium(V) complex with a chiral terpenoid ligand. J. Mol. Catal. A: Chem. 2003;194:79–88. doi: 10.1016/S1381-1169(02)00527-7. [DOI] [Google Scholar]
- 26.Phutdhawong W., Donchai A., Korth J., Pyne S. G., Picha P., Ngamkham J., Buddhasukh D. The components and anticancer activity of the volatile oil from Streblus asper. Flav. Frag. J. 2004;19:445–447. doi: 10.1002/ffj.1342. [DOI] [Google Scholar]
- 27.Roberta R., Nicoletta P., Yang M., Rice-Evans C. Antioxidant activity applying an improved ABTS radical catin decolorization assay. Free Rad. Biol. Med. 1999;26:1231–1237. doi: 10.1016/S0891-5849(98)00315-3. [DOI] [PubMed] [Google Scholar]
- 28.Benzie I. F. F., Strain J. J. The Ferric Reducing Ability of Plasma (FRAP) as a Measure of Antioxidant Power: The FRAP Assay. Anal. Biochem. 1996;239:70–76. doi: 10.1006/abio.1996.0292. [DOI] [PubMed] [Google Scholar]
- 29.Jiang P., Burczynski F., Campbell C., Pierce G., Austria J. A., Briggs C. J. Rutin and flavonoid contents in three buckwheat species Fagopyrum esculentum, F. tataricum, and F. homotropicum and their protective effects against lipid peroxidation. Food Res. Int. 2007;40:356–364. doi: 10.1016/j.foodres.2006.10.009. [DOI] [Google Scholar]
- 30.Miller N. J. A simplified method for the evaluation of antioxidants. J. Am. Oil. Chem. Soc. 1971;48:91. doi: 10.1007/BF02635693. [DOI] [Google Scholar]