

Abstract
Interactions between herbivorous insects and their host-plants are a central component of terrestrial food webs and a critical topic in agriculture, where a substantial fraction of potential crop yield is lost annually to pests. Important insights into plant-insect interactions have come from research on specific plant defenses and insect detoxification mechanisms. Yet, much remains unknown about the molecular mechanisms that mediate plant-insect interactions. Here we use multiple genome-wide approaches to map the molecular basis of herbivory from both plant and insect perspectives, focusing on butterflies and their larval host-plants. Parallel genome-wide association studies in the Cabbage White butterfly, Pieris rapae, and its host-plant, Arabidopsis thaliana, pinpointed a small number of butterfly and plant genes that influenced herbivory. These genes, along with much of the genome, were regulated in a dynamic way over the time course of the feeding interaction. Comparative analyses, including diverse butterfly/plant systems, showed a variety of genome-wide responses to herbivory, yet a core set of highly conserved genes in butterflies as well as their host-plants. These results greatly expand our understanding of the genomic causes and evolutionary consequences of ecological interactions across two of nature’s most diverse taxa, butterflies and flowering plants.
Introduction
Butterflies and moths, and the host-plants that their larvae feed on, comprise two of the largest groups of species on earth, the Lepidoptera and Angiosperms. The extreme diversity of these two groups has arisen from the co-evolutionary interactions between them, wherein evolution proceeded via reciprocal adaptations as each clade evolved in response to changes in the other 1,2. In their seminal paper, Ehrlich and Raven3 used the diffuse evolutionary relationships among butterflies and their host-plants to formally introduce the concept of co-evolution. In the 50 years since Ehrlich and Raven3, research on the molecular basis of herbivory between butterflies and their host-plants has revealed important insights into specific chemicals and defense pathways utilized by plants, such as furanocoumarins, glucosinolates, and cardenolides4. Research on butterflies, in turn, has revealed specific genes and gene families involved in host-plant detoxification, such as cytochrome P450 enzymes5,6 , the nitrile-specifier protein of pierid butterflies7, and the Na(+) ,K(+) –ATPase of the monarch butterfly8. Yet, despite these important advances, our understanding of the molecular genetic basis of these co-evolutionary interactions remains limited. Genomic approaches, including genome-wide association (GWA) studies and transcriptomics, provide a means to move beyond candidate genes and pathways to uncover the molecular determinants of this fundamental ecological interaction in an unbiased way9,10. Here we utilize these methods in a diversity of butterflies and their respective host-plant to uncover the genetic basis of herbivory.
Results
Genome-wide associations with herbivory: the host-plant, Arabidopsis thaliana
First, we investigated the genetics of butterfly/host herbivory by mapping associated variants in parallel GWA studies focused on the flowering plant Arabidopisis thaliana and its natural insect herbivore, the Cabbage White butterfly, Pieris rapae. For these experiments, we used either 96 natural accessions of A. thaliana (Supplementary Table 1) and a single lab strain of P. rapae (for the plant GWAS), or the offspring of 96 field-caught females of P. rapae (Supplementary Table 2) and a single accession of A. thaliana (for the butterfly GWAS). For both experiments, we measured herbivory as the amount of weight gained and the amount of leaf surface area eaten by second instar larvae over a period of 72 hours. Arabidopisis thaliana GWA resulted in a total of 90 associated SNPs that were in linkage-disequilibrium with 389 genes. Analysis of gene enrichment, using DAVID11, revealed plant defense as a prominent annotation cluster among these 389 genes (Supplementary data 1). While plant defense was not significantly enriched after correcting for multiple comparisons, this category had an enrichment score of 1.43, which is above the value (1.3) indicative of enrichment11. A subset of 12 well-supported candidate genes contained three or more associated SNPs each (Fig. 1a). Eight of these genes were functionally validate using SALK T-DNA mutants, showing increased larval weight gain and/or increased plant material eaten in knock outs vs. control (Fig. 1c, Supplementary Table 3). This validated gene set includes well-known and novel defense genes. For instance, the cytochrome P450 gene CYP79B2 is involved in the conversion of tryptophan to indole-3-acetaldoxime, a precursor of indole glucosinolates and indole-3-acetic acid12. Indole glucosinolates are important secondary metabolites used for defense by Arabidopsis, and other species in the plant family Brassicaceae, and they have been shown to deter herbivores and pathogens13,14. Insects that feed on Brassicaceae have evolved various physiological strategies against toxic effects of glucosinolates15. The larvae of P. rapae, for instance, redirect the hydrolysis pathway catalyzed by myrosinase7, and instead of producing toxic isothiocyanates, hydrolysis is redirected toward the formation of nitriles by the butterfly’s nitrile specifier protein (NSP)16,17. Another functionally validated gene, phosphoribosylanthranilate isomerase 3 (PAI3), catalyzes a step in the L-tryptophan synthesis pathway18, to produce the precursor of indole glucosionolates, although there is no direct evidence that it influences glucosinolates. The genes PROPEP1 and PROPEP3 belong to the AtPep (endogenous danger peptides) gene family. They are associated with activation of danger- or damage-associated molecular patterns (DAMPs) immunity in plants against both pathogen and herbivore attack19. The other four genes validated in our study do not have known roles in defense and include an uncharacterized cytochrome P450 (CYP705A33), Importin alpha (IMPA-1), a CTP synthase (AT4G20320), and an S-adenosyl-L-methionine-dependent methyltransferase (AT1G29470). The A. thaliana genes we found associated with herbivory did not overlap with those from a recent genome-wide association study of methionine-derived glucosinolates in A. thaliana20. This is expected, perhaps, because these aliphatic glucosinolates do not appear to negatively impact P. rapae larvae21.
Figure 1:
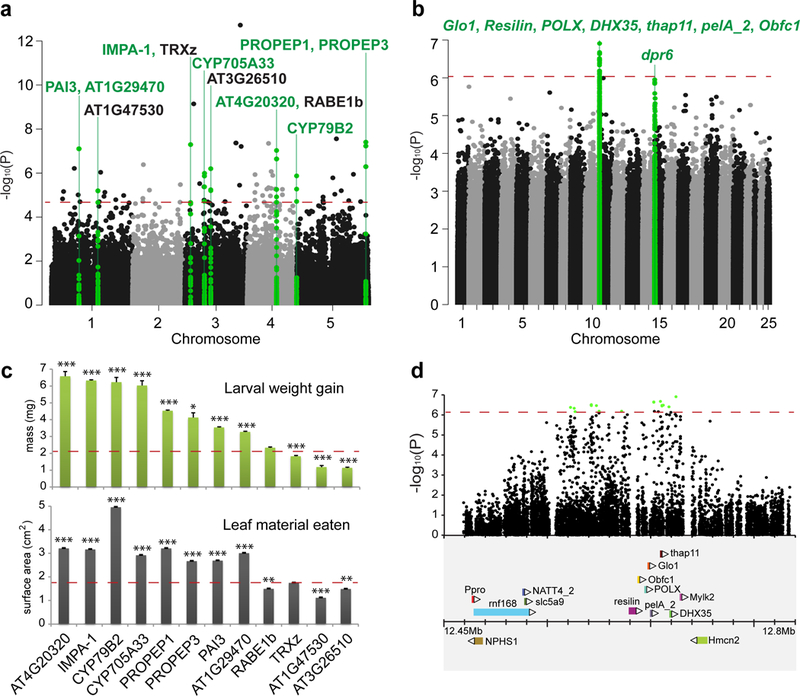
Butterfly and host-plant herbivory candidate genes identified by parallel genome-wide association studies. (a) Manhattan plot with the associated SNPs and their corresponding p-values from the herbivory GWAS based on 96 accessions of Arabidopsis thaliana. The eight genes that were functionally validated among the primary 12 candidate genes are highlighted in green. (b) Manhattan plot with the associated SNPs and their corresponding p-values from the herbivory GWAS based on 96 Pieris rapae samples. The genes contained in the two association peaks are listed. The significance threshold for each GWAS is indicated with a dashed red line. (c) Average larval weight gain (top panel) and leaf surface area eaten (bottom panel) on the SALK T-DNA mutants for the 12 candidate genes from the Arabdiopsis GWAS (* p < 0.05, ** p < 0.01, *** p < 0.001). Plots show mean ± s.e.m. and the corresponding mean for the wild type plant (col-0) is represented as a line across each histogram. Individual values, means, and statistical tests are in Supplementary Table 3. (d) Zoom-in on the chromosome 10 region associated with herbivory in the P. rapae genome. Significantly associated SNPs are indicated in green with a gene map below.
Pieris rapae genome sequencing
We began our butterfly GWAS by assembling a high-quality reference genome sequence for the Cabbage White butterfly, Pieris rapae. For the reference genome, we combined next-generation DNA sequencing data from one PCR-free paired-end Illumina library, three mate-pair libraries (3 kbp, 7 kbp, 40 kbp), a Dovetail Chicago library, and information from the genome assembly of a closely related species, Pieris napi22, to generate an assembly of 323 Mbp, with a N50 of 11.5 Mbp, spanning the 25 P. rapae chromosomes23 (Supplementary Figs. 1 and 2). Annotation was performed using a diverse set of RNA-seq data. For subsequent analyses, we removed unplaced scaffolds from the assembly which represented redundant haplotypes, yielding a chromosomal assembly of 272 Mbp. K-mer size estimation of the P. rapae genome was 289.8 Mbp. This haploid, chromosomal-level assembly of the butterfly genome provided an essential genomic foundation with which to explore the insect side of the plant-insect interaction.
Genome-wide associations with herbivory: the herbivore, Pieris rapae
For GWA, we performed our larval feeding trials with the offspring of wild-caught females and used whole genome resequencing to genotype 96 unrelated larvae at genome-wide single-nucleotide polymorphisms (SNPs). Pieris rapae GWA revealed just two strongly associated regions in the genome (Fig. 1b), the largest of which encompassed a total of 16 significantly associated SNPs distributed across 98 kbp on chromosome 10, spanning seven genes (Fig. 1d). One of these genes, Glyoxalase 1 (Glo1), a lactoylglutathione lyase, is a central enzyme in the glyoxalase pathway present in all organisms. Glyoxalase detoxifies cytosolic methyglyoxyl, a toxic by-product of metabolism, in a two-step process that utilizes glutathione. Glutathione itself is an important metabolite involved in multiple biological processes in plants and animals24. In addition to Glo1, the 98 kbp associated region also contained the following genes: Resilin, a Retrovirus-related Pol polyprotein POLX, the ATP-dependent RNA helicase DHX35, a THAP domain-containing protein, and two uncharacterized genes. A second associated region in the genome was prominent and close to the significance threshold so we explored it further. This region on chromosome 15 contained just one gene, Defective proboscis extension response 6 (dpr6) (Supplementary Fig. 3). The dpr gene family is involved in the sensory physiology of adult Drosophila melanogaster, specifically response to gustatory cues25. Our results suggest this gene plays a role in butterfly larval growth, possibly by mediating gustatory response to host-plant material or in the context of host-plant detoxification. The GWA regions on chromosomes 10 and 15 exhibited evidence of balancing selection, including elevated nucleotide diversity (π) and Tajima’s D values compared to the flanking 100 kbp upstream and the downstream, as well as significant HKA tests for a number of genes within the chromosome 10 GWA interval (Supplementary Figs. 4 and 5).
Herbivory-induced differential gene expression experiments
To further interrogate the GWA genes, and explore genome-wide patterns of gene expression throughout the plant-insect interaction, we next used transcriptomics to measure differential gene expression in P. rapae and A. thaliana over the time course of their interaction. For these experiments, butterfly and plant were allowed to interact for a period of time, after which we harvested tissue from both organisms, as well as controls, for RNA-seq (Fig. 2a-f, Supplementary Table 4). For these experiments, the plant and butterfly genome sequences were used as references to analyze RNA-seq data. The genes identified in our butterfly and plant GWA experiments showed patterns of expression consistent with a role in herbivory (Fig. 3). In A. thaliana, eight of 12 genes identified in the GWA study had the highest expression in leaves exposed to eggs and larvae and/or leaves with larvae only, including both cytochrome P450 genes and PROPEP3. PROPEP1 and PAI3 showed elevated expression in leaves with eggs only, followed by leaves with larvae only. The remaining two genes, IMPA-1 and RABE1b (one of the four genes that was not validated) had other expression patterns, showing elevated expression in mechanically wounded leaves and the leaf control, respectively (Fig. 3a). Statistical comparisons, using ANOVA and Tukey’s HSD post-hoc tests, supported a number of these expression differences, notably for the two cytochrome P450 genes as well as PROPEP1 (Supplementary Table 5). In P. rapae, a number of the associated genes had the highest expression in larval treatments—the larva is the herbivorous life stage of the butterfly—and were expressed at lower levels in eggs, pupae and adults. Notably, this pattern of expression was distinct from physically adjacent genes that were located outside the associated regions (Fig. 3b and 3c). Resilin expression was elevated specifically in larvae feeding on leaves, as opposed to those eating artificial diet, indicative of a role directly in the plant-insect interaction (Supplementary Table 5).
Figure 3:
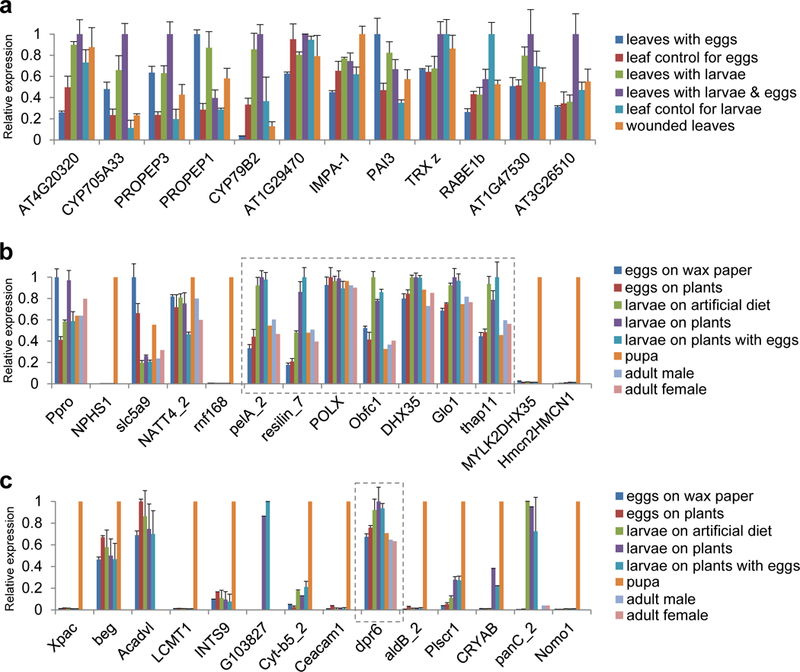
Temporal expression patterns of herbivory candidate genes in Arabidopsis thaliana and Pieris rapae. (a) Expression patterns from various RNA-seq treatments of the 12 Arabidopsis thaliana candidate genes identified in the herbivory GWAS. (b) Expression patterns from various RNA-seq treatments of the seven Pieris rapae genes contained in the associated region on chromosome 10 (boxed), identified in the Pieris rapae herbivory GWAS, as well as genes from the adjoining 100kb regions upstream and downstream. (c) Expression patterns from the various RNA-seq treatments of the dpr6 gene identified in the Pieris rapae herbivory GWAS, as well as genes from the adjoining 100kb regions upstream and downstream. Plots show mean ± s.e.m of gene expression values, fragments per kilobase of transcript per million fragments mapped (FPKM), scaled to the highest value treatment for each gene. Individual values, means, and statistical tests are in Supplementary Table 5.
The genome-wide responses of Arabidopisis and Pieris, in terms of differential gene expression, were also striking. In particular, A. thaliana showed a massive response to butterfly oviposition with approximately 50% of the plant’s genes changing expression, mostly up-regulated, in response to a female butterfly laying eggs (Fig. 2g). The response of the plant to larval feeding, in contrast, was more modest. The egg is the first life stage of the herbivore that is in contact with the plant and previous studies have shown that plants launch various defenses against insects even before larval hatching26. Egg-induced plant defense strategies include plant-mediated desiccation of eggs, egg dropping, egg crushing, and egg killing26. We found that a total of 14,563 genes were differentially expressed in Arabidopsis leaves after P. rapae egg deposition (Fig. 2i). Genes belonging to defense and stress responses were enriched in all treatments (Supplementary data 2). In addition to genes involved in production of glucosinolates and glutathiones, these include protein kinases, proteolytic enzymes, oxidoreductases, peroxidases, NBS-LRR and defense signaling transcription factors that are known to be involved in release of reactive oxygen species, production of pathogenicity-related proteins, activation of systemic acquired resistance, cell wall modification, and programmed cell death in pathogens27. These results mirror, and greatly extend, previous work with microarrays and a different species of Pieris, P. brassicae showing an elevated response of A. thaliana to insect oviposition with induction of genes involved in the hypersensitive-like response and pathogenesis-related genes as well as callose and reactive oxygen species accumulation28–30.
Figure 2:
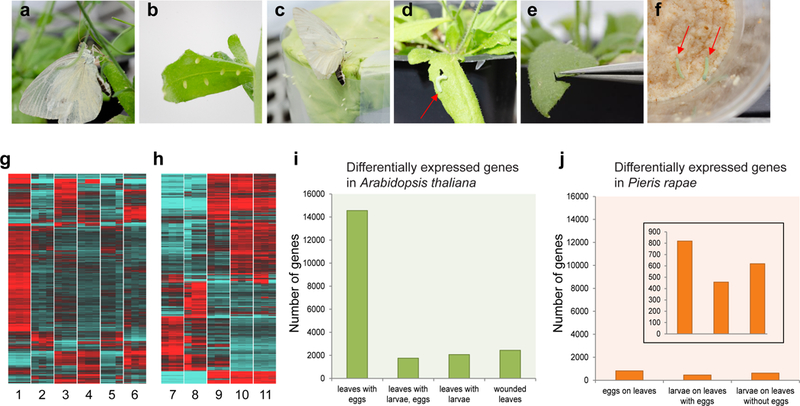
Genome-wide responses to herbivory in Arabidopsis thaliana and Pieris rapae over the time course of their interaction. Samples collected from various stages of interaction: (a, b) eggs laid on the leaves, (c) eggs laid on the wax paper, (d) larva feeding on a leaf, (e) mechanical wounding of the leaf, (f) larvae feeding on artificial diet. Heat maps of differentially expressed genes in (g) Arabidopsis thaliana and (h) Pieris rapae. Color scale ranges from ≤ –1.5 log fold change (blue) to ≥1.5 log fold change (red). Individual treatments correspond to: 1) leaves with eggs 72h after oviposition, 2) leaves with no eggs, control for 72h oviposition treatment, 3) leaves after 24h larval (48h old) feeding, 4) leaves with eggs after 24h larval (48h old) feeding, 5) leaves with no eggs and larvae, control for larval feeding and wounding, 6) leaves 24h after wounding, 7) eggs 72h after oviposition on wax paper, 8) eggs 72h after oviposition on leaves, 9) 48h old larvae after 24h feeding on artificial diet, 10) 48h old larvae after 24h feeding on leaves with eggs, 11) 48h old larvae after 24h feeding on leaves. (i) Number of differentially expressed Arabidopsis thaliana genes across treatments. (j) Number of differentially expressed Pieris rapae genes across treatments - the inset shows the same results plotted with a reduced y-axis scale. Details of treatment and control conditions are in Supplementary Table 4.
Similar to the plant, P. rapae eggs exhibited an elevated response in comparison to the larval stages (Fig. 2h). Of all the genes differentially expressed between eggs oviposited on leaves and wax paper, approximately 50% of them were uncharacterized. Among the genes with predicted functions, genes responding to stress, oxidoreductases, and proteolytic enzymes were abundant (Supplementary data 3). In the next stage of their interaction, larvae feeding on the plant, we analyzed gene expression patterns in larvae feeding on plants exposed to eggs as well as those feeding on plants not previously exposed to eggs. We found that larvae feeding on plants exposed to eggs had a smaller number of differentially expressed genes compared to larvae feeding on plants that were not exposed to eggs (Fig. 2j). This mirrored gene expression patterns in the plant (Fig. 2i), in which the added effect of herbivory after oviposition was less than the effect of herbivory alone. There were 120 differentially expressed larval genes in common between the two treatments, and among the genes with putative functions, genes involved in defense, stress response and proteolysis were overrepresented (Supplementary data 4). Interestingly, glutathione s-transferases (GSTs) frequently show elevated expression in the larvae of Lepidoptera31–33 and other insects34, but we did not detect differential expression of GSTs in P. rapae.
Comparative transcriptomics across butterflies and plants
The strong response of A. thaliana to butterfly eggs was a particularly striking result. To determine if this was a general property of butterfly-plant interactions, we expanded our analysis of gene expression to three additional plant/insect systems: Medicago sativa/Colias eurytheme, Citrofortunella microcarpa/Papilio polytes and Passiflora oerstedii/Heliconius cydno (Fig. 4a). For this comparative analysis, we generated de novo transcriptomes for all organisms to use as references to analyze patterns of differential gene expression using RNA-seq data. We also reanalyzed our A. thaliana/P. rapae data using de novo reference transcriptomes, as opposed to the genome sequences, for consistency. Interestingly, we found substantial variation in the responses of both plants and butterflies, and no plant showed the same, elevated response to oviposition that we saw in A. thaliana (Fig. 4b, c). To investigate which, if any, components of the defense network were conserved among plants or among butterflies, we extracted and compared common differentially expressed genes across the four systems. Among plants, common gene families that were differentially expressed in leaves after oviposition were protein kinases, proteases, heat shock proteins, esterases, MYB transcription factors and NAC transcription factors (Supplementary data 5), all of which have previously been implicated in defense against pathogens35,36. During larval feeding, leaves of the four plant species shared protein kinases, proteases, heat shock proteins, esterases, MYB transcription factors, NAC transcription factors, and in addition, glutathione s-transerfases, cytochrome P450s, xyloglucan endotransglycosylases, Dof zinc finger proteins and WRKY transcription factors (Supplementary data 6). Based on protein alignments, we identified a small set of true orthologs among the differentially expressed genes, 11 in total, which were shared across all plant species. Leaves after oviposition did not have any shared orthologs but the larval feeding treatments had 4 or 5 orthologs each (Supplementary data 7), the majority of which have putative roles in defense.
Figure 4:
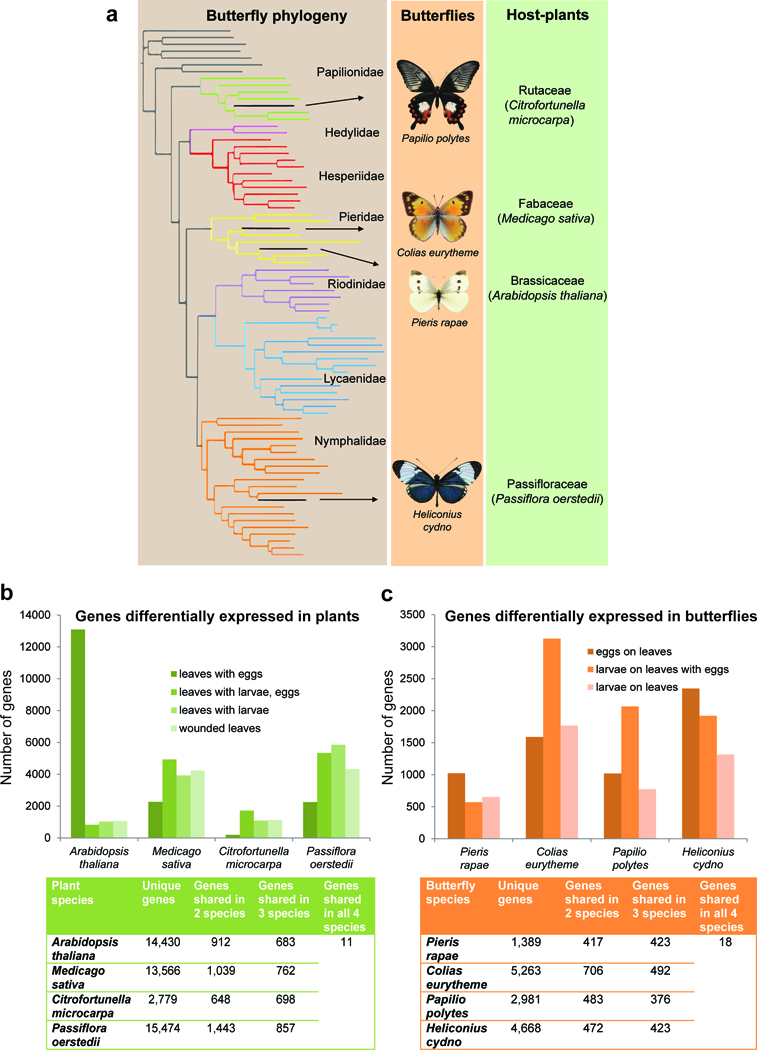
Comparative transcriptomics of herbivory across four diverse butterflies and their host-plants. (a) Butterfly phylogeny, adapted from ref. 91 displaying the four butterfly species included in the comparative transcriptomics analysis and their respective host-plants. (b) Number of differentially expressed genes across treatments for each host-plant species (above), and counts of differentially expressed genes that are unique or shared across species (below). (c) Number of differentially expressed genes across treatments for each butterfly species (above), and counts of differentially expressed genes that are unique or shared across species (below).
Among butterfly species, the common gene families that were differentially expressed in eggs 72 h following oviposition were proteases, heat shock proteins, esterases, chemosensory proteins and cuticular proteins (Supplementary data 8), all of which have previously been implicated in detoxification pathways37,38. In larvae feeding on plants, the genes differentially expressed belonged to gene families such as chitinases, proteases, cuticular proteins, lipases and Osiris (Supplementary data 9). There was a small set of differentially expressed genes, 17 in total, that were true orthologs shared among all butterfly species (Supplementary data 10). Among this set of conserved genes, Osiris 9 (Osi9) stood out because it was up-regulated in all four butterfly species specifically in larvae feeding on leaves with previous exposure to eggs. Osi9 is a transmembrane protein that is a member of the 24 gene Osiris family that is unique to insects39. The function of Osiris genes has been mysterious39,40 but the Osiris gene cluster was recently associated with detoxification of the Morinda citrifolia host-plant in both Drosophila sechellia41,42 and Drosophila yakuba43. Previous work has shown that Lepidoptera appear to have multiple copies of Osi939 and we detected multiple paralogs of Osi9 in the P. rapae genome and across our assembled transcriptomes (Supplementary Fig. 6). However, the copy of Osi9 that was up-regulated in larvae on all four butterfly species was the same, Osiris 9E (Supplementary Fig. 3). To further explore the potential role of Osi9E in butterflies, we surveyed spatial and temporal patterns of Osi9E expression in the four butterfly species and found elevated expression during larval stages, as opposed to pupae and adult, and expression specifically in the larval gut (Supplementary Figs. 7 and 8, Supplementary Tables 6 and 7). These results suggest Osi9E expression is up-regulated in response to contact with host-plant tissue.
Discussion
Butterflies and their larval host-plants provide a historically significant example of co-evolution and important prior research has explored the molecular genetic basis of this ubiquitous ecological interaction5–8,16,17,32,33. Here we used two genome-wide approaches, genome-wide association studies and transcriptomics, to characterize the genetic basis of herbivory in butterflies and plants simultaneously. Our genome-wide association studies uncovered a relatively small number of well-supported herbivory genes in both A. thaliana and P. rapae. The A. thaliana GWAS yielded 12 genes that contained three or more associated variants, eight of which we were able to validate with knock-out lines, and this gene set contained both established and novel plant defense genes. P. rapae GWAS revealed just one strongly associated region of the genome that contained only seven genes. One of these genes, Glyoxalase 1, stands out as a particularly good candidate gene because it utilizes glutathione to detoxify methylglyoxal, a toxic by-product of cellular metabolism. Glutathione is a defensive compound in plants: glutathione concentrations increase in plants during oxidative stress44–47 and it has been shown to be involved in defense against pathogens48 and insect feeding49. Furthermore, glutathione levels are known to vary among Arabidopsis accessions50 and we found natural sequence variation in the GST genes in our mapping panel (Supplementary data 11). It is intriguing to consider the possibility that P. rapae may somehow be using the glyoxalase pathway to detoxify host-plant glutathione. A second P. rapae GWA peak, while not statistically significant, was notable and contained just a single gene, dpr6. Defective proboscis extension response (dpr) genes are a subclass of immunoglobulin-domain containing proteins that interact with Dpr-interacting proteins (DIPs) to specify synaptic connections51. Functionally, dpr6 is an intriguing herbivory candidate gene because the dpr gene family is known to influence the response of Drosophila melanogaster to gustatory cues25. The combined P. rapae GWA results suggest that we may have identified genes related to host-plant detoxification and/or larval metabolism and growth.
Previous research on Pieris rapae and related butterflies has identified the nitrile-specifier protein (NSP) as a critical component in host-plant detoxification7. The origin of NSP appears to be the key innovation that allowed the ancestor of the butterfly subfamily Pierinae to colonize and detoxify host-plants in the order Brassicales, all of which produce glucosinolates (i.e., mustard oils)16,17. Prior to our experiments, we hypothesized that genetic variation at NSP may also influence detoxification capacity in contemporary populations and that we may see associations with NSP in our butterfly GWA study. This was not the case, but it is possible that a butterfly GWA study that incorporated other phenotypes, such as larval survival or development time, might detect associations with NSP. Furthermore, NSP was not differentially expressed in our transcriptomic experiments with P. rapae, although it was expressed in all larvae. In contrast, glucosinolate pathway genes did emerge in the Arabidopsis GWAS and many were differentially expressed in our transcriptomic studies, suggesting that glucosinolate defense remains an active front in this co-evolutionary arms race, at least on the plant side of the interaction.
Expanding our analysis to four diverse butterfly/plant systems, we found that over evolutionary time, the molecular dynamics of the plant-insect interaction change dramatically. The number, timing, and identity of genes expressed in plants and butterflies throughout the plant/insect interaction differed considerably across the four systems we studied. This finding highlights that the specific genetic underpinnings of herbivore-plant dynamics derived from a model system like Arabidopsis may not always be generalizable to other systems. However, such a result is also expected because the process of co-evolution should drive each system along a very different evolutionary trajectory, especially over the long times scales that separate the butterfly and plant species we are studying. What is surprising, then, is that we did identify a core set of orthologous genes that were differentially expressed in response to herbivory in all butterfly/plant systems. A total of 11 orthologs were differentially expressed in all plant species and 17 in butterflies, but this summary belies further complexity as a number of these genes, while all differentially expressed, were regulated in opposite directions among systems, being upregulated in one species and downregulated in another. One gene that stood out among the conserved orthologs in butterflies was Osiris 9E, both because it was consistently expressed at high levels in larvae of all four butterfly species and because Osiris genes were recently implicated in host-plant detoxification in Drosophila41–43. Furthermore, Osiris 9 and a number of other Osiris genes have been shown to be differentially expressed in the larvae of the fly Scaptomyza flava in response to feeding on glucosinolates52. These results suggest that Osiris genes are ancient players in insect/plant interactions.
As a whole, our results provide a comprehensive portrait of the molecular genetic dynamics mediating insect/host interactions. Importantly, we are able to not only corroborate previous findings related to the identity of particular defense genes and pathways in Arabidopsis, but also expand this to other plants as well as the insect herbivores. The results from Pieris and other butterflies pinpoint specific genes that appear important for herbivory and these results further hint at even deeper evolutionary ties among herbivorous insects as a whole. Important next steps will be to functionally characterize these genes and identify the molecular and cellular mechanisms by which they impact herbivory. A complete understanding of the molecular genetic basis of the pervasive, antagonistic relationship between caterpillars and their host-plants promises to inform our understanding of ecology, evolution, and human agriculture.
Methods
Genome assembly and annotation.
Genomic DNA was isolated from single, seventh-generation inbred female Pieris rapae pupae, using ethanol precipitation53. This inbred line of P. rapae was established using a singly-mated wild female caught in August 2013 near Rochester, Pennsylvania, USA (+40° 44’ 45”, –80° 9’ 45”). Three Illumina libraries were prepared, one PCR-free DNA library (180bp) and two mate-pair libraries (3kb and 7kb).The 180bp library was sequenced in two lanes and the two mate-pair libraries were sequenced in one lane each on Illumina HiSeq High Output mode, PE 100bp. Genomic DNA was isolated as described above from another inbred P. rapae pupae, a sibling of the first sample described above, and a 40kb mate pair library was constructed and sequenced by using a Lucigen NxSeq 40 kbp Mate-Pair Cloning Kit. For the final scaffolding step variable insert size libraries of 100bp – 100,000bp, using DNA from a third sibling, were generated using the Chicago and HiRise methods54 and these were sequenced by Centrillion Biosciences Inc. (Palo Alto, CA, USA), Illumina HiSeq High Output mode, PE100bp.
Genome size was estimated at 289.8 Mbp from unique k-mer distribution of the raw data using Jellyfish (version 2.1.3)55 and a custom R script. For the 3kb and 7kb libraries, Nextclip (version 0.8)56 was used to look for the absence of linker sequence in either read in a pair and discard those reads as potential contamination of non-mate pair sequence. All read sets were then quality filtered, the ends trimmed of adapters and low quality bases, and screened of common contaminants using bbduk (version 34.94, https://github.com/BioInfoTools/BBMap/blob/master/sh/bbduk.sh). For contig generation and scaffolding, the 180bp, 3kb, and 7kb reads were assembled using AllpathsLG (version 50960)57. The best assembly was obtained by using a random subset of 56 (33%) million reads from the initial 3kb and 7kb libraries with the full set of 162 million reads from the 180bp library, more input data resulted in reduced performance and quality of assembly. AllpathsLG was run with haploidify = true option to compensate for the high degree of heterozygosity present in the P. rapae data. The assembly was composed of 318 million bases contained in 15,799 scaffolds with a N50 of 54,044 bp. Complete conserved single copy ortholog content was assessed at 87% by CEGMA (version 2.5)58. A second scaffolding step using SSPACE v259 and the 3kb, 7kb, and 40kb libraries together brought up the assembly size and the N50 to 143, 392 bp. A final scaffolding step was undertaken by Dovetail genomics using the custom library and the HiRise scaffolding pipeline54, which improved the N50 to 3,706,409 bp.
The HiRise assembly was assessed with BUSCO (version 3.0.2)60 and found to contain 95.9% of complete single copy orthologs in the insecta protein set and 9.5% gene duplication (Supplementary Table 8). This was reduced to 5.5% duplication in the chromosomal assembly due to the selection of a single duplicated region for inclusion in the assembly from those that aligned to Pieris napi (Supplementary Table 8). The unplaced scaffolds of the full assembly account for 4.1% of the duplicated and 8% of the missing orthologs in the chromosome only assembly. Scaffolds that aligned to P. napi were left unplaced if the fell completely within a larger aligned scaffold. If there was a partial overlap, the smaller scaffold was broken and the unique fragment was placed adjacent to the larger scaffold (details in Supplementary Material finalPrapae.agp). Chromosomal relationships of scaffolds were inferred from alignment using LAST (version 714)61 to the chromosomal structure of a closely related species Pieris napi22 (Supplementary Fig. 1), and validated with scaffold junction spanning mate pair and syntenic blocks (Supplementary Fig. 2). The final P. rapae assembly contained 323,179,347 bp in 25 chromosomes and 2,747 unplaced scaffolds and a N50 of 11,535,178 bp. After excluding the unplaced scaffolds, a 272,024,799 bp haploid, chromosomal assembly was generated that served as the reference Pieris rapae genome for further analyses.
Our genome sequencing project was independent of another recently published Pieris rapae genome sequence62. The previously published genome sequence of Pieris rapae was 246 Mbp, compared to our chromosomal assembly of 272 Mbp, which prompted us to investigate the differences between the genomes. We found that 95% of the previously published assembly was contained in our HiRise scaffolds, covering an average of 56% of the HiRise scaffolds. Only 594 HiRise scaffolds had no coverage at all from the previous assembly, accounting for 5 Mbp.
The HiRise assembly was annotated using RNA-seq derived from Pieris rapae which were sampled in September 2013 from a continuous, inbred lab colony established in October 2012 from 4 females collected in Rochester, Pennsylvania. All pooled individuals were full siblings. Tissue specific samples included 3 individuals for the male reproductive tract, 5 individuals for the female reproductive tract, and 80 salivary glands from 40 individuals. Subsequent genome annotation involved the following pipeline: a) Collection of reference proteins from Uniprot database63 and assembly of high-confidence transcript sequences from previously published RNA-seq data64 using tophat2 (version 2.0.9)65,66 and the cufflinks package (version 2.2.1)67, b) Modelling of repeat sequences to mask the genome using RepeatMasker package (version 4.0.3, Smit, AFA, Hubley, R & Green, P. RepeatMasker Open-3.0.1996–2010 <http://www.repeatmasker.org>) and RepeatModeler package (version 1.0.8, Smit, AFA, Hubley, R. RepeatModeler Open-1.0.2008–2015 <Error! Hyperlink reference not valid. ), c) Evidence-based gene build to generate training models for ab-initio gene finders using the Maker package (version 2.31–6)68, d) Manual curation of gene models and training of the Augustus gene finder (version 2.7)69, e) Re-annotation of the evidence-based annotation using ab-initio predictions, and finally, f) Functional annotation of the refined gene build using Blast matches against Uniprot/Swissprot and results from InterproScan, condensed and reconciled using ANNotation Information Extractor (Annie)( Tate, R., Hall, B., DeRego, T., & Geib, S. (2014). Annie: the ANNotation Information Extractor (Version 1.0)). This process produced a gene build with a final count of 15,047 genes and 35,824 mRNAs.
Sample collection and data analysis for GWAS.
Host-plant GWAS: The growth chamber conditions for growing Pieris rapae and Arabidopsis thaliana were 23°C day/21oC night and 60% relative humidity on a 16 hour photoperiod. Three replicates of each of the 96 accessions of Arabidopsis thaliana that are listed in Supplementary Table 1 were grown until they were almost ready to bolt. After taking a picture of each plant, two 5 day old, lab-grown P. rapae larvae were weighed and placed on each plant and then the plant was enclosed in a plastic sleeve bag. After 72 h, the larvae were weighed and a new picture of the plant was taken in order to record plant surface area eaten by the larvae. The weight gained by the larvae and the total surface area eaten were calculated and used as the phenotype data for GWAS (Supplementary data 12). The SNP information for the 96 accessions from the 250K SNP data70 and the phenotype data were fit using a multivariate linear mixed model in GEMMA (version 0.94.1) for association studies71. The pipeline involves converting the SNP file into a PLINK binary PED file and generating a relatedness matrix file using default parameters. The average initial weight of the larvae was used as a covariate for the analysis. For the functional validation of the candidate genes, knock out T-DNA mutants from SALK and wild type plants were grown and assayed as described above.
Herbivore GWAS: 96 P. rapae females were collected from various locations across the US Midwest during June–July 2014 and raised in the lab green house. These butterflies included 57 from around the University of Chicago campus, 19 from Schaumburg, IL, and 16 from North Dakota, 3 from downtown Chicago, and 1 from Carolina Biological Supply. The growth chamber conditions for growing P. rapae and A. thaliana were 23°C day/21oC night and 65% relative humidity on a 16 hour photoperiod. Eggs were collected from each female and two 5 day old larvae from each family were weighed and placed together on a Col-0 A. thaliana plant and this was performed in triplicate. Leaf area eaten and weight gain phenotypes were assayed as described for the Arabidopsis GWAS (Supplementary data 13).
For genotyping, we selected one random GWAS larva from each family (a total of 96 larvae), sequenced the genome of each sample to a mean coverage of 13.8X (Supplementary Table 2), and analyzed the data using the P. rapae genome assembly. Medium coverage sequencing depth (5–20X) is considered the most cost-effective choice for sample sizes close to 100 individuals72 and variation in coverage did not introduce spurious population structure. The PCA plot was generated using the --pca option in PLINK (version 2.0)73 (Supplementary Fig. 9) and the admixture plot was generated using the default options of ADMIXTURE (version 1.3)74 (Supplementary Fig.10).
Genomic DNA was extracted from skin tissue of the larvae using the VDRC Drosophila genomic extraction (https://stockcenter.vdrc.at/images/downloads/GoodQualityGenomicDNA.pdf ) and sequencing libraries were prepared using KAPA Hyper Prep Kits (KR0961 – v1.14). Barcoded libraries were pooled and sequenced on an Illumina HiSeq 2500 to generate paired-end 100 bp data. The average QPhred score of the 96 samples was 36.2 and no sample had less than 35 (a Phred score of 20 corresponds to a 1% error rate). Raw reads were further passed through additional quality control software Trimmotatic (version 0.36)75, where all bases in the reads that were below a quality score of 15 were trimmed using a sliding window of 4 bp and all reads less than 36 bp in length were discarded. Trimmed reads were aligned to the P. rapae reference genome using bowtie276, using a –very-sensitive-local option, and the aligned SAM files were prepared for calling SNPs using PICARD tools (version 1.141, http://broadinstitute.github.io/picard). Three PICARD tools were used with default parameters in the following order: SortSam, AddOrReplaceReadGroups, and MarkDuplicates. GATK (version 3.4)77,78 was then used to call SNPs. During this process, we used BaseRecalibrator to readjust and correct any errors in the quality scores assigned by the sequencing machine, RealignerTargetCreator to locally realign reads reducing the number of mismatches, and IndelRealigner. Finally, UnifiedGenotyper was used to call SNPs jointly for all 96 samples with a minimum phred-scaled confidence threshold of 50. The accuracy of SNP calling was increased by providing all the samples jointly because the variant caller estimates posterior probability likelihoods at each site using the information from all samples and then proceeds to assign SNP genotypes. We used –dcov (downsampling of coverage) of 150 for uniformity among samples.
The SNP information for the 96 larvae (a total of 18,603,675 SNPs) and the phenotype data were fit using a multivariate linear mixed model in GEMMA (version 0.94.1) for association studies71. SNPs with missingness > 5% or a minor allele frequency < 5% were not be included in the analysis. The pipeline involves converting the SNP file into a PLINK binary PED file and generating a relatedness matrix file using default parameters. Again, the average initial weight of larvae was used as a covariate for the analysis. The statistical significance thresholds for both the Arabidopsis and Pieris GWAS were determined using simpleM79, a multiple testing correction method for genetic association studies using correlated SNPs. To test the robustness of the GWAS results, the association tests were repeated 1000 times with randomized sample names in the phenotype file using custom scripts. 99% of the randomized datasets yielded ≤ 3 significant SNPs at our empirical significance threshold and only one randomized dataset yielded more significant SNPs (18 compared to our empirical result of 16, Supplementary Figure 11).
Selection scans
Nucleotide diversity (π) and Tajima’s D values were estimated using a window size of 1 kbp and the --window-pi and --TajimaD options of the VCFtools (version 0.1.13). We analyzed Pieris rapae SNPs across the herbivory associated genomic regions, as well as the neighboring 100 kbp upstream and downstream (Supplementary Figs. 4 and 5). HKA tests80 were used to compare polymorphism and divergence (compared to Pieris napi) among genes in the herbivory associated genomic regions and flanking regions. HKA tests were performed using DnaSP (version 6)81.
Arabidopsis and Pieris RNA-seq.
Pieris rapae was raised on Arabidopsis thaliana at 23°C and 65% relative humidity on a 16 hour photoperiod. Samples at different time points were collected from both the plant and insect in TRIzol Reagent (Supplementary Table 4). To capture gene expression changes in response to oviposition, we compared gene expression profiles of (a) leaves with eggs (Fig. 2a,b) vs. leaves without eggs 72 h after oviposition, and (b) eggs deposited on leaves vs. eggs deposited on wax paper 72 h after oviposition (Fig. 2c). To capture gene expression changes of the plant in response to larval feeding, we compared (c) leaves after 24 h of larval feeding from plants previously exposed to eggs vs. leaves after 24 h of larval feeding from plants not previously exposed to eggs (Fig. 2d) vs. control leaves never exposed to eggs or larvae. We also analyzed leaves 24 h after mechanical wounding (Fig. 2e) to compare gene expression changes associated with larval feeding vs. wounding. To identify gene expression changes in the insect associated with feeding, we compared (d) larvae after 24 h of feeding on leaves from plants exposed to eggs vs. larvae after 24 h of feeding on leaves from plants not exposed to eggs vs. larvae after 24 h of feeding on artificial diet (Fig. 2f). We also measured gene expression in butterfly pupae and adults, for comparison.
RNA was extracted using TRIzol Reagent and the RNA-seq libraries were generated using an Illumina TruSeq Stranded Total RNA Kit with Ribo-Zero Plant for Arabidopsis and Illumina TruSeq Stranded mRNA Library Prep Kit for Pieris. These libraries were sequenced on an Illumina HiSeq 2500 to generate SR50bp reads. The DNA sequence data from all three replicates of each treatment were processed with the Trimmomatic (version 0.36)75 QC pipeline, where all the bases in the reads that were below the quality score of 15 were trimmed using a sliding window of 4bp. All reads that were less than 25 bp in length were discarded. The trimmed SR50bp reads were aligned to their respective genomes using STAR (version 2.4.2)82 software using default parameters, except for Intron minimum size, which was set to 0 bp for both Arabidopsis and Pieris. In addition, the non-canonical junctions were removed for compatibility with Cufflinks using the RemoveNoncanonical option. After alignment, using the Cufflinks program within the Cufflinks suite (version 2.2.1)67, each library was assembled as an individual transcriptome and expression values were quantified using default parameters. The library type was set to fr-firststrand. Then all GTF files generated by Cufflinks were merged together into a single GTF using Cuffmerge. The differential expression testing between all pairs of treatments was performed using Cuffdiff tool with all the default parameters and an FDR-corrected p-value set to 0.001. The differentially expressed set of genes in Arabidopsis thaliana and Pieris rapae were annotated using the annotations available for their respective genomes.
Comparative transcriptome analysis.
Each butterfly species was reared on its natural host-plant under favorable environmental conditions in the laboratory greenhouse. Pieris rapae was raised on Arabidopsis thaliana at 23°C and 65% relative humidity on a 16 hour photoperiod. Colias eurytheme was raised on Medicago sativa at 26°C and 65% relative humidity on a 16 hour photoperiod. Papilio polytes was raised on Citrofortunella microcarpa at 26°C and 65% humidity on a 16 hour photoperiod. Heliconius cydno was raised on Passiflora oerstedii at 26°C and 65% relative humidity for a 13 hour photoperiod. Samples at different time points were collected from both plants and herbivores in TRIzol Reagent. These are the same time points as those listed in Supplementary Table 4. RNA was extracted using TRIzol Reagent, and RNA-seq libraries were generated using Illumina TruSeq Stranded Total RNA Kit with Ribo-Zero Plant for the four plant species and Illumina TruSeq Stranded mRNA Library Prep Kit for the four butterfly species. These libraries were sequenced on an Illumina HiSeq 2500 to generate SR50bp reads. In addition, one library from each of the host-plant species and two libraries from of each butterfly species (the egg and a larval stage) were sequenced with PE100bp in order to generate a reference transcriptome for each species. The number of reads generated from each library is listed in Supplementary Table 9.
The SR50bp and PE100bp reads were processed with the Trimmomatic QC pipeline (version 0.36)75. All trimmed PE100bp reads and the trimmed SR50bp reads were assembled together for each host-plant and herbivore system to generate a reference transcriptome with the Trinity package (version r20140717)83. After excluding different isoforms, as predicted by Trinity, the number of unique coding sequences found in each plant species was: A. thaliana, 18,105; M. sativa, 43,843; C. microcarpa, 20,367; and P. oerstedii, 22,073. The number of unique coding sequences in each butterfly species was: P. rapae, 12,037; C. eurytheme, 16,277; P. polytes, 15,176; and H. cydno, 19,914. Trimmed SR50bp reads from each species were aligned to their respective reference transcriptome and differential expression analyses were performed with the Trinity package, which includes bowtie276 for alignment, RSEM84 for transcript quantification and edgeR85 for differential expression analysis. Default parameters and an FDR-adjusted p-value of 0.001 were used for analyses.
We visualized the correlation among samples and treatments by generating PCA plots of the RNA-seq expression data from all four plant and butterfly species using the PtR option in the Trinity package83 (Supplementary Figs.12 and 13). These analyses showed that the RNA-seq expression data clustered in biologically meaningful ways. For instance, we found that biological replicate samples grouped together, as did samples from similar experimental treatments (butterfly eggs vs. larvae, leaf tissue exposed to eggs vs. leaf tissue exposed to larvae).
For both Pieris rapae and Arabidopsis thaliana, we analyzed the RNA-seq data two different ways, using Cuffdiff67 and Trinity83. These different analyses produced similar numbers of differentially expressed genes, as revealed by comparing Fig. 2i & j to Fig 4.b & c. We explored this further by comparing the identity of genes across the two datasets and we found that while some differentially expressed genes were unique to one dataset or the other, many were found to be differentially expressed by both methods (Supplementary Table 10).
The differentially expressed set of genes from all four host-herbivore systems were annotated using Blast2GO (version 3.3)86 using default settings. The annotated genes were manually parsed to find the common gene families. To detect true orthologs that were differentially expressed across all plants or all butterflies, we analyzed the protein sequences of the differentially expressed genes with Proteinortho (version 5.11)87,88 using a p-value of 0.001.
Osiris 9 paralog detection.
All Osiris 9 protein sequences from Bombyx mori, Drosophila melanogaster and Danaus plexippus were extracted from the Osiris protein database39. Using them as the query, the homologous sequences were extracted from the de novo transcriptomes of all the four butterfly species and the P. rapae genome using tblastn similarity searches. A multiple sequence alignment of all extracted Osiris 9 protein sequences, including the Osiris 11 and Osiris 14 from B. mori, was generated using MAFFT (version 6.847b)89 with the L-INS-i algorithm. A phylogenetic tree was reconstructed using FastTree (version 2.1.10)90, which infers approximately maximum likelihood phylogenies from alignments of protein sequences. All the default options and the JTT+CAT model were used for tree inference.
Osiris 9E gene spatial and temporal assay.
For the temporal assay of Osiris 9E expression in butterflies, whole body tissue samples were collected at ten stages of development for all four butterfly species, except for adults where wings and eyes were excluded. Multiple individuals were pooled at early instar stages (24 hours to 72 hour instar) because of their small size. For the rest of the stages, a single individual was used for each RNA extraction. The developmental stages were: 24 hour larvae, 48 hour larvae, 72 hour larvae, 2nd instar larva, 3rd instar larva, 4th instar larva, 5th instar larva, pre-pupa, 72 hour pupa, and adult. For the spatial assay of Osiris 9E expression in larvae, the head, foregut, midgut, and skin were dissected out for each individual. RNA was extracted using TRIzol Reagent, and cDNA for each sample was synthesized using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) using 500 ng/ul of RNA. RT-qPCR for the temporal and spatial samples of each species was performed using SsoAdvanced SYBR Green Supermix (Bio-Rad) and a CFX96 Optical Reaction Module thermocycler (Bio-Rad). Elongation Factor 1 alpha (EF1α) served as a normalizing gene to measure the relative expression of Osiris 9E in each sample for all four butterfly systems, the primer sequences for each species are in Supplementary Table 11.
Code availability
All custom codes are available from the authors upon request.
Data availability
All the DNA-seq and RNA-seq raw reads generated during this study have been deposited in the NCBI SRA database under the study accession: SRP134094. The assembled Pieris rapae genome (v1_HIRISE) is available on the Lepidopteran genome database, Lepbase, and all eight de novo transcriptome assemblies have been deposited in the DDBJ/EMBL/GenBank TSA database, under the following accession numbers: Arabidopsis thaliana (GGJX00000000), Medicago sativa (GGKA00000000), Citrus x microcarpa (GGJQ00000000), Passiflora oerstedii (GGJO00000000), Pieris rapae (GGJY00000000), Colias eurytheme (GGJZ00000000), Papilio poytes (GGKB00000000) and Heliconius cydno (GGKC00000000).
Supplementary Material
Acknowledgments
We thank E. Westerman, R. Marquez, L. Southcott, A. Russell and G. Garcia for assistance with experiments and D. Samac for generously providing Medicago sativa seeds. N. Saleh helped with generating the inbred P. rapae material for the sequencing the initial genome assembly. We also thank Cristina Sahagun for assisting with the butterfly photography, and we thank reviewers for very helpful feedback on the manuscript. This project was funded by NIH grant GM108626 and NSF grant IOS-1452648 to MRK, funds from and University of Pittsburgh, the Pew Charitable Trust and Neubauer funds from the University of Chicago.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
Contributor Information
Sumitha Nallu, Department of Ecology and Evolution, University of Chicago, Chicago, IL, USA.
Kristine Don, Department of Ecology and Evolution, University of Chicago, Chicago, IL, USA.
Carlos Sahagun, Department of Ecology and Evolution, University of Chicago, Chicago, IL, USA.
Wei Zhang, Department of Ecology and Evolution, University of Chicago, Chicago, IL, USA.
Joy Bergelson, Department of Ecology and Evolution, University of Chicago, Chicago, IL, USA.
Marcus R. Kronforst*, Department of Ecology and Evolution, University of Chicago, Chicago, IL, USA
Jason A. Hill, Department of Zoology, Stockholm University, S-10691 Stockholm, Sweden
Christopher W. Wheat, Department of Zoology, Stockholm University, S-10691 Stockholm, Sweden
Camille Meslin, Department of Computational and Systems Biology, University of Pittsburgh, Pittsburgh, PA, USA.
Nathan L. Clark, Department of Computational and Systems Biology, University of Pittsburgh, Pittsburgh, PA, USA
Emilie Snell-Rood, Department of Ecology, Evolution and Behavior, University of Minnesota, Saint Paul, MN, USA.
Nathan I. Morehouse, Department of Biological Sciences, University of Cincinnati, Cincinnati, OH, USA
References
- 1.Futuyma DJ Some current approaches to the evolution of plant-herbivore interactions. Plant Species Biology 15, 1–9 (2000). [Google Scholar]
- 2.Futuyma DJ & Agrawal AA Macroevolution and the biological diversity of plants and herbivores. Proc. Natl. Acad. Sci. USA 106, 18054–18061 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ehrlich PR & Raven PH Butterflies and plants: A study in coevolution. Evolution 18, 586–608 (1964). [Google Scholar]
- 4.Howe GA & Jander G Plant immunity to insect herbivores. Annu. Rev. Plant. Biol 59, 41–66 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Berenbaum MR Postgenomic chemical ecology: from genetic code to ecological interactions. J. Chem. Ecol 28, 873–896 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Li W, Schuler MA & Berenbaum MR Diversification of furanocoumarin-metabolizing cytochrome P450 monooxygenases in two papilionids: Specificity and substrate encounter rate. Proc. Natl. Acad. Sci. USA 100 Suppl 2, 14593–14598 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wittstock U et al. Successful herbivore attack due to metabolic diversion of a plant chemical defense. Proc. Natl. Acad. Sci. USA 101, 4859–4864 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petschenka G et al. Stepwise evolution of resistance to toxic cardenolides via genetic substitutions in the Na+/K+ -ATPase of milkweed butterflies (Lepidoptera: Danaini). Evolution 67, 2753–2761 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Groen SC & Whiteman NK Using Drosophila to study the evolution of herbivory and diet specialization. Curr. Opin. Insect Sci 14, 66–72 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Schuman MC & Baldwin IT The layers of plant responses to insect herbivores. Annu. Rev. Entomol 61, 373–394 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Huang DW, Sherman BT & Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Mikkelsen MD, Hansen CH, Wittstock U & Halkier BA Cytochrome P450 CYP79B2 from Arabidopsis catalyzes the conversion of tryptophan to indole-3-acetaldoxime, a precursor of indole glucosinolates and indole-3-acetic acid. J. Biol. Chem 275, 33712–33717 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Kim JI, Dolan WL, Anderson NA & Chapple C Indole glucosinolate biosynthesis limits phenylpropanoid accumulation in Arabidopsis thaliana. Plant Cell 27, 1529–1546 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajniak J, Barco B, Clay NK & Sattely ES A new cyanogenic metabolite in Arabidopsis required for inducible pathogen defence. Nature 525, 376–379 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hopkins RJ, van Dam NM & van Loon JJ Role of glucosinolates in insect-plant relationships and multitrophic interactions. Annu. Rev. Entomol 54, 57–83 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Wheat CW et al. The genetic basis of a plant-insect coevolutionary key innovation. Proc. Natl. Acad. Sci. USA 104, 20427–20431 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edger PP et al. The butterfly plant arms-race escalated by gene and genome duplications. Proc. Natl. Acad. Sci. USA 112, 8362–8366 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Zhao J, Rose AB, Schmidt R & Last RL Arabidopsis phosphoribosylanthranilate isomerase: molecular genetic analysis of triplicate tryptophan pathway genes. Plant Cell 7, 447–461 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartels S et al. The family of Peps and their precursors in Arabidopsis: differential expression and localization but similar induction of pattern-triggered immune responses. J. Exp. Bot 64, 5309–5321 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Brachi B et al. Coselected genes determine adaptive variation in herbivore resistance throughout the native range of Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 112, 4032–4037 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Müller R et al. Differential effects of indole and aliphatic glucosinolates on lepidopteran herbivores. J. Chem. Ecol 36, 905–913 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Hill J et al. Cryptic, extensive and non-random chromosome reorganization revealed by a butterfly chromonome. bioRxiv doi: 10.1101/233700</italic>. [DOI] [Google Scholar]
- 23.Maeki K & Remignton C Studies of the chromosomes of North American Rhopalocera. J. Lep. Soc 14, 37–56 (1960). [Google Scholar]
- 24.Noctor G, Queval G, Mhamdi A, Chaouch S & Foyer CH Glutathione. Arabidopsis Book 9, e0142 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakamura M, Baldwin D, Hannaford S, Palka J, Montell C Defective proboscis extension response (DPR), a member of the Ig superfamily required for the gustatory response to salt. J. Neurosci 22, 3463–3472 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hilker M & Fatouros NE Plant responses to insect egg deposition. Annu. Rev. Entomol 60, 493–515 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Jones JD & Dangl JL The plant immune system. Nature 444, 323–329 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Little D, Gouhier-Darimont C, Bruessow F & Reymond P Oviposition by pierid butterflies triggers defense responses in Arabidopsis. Plant Physiol 143, 784–800 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gouhier-Darimont C, Schmiesing A, Bonnet C, Lassueur S & Reymond P Signalling of Arabidopsis thaliana response to Pieris brassicae eggs shares similarities with PAMP-triggered immunity. J. Exp. Bot 64, 665–674 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmiesing A, Emonet A, Gouhier-Darimont C & Reymond P Arabidopsis MYC transcription factors are the target of hormonal salicylic acid/jasmonic acid cross talk in response to Pieris brassicae egg extract. Plant Physiol 170, 2432–2443 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Zou X et al. Glutathione s-transferase SlGSTE1 in Spodoptera litura may be associated with feeding adaptation of host plants. Insect Biochem. Mol. Biol 70, 32–43 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Yu Q et al. The transcriptome response of Heliconius melpomene larvae to a novel host plant. Mol. Ecol 25, 4850–4865 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Schweizer F et al. Arabidopsis glucosinolates trigger a contrasting transcriptomic response in a generalist and a specialist herbivore. Insect Biochem. Mol. Biol 85, 21–31 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Gloss AD et al. Evolution in an ancient detoxification pathway is coupled with a transition to herbivory in the Drosophilidae. Mol. Biol. Evol 31, 2441–2456 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agrios G Plant Pathology, 952 (Academic Press; 5 edition, 2005). [Google Scholar]
- 36.Tsuda K & Somssich IE Transcriptional networks in plant immunity. New Phytol 206, 932–947 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Roitberg BD & Isman MB Insect Chemical Ecology, 360 (Springer US, 1992). [Google Scholar]
- 38.Gerardo NM et al. Immunity and other defenses in pea aphids, Acyrthosiphon pisum. Genome Biol 11, R21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shah N, Dorer DR, Moriyama EN & Christensen AC Evolution of a large, conserved, and syntenic gene family in insects. G3 2, 313–319 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dorer DR, Rudnick JA, Moriyama EN & Christensen AC A family of genes clustered at the triplo-lethal locus of Drosophila melanogaster has an unusual evolutionary history and significant synteny with Anopheles gambiae. Genetics 165, 613–621 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hungate EA et al. A locus in Drosophila sechellia affecting tolerance of a host plant toxin. Genetics 195, 1063–1075 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andrade López JM et al. Genetic basis of octanoic acid resistance in Drosophila sechellia: functional analysis of a fine-mapped region. Mol. Ecol 26, 1148–1160 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yassin A et al. Recurrent specialization on a toxic fruit in an island Drosophila population. Proc. Natl. Acad. Sci. USA 113, 4771–4776 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vanderauwera S et al. Genome-wide analysis of hydrogen peroxide-regulated gene expression in Arabidopsis reveals a high light-induced transcriptional cluster involved in anthocyanin biosynthesis. Plant Physiol 139, 806–821 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Queval G & Noctor G A plate reader method for the measurement of NAD, NADP, glutathione, and ascorbate in tissue extracts: Application to redox profiling during Arabidopsis rosette development. Anal. Biochem 363, 58–69 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Queval G et al. H2O2-activated up-regulation of glutathione in Arabidopsis involves induction of genes encoding enzymes involved in cysteine synthesis in the chloroplast. Mol. Plant 2, 344–356 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Chaouch S & Noctor G Myo-inositol abolishes salicylic acid-dependent cell death and pathogen defence responses triggered by peroxisomal hydrogen peroxide. New Phytol 188, 711–718 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Parisy V et al. Identification of PAD2 as a gamma-glutamylcysteine synthetase highlights the importance of glutathione in disease resistance of Arabidopsis. Plant J 49, 159–172 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Schlaeppi K, Bodenhausen N, Buchala A, Mauch F & Reymond P The glutathione-deficient mutant pad2–1 accumulates lower amounts of glucosinolates and is more susceptible to the insect herbivore Spodoptera littoralis. Plant J 55, 774–786 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Loudet O et al. Natural variation for sulfate content in Arabidopsis thaliana is highly controlled by APR2. Nat. Genet 39, 896–900 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Carrillo RA et al. Control of synaptic connectivity by a network of Drosophila IgSF cell surface proteins. Cell 163, 1770–1782 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Whiteman NK et al. Genes involved in the evolution of herbivory by a leaf-mining, Drosophilid fly. Genome Biol. Evol 4, 900–916 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Green M & Sambrook J Molecular Cloning: A Laboratory Manual (Fourth Edition), 2028 (Cold Spring Harbor Laboratory Press, 2012). [Google Scholar]
- 54.Putnam NH et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res 26, 342–350 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marçais G & Kingsford C A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leggett RM, Clavijo BJ, Clissold L, Clark MD & Caccamo M NextClip: an analysis and read preparation tool for Nextera Long Mate Pair libraries. Bioinformatics 30, 566–568 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gnerre S et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. USA 108, 1513–1518 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parra G, Bradnam K & Korf I CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Boetzer M, Henkel CV, Jansen HJ, Butler D & Pirovano W Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011). [DOI] [PubMed] [Google Scholar]
- 60.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV & Zdobnov EM BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015). [DOI] [PubMed] [Google Scholar]
- 61.Kiełbasa SM, Wan R, Sato K, Horton P & Frith MC Adaptive seeds tame genomic sequence comparison. Genome Res 21, 487–493 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen J et al. Complete genome of Pieris rapae, a resilient alien, a cabbage pest, and a source of anti-cancer proteins. F1000 Res 5, 2631 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Magrane M & Consortium U UniProt Knowledgebase: a hub of integrated protein data. Database (Oxford) 2011, bar009 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meslin C et al. Digestive organ in the female reproductive tract borrows genes from multiple organ systems to adopt critical functions. Mol. Biol. Evol 32, 1567–1580 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trapnell C et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc 7, 562–578 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim D et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Trapnell C et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol 28, 511–515 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Holt C & Yandell M MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stanke M, Diekhans M, Baertsch R & Haussler D Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008). [DOI] [PubMed] [Google Scholar]
- 70.Horton MW et al. Genome-wide patterns of genetic variation in worldwide Arabidopsis thaliana accessions from the RegMap panel. Nat. Genet 44, 212–216 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou X & Stephens M Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat. Methods 11, 407–409 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nielsen R, Paul JS, Albrechtsen A & Song YS Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet 12, 443–451 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chang CC et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 1–16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Alexander DH, Novembre J, & Lange K Fast model-based estimation of ancestry in unrelated individuals. Genome Research 19,1655–1664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bolger AM, Lohse M & Usadel B Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.DePristo MA et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Van der Auwera GA et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–11.10.33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gao X, Starmer J & Martin ER A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet. Epidemiol 32, 361–369 (2008). [DOI] [PubMed] [Google Scholar]
- 80.Hudson RR, Kreitman M & Aguade M A test of neutral molecular evolution based on nucleotide data. Genetics 116, 153–159 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rozas J et al. DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol. Biol. Evol 34, 3299–3302 (2017). [DOI] [PubMed] [Google Scholar]
- 82.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Haas BJ et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc 8, 1494–1512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li B & Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Conesa A et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005). [DOI] [PubMed] [Google Scholar]
- 87.Lechner M et al. Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics 12, 124 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lechner M et al. Orthology detection combining clustering and synteny for very large datasets. PLoS One 9, e105015 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Katoh K &Toh, H. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform 9, 286–298 (2008). [DOI] [PubMed] [Google Scholar]
- 90.Price MN et al. FastTree: Computing large minimum-evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol 26, 1641–1650 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Heikkilä M, Kaila L, Mutanen M, Peña C & Wahlberg N Cretaceous origin and repeated tertiary diversification of the redefined butterflies. Proc. R. Soc. Lond. B 279, 1093–1099 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the DNA-seq and RNA-seq raw reads generated during this study have been deposited in the NCBI SRA database under the study accession: SRP134094. The assembled Pieris rapae genome (v1_HIRISE) is available on the Lepidopteran genome database, Lepbase, and all eight de novo transcriptome assemblies have been deposited in the DDBJ/EMBL/GenBank TSA database, under the following accession numbers: Arabidopsis thaliana (GGJX00000000), Medicago sativa (GGKA00000000), Citrus x microcarpa (GGJQ00000000), Passiflora oerstedii (GGJO00000000), Pieris rapae (GGJY00000000), Colias eurytheme (GGJZ00000000), Papilio poytes (GGKB00000000) and Heliconius cydno (GGKC00000000).